
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemically dealloyed platinum with hierarchical pore structure as highly active catalytic coating
Ralph
Kraehnert
*a,
Erik
Ortel
a,
Benjamin
Paul
a,
Bjoern
Eckhardt
a,
Michael
Kanis
b,
Ran
Liu
c and
Antonia
Antoniou
*c
aDepartment of Chemistry, Technische Universität Berlin, 10623 Berlin, Germany. E-mail: ralph.kraehnert@tu-berlin.de
bInstitute for Solar Fuels, Helmholtz-Zentrum Berlin für Materialien und Energie GmbH, 14109 Berlin, Germany
cThe Woodruff School of Mechanical Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA. E-mail: antonia.antoniou@me.gatech.edu
First published on 26th September 2014
Abstract
Micro structured reactors are attractive candidates for further process intensification in heterogeneous catalysis. However, they require catalytic coatings with significantly improved space-time yields compared to traditional supported catalysts. We report the facile synthesis of homogeneous nanocrystalline Pt coatings with hierarchical pore structure by electrochemical dealloying of amorphous sputter-deposited platinum silicide layers. Thickness, porosity and surface composition of the catalysts can be controlled by the dealloying procedure. XPS analysis indicates that the catalyst surface is primarily composed of metallic Pt. Catalytic tests in gas-phase hydrogenation of butadiene reveal the typical activity, selectivity and activation energy of nanocrystalline platinum. However, space time yields are about 13 to 200 times higher than values reported for Pt-based catalysts in literature. The highly open metallic pore structure prevents heat and mass transport limitations allowing for very fast reactions and reasonable stability at elevated temperatures.
1. Introduction
The intensification of catalytic processes requires converters that provide optimal activity while minimizing the amount of utilized catalyst material and reactor volume. Superior space-time yields can be achieved in so-called micro-structured reactors. Such devices are typically characterized by flow channels with at least one dimension between 10 and 500 μm, laminar flow conditions and surface areas as high as 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 50000 m2 m−3.1,2 The small channel dimensions facilitate extremely fast mass and heat transfer (up to 10 kW m−2 K−1 for liquids),3 which significantly reduces transport limitations and hot spot formation. Hence, very fast reactions with large associated heat release can be performed under isothermal conditions. Moreover, the small channel dimensions efficiently quench gas phase radical chemistry, which allows reactor operation in the explosive regime of feed gas mixtures4 (e.g. C2H4/O2 in ethylene epoxidation on Ag,5 and H2/O2 on Pt/Al2O36). The excellent transfer capabilities, low catalyst inventory and increased safety allow operation under unconventional harsh reaction conditions, i.e. high temperature, pressure and reactant concentration. Therefore, new process windows for industrially relevant reactions can be utilized,7e.g. the direct synthesis of hydrogen peroxide from H2 and O2 at a pressure of 2 MPa on supported Pd catalysts.8 However, the full potential of micro structured reactors can be utilized only with catalytic coatings that are about 1000× more active than classical catalysts.9,10
000 to 50000 m2 m−3.1,2 The small channel dimensions facilitate extremely fast mass and heat transfer (up to 10 kW m−2 K−1 for liquids),3 which significantly reduces transport limitations and hot spot formation. Hence, very fast reactions with large associated heat release can be performed under isothermal conditions. Moreover, the small channel dimensions efficiently quench gas phase radical chemistry, which allows reactor operation in the explosive regime of feed gas mixtures4 (e.g. C2H4/O2 in ethylene epoxidation on Ag,5 and H2/O2 on Pt/Al2O36). The excellent transfer capabilities, low catalyst inventory and increased safety allow operation under unconventional harsh reaction conditions, i.e. high temperature, pressure and reactant concentration. Therefore, new process windows for industrially relevant reactions can be utilized,7e.g. the direct synthesis of hydrogen peroxide from H2 and O2 at a pressure of 2 MPa on supported Pd catalysts.8 However, the full potential of micro structured reactors can be utilized only with catalytic coatings that are about 1000× more active than classical catalysts.9,10
The most common method of immobilizing catalysts in micro-structured reactors is the filling or wash-coating of reactor walls with a slurry prepared from conventional powder catalysts. Typically employed powders are micron sized (Pt/Al2O3, Rh/Al2O3,11 Al2O312) or ball-milled (Al2O3,13 1.5 wt% Pd/Na–Al–Si–Ox14) commercial catalysts or catalyst supports. Smaller particle sizes of the deposited powders were obtained with self-made small sols of oxides (Al2O3,15,16 SiO2,15 TiO2,15 CeO217). Supported catalysts were realized by adding an active metal to the sol either prior to deposition16 or after deposition15,17 and subsequent calcination/reduction.
When the sols are deposited in the presence of pore templates such as polymer micelles wall-coated catalyst layers with ordered porosity and high internal surface area can be obtained. The active metal has been introduced either after the synthesis of the support oxide using classical impregnation (PtRu/SiO218) or via codeposition of the sol and pore template together with colloidal metal nanoparticles (e.g. PdNP/TiO219).
Unfortunately, all of the catalytic coatings reported so far resemble in their structure and composition classical supported catalysts that had been designed for conventional packed-bed reactors. Hence, these catalytic coatings inherit the typical activity of the respective catalyst powders. As a consequence, they do not meet the requirement put forward by Hessel et al.10 for the efficient utilization of microstructured reactors, i.e. a performance increase by an order of magnitude.
Nanoporous platinum-group metals could combine the high surface area of a porous material and excellent mechanical properties20,21 with enhanced catalytic properties. Due to the fact that their pore system consists entirely of the catalytic material, a large number of active surface sites can be packed in a small volume. If all of the surface sites were accessible and active, catalysts that achieve space time yields about one order of magnitude higher than conventional supported catalysts should be feasible. Moreover, the high intrinsic heat conductivity of metals should also facilitate very rapid heat transfer preventing runaway and hot spots even for fast reactions with high exothermicity.
Nanoporous metal powders with controlled porosity have been generated by a number of chemical and electrochemical synthesis methods. Chemical methods include reduction of a metal precursor in presence of a porous hard template (e.g. mesoporous Pt templated by KIT-622), reduction in presence of a surfactants lyotropic phase23 and self-assembly of ligand-stabilized colloidal metal nanoparticles, e.g. in the presence of block copolymers.24 However, these syntheses typically produce mechanically fragile agglomerates and powders, not extended homogeneous and mechanically stable films coated to a substrate material from which a catalytic micro reactor could be manufactured.
Electro-chemical synthesis methods typically involve already the presence of a substrate (the electrode) on which a porous metal film forms. Respective preparation methods include electrodeposition, galvanic replacement, reduction of a metal salt in the presence of structure-directing surfactant25–28 as well as electro-chemical dealloying of metal alloys (see e.g. reviews29,30 and31 for further details). In particular electrochemical dealloying of a pre-deposited dense binary metal alloy can produce homogeneous 3D structured noble metal coatings with open porosity.32–38 In this procedure a binary alloy is deposited on a substrate by e.g. sputtering or electron-beam evaporation. The alloy is subsequently immersed into an etching solution while applying an electrical potential, which leads to selective dissolution of the less noble element. The more noble element is thus left behind and can assemble into struts with characteristic dimensions in the range of 2 to 100 nm. The synthesis of nanoporous metal film by electrochemical dealloying has been reported for systems including Cu–Au,39 Zn–Cu, Al–Cu, Mn–Cu,40 Ni–Cu,41 Ag–Au,42 Pt–Cu,43 Cu–Si44 and Pt–Si.38
We recently reported improved structural control in nanoporous Pt films.20,21,45 Amorphous Pt–Si alloys were etched in HF solutions applying potentials in the range of 0.3–0.9 V.45 Porous Pt films with typically 1 cm2 planar dimensions, pore diameters between 10 and 25 nm and a thickness of ca. 250 nm were obtained. Respective surface areas amounted to about 41 m2 g−1 Pt, which corresponds to a surface area enhancement of about 22 m2 Pt per m2 substrate. Following a similar procedure Jung et al. obtained nanoporous metal Pt–Si foam electrodes with ca. 1 cm2 planar dimensions, pore diameters between 5 and 22 nm (depending on Pt content) and a thickness of about 160–180 nm. The foams were concluded to be active in electrocatalysis (MeOH oxidation37). (It should be noted that employed substrate (30 nm thick Pt layers) might have also contributed to the reported activity.)
Due to fact that nanoporous platinum films possess a high surface area constituted of an active metal, excellent mechanical stability and heat conductivity, they have the potential to increase space-time-yields in heterogeneous catalysis significantly. However, nanoporous Pt as a catalytic reactor coating and its behavior in gas-phase catalysis have not been described so far.
We report the synthesis of nanoporous Pt coatings and their application in catalytic micro-structured reactors. The coatings were produced by electro-chemical dealloying of amorphous Pt–Si films. Porous Pt layers as thick as 1.1 μm could be prepared on reactor plates up to 10 cm2 in size. N2 sorption analysis in combination with electron microscopy reveals that the Pt coatings feature a bimodal hierarchical open mesopore structure, which facilitates excellent diffusional access to the catalytic sites. The coatings catalytic performance was studied in the very fast and highly exothermic gas phase reaction of butadiene hydrogenation. The coatings allowed isothermal reactor operation and showed a significantly higher activity than all supported catalysts reported in literature. Moreover, the catalyst geometry permits several plates to be mounted as a stack, which allows a further upscaling of the reactor by numbering up.
The following sections of this paper describe first the catalyst synthesis, applied analytical methods and the catalytic tests (2.). Thereafter, typical properties of a dealloyed porous Pt catalyst are reported (3.1) followed by the investigation of the evolution of pore structure, crystallinity and surface composition during the electro-chemical dealloying (3.2). Finally, the performance of differently dealloyed catalysts in the gas-phase hydrogenation of butadiene is studied and discussed in context of available literature data (3.3).
2. Experimental
2.1 Catalyst synthesis
Nanoporous platinum thin films were synthesized following a two-step process. First, an amorphous platinum silicide of nominal composition Pt0.1Si0.9 was co-sputtered under 100 V bias on one side of (100) silicon plates. The plate average geometric surface area was ca. 9 cm2 and the thickness of the amorphous silicide amounted to ca. 3.5 μm. The film composition was verified by XPS (Pt0.1Si0.9) and EDX analysis (Pt0.08Si0.92). The samples were subsequently electrochemically dealloyed at 0.1 V (vs. SCE) in an electrolyte of 3% HF in deionized water for three different time intervals. The dealloying was performed in atmospheric conditions without purging. During dealloying, silicon dissolves in the electrolyte leaving behind platinum that self assembles into a three dimensional network of struts. The total amount of charge C passed through the system can therefore be thought of as a measure of the amount of silicon removed. Pt–Si films were dealloyed with total charges of 2.0 C mm−2 (denoted as “Pt–A”), 6.5 C mm−2 (“Pt–B”) and 23.8 C mm−2 (“Pt–C”). The three electrode system used for dealloying consisted of the initial alloy as the working electrode, a Pt counter electrode and a saturated calomel electrode (SCE) that acted as reference electrode. To eliminate film delamination all but one side of each sample was coated with a protective polymeric film that was inactive in the electrolyte. This polymer film was completely removed prior to the catalysis measurements.2.2 Sample characterization
Micrographs were obtained on a JEOL 7401F Scanning Electron Microscope (SEM). To determine the film thickness, coated silicon wafers were split into two pieces and imaged at the cross-section. Image J Version 1.39u46 (http://rsbweb.nih.gov/ij) was employed to determine the average thickness of the formed porous layers. This average thickness was used to calculate the overall volume of the porous Pt layer as a product of thickness multiplied the coated surface area. Nitrogen adsorption was measured at 77 K on Quantachrome Autosorb-1-C. From this data the surface area of the coatings was calculated via Brunauer–Emmett–Teller (BET) method. The specific surface area of the porous Pt was estimated in terms of m2 m−3porous layer as the ratio between the total surface area from BET and the volume of the porous Pt layer obtained from cross-section SEM, and in terms of m2 gPt−1 considering the mass of Pt contained in the same volume. The pore size distribution was calculated from N2 sorption data by evaluation with the NLDFT equilibrium model for N2 at 77 K on silica for cylindrical pores (Quantachrom). Film morphology and crystallinity of film fragments removed from the substrates was studied by Transmission Electron Microscopy (TEM) using a FEI Tecnai G2 20 S-TWIN instrument operated at 200 kV. X-Ray Diffraction (XRD) data were collected on Bruker D8 Advance instrument (Cu Kα radiation). The average crystallite size was calculated applying the Scherrer equation. XPS spectra of deposited films were acquired on two different spectrometers, thermal K-Alpha XPS with monochromated, micro-focused Al K-alpha X-ray source as well as SPECS with Mg K-alpha X-ray source (slit 3). The recorded spectra were fitted with Unifit2013 software version 2013 Revision H. The hydrocarbon C 1s signal at 285.0 eV was used as the energy reference to correct for charging. Scanning Auger Microscopy SAM was recorded on a PHI 700 instrument (ULVAC-PHI Inc.). Auger electrons were excited by a primary electron beam of 25 keV at 1 nA.2.3 Catalytic tests in butadiene hydrogenation
The catalytic performance of porous Pt catalyst films in the gas-phase hydrogenation of 1,3-butadiene was studied at temperatures between 35 and 80 °C. The porous Pt films were coated on one side of silicon plates (size 27 × 30 mm). For each catalytic run, either single plates or two identical plates were stacked into the reactor housing. A test setup and procedure similar to the one described by Cukic et al.47 was used. A reaction mixture consisting of 5% butadiene (2.5 purity), 10% hydrogen (5.0 purity) and 85% nitrogen (5.0 purity) was passed through the reactor at a flow rate of 30 ml min−1 (STP) at 1.05 bar. The catalyst was initially equilibrated to reaction conditions under reactive gas flow for 30 min at 35 °C. Thereafter, the temperature was increased stepwise to 80 °C with a dwell time of 30 min for each temperature set point. Analysis of the gas products was performed continuously every 7 min by online gas chromatograph (Agilent GC 7890 equipped with FID, TCD and columns HP Plot Al2O3, Molsieve 5A, HP Plot Q and DB FFAP.) The space-time yield [mol s−1 kg−1] was calculated as produced moles of butenes per second per kg of the catalyst (STYbutenes) and as the sum of produced butenes and butane (STYbutenes+butane).3. Results and discussion
3.1 Morphology, porosity and phase composition
The dealloyed Pt–Si coatings were analyzed by Scanning Electron Microscopy (SEM), Transmission Electron Microscopy (TEM), SAED and N2 sorption in order to reveal the materials morphology and pore structure. Fig. 1 shows the characterization of the sample Pt–C dealloyed with the highest total charge (23.8 C mm−2) with high magnification SEM images in top-view (A1, A2) and cross-section view (B1, B2), a TEM image (C), SAED pattern (D), N2-sorption data (E) and the pore-size distribution calculated from the sorption curve (F). Additional compositional information obtained by Scanning Auger Microscopy (SAM) is provided in Fig. 1B1. The inset in A2 shows the sample surface before dealloying.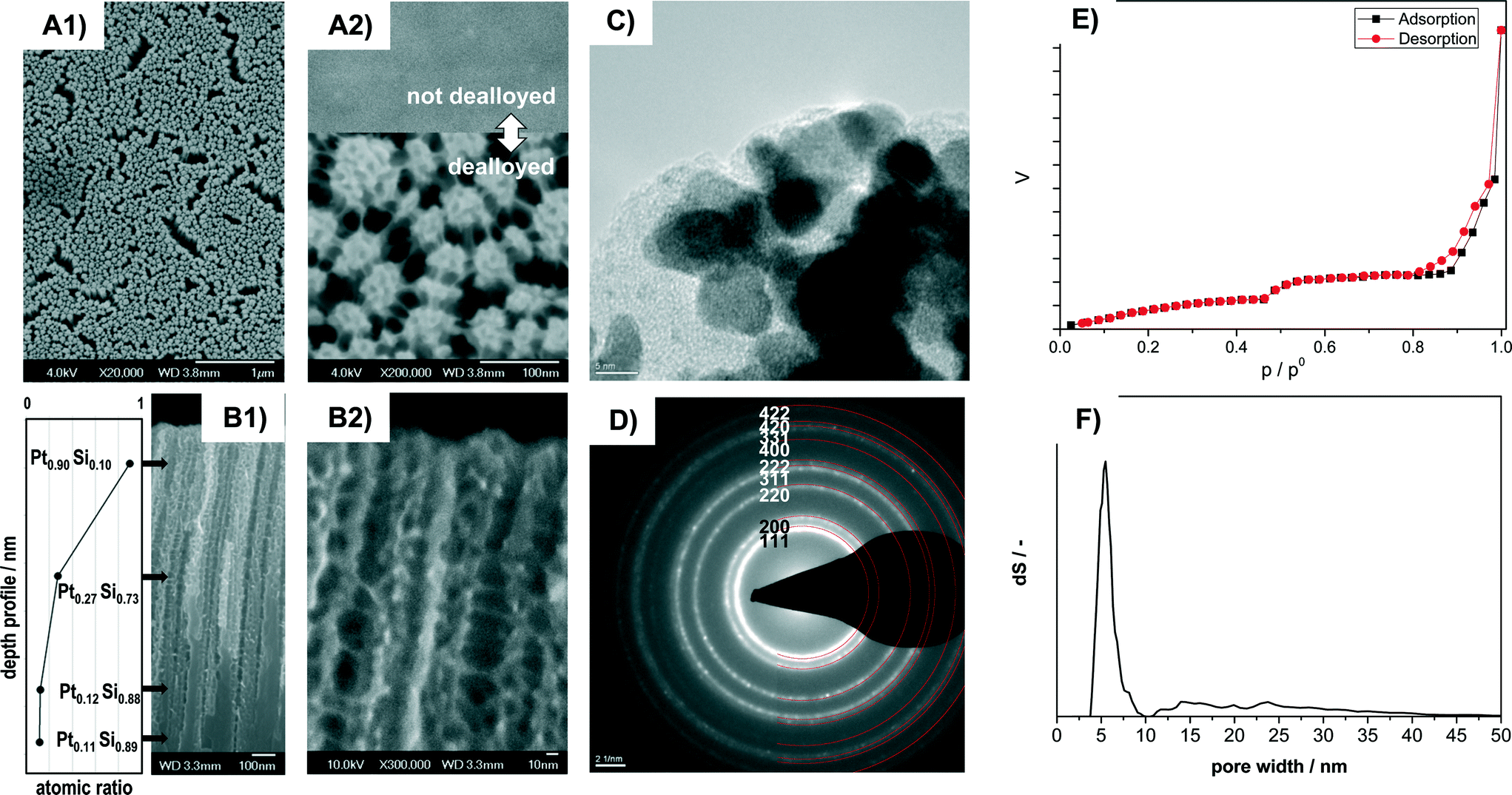 | ||
| Fig. 1 Analysis of morphology, crystallinity, porosity and local composition of dealloyed porous Pt sample Pt–C by A) SEM in top-view (A1 ×20000, A2 ×200000). Inset in A2) shows for comparison the sample surface before dealloying. B) Cross-section SEM of the porous film section (B1 ×60000, B2 ×300000) and depth profile of local Pt concentration. The provided local Pt content at different sample positions in B1 was obtained by SAM analysis. C) TEM, D) SAED. The marking and indexing of diffraction rings refers to crystalline Pt (PDF# 00-004-0802), E) adsorption and desorption branch of the N2 sorption curve and F) pore size distribution obtained from the N2 sorption data by evaluation with the NLDFT equilibrium model for N2 at 77 K on silica. | ||
The SEM images recorded in top view at different magnifications indicate that the entire surface is homogeneously structured (Fig. 1A1) and highly porous (Fig. 1A2). It consists of numerous domains of smaller pores of about 6 nm apparent diameter. All the domains are also connected by thin pore walls forming larger channels of about 15 to 40 nm diameter (Fig. 1A2). Cross section SEM images indicate that the pore system consists of porous columnar structures (Fig. 1B1) and that the columnar structures are connected by thin pore walls (Fig. 1B2). The average thickness of the porous layer amounts to 1.1 micron (from cross-section SEM).
HR-TEM images (Fig. 1C) reveal that the walls are composed of crystallites of about 4 to 8 nm size. The crystallites show lattice fringes that can be assigned to crystalline Pt. SAED analysis of the sample (Fig. 1D) indicates numerous diffraction rings. All the diffraction rings can be assigned to lattice planes of crystalline Pt (PDF 00-004-0802). However, some features observed in HR-TEM (Fig. 1C) appear to be of amorphous nature and could be related to remnants of the original amorphous Pt–Si phase.
Remaining Si was confirmed by Scanning Auger Microscopy (SAM), a method that can provide compositional information with about 10 nm spatial resolution. SAM spectra were recorded at different positions along the films cross section (see values in Fig. 1B1). At a depth of 0.18 μm from the outer film surface the film contains primarily Pt (90%) and small amounts of Si (10%). At about the middle of the porous part (0.65 μm) significantly less Pt (27%) is detected. The Pt content in the nonporous lower section (1.15 μm: 12% Pt; 1.5 μm: 11%) corresponds to the composition of the not-dealloyed sample. The dealloying procedure therefore produces porous Pt with low Si content near the external surface, but retains higher Si amounts towards the bulk of the film.
The N2 sorption isotherms contain two distinct steps suggesting a bimodal pore-size distribution (Fig. 1E). The corresponding pore size (from NLDFT) shows a narrow distribution at about 4 to 7 nm and a wider distribution at about 12 to 40 nm (Fig. 1F). The obtained pore sizes correspond well with the morphology observed in SEM images, where domains of smaller mesopores are separated by larger mesopores (Fig. 1A, B). The combined sorption and microscopy analysis therefore indicates that the electro-chemical dealloying of Pt–Si can form a bimodal and hierarchically organized Pt pore structure where larger mesopores connect domains of smaller mesopores.
The resulting surface area corresponds to about 350 m2 porous film per m2 of coated reactor wall. This value is significantly higher than e.g. the 22 mPt2 m−2 substrate reported by Thorp et al.38 The higher Pt surface area obtained in the present study can be attributed to the fact that significantly thicker porous Pt layers could be synthesized. We note that even thicker films could be synthesized with this technique.
Taking the volume of the porous layer and the initial molar stoichiometry of the Pt–Si alloy (0.1:0.9) into account a specific surface area of about 190 m2 gPt−1 can be estimated for the porous film section. This value is higher than the specific surface area reported previously by e.g. Thorp et al. (41 m2 gPt−1, from Cu underpotential deposition) and Jung et al. (49 m2 gPt−1, from hydrogen adsorption and desorption measured by CVs in H2SO4 and conversion of the charge into a Pt surface area with a conversion factor of 0.21 mC cm−2). However, it should be taken into account that the previously reported values were measured by different electro-chemical techniques, not gas sorption, hence not all parts of the surface might have been accessible. Moreover, the reported thin films were prepared under different film-deposition and dealloying conditions, and remaining Si and Pt–Si could contribute to the surface area derived in the present study from gas physisorption.
The combined data indicate that the dealloying of Pt–Si produces porous layers consisting of hierarchically organized bimodal pores formed by Pt crystals with a very high surface area of up to 350 m2 film per m2 of reactor wall. In terms of catalyst volume the surface area of the dealloyed Pt catalyst amounted to ca. 900000 m2 m−3 porous layer, which is about 20 to 90 times higher than the specific surface area provided by typical micro-structured reactors.2
3.2 Evolution of pore system, crystallinity and surface composition during dealloying
The amorphous Pt–Si films were dealloyed for different times to produce porous films with different thickness of the porous layer. Fig. 2 shows the cross-section SEM analysis of Pt–Si films dealloyed with total charges of (A) 2.0, (B) 6.5 and (C) 23.8 C mm−2, respectively. Moreover, the calculated volume of the porous sections of the films is plotted vs. the applied charge in Fig. 2D. Fig. 2E depicts X-ray diffractograms of the corresponding samples.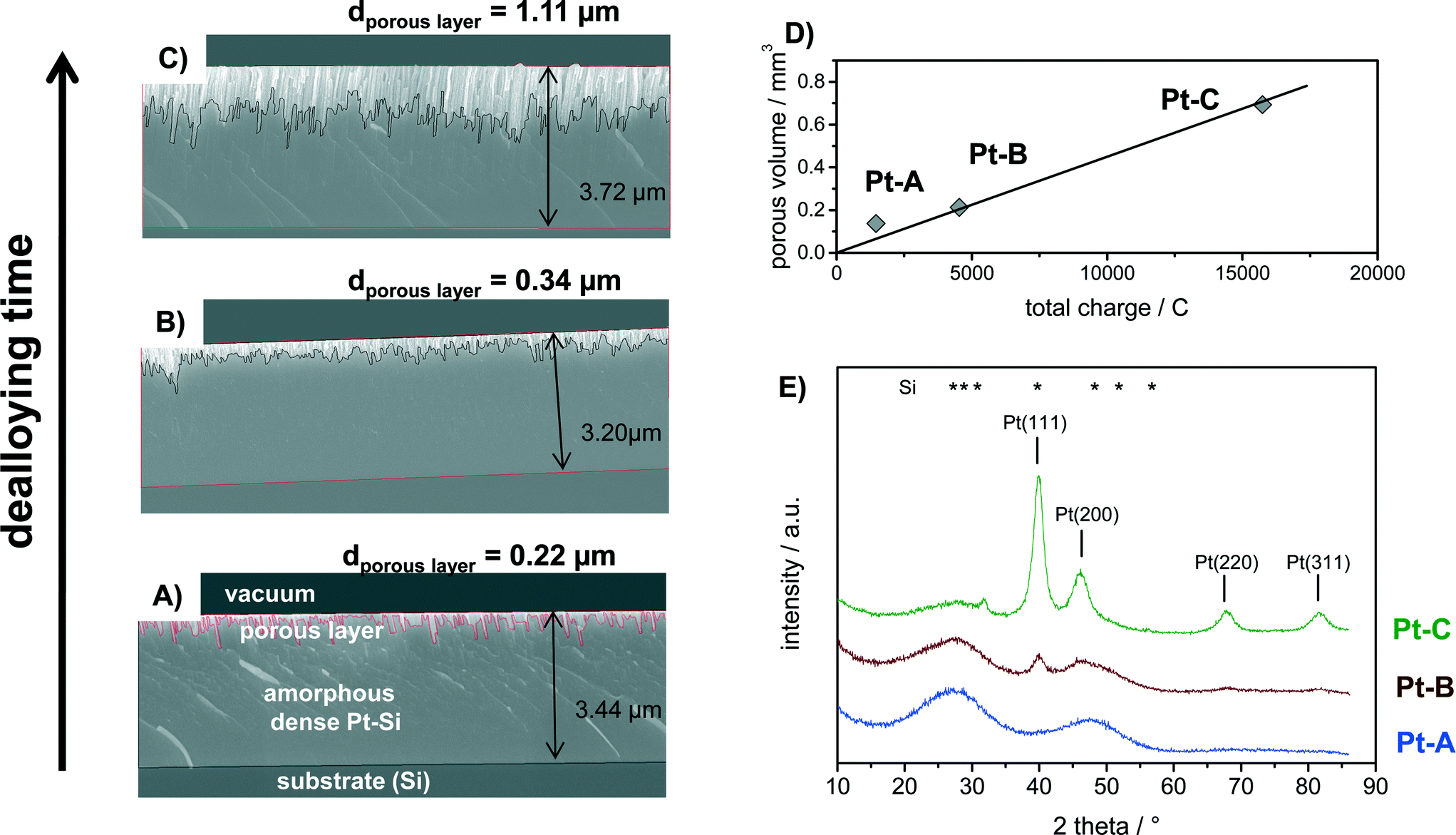 | ||
| Fig. 2 Analysis of (A–C) morphology, D) volume and E) crystallinity of Pt–Si coatings dealloyed with total charges of (Pt–A) 2.0, (Pt–B) 6.5 and (Pt–C) 23.8 C mm−2. (A–C) SEM cross-section images of each sample marking also the total thickness of each respective Pt–Si-film and of the formed porous layer, D) calculated total volume of the porous layer (film thickness from SEM analysis multiplied with the coated substrate area) vs. the total charge applied during dealloying, E) XRD analysis of sample phase composition showing the gradual transition from amorphous Pt–Si towards crystalline Pt. Positions of the reflections of Pt (PDF 00-004-0802) and Si (PDF 01-080-0005) are provided for comparison. | ||
Cross-section SEM images show that the sputtered amorphous films were about 3.2 to 3.7 micron thick (Fig. 2A–C). The upper section in each image represents the porous Pt layer produced by dealloying, followed by the remaining amorphous Pt–Si layer and the substrate (Si). It is evident from the images that the thickness of the porous film section increases with increasing total charge applied during dealloying from 0.22 to 0.34 and finally 1.11 micron average thickness. The produced porous layers are therefore significantly thicker than previously films reported by e.g. Thorp et al. (~250 nm38) or Jung et al. (<200 nm37), confirming the success of the upscaled synthesis procedure.
Fig. 2D plots the volume of the porous film section (estimated from average layer thickness times the coated surface area) vs. the applied charge. It can be seen that this relation follows a linear trend. Previous experimental studies have shown that the dealloying front propagates self similarly from the free surface in the through-thickness direction thus allowing for the synthesis of large scale porous structures.48,49 It should be noted that the dealloying was performed in atmospheric conditions without purging. At the applied potential the oxygen reduction reaction (ORR) is a well-known phenomenon. A part of the total measured charge can therefore be related to the ORR occurring in parallel. The respective ORR current is expected to reach a constant value after longer dealloying times due to diffusion limitation of O2. However, the oxygen content in the fresh dealloying solution might be higher than under diffusion limited conditions. In this case also the initial ORR current would be higher. Indeed, the total current measured during dealloying remained nearly constant after short initial transients. The transient, possibly due to ORR current, may explain why the sample with the smallest charge (Pt–A) deviates slightly from a linear fit.
The phase composition of the bulk of the dealloyed films can be deduced from corresponding X-ray diffractograms plotted in Fig. 2E. Sample “Pt–A” which was dealloyed for the shortest time shows two broad reflections that can be assigned to the initially present platinum silicide. With increasing volume of the porous layer the broad reflections corresponding to Pt–Si progressively decrease in intensity, while narrow reflections at 2 theta positions of 39.9°, 46.4°, 67.8° and 81.6° appear and increase in intensity with increasing dealloying charge (Fig. 1E, samples Pt–B, Pt–C). The signal positions correspond well with the (111), (200), (220) and (311) reflection reported for Pt in literature (PDF 00-004-0802). An additional small signal observed only for Pt–C at 2 theta of 31.8° could originate from crystalline Pt–Si (PDF# 00-007-025). Crystallite size estimates for sample Pt–C by Debye–Scherrer equation applied to the reflections assigned to Pt(111), (200), (220) and (311) amounted to 5.1, 3.1, 4.6 and 4.4 nm, respectively. Hence, the electrochemical dealloying of Pt–Si produces porous polycrystalline Pt layers with a about 4.5 nm crystallite size and a layer thickness that is controlled by the respective dealloying procedure (applied voltage and time).
The surface composition of the dealloyed samples Pt–B and Pt–C as well as of a fresh (not-dealloyed) sample Pt–Si was studied by XPS analysis. Fig. 3 shows the recorded spectra in A) the Pt 4f region and B) the Si 2p region of binding energies. The Pt 4f region of the fresh (not dealloyed) Pt–Si sample features only one doublet signal with a binding energy of 72.8 eV obtained for the Pt 4f7/2 peak (Fig. 3A, “Pt–Si”). The signal position is typically assigned to intermetallic Pt–Si phases.50,51 The surface composition indicated by XPS (Pt0.1Si0.9) is consistent with the composition derived from SEM-EDX analysis (Pt0.08Si0.92). It also agrees well with the composition obtained locally by SAM in the not-dealloyed region of sample Pt–C (Pt0.11Si0.89, see Fig. 1B1).
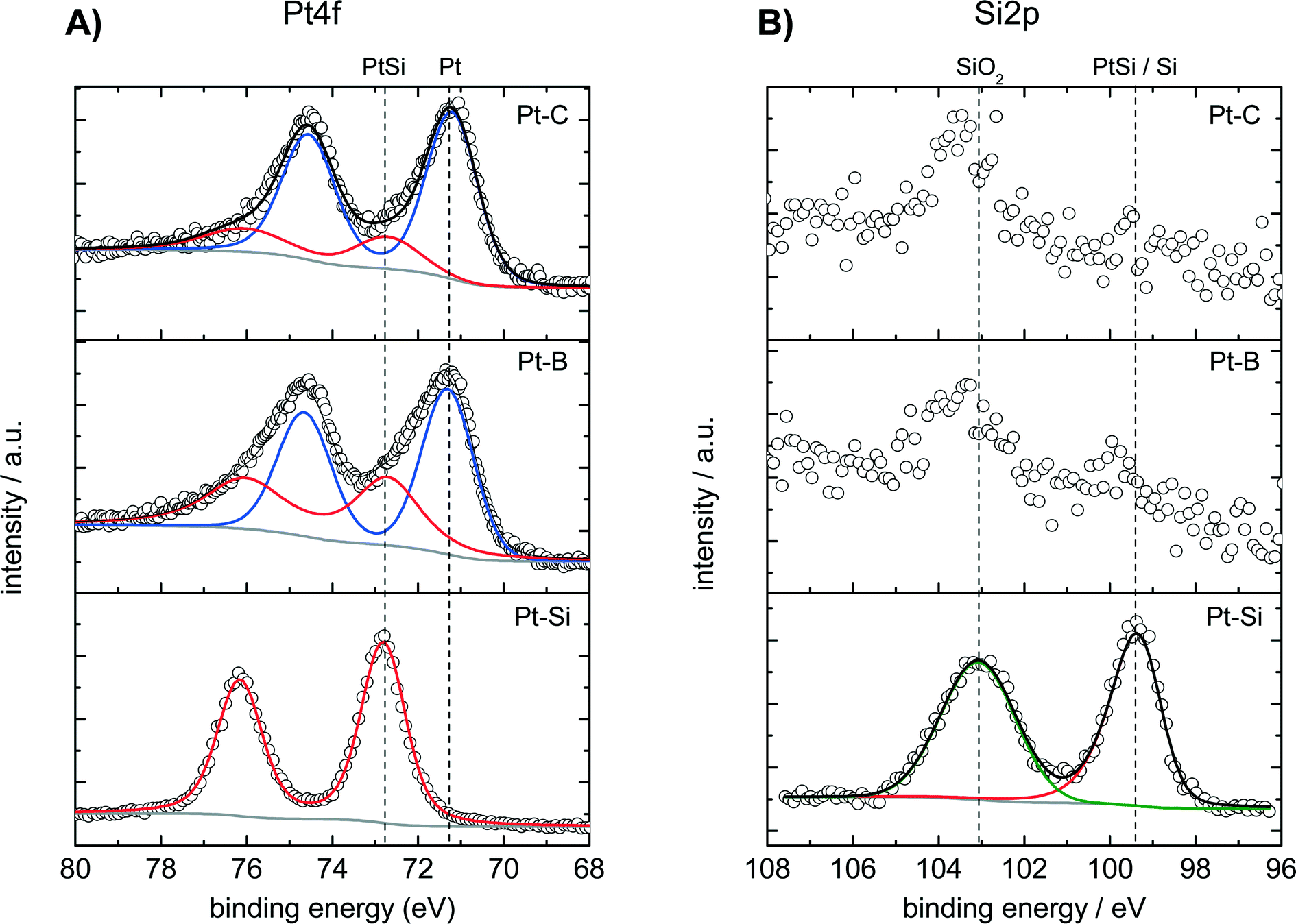 | ||
| Fig. 3 XPS analysis in the region of A) Pt 4f and B) Si 2p binding energies of the surface composition of not-dealloyed sample Pt–Si and samples dealloyed with total charges of (Pt–B) 6.5 and (Pt–C) 23.8 C mm−2. Respective experimental data are indicated by symbols, deconvoluted fitted individual components and sum spectra by lines. | ||
The Pt 4f signal shifts by about −1.6 eV for the dealloyed sample Pt–B indicating the formation of metallic Pt (Fig. 3A, Pt–B). The corresponding experimental spectra can be described well by two species with binding energies of 72.8 eV and of 71.2 eV. The species can be assigned to PtSi (about 39 at%, from peak deconvolution) and metallic Pt (ca. 61 at%), respectively. XPS recorded for the further dealloyed sample Pt–C shows a similar Pt 4f spectrum dominated by an increased amount of metallic Pt (Fig. 3A, Pt–C). Peak deconvolution indicates the presence of ca. 21 at% PtSi and 79 at% Pt. Dealloying therefore converts the surface of the initial intermetallic PtSi into metallic Pt. However, small amounts of PtSi remain also for the sample dealloyed with the highest total charge (Pt–C).
The Si 2p region of the fresh Pt–Si sample shows two signals located at 99.5 and 103.0 eV, respectively (Fig. 3B, “Pt–Si”). The species at 99.5 eV could be related to either PtSi (typical BE: 99.7–100.2 eV)51 or metallic Si (typical BE: 98.4–99.6 eV).52,53 The signal at 99.5 eV could originate thus either from PtSi or Si, however, PtSi is more likely in the context of the species observed in the Pt 4f spectrum. The signal observed at 103.0 eV can be attributed to SiO2 and is assigned to silicon in the deposited amorphous film that does not contribute to the PtSi formation.
The Si 2p spectra recorded for the dealloyed samples Pt–B indicate the presence of the same two Si species (Fig. 3B). The dealloying procedure significantly decreases the signal at 99.5 eV (PtSi), whereas the signal at 103.0 eV (SiO2) remains high. Longer dealloying (sample Pt–C) further decreases the signal at 99.5 eV. The species and changes observed in the Si 2p region are consistent with the Pt 4f spectra: dealloying converts the majority of an initially present PtSi phase into metallic Pt. However, samples dealloyed for different times differ in their surface composition, and small amounts of PtSi and SiO2 remain in all cases. Since XRD did not reveal crystalline SiOx phases, EDX analysis indicates a low oxygen content and the films show a very high electrical conductivity the formation of an SiOx bulk phase is unlikely. The SiO2 observed in XPS analysis is therefore attributed to Si in the near-surface region that becomes oxidized under ambient conditions.
Estimates of the Pt and Si content of the samples obtained by XPS and by SEM-EDX after different dealloying times are given in Table 1. Both methods provide very similar values for the nonporous fresh sample (XPS Pt0.1Si0.9; SEM-EDX Pt0.08Si0.92). Both methods also show that the Pt content increases with increasing dealloying charge. However, exact values differ between both methods for the porous samples, and both methods deliver only semiquantitative composition estimates for porous samples with local concentration gradients as is the case here (see Fig. 1B1). The differences can be understood from the fact that XPS and EDX probe different sample volumes and cannot account for the observed concentration gradients. Analysis by EDX coupled to SEM evaluates X-rays emitted from at least 100 nm of the layer depth of the Pt–Si films (at 4 keV). Moreover, the evaluation algorithm is not capable of correcting for the gradient present in this volume for the porous dealloyed films. In the case of XPS the information results from electrons emitted from a few atomic layers of the local Pt–Si surface. However, the samples Pt–A, Pt–B and Pt–C are porous and electrons can escape from pore surfaces positioned much deeper within the Pt–Si layer. Therefore, XPS probes for the porous Pt–Si the local pore surfaces positioned at different depths of the Pt–Si layer. In consequence the composition analysis via XPS, SAM and EDX agrees well for the non porous sample, however for the study of the local composition of porous Pt–Si samples SAM recorded on the films cross section remains the method of choice.
| Molar composition | ||||
|---|---|---|---|---|
| From XPSa | From EDXb | |||
| Pt | Si | Pt | Si | |
| a Pt provided as a sum of metallic Pt and the Pt in PtSi, Si as the sum of Si in PtSi and SiO2. Note that Pt–B and Pt–C are porous and that the local depth within these porous films from which the XPS information results cannot be determined exactly. b Note that EDX data was recorded by SEM-EDX in top view, which probes a sample depth of at least 100 nm. Due to the fact that the Pt shows a concentration gradient in the probed volume the model applied for quantification is not strictly valid. | ||||
| Fresh | 0.1 | 0.9 | 0.08 | 0.92 |
| Pt–A | — | — | 0.10 | 0.90 |
| Pt–B | 0.5 | 0.5 | 0.14 | 0.86 |
| Pt–C | 0.5 | 0.5 | 0.54 | 0.46 |
The applied dealloying potential amounted to 0.34 V vs. RHE. According to the standard reduction potential54 Si dissolution is likely to occur in the presence of HF and under acidic conditions by the oxidation of Si via the following reaction
| Si + 6F− ↔ SiF62− + 4e− → E0 = − 1.24 V |
According to the active species for Si dissolution55 in acidic electrolytes in presence of fluoride are HF, (HF)2 and (HF)2−. At low current densities the reaction
| Si + 4HF2− + h+ → SiF62− + H2 + e− |
3.3 Activity, selectivity and stability in catalytic butadiene hydrogenation
Butadiene hydrogenation carried out in the gas-phase represents a very challenging test reaction. The reaction is highly exothermal and can proceed very fast on metals like Pd and Pt. Moreover, it can form different total (butane) and partially hydrogenated products (1-butene, trans-2-butene, cis-2-butene). Butadiene hydrogenation was therefore used to assess activity, selectivity as well as limitations of mass- and heat transfer. Pt coatings with three different thicknesses of the porous Pt layer (Pt–A, Pt–B, Pt–C) were tested passing a continuous flow of 30 ml min−1 containing 5% 1,3-butadiene and 10% hydrogen in N2 over the catalyst while the reactor temperature was increased in several step from 35 to 80 °C. Fig. 4A reports the concentration of gaseous reaction products at different temperatures for catalyst Pt–C. Fig. 4B compares the butadiene conversion measured for the catalysts Pt–A, Pt–B and Pt–C as well as for catalyst Pt–C after an additional catalyst treatment in O2 for 10 min at 200 °C. Fig. 4C provides Arrhenius plots for Pt–A and Pt–B, i.e. the catalysts with the lower thickness of the porous Pt layer, which show lower butadiene conversions and thus allow the Arrhenius evaluation in the differential regime.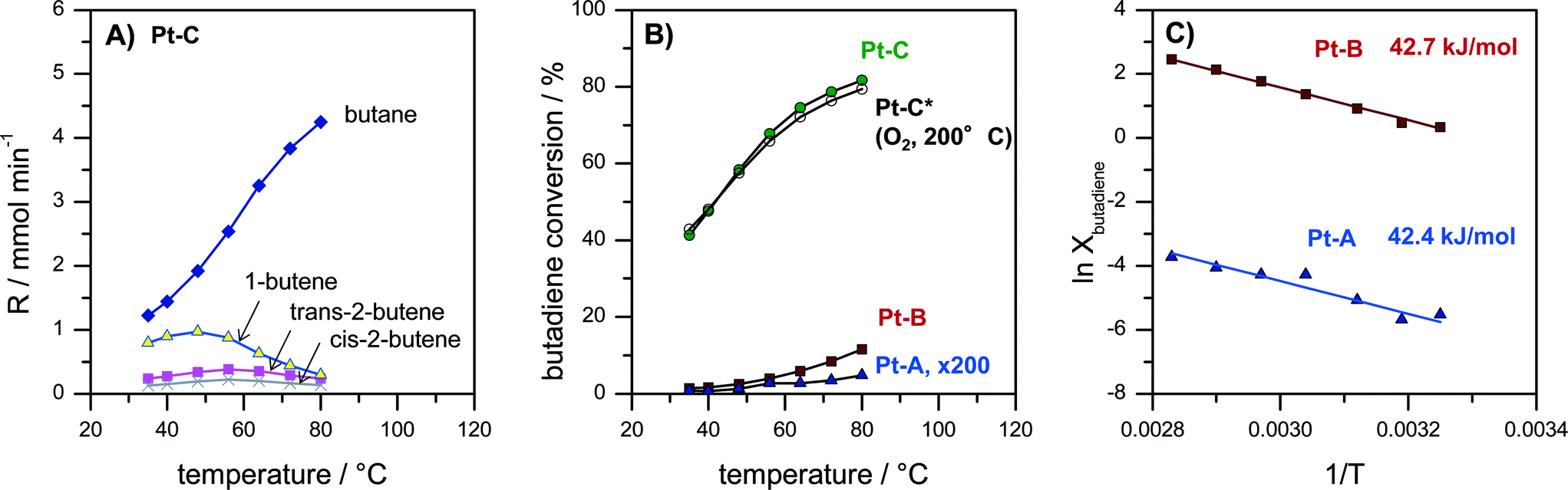 | ||
| Fig. 4 Catalytic evaluation of catalysts Pt–A, Pt–B and Pt–C in the gas-phase hydrogenation of 1,3-butadiene. A) product distribution vs. temperature measured on catalyst Pt–C. B) Comparison of butadiene conversion vs. temperature for Pt–A (scaling factor ×200), Pt–B, Pt–C and repeated analysis for Pt–C after treatment for 10 min at 200 °C in oxygen (“Pt–C*–O2-200 °C”). C) Arrhenius plot as ln(conversion) vs. 1/T for samples Pt–A and Pt–B (low butadiene conversion, differential regime). Apparent energy of activation for Pt–A (42.4 kJ mol−1) and Pt–B (42.7 kJ mol−1) calculated from the slope of the Arrhenius plot. All catalytic data measured on plates coated with 2.5 × 2.5 cm2 catalyst in a continuous flow reactor at a flow rate of 30 ml min−1 containing 5% butadiene and 10% hydrogen in N2 at temperatures increased stepwise from 35 to 40, 48, 56, 64, 72 and finally 80 °C. | ||
The observed reaction products were in all cases butane, 1-butene, trans-2-butene as well as cis-2-butene. The selectivity obtained e.g. for sample Pt–C at 35 °C amounted to 53% butane, 34% 1-butene, 10% trans-2-butene and 5% cis-2-butene (Fig. 4A). With increasing temperature the product distribution shifts towards more butane, which is the total hydrogenation product. The selectivity to butane increased for Pt–C from 53% at 35 °C to 87% at 80 °C.
Activity and selectivity in butadiene hydrogenation strongly depend on the employed catalyst support as well as the modification of Pt with a second metal as reported by Chen and co-workers for different Pt-based catalysts.56,57 They observed for e.g. 1.7 wt% Pt/Al2O3 that selectivity scaled in the order 1-butene > n-butane > cis-2-butene > trans-2-butene (35 °C, 2× H2 excess). Yoon et al. observed for polycrystalline Pt foil (unsupported catalyst) butane and 1-butene as the main products and smaller amounts of trans-2-butene and then cis-2-butene (25 °C, 10× H2 excess).58 More recently, the effect of the size of colloidal PVP-capped platinum nanoparticles deposited on flat silica surfaces in butadiene hydrogenation was studied by Somorjai and co-workers.59 They reported selectivities of 50% butane, 30% 1-butene, 15% trans-2-butene and 8% cis-2-butene for Pt nanoparticles below 2 nm diameter (75 °C, 10× H2 excess).59 The selectivities obtained on our porous Pt samples Pt–A, Pt–B and Pt–C (Fig. 4A) are almost identical to those observed by Somorjai and co-workers,59 which supports the interpretation that unsupported small Pt particles form the active phase of the developed porous Pt foam catalyst.
Fig. 4B compares the activity of the different catalysts Pt–A, Pt–B and Pt–C plotting butadiene conversion vs. temperature. It can be seen that all three catalysts are active. Butadiene conversion increases with increasing temperature. Moreover, the catalytic activity also scales with the volume of the porous Pt layer (see also Fig. 2D). Butadiene conversion measured at 80 °C increases from 0.03% (Pt–A) to 11.6% (Pt–B) and finally 81.7% (Pt–C). Longer dealloying times therefore result in more active catalysts, which can be rationalized by an increased amount of accessible Pt sites for the thicker porous layers. However, the dependence of measured catalyst activity on the thickness of the porous layer is faster than linear. Hence, also the surface composition of the porous layer changes with prolonged dealloying as indicated by XPS analysis (Fig. 3), i.e. increasing the surface Pt content.
Additional tests were carried out to study the thermal stability of the catalysts also under oxidizing conditions which are typical for Pt-catalyzed combustion reactions. Fig. 4B compares the activity of sample Pt–C before and after an additional treatment of the catalyst for 10 min at 200 °C in oxygen (“Pt–C*–O2-200 °C”). It is evident that the recorded butadiene conversions deviate less than 3% in the whole temperature range (Fig. 4B, Pt–C vs. Pt–C*–O2-200 °C). Hence, the produced porous Pt catalysts are also sufficiently stable in oxidizing atmosphere up to at least 200 °C.
In order to compare the intrinsic catalytic behavior to literature reports Arrhenius plots were constructed in the differential regime for the two catalysts with lower butadiene conversion Pt–A and Pt–B (Fig. 4C). The curves follow a straight line without any change of slope, indicating the absence of external and internal diffusion limitation. The deduced values of apparent activation energy are almost identical (Pt–A 42.4 kJ mol−1, Pt–B 42.7 kJ mol−1) suggesting the same active sites to be present on both catalysts. Yoon et al. reported values of 41, 39 and 41 kJ mol−1 for polycrystalline Pt foil, single crystals Pt(111) and Pt(755), respectively.58 The values agree very well with the activation energies observed in the present study (Fig. 4C). It can be concluded that the dealloyed porous Pt performs like nanocrystalline Pt with very high surface area provided by the pore system.
A significant increase in the catalytic performance of the developed catalyst can be demonstrated by comparing the space time yields (STYbutenes+butane) with literature data. For Pt-based catalysts 0.006 mol s−1 kg−1 (electrodeposited Pt black, 45 °C),60 0.03 mol s−1 kg−1 (1.7 wt% Pt/Al2O3, 35 °C)57 and 0.002 mol s−1 kg−1 (0.1 wt% Pt/Al2O3, 25 °C)61 have been reported in literature (all STYbutenes+butane are given as sum of production of butenes and butane). The corresponding STYbutenes+butane for catalyst Pt–C at 35 °C (Fig. 4A) amounts to 0.4 mol s−1 kg−1, i.e. approx. 13 times57 to 200 times61 higher when compared based on the catalysts total mass.
Catalysts based on palladium typically show better performance and higher selectivity in butadiene hydrogenation than Pt. Therefore the obtained STYbutenes to butenes is compared also to Pd catalysts. Values reported in literature for the reaction at 50 °C amount to 0.019 mol s−1 kg−1 for 0.5 wt% Pd-NP/TiO2,19 0.016 mol s−1 kg−1 for 0.5 wt% Pd–Al2O362 and 0.066 mol s−1 kg−1 for a TiO2-modified 1 wt% Pd–SiO2.63 Approximately 0.25 mol s−1 kg−1 can be obtained with the catalyst Pt–C at 48 °C. The porous Pt catalyst shows thus about 4 to 15 times higher space time yields to butenes than supported Pd catalysts.
Hence, the dealloyed porous (unsupported) Pt catalyst reaches the highest activity for butadiene hydrogenation reported in literature when compared on the basis of the catalyst mass. However, it should be kept in mind that also the density of our Pt catalyst is approximately 5 to 8 times higher than for catalysts based on alumina or silica. Hence, the volume-based activity should be at least 50 times higher than for supported Pt catalyst.
Butadiene hydrogenation is a very fast and highly exothermic reaction. The significant amount of heat generated by the reaction can therefore lead to local catalyst heating and thermal runaway if the heat removal is transport limited. Despite the high catalytic activity of the porous Pt films no signs of ignition were detected, neither a sudden increase in conversion (Fig. 4B) nor a deviation from a straight slope in the Arrhenius plot. Moreover, the differences between the gas inlet temperature and the temperature measured at the end of the catalyst plates never exceeded 0.4 K proofing that isothermal conditions can be realized. Despite the very high activity of the developed porous Pt catalyst, heat transport in the reactor can be efficiently controlled due to the high intrinsic heat conductivity of the catalyst and the chosen configuration as wall-coated catalyst layer.
