
Arene C–H activation using Rh(I) catalysts supported by bidentate nitrogen chelates†
Michael S.
Webster-Gardiner
a,
Ross
Fu
b,
George C.
Fortman
a,
Robert J.
Nielsen
b,
T. Brent
Gunnoe
*a and
William A.
Goddard III
*b
aDepartment of Chemistry, University of Virginia, Charlottesville, Virginia 22904, USA. E-mail: tbg7h@virginia.edu
bMaterials and Process Simulation Center, Dept of Chemistry, California Institute of Technology, Pasadena, California 91125, USA. E-mail: wag@wag.caltech.edu
First published on 20th October 2014
Abstract
The Rh(I) complexes [(FlDAB)Rh(coe)(TFA)] (1) and [(BOZO)Rh(coe)(TFA)] (2) [FlDAB = N,N-bis-(pentafluorophenyl)-2,3-dimethyl-1,4-diaza-1,3-butadiene, coe = cyclooctene, TFA = trifluoroacetate, BOZO = bis(2-oxazolin-2-yl)] are efficient catalyst precursors for H/D exchange between arenes and DTFA. Catalyst precursor 1 exhibits a TOF of 0.06 s−1 at 150 °C for benzene H/D exchange. DFT calculations revealed that H/D exchange through reversible oxidative addition or internal electrophilic substitution of benzene is a viable pathway.
The synthesis of catalysts for the selective and efficient functionalization of C–H bonds remains a challenge.1–9 In their seminal work, Shilov and co-workers found that Pt salts can activate and functionalize the C–H bonds of methane, resulting in oxidation to methanol and methyl halides when PtIV is used as oxidant.10–12 Numerous studies have sought to understand and improve the Shilov catalyst,13–18 and several electrophilic late transition metal and main group reagents have been developed for light alkane functionalization.19–25 These electrophilic catalysts are often inhibited by Lewis bases and, as a result, typically require superacidic media (e.g., oleum). The use of strong acid (HX) can lead to the formation of RX, and the electron withdrawing group “X” can protect the functionalized product from over-oxidation (e.g., MeOSO3H formation in H2SO4); but, product extraction from strong acids can be problematic.
Less electronegative metal centres should be less susceptible to inhibition by Lewis bases. Thus, moving to the left in the transition metal series provides a strategy to attenuate inhibition of catalysis in weaker acids. Earlier transition metal complexes can activate hydrocarbons.26–33 However, transition metals earlier than group 10 are likely to be more susceptible to oxidation in acidic media, which could place the catalyst in an oxidation state that is incapable of C–H activation (Scheme 1). Thus, a desirable but challenging aspect of developing catalysts using acidic solvents is maintaining efficient C–H activation. Rh catalysts provide a possible alternative to later metal and main group counterparts since C–H activation by Rh(I) complexes has been reported,34–37 but oxidation to Rh(III) can be facile38 and, thus, rapid C–H activation in acidic media is potentially challenging.
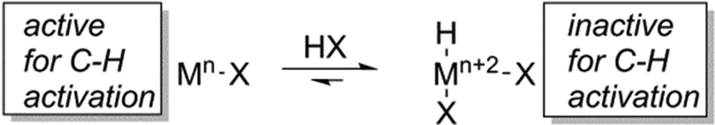 | ||
| Scheme 1 Metal oxidation in acidic media could lead to high oxidation state complexes (i.e., Mn+2) that are less active for or incapable of C–H activation. | ||
A major focus of C–H activation has been on aromatic hydrocarbons. For example, benzene is used to generate styrene and phenol.39–42 Direct oxidation of benzene to phenol and oxidative conversion of benzene and ethylene to styrene are desirable processes.41,42 One method to probe for benzene C–H activation is to study H/D exchange between benzene and a deuterium source. A Rh complex with a PNP pincer ligand catalyses H/D exchange of benzene with D2O with a turnover frequency (TOF) of 2.8 × 10−5 s−1 at 100 °C.43,44 The H/D exchange of benzene with deuterated trifluoroacetic acid at 100 °C with a Rh complex has also been reported.32 A Rh(III) precursor leads to catalytic benzene C–H activation with a TOF of 4.7 × 10−4 s−1 in acetic acid at 150 °C.27 A (hfac)rhodium complex (hfac = hexafluoroacetylacetonate) catalyses H/D exchange of benzene with a TOF of 2.8 × 10−3 s−1 at 190 °C.33 Despite the success of these Rh catalysts for C–H activation of arenes, the reaction rates are slower, typically by an order of magnitude or more, than Pt- or Pd-based catalysts.13,30,45,46 Bidentate nitrogen chelates have been used successfully for H/D exchange with Pt and Pd systems but have not been as thoroughly examined with Rh.13,15,16,18,47–49 Herein, we report the synthesis and reactivity of two new Rh complexes with bidentate nitrogen ligands that exhibit rates of arene H/D exchange with trifluoroacetic acid that are comparable to Pt and Pd catalysts.
The complexes (FlDAB)Rh(COE)(TFA) (1) and (BOZO)Rh(COE)(TFA) (2) [FlDAB = N,N-bis-(pentafluorophenyl)-2,3-dimethyl-1,4-diaza-1,3-butadiene, COE = cyclooctene, TFA = trifluoroacetate, BOZO = bis(2-oxazolin-2-yl)] were synthesized by treatment of [(COE)2Rh(TFA)]2 with two equivalents of ligand in THF at room temperature (Scheme 2). Complexes 1 and 2 are isolated as purple solids in 58% and 73% yield, respectively.
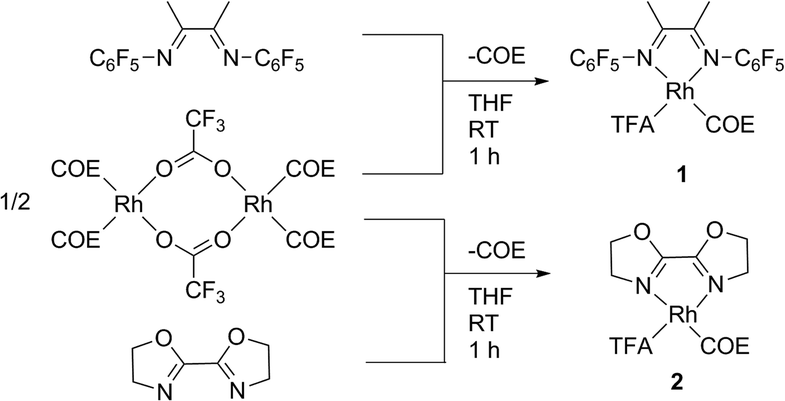 | ||
| Scheme 2 Synthesis of rhodium catalyst precursors 1 and 2. (COE = cyclooctene, TFA= trifluoroacetate). | ||
We examined Rh complexes 1 and 2 for H/D exchange between DTFA and benzene (eqn (1)). Under our conditions (1.6 mol% Rh in C6H6 with 17.5 equiv. of [D1] trifluoroacetic acid relative to C6H6, 130 °C), both 1 and 2 gave 82(8) and 91(11) turnovers (TO) of H/D exchange products, respectively, after 2 hours. Using the turnover number (TON) after 2 hours results in calculated turnover frequencies (TOFs) of ~0.01 s−1. Note that the possibility of some catalyst deactivation means that the TOFs (and others herein) are lower limits of catalyst activity. Importantly, minimal TOs were detected for the reaction of [(COE)2Rh(TFA)]2 in [D1] trifluoroacetic acid under these conditions. Thus, the bis-imine ligands play a role in the enhancement of catalytic H/D exchange.
 | (1) |
Measuring the effect of temperature on catalysis reveals that the highest TONs after 2 hours occur at 150 °C for complex 1. Using complex 1 as a catalyst precursor results in a decrease in TONs at temperatures above 150 °C, which is most likely due to catalyst decomposition (Fig. 1). After 2 hours of reaction, complex 2 exhibits the highest TON at 130 °C. The decrease in TON at higher temperatures for 2 suggests reduced stability compared to 1.
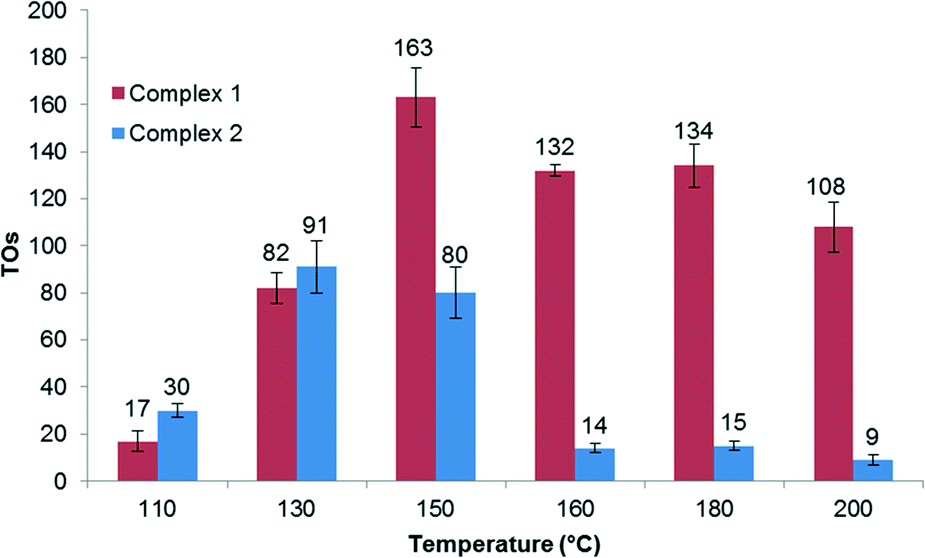 | ||
| Fig. 1 TOs as a function of temperature for complexes 1 and 2. Reactions performed using 1.6 mol% catalyst in C6H6 with 17.5 equiv. DTFA relative to C6H6, 2 hours. TOs are average of at least two runs with standard deviation. TOs are defined as moles of deuterium incorporated into benzene per mole of catalyst, determined by GC-MS. | ||
Experiments were performed to probe the H/D exchange as a function of catalyst concentration. By lowering the catalyst loading relative to benzene, an increase in TONs was observed for complex 1 (Fig. 2). After 2 hours at 150 °C, complex 1 shows 456 TONs to give a calculated TOF of 0.06 s−1. However, no such increase was observed for complex 2. We suspect that complex 1 may undergo a binuclear decomposition; however, definitive conclusions cannot be drawn without more detailed kinetic analysis.
