
Amidinate group 4 complexes in the polymerization of olefins
Tatyana
Elkin
and
Moris S.
Eisen
*
Schulich Faculty of Chemistry, Technion – Israel Institute of Technology, Haifa 32000, Israel. E-mail: chmoris@tx.technion.ac.il
First published on 2nd October 2014
Abstract
The current perspective will present the use of amidinate group 4 complexes in α-olefin polymerizations. We will present the structural studies of the complexes bearing various numbers of amidinates as spectator ligands, with a special emphasis on the bis(amidinate) group 4 systems. The mechanistic studies elucidate the influence of various reaction conditions on the behaviour of the reactive species. