
Highly efficient and chemoselective hydrogenation of α,β-unsaturated carbonyls over Pd/N-doped hierarchically porous carbon†
Zhongzhe
Wei
,
Yutong
Gong
,
Tianyi
Xiong
,
Pengfei
Zhang
,
Haoran
Li
and
Yong
Wang
*
Carbon Nano Materials Group, ZJU-NHU United R&D Center, Department of Chemistry, Zhejiang University, Hangzhou 310028, P.R. China. E-mail: chemwy@zju.edu.cn; Fax: +86 571 87951895
First published on 18th August 2014
Abstract
Palladium nanoparticles supported on N-doped hierarchically porous carbon, Pd/CNx, has been developed as a highly efficient, reusable and environmentally benign heterogeneous catalyst for the selective hydrogenation of various α,β-unsaturated carbonyls to their corresponding saturated carbonyls under mild conditions (303 K, 1 bar H2). Complete conversion of a series of α,β-unsaturated carbonyls was achieved with excellent selectivity (>99%) within 4 h. Moreover, the catalyst can be easily recovered by centrifugation and withstands recycling up to 8 times without apparent loss of activity and selectivity. The considerable catalytic performance is attributed to the hierarchically porous network and incorporation of nitrogen atoms. This catalytic system opens up an efficient, selective, recyclable and sustainable method for selective hydrogenation.
Introduction
Chemoselective hydrogenation of α,β-unsaturated carbonyls is fundamentally important as it has extensive applications in the synthesis of fine chemicals, pharmaceuticals and functional materials.1–4 The selectivity varies due to the possibility of hydrogenation of either the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond or the CO bond, or both of them (Scheme 1). The selective hydrogenation of the CC bond is an important industrial process in which heterogeneous catalysts, especially supported precious metal catalysts, have attracted much attention due to the ease of their separation and recycling together with their outstanding performances.5–7 Supported palladium catalysts are widely used for the reaction. For example, Kantam et al. reported that layered double hydroxide-supported Pd was used as a heterogeneous catalyst for olefinic bond hydrogenation with high selectivity.8 85%–97% yield was achieved by Bandoo et al. for the hydrogenation of CC bond in functional conjugated alkenes over polysiloxane-encapsulated Pd nanoclusters.9 Bhanage et al. also applied a polymer-supported catalytic system, polymer-supported Pd-N-heterocyclic carbine, for the chemoselective conjugate reduction of α,β-unsaturated carbonyls at 100 °C.10 The advantages of both homogeneous and heterogeneous catalysts were achieved when supported catalysts with the aid of ionic liquids were used in the selective reduction of the CC bond.11–14 To get better performance from catalysts, the use of additives is sometimes inevitable. The use of Pd/C together with various additives like pyridines, amines and diphenylsulfide, which resulted in the selective hydrogenation of the CC bond, has been reported.15 Bimetallic catalysts have also received intense interest because of a possible synergistic effect which makes selective hydrogenation reactions easier. Xu and co-workers disclosed that a small amount of fully dispersed Pt entities loaded on Au nanoparticles provided dramatic activity enhancement for the chemoselective hydrogenation of α,β-unsaturated carbonyls.16 A similar effect was also illustrated in Ni/TiO2 doping with a tiny amount of Ir for the hydrogenation of cinnamaldehyde (CAL) to hydrocinnamaldehyde (HCAL) under 2 MPa H2 at 80 °C.17 Li et al. applied water-soluble PtxNi1–x (0 < x < 1) to the hydrogenation of the CC bond in benzalacetone, and obtained an excellent yield.18 Despite all of these well-established methodologies, the development of a highly effective, selective and environmentally benign heterogeneous catalyst usable under mild conditions is highly desirable.
C bond or the CO bond, or both of them (Scheme 1). The selective hydrogenation of the CC bond is an important industrial process in which heterogeneous catalysts, especially supported precious metal catalysts, have attracted much attention due to the ease of their separation and recycling together with their outstanding performances.5–7 Supported palladium catalysts are widely used for the reaction. For example, Kantam et al. reported that layered double hydroxide-supported Pd was used as a heterogeneous catalyst for olefinic bond hydrogenation with high selectivity.8 85%–97% yield was achieved by Bandoo et al. for the hydrogenation of CC bond in functional conjugated alkenes over polysiloxane-encapsulated Pd nanoclusters.9 Bhanage et al. also applied a polymer-supported catalytic system, polymer-supported Pd-N-heterocyclic carbine, for the chemoselective conjugate reduction of α,β-unsaturated carbonyls at 100 °C.10 The advantages of both homogeneous and heterogeneous catalysts were achieved when supported catalysts with the aid of ionic liquids were used in the selective reduction of the CC bond.11–14 To get better performance from catalysts, the use of additives is sometimes inevitable. The use of Pd/C together with various additives like pyridines, amines and diphenylsulfide, which resulted in the selective hydrogenation of the CC bond, has been reported.15 Bimetallic catalysts have also received intense interest because of a possible synergistic effect which makes selective hydrogenation reactions easier. Xu and co-workers disclosed that a small amount of fully dispersed Pt entities loaded on Au nanoparticles provided dramatic activity enhancement for the chemoselective hydrogenation of α,β-unsaturated carbonyls.16 A similar effect was also illustrated in Ni/TiO2 doping with a tiny amount of Ir for the hydrogenation of cinnamaldehyde (CAL) to hydrocinnamaldehyde (HCAL) under 2 MPa H2 at 80 °C.17 Li et al. applied water-soluble PtxNi1–x (0 < x < 1) to the hydrogenation of the CC bond in benzalacetone, and obtained an excellent yield.18 Despite all of these well-established methodologies, the development of a highly effective, selective and environmentally benign heterogeneous catalyst usable under mild conditions is highly desirable.
 | ||
| Scheme 1 Reaction pathways for the hydrogenation of α,β-unsaturated carbonyls. | ||
In the construction of heterogeneous catalysts to selectively reduce the CC bond in α,β-unsaturated carbonyls, Pd is conventionally used as the catalytic center, which is confirmed by experimental results8,19,20 and theoretical work.21–23 It is well recognized that the nature of the support has a significant effect on the activity and selectivity behavior of the catalysts.24,25 The catalytic activity of a heterogeneous catalyst can be enhanced by suitably controlling and selecting the support.26 Hence, the choice of support is vital. Carbon materials, including activated carbon, carbon nanofibers, hydrophilic carbon, etc., are good supports for heterogeneous catalysts because of their specific properties.27–30 Commercial Pd/C can catalyze the selective hydrogenation of CAL, but the apparent rate of the reaction is low and the selectivity towards HCAL remains poor.27,31 This could be explained by the fact that activated microporous carbon materials may depress the mass transport of molecules because of the space confinement imposed by small pore sizes.32 To overcome these limitations, efforts have been directed towards mesoporous carbon materials, especially hierarchically structured porous materials.33 Thanks to the much reduced diffusion restriction for molecules in mesoporous structures and the greatly shortened diffusion distances in the channels, catalysts with hierarchically porous structures exhibit high catalytic behavior.34 However, noble metals deposited on carbons easily leach during catalytic processes because the interaction between the metal nanoparticles and the carbon surface is always weak, and hence modification of the carbon is necessary.35 In this respect, doping the carbon with heteroatoms is regarded as an impressive strategy. Nitrogen-doped carbon materials have attracted worldwide attention recently due to having an even wider range of applications than carbon materials, such as oxygen reduction reactions,36 catalyst support,37etc. The incorporation of nitrogen atoms may not only enhance the electronic density of supported metal nanoparticles, but may also provide more structural defects.
Very recently, we successfully designed fascinating N-doped carbon (CN) materials with hierarchical structures from glucose using a hydrothermal carbonization procedure, which is a green and sustainable process.37,38 In the search for a suitable catalyst for the chemoselective hydrogenation of α,β-unsaturated carbonyls, we anticipated that Pd/CN could exhibit an interesting catalytic activity due to the hierarchically porous structure of the CN and the special catalyst support–metal heterojunction. Herein, we employed Pd/CN as an efficient catalyst for the selective hydrogenation of the CC bond using benign H2 as the reduction agent. The activity of the catalyst under different reaction conditions, the hydrogenation kinetics, and the recycling of the catalyst as well as the substrate scope were explored.
Experimental section
Materials and methods
Unless otherwise stated, all solvents and chemicals used were of commercially available analytical grade and used without further treatment. The surface areas of CN and catalyst Pd/CN were determined using an AUTOSORB-1 instrument. The BET equation was used to calculate the surface area and pore volume. X-ray power diffraction (XRD) data was collected using an Ultima TV X-ray diffractometer with Cu Kα radiation (1.54 Å). X-ray photoelectron spectroscopy (XPS) was carried out using an ESCALAB MARKII spherical analyzer with an aluminum–magnesium binode (Al 1486.6 eV, Mg 1253.6 eV) X-ray source. Transmission election microscopy (TEM) characterization was carried out on a Hitachi HT-7700 microscope. High-resolution TEM (HRTEM) was performed on a Tecnai G2 F30 S-Twin at an acceleration voltage of 300 kV. The particle morphology was visualized by scanning electron microscopy (SEM) using a Gemini scanning electron microscope. The elemental analysis was carried out on a Vario El elemental analyzer. The Pd content was determined using a PerkinElmer Optima OES 8000.Preparation of catalysts
Synthesis of the support material CNx: 9.0 g D-glucose, 1.2 g additive (poly(ionic liquid)s)39 and 0.75 g Borax were dissolved in 30 mL water. The mixture was loaded into a PTFE lined autoclave and treated at 200 °C for 8 h. The resulting black solid was filtered and washed several times with deionized water and ethanol. Then, the solid material was carbonized at 550 °C (heating rate: 3 K min−1, holding time: 4 h) under an inert N2 atmosphere. After cooling to the room temperature, the black carbon material can be obtained. The process to synthesize spherical carbon without nitrogen (Cs) was almost the same as for CNx, but in absence of poly(ionic liquid)s.Synthesis of the supported palladium catalysts: we synthesized 1.5% Pd/CN and 1.5% Pd/Cs using an ultrasound-assisted deposition method.40 1.5% Pd/MgO and 1.5% Pd/TiO2 were prepared by wet impregnation according to the literature.41
Typical procedure for selective hydrogenation and recycling
In a typical reaction, 104.2 mg (0.5 mmol) chalcone, 10 mg Pd/CNx and 5 mL ethanol were precharged in a three-neck flask. The flask was purged with H2 three times to remove the air, and the mixture was then magnetically stirred at 1000 rpm under 1 bar of H2 at room temperature. After the reaction, the contents of the mixture were separated and analyzed by GC (using a Shimadzu GC-2014 equipped with a Rtx-1071 column). Phenethyl phenylketone was used as an internal standard. The catalyst used above was recovered by centrifugation, washed 3 times with ethanol and dried at 80 °C overnight, then used for the next run without any further treatment. In order to obtain the intrinsic kinetic data, the reaction was controlled to give low conversion of chalcone.Results and discussion
Catalyst characterization