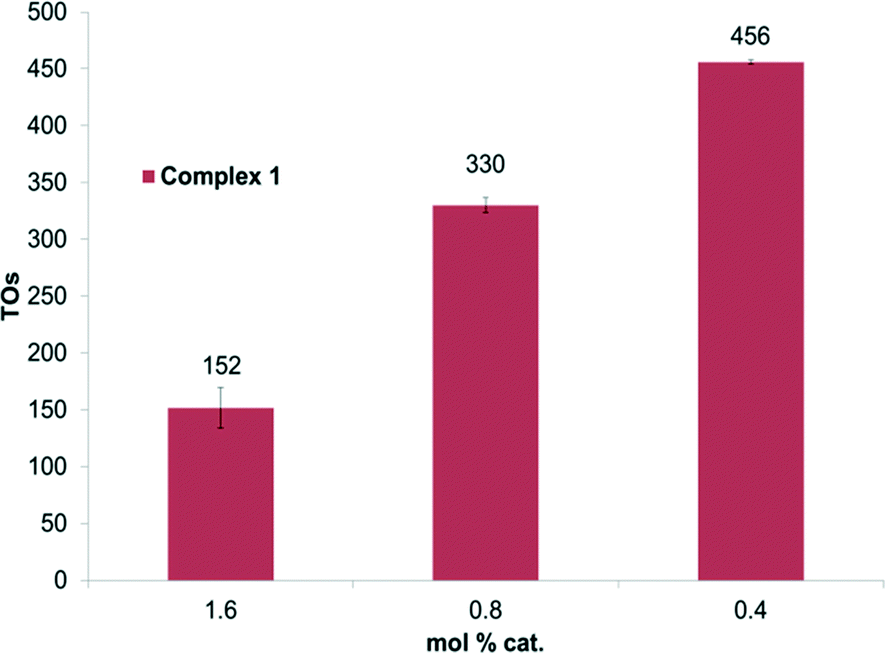 | ||
| Fig. 2 TOs as a function of mol% of 1 for benzene H/D exchange. Reactions were performed at 150 °C under conditions outlined in Fig. 1. | ||
The selectivity of the reaction was determined by examining the H/D exchange of toluene in trifluoroacetic acid after 5% H/D exchange. The ortho![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :meta:para selectivity is 6.9:1:6.4 and 5.7:1:5.2 for 1 (150 °C) and 2 (130 °C), respectively (Scheme 3). Deuteration of the methyl fragment of toluene was not observed, which is evidence against a radical mechanism. The selectivity is similar to electrophilic aromatic substitution and leads us to tentatively conclude that Rh is acting as an electrophile in the C–H bond breaking step.
:meta:para selectivity is 6.9:1:6.4 and 5.7:1:5.2 for 1 (150 °C) and 2 (130 °C), respectively (Scheme 3). Deuteration of the methyl fragment of toluene was not observed, which is evidence against a radical mechanism. The selectivity is similar to electrophilic aromatic substitution and leads us to tentatively conclude that Rh is acting as an electrophile in the C–H bond breaking step.
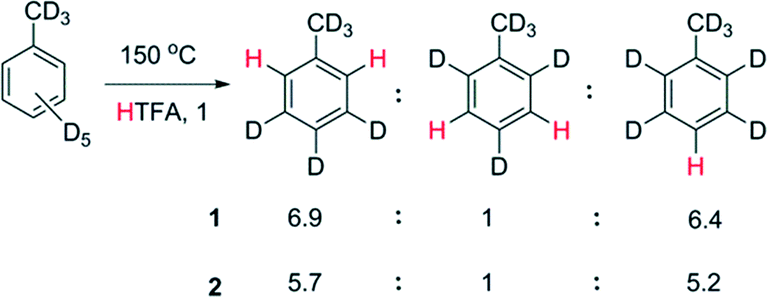 | ||
| Scheme 3 H/D exchange selectivity with [D8] toluene in [H1] trifluoroacetic acid catalysed 1 and 2 by (0.4 mol%) at 5% conversion. | ||
We also explored the recyclability of complex 1. Complex 1, at 0.4 mol% relative to benzene, was dissolved in trifluoroacetic acid and C6D6 in thick-walled (high pressure) glass tubes and heated to 150 °C in an oil bath. After 24 hours, the reactions were sampled and analyzed by GC-MS, and then the volatiles were removed in vacuo. Fresh trifluoroacetic acid and benzene were added to the reaction vessels, and the experiments were repeated. For complex 1, this was successfully done 3 times for a period of over 72 hours with H/D exchange observed each time (Fig. 3).