4. Conclusions
Electrochemical dealloying of sputter-deposited amorphous Pt–Si layers produces homogeneous layers of Pt catalysts with hierarchical pore structure. The layers show a very high surface area (about 350 m2 catalyst per m2 substrate) composed primarily of metallic Pt. Tests in butadiene hydrogenation indicate the typical catalytic activity, selectivity and activation energy of polycrystalline platinum, but with about 13 to 200 times higher space time yields than reported for Pt-based catalysts in literature. The highly open metallic foam structure alleviates heat and mass transport limitations allowing for very fast reactions. Moreover, the porous Pt catalysts are stable also when exposed to oxygen atmosphere at temperatures as high as 200 °C.The catalytic application is demonstrated successfully for a reactor constructed from planar substrates. Future work could allow further upscaling of the synthesis also to complex three dimensional channel geometries. Moreover, the combined reactor and catalyst concept can be used for kinetic studies of very fast and highly exothermic reactions. Transferring the synthesis approach to other noble metals could provide access to a wide range for porous materials for applications in heterogeneous catalysis.
Acknowledgements
The authors thank ZELMI (TU-Berlin) for TEM analysis. M. Bernicke and R. Kraehnert appreciate generous funding from BMBF under contract FKZ 03EK3009 and Einstein Stiftung Berlin (EJF-2011-95). B.E. is thankful for financial support from the German Cluster of Excellence in Catalysis (UNICAT) funded by the German National Science Foundation (DFG) and managed by the Technical University of Berlin (TU Berlin). RL AA are grateful to the National Science Foundation for support through grant no. CMMI-1351705.References
- L. Kiwi-Minsker and A. Renken, Microstructured reactors for catalytic reactions, Catal. Today, 2005, 110, 2–14 CrossRef PubMed.
- B. Bromley, V. Hessel, A. Renken and L. Kiwi-Minsker, “Sandwich Reactor” for heterogeneous catalytic processes: N2O decomposition as a case study, Chem. Eng. Technol., 2008, 31, 1162–1169 CrossRef CAS.
- W. Ehrfeld, V. Hessel and V. Haverkamp, Microreactors, in Ullmann's encyclopedia of industrial chemistry, VCH, Weinhein, 1999 Search PubMed.
- C. Liebner, J. Fischer, S. Heinrich, T. Lange, H. Hieronymus and E. Klemm, Are micro reactors inherently safe? An investigation of gas phase explosion propagation limits on ethene mixtures, Process Saf. Environ. Prot., 2012, 90, 77–82 CrossRef CAS PubMed.
- A. Kursawe and D. Hönicke, Ethene epoxidation in Ag/Al microchannel reactors: Effects of NO2 and Cs, Catal. Commun., 2001, 2, 347–351 CrossRef CAS.
- M. T. Janicke, H. Kestenbaum, U. Hagendorf, F. Schuth, M. Fichtner and K. Schubert, The controlled oxidation of hydrogen from an explosive mixture of gases using a microstructured reactor/heat exchanger and Pt/Al2O3 catalyst, J. Catal., 2000, 191, 282–293 CrossRef CAS.
- V. Hessel, B. Cortese and M. de Croon, Novel process windows - Concept, proposition and evaluation methodology, and intensified superheated processing, Chem. Eng. Sci., 2011, 66, 1426–1448 CrossRef CAS PubMed.
- T. Inoue, M. A. Schmidt and K. F. Jensen, Microfabricated multiphase reactors for the direct synthesis of hydrogen peroxide from hydrogen and oxygen, Ind. Eng. Chem. Res., 2007, 46, 1153–1160 CrossRef CAS.
- R. Zapf and V. Hessel, Nanoporous catalyst layers for use in microreactors, Chem. Ing. Tech., 2004, 76, 513–514 CrossRef.
- V. Hessel, S. Hardt and H. Löwe, A Multi-Faceted, Hierarchic Analysis of Chemical Micro Process Technology: Sections 1.6–1.9, in Chemical Micro Process Engineering, Wiley-VCH Verlag GmbH & Co. KGaA, 2005, pp. 66–124 Search PubMed.
- R. Zapf, G. Kolb, H. Pennemann and V. Hessel, Basic study of adhesion of several alumina-based washcoats deposited on stainless steel microchannels, Chem. Eng. Technol., 2006, 29, 1509–1512 CrossRef CAS.
- E. Simsek, A. K. Avci and Z. I. Onsan, Investigation of catalyst performance and microstructured reactor configuration for syngas production by methane steam reforming, Catal. Today, 2011, 178, 157–163 CrossRef PubMed.
- S. A. Schmidt, N. Kumar, B. Zhang, K. Eranen, D. Y. Murzin and T. Salmi, Preparation and characterization of alumina-based microreactors for application in methyl chloride synthesis, Ind. Eng. Chem. Res., 2012, 51, 4545–4555 CrossRef CAS.
- H. Qiu, L. Bednarova and W. Y. Lee, Infiltration and immobilization of catalyst particles into the confined space of microstructured reactors via layer-by-layer self-assembly, Appl. Catal., A, 2006, 314, 200–207 CrossRef CAS PubMed.
- K. Haas-Santo, M. Fichtner and K. Schubert, Preparation of microstructure compatible porous supports by sol–gel synthesis for catalyst coatings, Appl. Catal., A, 2001, 220, 79–92 CrossRef CAS.
- H. Chen, L. Bednarova, R. S. Besser and W. Lee, Surface-selective infiltration of thin-film catalyst into microchannel reactors, Appl. Catal., A, 2005, 286, 186–195 CrossRef CAS PubMed.
- A. Ates, P. Pfeifer and O. Görke, Thin-film catalytic coating of a microreactor for preferential CO oxidation over Pt catalysts, Chem. Ing. Tech., 2013, 85, 664–672 CrossRef CAS.
- O. Muraza, E. V. Rebrov, T. Khirnyak, B. F. G. Johnson, P. J. Kooyman, U. Lafont, M. De Croon and J. C. Schouten, Mesoporous silica films as catalyst support for microstructured reactors: Preparation and characterization, Chem. Eng. J., 2008, 135, 99–103 CrossRef PubMed.
- E. Ortel, S. Sokolov, C. Zielke, I. Lauermann, S. Selve, K. Weh, B. Paul, J. Polte and R. Kraehnert, Supported mesoporous and hierarchical porous Pd/TiO2 catalytic Coatings with controlled particle size and pore structure, Chem. Mater., 2012, 24, 3828–3838 CrossRef CAS.
- L. Yuan and A. Antoniou, Synthesis of transversely isotropic nanoporous platinum, Scr. Mater., 2012, 66, 503–506 CrossRef PubMed.
- L. Ran and A. Antoniou, A relationship between the geometrical structure of a nanoporous metal foam and its modulus, Acta Mater., 2013, 61, 2390–2402 CrossRef PubMed.
- H. J. Wang, H. Y. Jeong, M. Imura, L. Wang, L. Radhakrishnan, N. Fujita, T. Castle, O. Terasaki and Y. Yamauchi, Shape- and size-controlled synthesis in hard templates: sophisticated chemical reduction for mesoporous monocrystalline platinum nanoparticles, J. Am. Chem. Soc., 2011, 133, 14526–14529 CrossRef CAS PubMed.
- G. S. Attard, S. A. A. Leclerc, S. Maniguet, A. E. Russell, I. Nandhakumar and P. N. Bartlett, Mesoporous Pt/Ru alloy from the hexagonal lyotropic liquid crystalline phase of a nonionic surfactant, Chem. Mater., 2001, 13, 1444–1446 CrossRef CAS.
- S. C. Warren, L. C. Messina, L. S. Slaughter, M. Kamperman, Q. Zhou, S. M. Gruner, F. J. DiSalvo and U. Wiesner, Ordered mesoporous materials from metal nanoparticle–block copolymer self-assembly, Science, 2008, 320, 1748–1752 CrossRef CAS PubMed.
- Y. Yamauchi, A. Sugiyama, R. Morimoto, A. Takai and K. Kuroda, Mesoporous platinum with giant mesocages templated from lyotropic liquid crystals consisting of diblock copolymers, Angew. Chem., Int. Ed., 2008, 47, 5371–5373 CrossRef CAS PubMed.
- H. J. Wang, L. Wang, T. Sato, Y. Sakamoto, S. Tominaka, K. Miyasaka, N. Miyamoto, Y. Nemoto, O. Terasaki and Y. Yamauchi, Synthesis of mesoporous Pt films with tunable pore sizes from aqueous surfactant solutions, Chem. Mater., 2012, 24, 1591–1598 CrossRef CAS.
- C. L. Li and Y. Yamauchi, Synthesis of Mesoporous Platinum-Copper Films by Electrochemical Micelle Assembly and Their Electrochemical Applications, Chem. – Eur. J., 2014, 20, 729–733 CrossRef CAS PubMed.
- C. L. Li, H. J. Wang and Y. Yamauchi, Electrochemical Deposition of Mesoporous Pt-Au Alloy Films in Aqueous Surfactant Solutions: Towards a Highly Sensitive Amperometric Glucose Sensor, Chem. – Eur. J., 2013, 19, 2242–2246 CrossRef CAS PubMed.