Elemental analysis indicated that the mesoporous N-doped carbon (denoted as CN,) had a N/C atom ratio of about 0.062 (Table S1†). SEM and TEM images of the CN and Cs demonstrated a hierarchically porous network with well-dispersed spherules (Fig. 2a, Fig. S1, S2†). The textural properties of the CN and Pd/CN were measured using N2 adsorption–desorption analysis (Fig. 1a). Application of the BET model resulted in a surface area of 424 m2 g−1 for the CN. The pore size analysis from nitrogen adsorption by application of density functional theory (DFT) is displayed in Fig. S3.† The pores in the CN have a well-defined micro-mesopore size distribution. The surface area decreased after loading with Pd nanoparticles, but still had a considerable value of 193 m2 g−1, suggesting that most of the pores were still accessible. The structures of the CN and Pd/CN were investigated using XRD and the patterns are shown in Fig. 1b. A broad diffraction peak at around 25° and a weak reflection around 44° were observed for the CN and are ascribed to the (002) and (100) reflections of the graphite-type lattice. The diffraction peaks at 40.0°, 46.6° and 68.1° in the XRD pattern could be assigned to the (111), (200) and (220) planes of the Pd nanoparticles, respectively.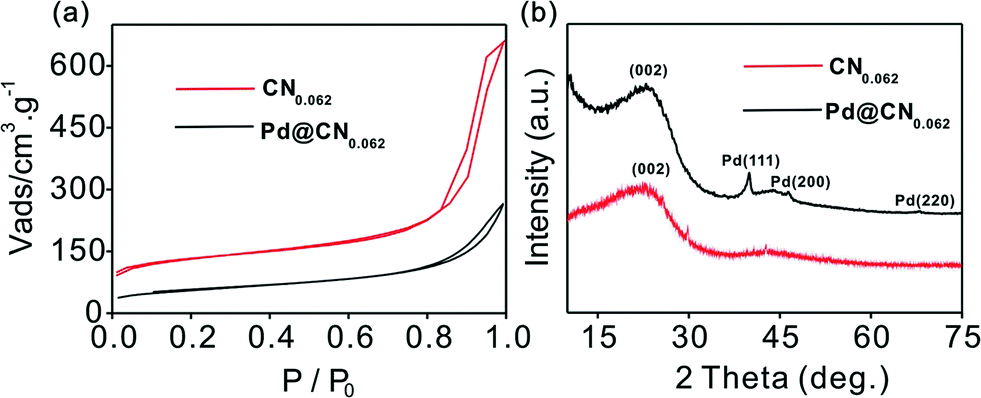 | ||
| Fig. 1 (a) N2 adsorption–desorption isotherms and (b) XRD patterns of the parent CN and the Pd/CN. | ||
The morphologies of the Pd/CN composite materials were also unambiguously characterized using TEM. The TEM images evidenced the high and homogeneous dispersion of spheroidal Pd particles with a mean size of 3.2 nm (Fig. 2b). Pd nanoparticles with well-defined lattice fringes in the HRTEM image confirmed the (111) crystal plane of Pd, as the crystal plane spacing was measured as 0.220 nm (Fig. 2c). To obtain further insight into the structure of the catalyst, XPS investigations were conducted. The Pd 3d XPS spectra in Fig. 2d consist of two asymmetric peaks assigned to the Pd 3d5/2 and Pd 3d3/2 core levels, which can be fitted using two doublets. The peaks around 335.7 and 340.9 eV were attributed to metallic Pd0, while those around 337.7 and 343.0 eV correspond to Pd2+ species. The percentage of the Pd0 and Pd2+ species was calculated from the relative areas of these peaks, which indicated that Pd0 was the main metal species on the surface of the as-prepared catalyst (~75%). The amine groups in the carbon carrier are suitable for stabilizing the highly dispersed Pd0 nanoparticles and prevent their oxidation.
 | ||
| Fig. 2 (a) SEM image of the N-doped carbon material, CN. (b) TEM image and particle size distribution, (c) HRTEM image and (d) XPS spectra of Pd/CN. | ||
Catalytic hydrogenation
To illustrate the usefulness of the nanohybrid catalyst Pd/CN, we use chemoselective hydrogenation of chalcone at room temperature under 1 bar of hydrogen pressure as a model reaction.Effect of solvents
Solvents play a critical role in heterogeneous catalytic hydrogenation and may alter the product distribution.42,43 Consequently, the influence of different solvents on the reaction was given priority in the investigation.Table 1 presents the corresponding conversions and selectivities at 35 °C and 1 bar H2 over Pd/CN, along with the physical and chemical properties of the solvents used including polarity and solubility of H2. Although the mechanistic basis of the solvent influence on the heterogeneous hydrogenation is not clear, some conclusions have been rationalized by correlating the reaction rate and selectivity with H2 solubility or solvent polarity. However, in our reaction system, we could readily achieve conversions of chalcone in the range 88–100%, with >99% selectivities toward dihydrochalcone in the various solvents listed in Table 1, which was not correlated with either the H2 solubility or solvent polarity. This phenomenon was also observed in the chemoselective hydrogenation of quinoline.44 Though chalcone is insoluble in water, we could also get a similar activity to those in other solvents. Doping with N atoms increased the hydrophilic property of the catalyst, which may enhance the catalyst dispersion in water and improve the exposure of the catalyst to the substrate, thereby increasing the catalytic performance. Among the solvents, both toluene and ethanol gave the highest activity. Considering the toxicity of toluene, we selected ethanol as the reaction medium in the subsequent research. It is also a cheap and versatile medium.
| Entry | Solvent | Conv.b (%) | Sel.b (%) | δ | p |
|---|---|---|---|---|---|
| a Reaction conditions: chalcone (0.5 mmol), Pd (0.28 mol% relative to substrate), solvent (5 mL), temperature (35 °C), H2 (1 bar pressure), time (2 h). b Determined by GC and GC-MS. c Solubility of H2 (10−3 mol L−1, taken from Hydrogen and Deuterium; C. L. Young; Pergamon Press: Oxford, U.K., 1981). d Solvent polarity. | |||||
| 1 | Toluene | >99 | >99 | 2.75 | 2.4 |
| 2 | Dioxane | 88 | >99 | 1.95 | 4.8 |
| 3 | Ethanol | >99 | >99 | 3.43 | 4.3 |
| 4 | DMF | 90 | >99 | — | 6.4 |
| 5 | Acetonitrile | 95 | >99 | — | 6.2 |
| 6 | Water | 93 | >99 | 0.81 | 10.2 |
Effect of reaction temperature
Variation of the reaction temperature had a considerable effect on the reaction conversion, as illustrated in Fig. 3a. Complete conversion was achieved with excellent selectivity within 1.7 h at an ambient hydrogen pressure and 35 °C. The reaction was accelerated at higher temperature without any loss of selectivity. For example, full conversion in 1.2 h was achieved at 45 °C with a high selectivity (>99%). However, even at a low temperature of 25 °C, outstanding performance (>99% yield) could be achieved after a slightly longer reaction time of 3 h. The smooth completion of the chemoselective hydrogenation under mild conditions may provide the possibility for industrial applications.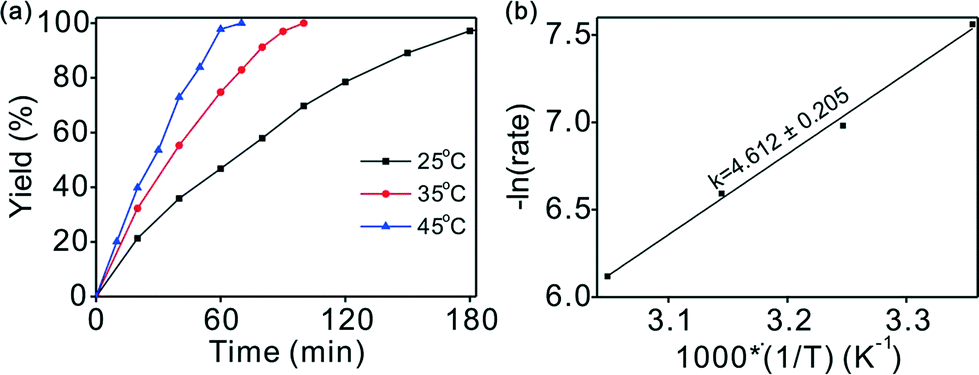 | ||
| Fig. 3 (a) The time–activity profile under different temperatures. Reaction conditions: chalcone (0.5 mmol), Pd (0.28 mol% relative to substrate). (b) Temperature dependence of the rate of reaction in the Arrhenius coordinates for Pd/CN. Reaction conditions: chalcone (1 mmol), Pd (0.28 mol% relative to substrate), CH3CH2OH (5 mL), H2 (1 bar pressure). | ||
Straight lines can be fitted at different temperatures using the data in Fig. 3a (conversion below 45%), indicating that the reaction is zero-order in chalcone when below 45% conversion (Fig. S4†). The influence of temperature on the kinetics of the reaction was studied at 25, 35, 45, and 55 °C. To determine the activation energy of the hydrogenation reaction, the data was obtained at low conversions. Based on the linear fit of the Arrhenius plot in Fig. 3b, the apparent activation energy (Ea) was found to be 38.3 ± 1.7 kJ mol−1, which is consistent with its high catalytic activity, suggesting an easily initiated reaction.
The comparison of various catalysts
To further study the effect of the support, the Pd/CN catalyst was compared with several other catalysts including Pd/Cs, Pd/TiO2, and Pd/MgO under identical conditions (Table 2). The textural properties of the different catalysts are listed in Table S2.† Among the different catalysts, Pd/CN was found to give the most active system (Table 2, entry 1). Oxide-supported Pd nanoparticles were not effective for this reaction, affording the target product in lower yields (Table 2, entries 3 and 4). The reasons for this may arise from the uneven distribution of active positions of Pd on the inorganic oxide surface. Particularly worth mentioning was that the performance of Pd/Cs was almost as good as Pd/CN (Table 2, entry 2), which may be attributed to the fast diffusion of the reactant or product in the hierarchically porous networks with well-dispersed small spherules of Cs and CN. Taking into account the above results, a conclusion may be drawn that the nature of the support has a marked influence on the activity of Pd catalysts for this reaction.| Entry | Catalyst | Conv.b (%) | Sel.b (%) | TOF (h−1) |
|---|---|---|---|---|
| a Reaction conditions: chalcone (0.5 mmol), Pd (0.28 mol% relative to substrate), CH3CH2OH (5 mL), temperature (35 °C), H2 (1 bar pressure), time (1.5 h). b Determined by GC. | ||||
| 1 | Pd/CN | >99 | >99 | 238.1 |
| 2 | Pd/Cs | 91 | >99 | 216.7 |
| 3 | Pd/TiO2 | 10 | >99 | 23.8 |
| 4 | Pd/MgO | 31 | >99 | 73.8 |
Reaction kinetics
Mass transfer can play an important role in liquid phase hydrogenations using porous catalysts.45 Therefore, it is necessary to make sure that mass effects are ruled out before obtaining reliable kinetic data.46 To check the true reaction kinetics were not controlled by any of the diffusion processes, a range of different catalyst masses were used to measure the conversion and the stirring speed was varied from 500 to 1000 rpm. No obvious difference in the rates of reaction at various stirring speeds could be observed.The straight line shown in Fig. 4 also elucidated that external diffusion was not rate-limiting under the reaction conditions. On the other hand, based on the very small particles of the catalyst used, the role of internal diffusion was estimated to be insignificant. Overall, the smallest catalyst particle size and highest stirring speed (1000 rpm) were adopted to minimize the possibility of mass transfer effects.
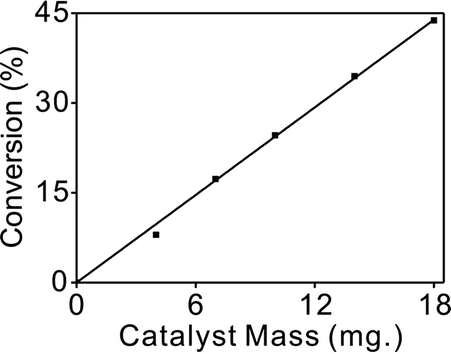 | ||
| Fig. 4 The catalytic activity of 1.5% Pd/CN as a function of catalyst mass, at 35 °C and 1 bar H2, 0.5 h reaction time. | ||
The overall rate of this reaction can be given by the following equation: 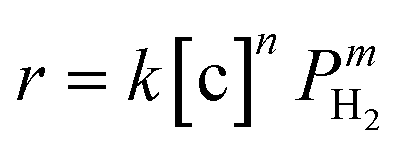 , where k is the overall rate constant, n is the order with respect to chalcone and m is the order corresponding to hydrogen. The reaction orders n and m were determined using double logarithmic plots of the initial TOF (TOF = [moles of reactants reacted] / [(moles of metal used) × (reaction time)]) against chalcone concentration or H2 pressure. As can be seen in Fig. 5a, the TOF remained static as the initial chalcone concentration was increased from 0.1 to 1.0 M, indicating a typical characteristic of a zero-order reaction with respect to chalcone. This conclusion coincides with Fig. 3a, namely, the reaction is zero-order in chalcone when below 45% conversion. The zero-order reaction with respect to chalcone implies a surface largely covered with reactive hydrocarbon intermediates.47 Over a hydrogen pressure range of 2–15 bar, the initial TOF showed a lower exponential dependence on PH2 of 0.38 (see Fig. 5b). As a result, a power law rate expression was derived:
, where k is the overall rate constant, n is the order with respect to chalcone and m is the order corresponding to hydrogen. The reaction orders n and m were determined using double logarithmic plots of the initial TOF (TOF = [moles of reactants reacted] / [(moles of metal used) × (reaction time)]) against chalcone concentration or H2 pressure. As can be seen in Fig. 5a, the TOF remained static as the initial chalcone concentration was increased from 0.1 to 1.0 M, indicating a typical characteristic of a zero-order reaction with respect to chalcone. This conclusion coincides with Fig. 3a, namely, the reaction is zero-order in chalcone when below 45% conversion. The zero-order reaction with respect to chalcone implies a surface largely covered with reactive hydrocarbon intermediates.47 Over a hydrogen pressure range of 2–15 bar, the initial TOF showed a lower exponential dependence on PH2 of 0.38 (see Fig. 5b). As a result, a power law rate expression was derived: 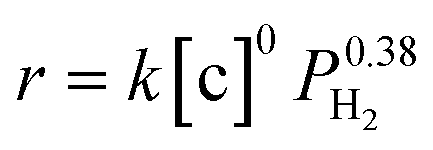 .
.
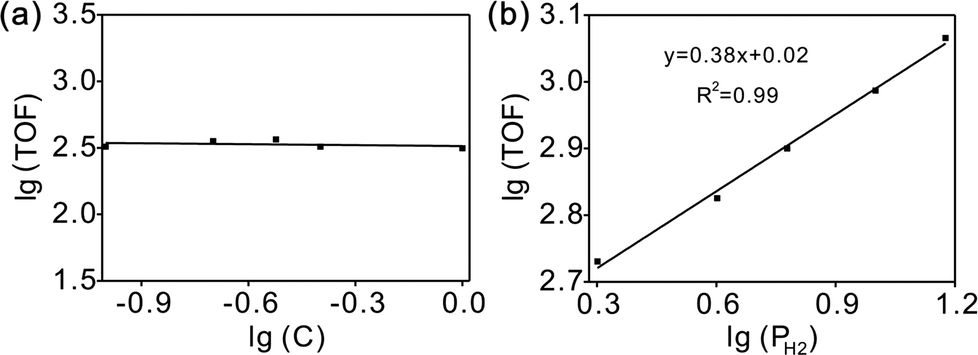 | ||
| Fig. 5 Dependence of initial TOF on chalcone concentration (a) and H2 pressure (b) for chalcone hydrogenation on Pd/CN. | ||
The reusability of the catalyst
For recycling studies, chemoselective hydrogenation of chalcone was conducted whilst maintaining the same reaction conditions as before, but using the recovered catalyst. We tested the catalytic activity of Pd/CN and Pd/Cs in successive runs and compiled the results in Fig. 6. To our delight, Pd/CN was recycled up to 8 times with little loss of activity, which is a prerequisite for practical applications. Nevertheless, the Pd/Cs displayed a noticeable inactivation during the recycling, indeed only giving a 24% conversion at the third cycle.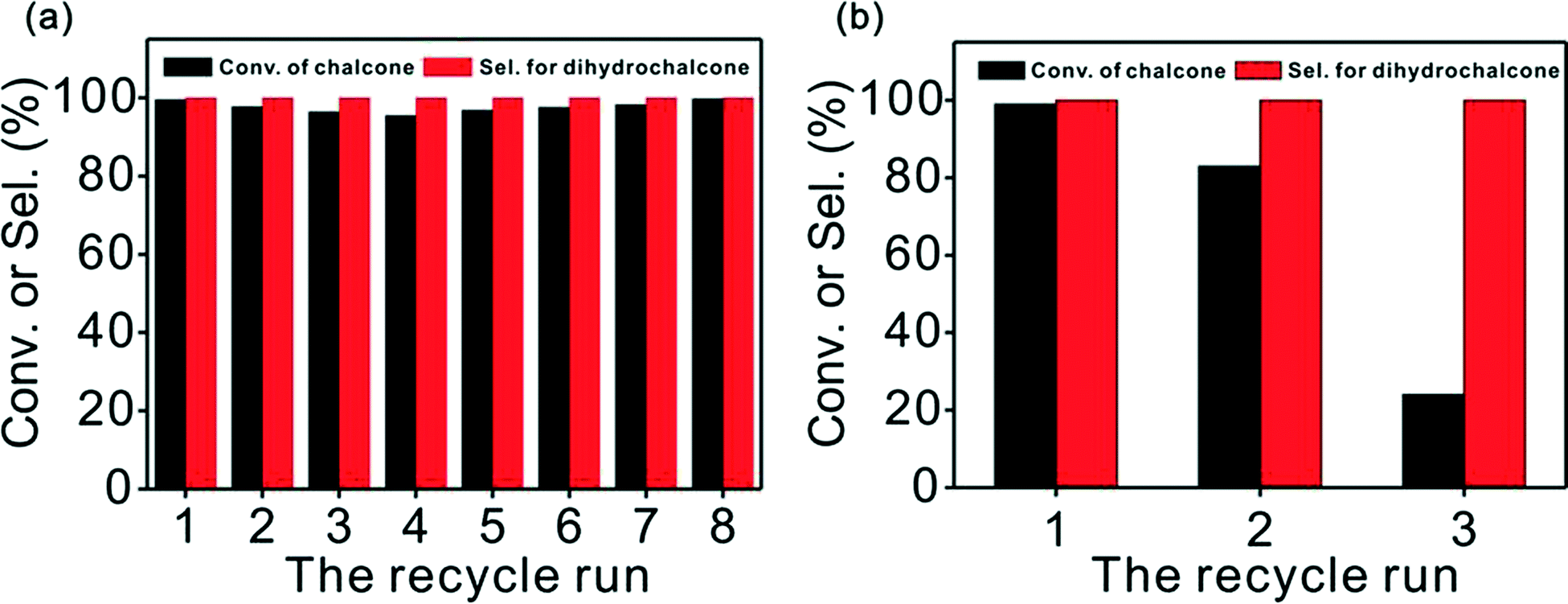 | ||
| Fig. 6 (a) Reuse of Pd/CN and (b) reuse of Pd/Cs. (35 °C, 2.5 h). | ||
The TEM images of the Pd/CN after 8 cycles revealed a particle size of ~3.5 nm, almost the same as that of the fresh catalyst (Fig. S5†), which indicated that it was aggregation-free after reuse. The slightly decreased activity of the Pd/CN was primarily due to minor metal leaching (as shown in Table S3†) during the separation and purification operations. A hot filtration experiment indicated that the leached species in the filtrate made no difference to the conversion of chalcone (Fig. S6†). Though the mean particle size of the reused Pd/Cs remained about the same as that of the fresh catalyst (Fig. S5†), the palladium content declined dramatically to 0.82 wt% from 1.29 wt% in the third cycle, leading to the conspicuous loss in activity.
Notably, the palladium content of the Pd/Cs dropped faster than that of Pd/CN, which was ascribed to the versatile nitrogen in the catalyst support. XPS spectra suggested that the nature and the content of the nitrogen species in the surface of the fresh and recycled catalyst are almost the same (Fig. S7†). As nitrogen is strongly electronegative and usually has a lone pair of electrons, the nitrogen functionalities on the surface might act as Lewis base sites and are expected to be more effective in retaining metal nanoparticles. Simultaneously, the N-doped carbon material enriches the electron density of the metallic Pd and accelerates the reaction as compared to Pd/Cs.
The possible reaction mechanism
More studies have focused on the reaction pathway and mechanism with acrolein and crotonaldehyde. Though there are no similar studies for chalcone, some generalizations can be made based on the experimental data and the theoretical results that have been reported. The extended Hückel calculations suggest that the narrower the width of the metal d-band, the greater the interaction of the metal surface with the conjugated olefinic bond compared to the carbonyl bond (width of the metal d-band: Pd < Pt < Os).21 DFT calculations48 and an HREELS study49 have shown that maleic anhydride interacts with the Pd (111) surface via the CC bond. On the other hand, the CC bond may be adsorbed almost parallel to the metal surface because the mean particle size of Pd is as small as 3.2 nm. Presumably, the close interaction between Pd and the CC bond benefited the high catalytic performance in our system. More importantly, the hierarchically structured porous materials may modify the adsorption properties of the metal surface itself and can provide large surface areas for reaction, interfacial transport, or dispersion of active sites at different length scales of pores, and can shorten diffusion paths or reduce the diffusion effect.33Scheme 2 illustrates the proposed step sequence of the chemoselective hydrogenation process. In the initial step, the substrate interacts with the Pd surface via the CC bond and H2 is dissociated from the electronically supported Pd. Subsequently, the CC bond is selectively hydrogenated by the attack of the activated hydrogen atoms. Because of the rapid diffusion pathways and shortened diffusion paths in the hierarchical mesoporous channels, the formed saturated carbonyls leave the surface of the catalyst quickly, being replaced by a new molecule and avoiding further hydrogenation to a saturated alcohol.
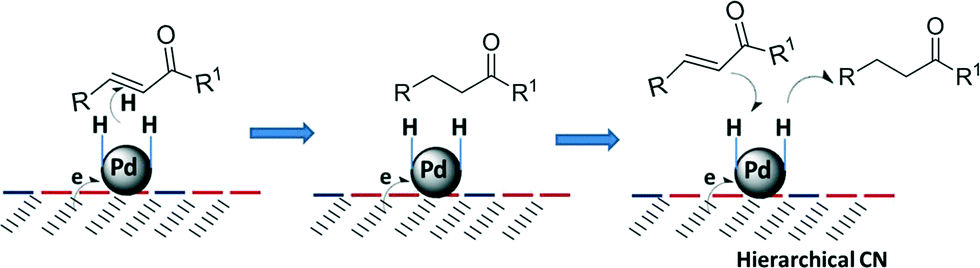 | ||
| Scheme 2 Possible reaction mechanism of the selective hydrogenation process over Pd/CN. | ||
Catalytic performance for various α,β-unsaturated carbonyls
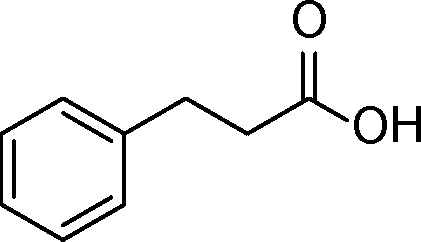
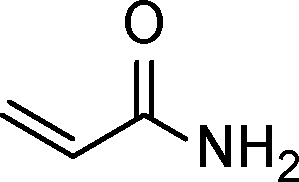
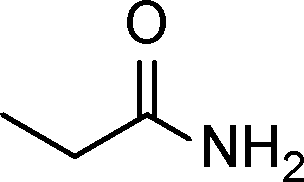
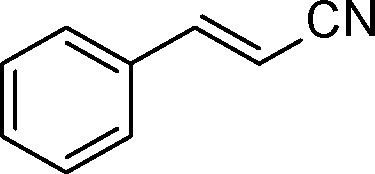
The general scope of the Pd/CN was tested in the reduction of a series of α,β-unsaturated carbonyls with the results summarized in Table 3. Gratifyingly, full conversion was achieved in all cases, and the selectivities towards the corresponding products were excellent (>99%). Apart from simple α,β-unsaturated aldehydes such as butenal and citral (Table 3, entries 1–2), α,β-unsaturated ketones, like benzalacetone and chalcone, were also selectively reduced (Table 3, entries 3–4). Furthermore, remarkable chemoselectivity was demonstrated in the hydrogenation of the CC bond in the presence of other reducible groups, such as ester, carboxyl, amino and nitrile groups, at room temperature with 1 bar hydrogen pressure (Table 3, entries 5–9). In view of the results, it appeared that steric effects imposed by substituent groups on the C atoms in the CC bond did not have an influence on the intramolecular selectivity, which was exactly in line with the conclusion reported by Utpal K. Singh et al.50
| Entry | Substrate | Product | Time (h) | Yield. (%)b | TOF (h−1) |
|---|---|---|---|---|---|
| a Reaction conditions: substrate (0.5 mmol), Pd (0.28 mol% relative to substrate), CH3CH2OH (5 mL), 30 °C, H2 (1 bar pressure). b Determined by GC and GC-MS. | |||||
| 1 |
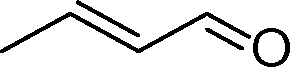
|

|
0.5 | >99 | 714.2 |
| 2 |
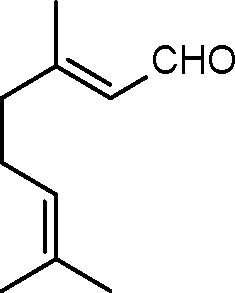
|
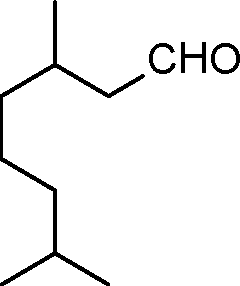
|
4 | >99 | 89.2 |
| 3 |
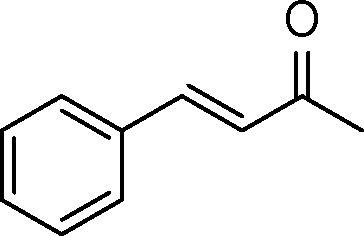
|
|
2 | >99 | 178.6 |
| 4 |
|
|
2.5 | >99 | 160 |
| 5 |
|
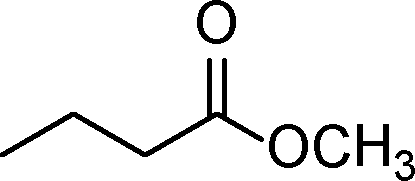
|
0.5 | >99 | 714.2 |
| 6 |
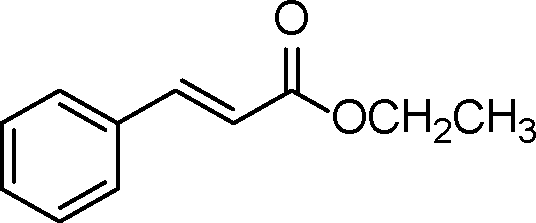
|
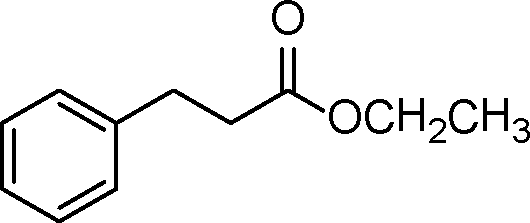
|
0.5 | >99 | 714.2 |
| 7 |
|
|
0.5 | >99 | 714.2 |
| 8 |
|
|
0.5 | >99 | 714.2 |
| 9 |
|
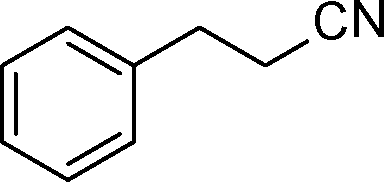
|
2 | >99 | 178.6 |
Conclusions
In summary, the Pd/CN shows distinctly high catalytic activity in the chemoselective hydrogenation of the CC bond of various α,β-unsaturated carbonyls under mild conditions (303 K, 1 bar H2). It can be reused 8 times without significant loss of catalytic activity and selectivity, which is much better than Pd/Cs. The considerable catalytic performance stems from the hierarchically porous network and incorporation of nitrogen atoms. The hierarchically porous network and mesoporous character enormously inhibit the limitation of mass transfer of the reactant to the active sites, promote the free diffusion of molecules and shorten the diffusion distances in the channels. The introduction of nitrogen atoms leads not only to a very stable and uniform dispersion of Pd but also to additional electronic activation of the metal nanoparticles. This synthesis strategy (by modulation of pore structure and introduction of heteroatoms in the catalyst support) can open routes for the design of many more powerful catalysts.
Acknowledgements
Financial support from the National Natural Science Foundation of China (21376208 & U1162124), the Zhejiang Provincial Natural Science Foundation for Distinguished Young Scholars of China (LR13B030001), the Specialized Research Fund for the Doctoral Program of Higher Education (J20130060), the Fundamental Research Funds for the Central Universities, the Program for Zhejiang Leading Team of S&T Innovation, and the Partner Group Program of the Zhejiang University and the Max-Planck Society is greatly appreciated.Notes and references
- G. Neri, L. Mercadante, A. Donato, A. M. Visco and S. Galvagno, Catal. Lett., 1994, 29, 379–386 CrossRef CAS.
- M. Englisch, A. Jentys and J. A. Lercher, J. Catal., 1997, 166, 25–35 CrossRef CAS.
- M. D. Bhor, A. G. Panda, S. R. Jagtap and B. M. Bhanage, Catal. Lett., 2008, 124, 157–164 CrossRef CAS.
- N. Mahata, A. F. Cunha, J. J. M. Orfao and J. L. Figueiredo, Chem. Eng. J., 2012, 188, 155–159 CrossRef CAS PubMed.
- Y. Wang, J. Yao, H. R. Li, D. S. Su and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 2362–2365 CrossRef CAS PubMed.
- Y. H. Niu, L. K. Yeung and R. M. Crooks, J. Am. Chem. Soc., 2001, 123, 6840–6846 CrossRef CAS.
- F. A. Khan, A. Vallat and G. Suss-Fink, J. Mol. Catal. A: Chem., 2012, 355, 168–173 CrossRef CAS PubMed.
- M. L. Kantam, T. Parsharamulu and S. V. Manorama, J. Mol. Catal. A: Chem., 2012, 365, 115–119 CrossRef PubMed.
- B. P. S. Chauhan, J. S. Rathore and T. Bandoo, J. Am. Chem. Soc., 2004, 126, 8493–8500 CrossRef CAS PubMed.
- D. B. Bagal, Z. S. Qureshi, K. P. Dhake, S. R. Khan and B. M. Bhanage, Green Chem., 2011, 13, 1490–1494 RSC.
- T. Gallert, M. Hahn, M. Sellin, C. Schmöger, A. Stolle, B. Ondruschka, T. F. Keller and K. D. Jandt, ChemSusChem, 2011, 4, 1654–1661 CrossRef CAS PubMed.
- S. Morrissey, I. Beadham and N. Gathergood, Green Chem., 2009, 11, 466–474 RSC.
- H. Hagiwara, T. Nakamura, T. Hoshi and T. Suzuki, Green Chem., 2011, 13, 1133–1137 RSC.
- Z. Baan, Z. Finta, G. Keglevich and I. Hermecz, Green Chem., 2009, 11, 1937–1940 RSC.
- A. Mori, Y. Miyakawa, E. Ohashi, T. Haga, T. Maegawa and H. Sajiki, Org. Lett., 2006, 8, 3279–3281 CrossRef CAS PubMed.
- Y. C. Hong, K. Q. Sun, G. R. Zhang, R. Y. Zhong and Q. Xu, Chem. Commun., 2011, 47, 1300–1302 RSC.
- W. W. Lin, H. Y. Cheng, L. M. He, Y. C. Yu and F. Y. Zhao, J. Catal., 2013, 303, 110–116 CrossRef CAS PubMed.
- Y. E. Wu, S. F. Cai, D. S. Wang, W. He and Y. D. Li, J. Am. Chem. Soc., 2012, 134, 8975–8981 CrossRef CAS PubMed.
- B. H. Zhao, J. G. Chen, X. Liu, Z. W. Liu, Z. P. Hao, J. L. Xiao and Z. T. Liu, Ind. Eng. Chem. Res., 2012, 51, 11112–11121 CrossRef CAS.
- N. Morimoto, S. Yamamoto, Y. Takeuchi and Y. Nishina, RSC Adv., 2013, 3, 15608–15612 RSC.
- F. Delbecq and P. Sautet, J. Catal., 1995, 152, 217–236 CrossRef CAS.
- V. Pallassana and M. Neurock, Chem. Eng. Sci., 1999, 54, 3423–3431 CrossRef CAS.
- H. G. Manyar, B. Yang, H. Daly, H. Moor, S. McMonagle, Y. Tao, G. D. Yadav, A. Goguet, P. Hu and C. Hardacre, ChemCatChem, 2013, 5, 506–512 CrossRef CAS PubMed.
- K. Q. Sun, Y. C. Hong, G. R. Zhang and B. Q. Xu, ACS Catal., 2011, 1, 1336–1346 CrossRef CAS.
- S. Handjani, E. Marceau, J. Blanchard, J.-M. Krafft, M. Che, P. Maeki-Arvela, N. Kumar, J. Waerna and D. Y. Murzin, J. Catal., 2011, 282, 228–236 CrossRef CAS PubMed.
- R. J. White, R. Luque, V. L. Budarin, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 481–494 RSC.
- J. P. Tessonnier, L. Pesant, G. Ehret, M. J. Ledoux and C. Pham-Huu, Appl. Catal., A, 2005, 288, 203–210 CrossRef CAS PubMed.
- P. Makowski, R. Demir Cakan, M. Antonietti, F. Goettmann and M.-M. Titirici, Chem. Commun., 2008, 999–1001, 10.1039/B717928F.
- P. Serp, M. Corrias and P. Kalck, Appl. Catal., A, 2003, 253, 337–358 CrossRef CAS.
- H. Bernas, I. Simakova, I. P. Prosvirin, P. Maki-Arvela, R. Leino and D. Y. Murzin, Catal. Lett., 2012, 142, 690–697 CrossRef CAS PubMed.
- C. Pham-Huu, N. Keller, L. J. Charbonniere, R. Ziessle and M. J. Ledoux, Chem. Commun., 2000, 1871–1872, 10.1039/b005306f.
- C. D. Liang, Z. J. Li and S. Dai, Angew. Chem., Int. Ed., 2008, 47, 3696–3717 CrossRef CAS PubMed.
- Y. Li, Z. Y. Fu and B. L. Su, Adv. Funct. Mater., 2012, 22, 4634–4667 CrossRef CAS PubMed.
- Y. Zhu, Z. L. Hua, Y. D. Song, W. Wu, X. X. Zhou, J. Zhou and J. L. Shi, J. Catal., 2013, 299, 20–29 CrossRef CAS PubMed.
- X. Xu, Y. Li, Y. Gong, P. Zhang, H. Li and Y. Wang, J. Am. Chem. Soc., 2012, 134, 16987–16990 CrossRef CAS PubMed.
- C. Han, J. Wang, Y. Gong, X. Xu, H. Li and Y. Wang, J. Mater. Chem. A, 2014, 2, 605–609 CAS.
- P. F. Zhang, Y. T. Gong, H. R. Li, Z. R. Chen and Y. Wang, Nat. Commun., 2013, 4, 1593 CrossRef PubMed.
- P. Zhang, J. Yuan, T.-P. Fellinger, M. Antonietti, H. Li and Y. Wang, Angew. Chem., Int. Ed., 2013, 52, 6028–6032 CrossRef CAS PubMed.
- T. P. Fellinger, R. J. White, M. M. Titirici and M. Antonietti, Adv. Funct. Mater., 2012, 22, 3254–3260 CrossRef CAS PubMed.
- Y. Li, X. Xu, P. Zhang, Y. Gong, H. Li and Y. Wang, RSC Adv., 2013, 3, 10973–10982 RSC.
- U. R. Pillai and E. Sahle-Demessie, Green Chem., 2004, 6, 161–165 RSC.
- R. J. Hou, T. F. Wang and X. C. Lan, Ind. Eng. Chem. Res., 2013, 52, 13305–13312 CrossRef CAS.
- S. Mukherjee and M. A. Vannice, J. Catal., 2006, 243, 108–130 CrossRef CAS PubMed.
- Y. T. Gong, P. F. Zhang, X. Xu, Y. Li, H. R. Li and Y. Wang, J. Catal., 2013, 297, 272–280 CrossRef CAS PubMed.
- G. Zhang, B. L. Scott and S. K. Hanson, Angew. Chem., Int. Ed., 2012, 51, 12102–12106 CrossRef CAS PubMed.
- S. Mahmoud, A. Hammoudeh, S. Gharaibeh and J. Melsheimer, J. Mol. Catal. A: Chem., 2002, 178, 161–167 CrossRef CAS.
- J. P. Breen, R. Burch, J. Gomez-Lopez, K. Griffin and M. Hayes, Appl. Catal., A, 2004, 268, 267–274 CrossRef CAS PubMed.
- V. Pallassana, M. Neurock and G. W. Coulston, J. Phys. Chem. B, 1999, 103, 8973–8983 CrossRef CAS.
- C. Xu and D. W. Goodman, Langmuir, 1996, 12, 1807–1816 CrossRef CAS.
- U. K. Singh and M. A. Vannice, J. Catal., 2001, 199, 73–84 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c4cy00946k |
| This journal is © The Royal Society of Chemistry 2015 |