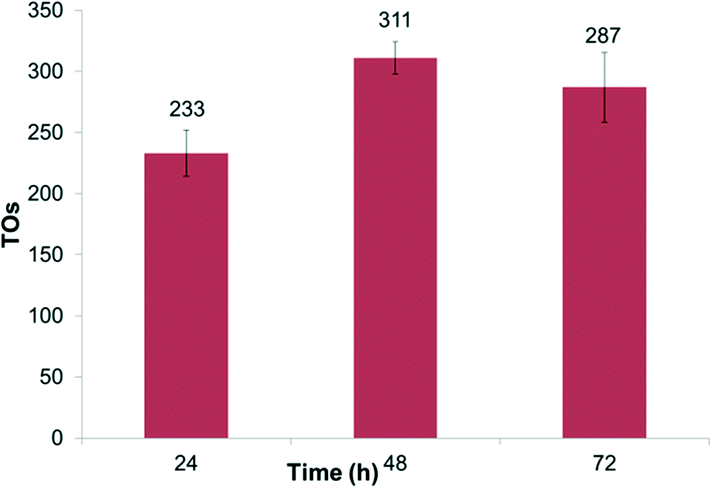 | ||
| Fig. 3 Recyclability of 1 with TOs after 24 hour periods. Reactions were performed at 150 °C in 2 mL of HTFA with C6D6 and 0.4 mol% 1. | ||
We probed the influence of cyclooctene on the H/D exchange reaction. The addition of free cyclooctene led to a decrease in turnovers for complex 2. For example, under our conditions (1.6 mol% 2 in C6H6 with 17.5 equiv. of [D1] trifluoroacetic acid relative to C6H6, 2 hours, 130 °C), the addition of one equivalent of cyclooctene relative to 2 led to a decrease from 91 TO to 32 TO over two hours. This could be due to cyclooctene binding to the metal centre and either suppressing formation of the active catalyst or inhibiting the coordination of benzene.
Monitoring the reaction of 2 in HTFA by 1H and 13C NMR spectroscopy revealed that the initially coordinated cyclooctene is converted to the cyclooctyl trifluoroactate. Indeed, Nordlander et al. reported that cyclooctene in trifluoroacetic acid reacts to form cyclooctyl trifluoroacetate.50 Thus, additional COE may bind to the metal centre until it is consumed to give cycloctyl trifluoroacetate and the active catalyst species.
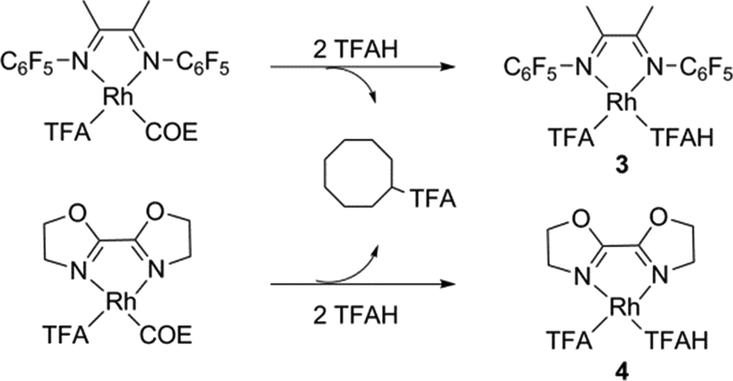
We explored the reaction mechanism by DFT calculations at the M06 level of theory. Our reference complexes were (L)Rh(TFA)(TFAH) (3 and 4 for L = F1DAB or BOZO, respectively); these being the presumed species after the COE is converted to cyclooctyl trifluoroacetate (eqn (2)).
We hypothesized that C–H activation of benzene could proceed through one of four routes. The first route is by direct oxidative addition to (L)RhI(TFA)(TFAH) to form (L)RhIII(TFA)(TFAH)(Ph)(H) (Schemes 4 and 5, top pathway). H/D exchange could then occur because the RhIII(H) bond can be reversibly reductively deprotonated: RhIII(TFA)(H) ⇌ RhI(TFAH). The second route we considered is by direct addition of benzene to (L)RhI(TFA)(TFAH) in a concerted intramolecular electrophilic substitution (IES) step, in which no RhIII intermediate is produced (Schemes 4 and 5, bottom pathway). The third route we considered is an internal proton transfer (L)RhI(TFA)(TFAH) → (L)RhIII(TFA)2(H), followed by either benzene coordination and deprotonation by TFA via a six-membered ring transition state or direct hydrogen exchange with the RhIII(H) hydride (Schemes S1 and S2 in the ESI†). The final route we considered is the oxidative addition of benzene as in the first route, but with isomerization to a RhIII(η2-H2) adduct leading to H/D exchange (Fig. S8 and S9 in the ESI†). We found that for both complexes 3 and 4 the first two scenarios (direct oxidative addition of benzene and IES) are the most likely, with very similar barriers.
 | (2) |
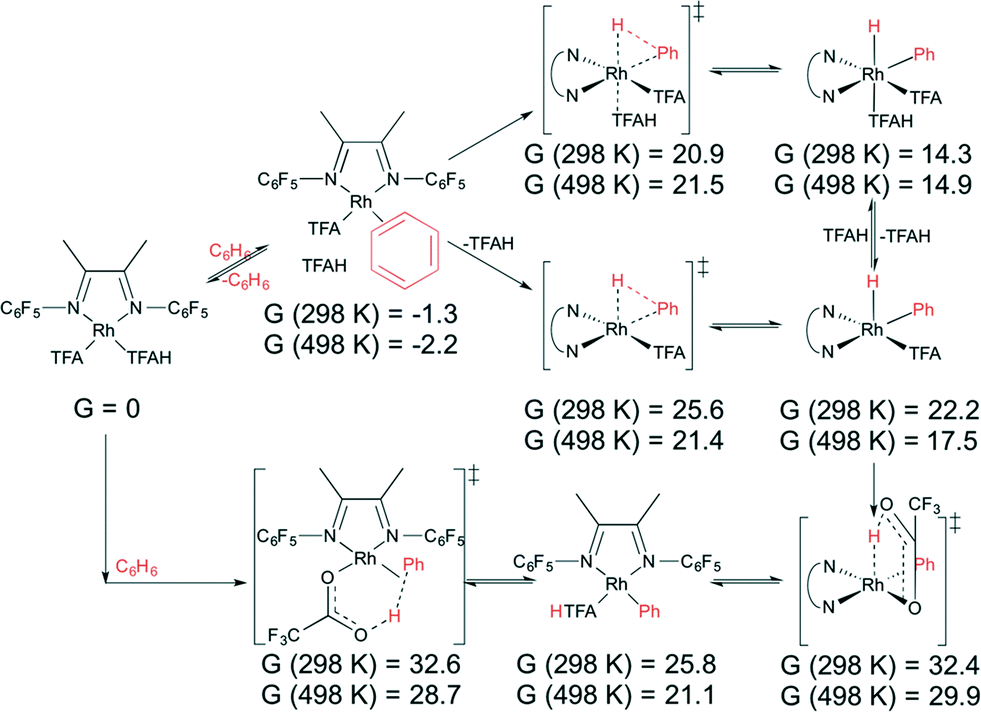 | ||
| Scheme 4 Oxidative addition of benzene to (FlDAB)Rh(TFA)(TFAH) (3) (top), followed by internal protonation, and intramolecular electrophilic substitution (bottom). Values given in kcal mol−1. | ||
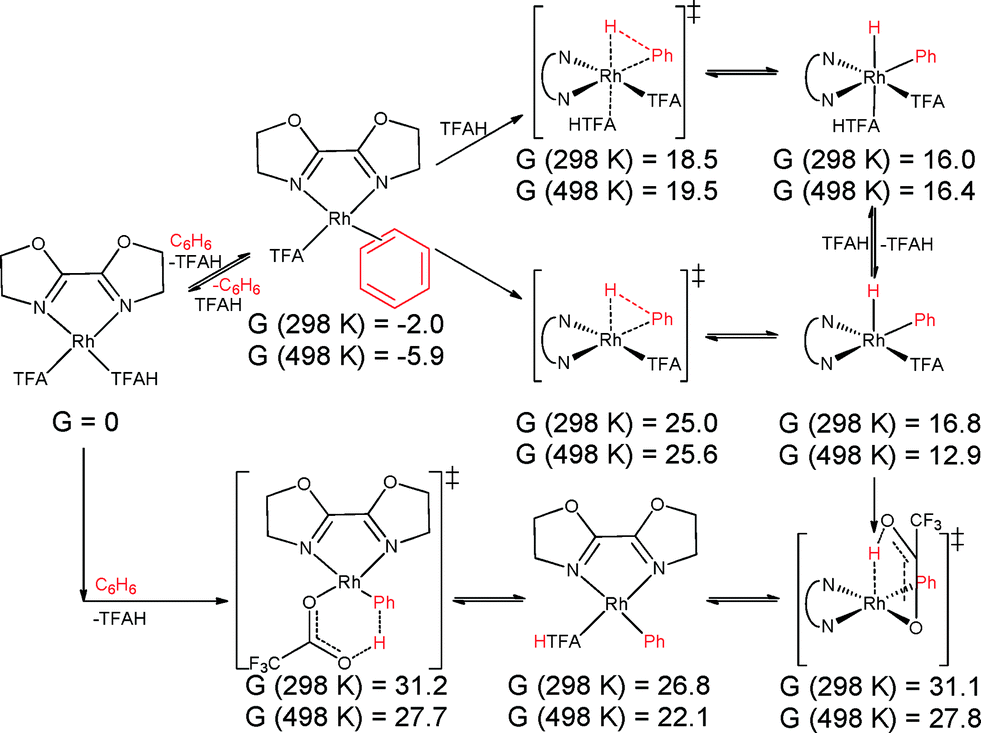 | ||
| Scheme 5 Oxidative addition of benzene by (BOZO)Rh(TFA)(TFAH) (4) (top), followed by internal protonation, and intramolecular electrophilic substitution (bottom). Values given in kcal mol−1. | ||
Direct oxidative addition and IES of benzene by 3 is shown in Scheme 4. The lowest transition state found for oxidative addition to 3 is 20.9 kcal mol−1 at 298 K and 21.5 kcal mol−1 at 498 K. Subsequent internal protonation has calculated free energies of 32.4 kcal mol−1 at 298 K and 29.9 kcal mol−1 at 498 K. In contrast, the IES pathway is calculated to occur with a transition state energy of 32.6 kcal mol−1 at 298 K and 28.7 kcal mol−1 at 498 K relative to the starting complex. This shows that IES is slightly preferred but that both pathways are viable. Note that displacement of TFAH with benzene is favourable by 1.3/2.2 kcal mol−1 at 298 K/498 K, so the actual overall barriers are slightly higher by that amount. The lowest barrier found for 4 is 18.5 kcal mol−1 at 298 K and 19.5 kcal mol−1 at 498 K for oxidative addition, and 31.1 kcal mol−1 at 298 K and 27.8 kcal mol−1 at 498 K for subsequent internal protonation, versus 31.2 kcal mol−1 at 298 K and 27.7 kcal mol−1 at 498 K for internal deprotonation (Scheme 5). However, displacement of TFAH with benzene in 4 is favourable by 2.0/5.9 kcal mol−1 at 298 K/498 K, so the actual overall barriers are higher by that amount. All of these values imply accessible benzene oxidative addition at the reaction temperatures investigated. A comparison of direct oxidative addition and benzene coordination/deprotonation (our third proposed route) is shown in Scheme S1 for complex 3 and Scheme S2 for complex 4; and isomerization to RhIII(η2-H2) adducts (our fourth proposed route) considered in Fig. S8 for complex 3 and Fig. S9 for complex 4 (see ESI†). The experimental data for complex 1 indicate a TOF of ~0.06 s−1, which gives an activation barrier of 27.4 kcal mol−1 (using the Eyring equation). Given the uncertainties in the experimental data (i.e., using TOs after 2 h rather than rigorous kinetic studies), the experimental activation barrier (27.4 kcal mol−1 at 423 K) and calculated barrier (30.9 kcal mol−1 at 498 K) are in reasonable agreement.
Conclusions
This work has demonstrated two efficient Rh catalyst precursors for H/D exchange between benzene and [D1] trifluoroacetic acid. Although caution is warranted for comparison of catalysts under different reaction conditions and solvents, catalyst 1 is among the most active Rh catalysts for benzene H/D exchange in [D1] trifluoroacetic acid (Table 1). Complex 1 exhibits similar activity to the most active Pt and Pd complexes reported for benzene H/D exchange in acidic media. In addition, complex 1 can be recycled at least three times without a significant decrease in activity. These results suggest that development of Rh catalysts for C–H functionalization is a promising target.| Entry | Catalyst | Additive | Solvent | Temp (°C) | TOF (s−1) |
|---|---|---|---|---|---|
| a (PNP = 2,6-bis[(di-tertbutylphosphino)methyl]pyridine). b (OAc = acetate). c (bdmpza = bis(3,5-dimethylpyrazol-1-yl)acetate, OTf = triflate). d (Py = pyridine). | |||||
| 1 | Rh(PNP)Me (ref. 43)a | D2O | 100 | 2.8 × 10−5 | |
| 2 | Rh(pyridinium)Cl3 (ref. 27)b | AgOAc | AcOD | 150 | 4.8 × 10−4 |
| 3 | Rh(bdmpza)Cl3 (ref. 32)c | AgOTf | TFAD | 100 | 1.0 × 10−3 |
| 4 | Rh(hfacac)2(Py)(Me) (ref. 33)d | CD3OD | 190 | 2.8 × 10−3 | |
| 5 | Rh(BOZO)(coe)(tfa) | TFAD | 130 | 2 × 10−2 | |
| 6 | Pd(pyridinium)Cl2 (ref. 46) | AgBF4 | AcOD | 150 | 5 × 10−2 |
| 7 | Pt(2,6 dichloro-DAB) (ref. 45) | AgOAc | TFAD | 150 | 5 × 10−2 |
| 8 | Rh(FlDAB)(coe)(tfa) | TFAD | 150 | 5 × 10−2 | |
| 9 | Pt(pyridinium)Cl2 (ref. 46) | AgBF4 | AcOD | 150 | 1 × 10−1 |
| 10 | Pt(2,6 dichloro-DAB) (ref. 45) | AgOAc | AcOD | 150 | 2 × 10−1 |
Acknowledgements
The authors acknowledge the Center for Catalytic Hydrocarbon Functionalization, an Energy Frontier Research Center funded by the US Department of Energy, Office of Basic Energy Sciences (DE-SC0001298). M.S.W-G. acknowledges support from AES for a graduate student fellowship.Notes and references
- R. H. Crabtree, Chem. Rev., 1985, 85, 245–269 CrossRef CAS.
- J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS PubMed.
- J. R. Webb, T. Bolano and T. B. Gunnoe, ChemSusChem, 2011, 4, 37–49 CrossRef CAS PubMed.
- J. A. Labinger, J. Mol. Catal. A: Chem., 2004, 220, 27–35 CrossRef CAS PubMed.
- R. A. Periana, G. Bhalla, W. J. 3. Tenn, K. J. H. Young, X. Y. Liu, O. Mironov, C. J. Jones and V. R. Ziatdinov, J. Mol. Catal. A: Chem., 2004, 220, 7–25 CrossRef CAS PubMed.
- K. I. Goldberg and A. S. Goldman, Activation and Functionalization of C-H Bonds, ACS Symposium Series 885, American Chemical Society, Washington, DC, 2004 Search PubMed.
- H. Schwarz, Angew. Chem., Int. Ed., 2011, 50, 10096–10115 CrossRef CAS PubMed.
- P. J. Perez, Alkane C-H Activation by Single-Site Metal Catalysis, Springer, Dordrecht, 2012 Search PubMed.
- M. E. O'Reilly, D. R. Pahls, J. R. Webb, N. C. Boaz, S. Majumdar, C. D. Hoff, J. T. Groves, T. R. Cundari and T. B. Gunnoe, Dalton Trans., 2014, 43, 8273–8281 RSC.
- N. F. Goldshlegger, M. B. Tyabin, A. E. Shilov and A. A. Shteinman, Zh. Fiz. Khim., 1969, 43, 2174–2175 Search PubMed.
- N. F. Goldshlegger, V. V. Eskova, A. E. Shilov and A. A. Shteinman, Zh. Fiz. Khim., 1972, 46, 1353–1354 Search PubMed.
- A. E. Shilov and G. B. Shul'pin, Activation and Catalytic Reactions of Saturated Hydrocarbons in the Presence of Metal Complexes, Kluwer Academic Publishers, Dordrecht, 2000 Search PubMed.
- H. A. Zhong, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2002, 124, 1378–1399 CrossRef CAS PubMed.
- S. S. Stahl, J. A. Labinger and J. E. Bercaw, Angew. Chem., Int. Ed., 1998, 37, 2180–2192 CrossRef.
- J. Lars, T. Mats, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2000, 122, 10846–10855 CrossRef.
- M. Lersch and M. Tilset, Chem. Rev., 2005, 105, 2471–2526 CrossRef CAS PubMed.
- F. Zhang, C. W. Kirby, D. W. Hairsine, M. C. Jennings and R. J. Puddephatt, J. Am. Chem. Soc., 2005, 127, 14196–14197 CrossRef CAS PubMed.
- L. M. Rendina and R. J. Puddephatt, Chem. Rev., 1997, 97, 1735–1754 CrossRef CAS PubMed.
- R. A. Periana, O. Mironov, D. J. Taube and S. Gamble, Chem. Commun., 2002, 2376–2377 RSC.
- R. A. Periana, D. J. Taube, E. R. Evitt, D. G. Loffler, P. R. Wentrcek, G. Voss and T. Masuda, Science, 1993, 259, 340–343 CAS.
- R. A. Periana, D. J. Taube, S. Gamble, H. Taube, T. Satoh and H. Fujii, Science, 1998, 280, 560–564 CrossRef CAS.
- B. G. Hashiguchi, M. M. Konnick, S. M. Bischof, S. J. Gustafson, D. Devarajan, N. Gunsalus, D. H. Ess and R. A. Periana, Science, 2014, 343, 1232–1237 CrossRef CAS PubMed.
- G. C. Fortman, N. C. Boaz, D. Munz, M. M. Konnick, R. A. Periana, J. T. Groves and T. B. Gunnoe, J. Am. Chem. Soc., 2014, 136, 8393–8401 CrossRef CAS PubMed.
- D. Munz and T. Strassner, Angew. Chem., Int. Ed., 2014, 53, 2485–2488 CrossRef CAS PubMed.
- C. J. Jones, D. Taube, V. R. Ziatdinov, R. A. Periana, R. J. Nielsen, J. Oxgaard and W. A. Goddard 3rd, Angew. Chem., Int. Ed., 2004, 43, 4626–4629 CrossRef CAS PubMed.
- D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed.
- J. B. Gary, T. J. Carter and M. S. Sanford, Top. Catal., 2012, 55, 565–570 CrossRef CAS.
- G. Bhalla, X. Y. Liu, J. Oxgaard, W. A. Goddard 3rd and R. A. Periana, J. Am. Chem. Soc., 2005, 127, 11372–11389 CrossRef CAS PubMed.
- Y. Feng, B. Jiang, P. A. Boyle and E. A. Ison, Organometallics, 2010, 29, 2857–2867 CrossRef CAS.
- A. J. Hickman, M. A. Cismesia and M. S. Sanford, Organometallics, 2012, 31, 1761–1766 CrossRef CAS.
- M. C. Lehman, J. B. Gary, P. B. Boyle, M. S. Sanford and E. A. Ison, ACS Catal., 2013, 3, 2304–2310 CrossRef CAS.
- J. L. Rhinehart, K. A. Manbeck, S. K. Buzak, G. M. Lippa, W. W. Brennessel, K. I. Goldberg and W. D. Jones, Organometallics, 2012, 31, 1943–1952 CrossRef CAS.
- W. J. Tenn 3rd, B. L. Conley, S. M. Bischof and R. A. Periana, J. Organomet. Chem., 2011, 696, 551–558 CrossRef PubMed.
- H. Chen, S. Schlecht, T. C. Semple and J. F. Hartwig, Science, 2000, 287, 1995–1997 CrossRef CAS.
- W. D. Jones, Inorg. Chem., 2005, 44, 4475–4484 CrossRef PubMed.
- S. R. Klei, K. L. Tan, J. T. Golden, C. M. Yung, R. K. Thalji, K. A. Ahrendt, J. A. Ellman, T. D. Tilley and R. G. Bergman, Carbon-Hydrogen Bond Activation by Iridium and Rhodium Complexes: Catalytic Hydrogen/Deuterium Exchange and Carbon-Carbon Bond-Forming Reactions, ACS Symposium Series 885, Activation and Functionalization of C-H Bonds, Washington D.C., 2004 Search PubMed.
- B. A. Arndtsen, R. G. Bergman, A. Mobley and T. H. Peterson, Acc. Chem. Res., 1995, 28, 154–162 CrossRef CAS.
- T. S. Teets and D. G. Nocera, Inorg. Chem., 2012, 51, 7192–7201 CrossRef CAS PubMed.
- P. J. Chenier, Survey of industrial chemistry, VCH Publishers, New York, 1992 Search PubMed.
- G. A. Olah and Á. Molnár, Hydrocarbon chemistry, Wiley & Sons, New York, 1995 Search PubMed.
- K. Weissermel and H. Arpe, Industrial Organic Chemistry, Wiley-VCH, Weinheim, 3rd edn, 1997 Search PubMed.
- J. R. Andreatta, B. A. McKeown and T. B. Gunnoe, J. Organomet. Chem., 2011, 696, 305–315 CrossRef CAS PubMed.
- S. M. Kloek, D. M. Heinekey and K. I. Goldberg, Angew. Chem., Int. Ed., 2007, 46, 4736–4738 CrossRef CAS PubMed.
- S. M. Kloek, D. M. Heinekey and K. I. Goldberg, Organometallics, 2008, 27, 1454–1463 CrossRef.
- A. J. Hickman, J. M. Villalobos and M. S. Sanford, Organometallics, 2009, 28, 5316–5322 CrossRef CAS.
- M. H. Emmert, J. B. Gary, J. M. Villalobos and M. S. Sanford, Angew. Chem., Int. Ed., 2010, 49, 5884–5886 CrossRef CAS PubMed.
- T. J. Williams, A. J. Caffyn, N. Hazari, P. F. Oblad, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2008, 130, 2418–2419 CrossRef CAS PubMed.
- M. W. Holtcamp, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 1997, 119, 848–849 CrossRef CAS.
- J. E. Bercaw, N. Hazari, J. A. Labinger and P. F. Oblad, Angew. Chem., Int. Ed., 2008, 47, 9941–9943 CrossRef CAS PubMed.
- J. E. Nordlander, K. D. D. Kotian, D. E. I. Raff, G. F. Njoroge and J. J. Winemiller, J. Am. Chem. Soc., 1984, 106, 1427–1432 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details, additional computational schemes and experimental details. See DOI: 10.1039/c4cy00972j |
| This journal is © The Royal Society of Chemistry 2015 |