- D. J. Guo and Y. Ding, Porous nanostructured metals for electrocatalysis, Electroanalysis, 2012, 24, 2035–2043 CrossRef CAS.
- J. T. Zhang and C. M. Li, Nanoporous metals: fabrication strategies and advanced electrochemical applications in catalysis, sensing and energy systems, Chem. Soc. Rev., 2012, 41, 7016–7031 RSC.
- A. Kloke, F. von Stetten, R. Zengerle and S. Kerzenmacher, Strategies for the fabrication of porous platinum electrodes, Adv. Mater., 2011, 23, 4976–5008 CrossRef CAS.
- D. Yi, K. Young-Ju and E. Jonah, Nanoporous gold leaf: “Ancient technology”/advanced material, Adv. Mater., 2004, 16, 4 CrossRef.
- J. Weissmuller, R. C. Newman, J. Hai-Jun, A. M. Hodge and J. W. Kysar, Nanoporous metals by alloy corrosion: formation and mechanical properties, MRS Bull., 2009, 34, 577–586 CrossRef CAS.
- S. Ye, K. P. Kucera, S. A. Burger and T. J. Balk, Microstructure, stability and thermomechanical behavior of crack-free thin films of nanoporous gold, Scr. Mater., 2008, 58, 1018–1021 CrossRef PubMed.
- E. Detsi, M. Van De Schootbrugge, S. Punzhin, P. R. Onck and J. T. M. De Hosson, On tuning the morphology of nanoporous gold, Scr. Mater., 2011, 64, 319–322 CrossRef CAS PubMed.
- A. Antoniou, D. Bhattacharrya, J. K. Baldwin, P. Goodwin, M. Nastasi, S. Picraux and A. Misra, Controlled nanoporous Pt morphologies by varying deposition parameters, Appl. Phys. Lett., 2009, 95, 073113 CrossRef PubMed.
- H. Y. Jung, D. H. Kim, H. K. Chun, S. H. Kim, C. S. Lim, J. Y. Byun and Y. J. Jung, Towards engineering nanoporous platinum thin films for highly efficient catalytic applications, Adv. Energy Mater., 2011, 1, 1126–1132 CrossRef CAS.
- J. C. Thorp, K. Sieradzki, L. Tang, P. A. Crozier, A. Misra, M. Nastasi, D. Mitlin and S. T. Picraux, Formation of nanoporous noble metal thin films by electrochemical dealloying of PtxSi1-x, Appl. Phys. Lett., 2006, 88, 033110 CrossRef PubMed.
- B. G. Ateya, J. D. Fritz and H. W. Pickering, Kinetics of Dealloying of a Copper-5 Atomic Percent Gold Alloy, J. Electrochem. Soc., 1997, 144, 2606–2613 CrossRef CAS PubMed.
- J. R. Hayes, A. M. Hodge, J. Biener, A. V. Hamza and K. Sieradzki, Monolithic nanoporous copper by dealloying Mn-Cu, J. Mater. Res., 2006, 21, 2611–2616 CrossRef CAS.
- M. J. Pryor and J. C. Fister, The Mechanism of Dealloying of Copper Solid Solutions and Intermetallic Phases, J. Electrochem. Soc., 1984, 131, 1230–1235 CrossRef CAS PubMed.
- J. Snyder, P. Asanithi, A. B. Dalton and J. Erlebacher, Stabilized Nanoporous Metals by Dealloying Ternary Alloy Precursors, Adv. Mater., 2008, 20, 4883–4886 CrossRef.
- D. V. Pugh, A. Dursun and S. G. Corcoran, Formation of nanoporous platinum by selective dissolution of Cu from Cu{sub 0.75}Pt{sub 0.25}, J. Mater. Res., 2003, 18, 216–221 CrossRef CAS.
- R. Liu, S. Zheng, J. Kevin Baldwin, M. Kuthuru, N. Mara and A. Antoniou, Synthesis and mechanical behavior of nanoporous nanotwinned copper, Appl. Phys. Lett., 2013, 103, 241907 CrossRef PubMed.
- A. Antoniou, D. Bhattacharrya, J. K. Baldwin, P. Goodwin, M. Nastasi, S. T. Picraux and A. Misra, Controlled nanoporous Pt morphologies by varying deposition parameters, Appl. Phys. Lett., 2009, 95, 0731161 CrossRef PubMed.
- M. D. Abramoff, P. J. Magalhaes and S. J. Ram, Image processing with imageJ, Biophotonics Intern., 2004, 11, 36–41 Search PubMed.
- T. Cukic, R. Kraehnert, M. Holena, D. Herein, D. Linke and U. Dingerdissen, The influence of preparation variables on the performance of Pd/Al2O3 catalyst in the hydrogenation of 1, 3-butadiene: Building a basis for reproducible catalyst synthesis, Appl. Catal., A, 2007, 323, 25–37 CrossRef CAS PubMed.
- E. Biermans, L. Molina, K. J. Batenburg, S. Bals and G. Van Tendeloo, Measuring Porosity at the Nanoscale by Quantitative Electron Tomography, Nano Lett., 2010, 10, 5014–5019 CrossRef CAS PubMed.
- W.-K. Chu, Backscattering spectrometry, Academic Press, New York, 1978 Search PubMed.
- L. Ley, Y. Wang, V. N. Van, S. Fisson, D. Souche, G. Vuye and J. Rivory, Initial stages in the formation of PtSi on Si(111) as followed by photoemission and spectroscopic ellipsometry, Thin Solid Films, 1995, 10, 561–566 CrossRef.
- S. J. Morgan, R. H. Williams and J. M. Mooney, An XPS study of thin Pt and Ir silicide overlayer formation on Si(100)2 X 1 surfaces, Appl. Surf. Sci., 1992, 56–58, Part 1, 493–500 CrossRef.
- F. Sirotti, M. De Santis and G. Rossi, Synchrotron-radiation photoemission and x-ray absorption of Fe silicides, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 8299–8306 CrossRef CAS.
- T. Aoyama, T. Sugii and T. Ito, Determination of band line-up in β-SiC/Si heterojunction for Si-HBT's, Appl. Surf. Sci., 1989, 41–42, 584–586 CAS.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, CRC press, Boca Raton, 82nd edn, 2002, pp. 8–25 Search PubMed.
- V. Lehmann, Electrochemistry of silicon: instrumentation, science, materials and applications, Wiley-VCH, Weinheim, 2002 ISBN 3-527-29321-3, p. 52 Search PubMed.
- T. Wang, W. Lonergan and J. G. Chen, Selection of oxide supports to anchor desirable bimetallic structures for ethanol reforming and 1,3-butadiene hydrogenation, Chin. J. Catal., 2013, 34, 2009–2017 CrossRef CAS.
- W. W. Lonergan, X. J. Xing, R. Y. Zheng, S. T. Qi, B. Huang and J. G. G. Chen, Low-temperature 1,3-butadiene hydrogenation over supported Pt/3d/gamma-Al2O3 bimetallic catalysts, Catal. Today, 2011, 160, 61–69 CrossRef CAS PubMed.
- C. Yoon, M. X. Yang and G. A. Somorjai, Hydrogenation of 1,3-butadiene on platinum surfaces of different structures, Catal. Lett., 1997, 46, 37–41 CrossRef CAS.
- W. D. Michalak, J. M. Krier, K. Komvopoulos and G. A. Somorjai, Structure Sensitivity in Pt Nanoparticle Catalysts for Hydrogenation of 1,3-Butadiene: In Situ Study of Reaction Intermediates Using SFG Vibrational Spectroscopy, J. Phys. Chem. C, 2013, 117, 1809–1817 CAS.
- J. Gaube and H. F. Klein, Kinetics and mechanism of butene isomerization/hydrogenation and of 1,3-butadiene hydrogenation on palladium, Appl. Catal., A, 2014, 470, 361–368 CrossRef CAS PubMed.
- A. Sarkany, G. Stefler and J. W. Hightower, Participation of support sites in hydrogenation of 1,3-butadiene over Pt/Al2O3 catalysts, Appl. Catal., A, 1995, 127, 77–92 CrossRef CAS.
- K. Pattamakomsan, E. Ehret, F. Morfin, P. Gélin, Y. Jugnet, S. Prakash, J. C. Bertolini, J. Panpranot and F. J. C. S. Aires, Selective hydrogenation of 1, 3-butadiene over Pd and Pd–Sn catalysts supported on different phases of alumina, Catal. Today, 2011, 164, 28–33 CrossRef CAS PubMed.
- D. C. Lee, J. H. Kim, W. J. Kim, J. H. Kang and S. H. Moon, Selective hydrogenation of 1,3-butadiene on TiO2-modified Pd/SiO2 catalysts, Appl. Catal., A, 2003, 244, 83–91 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |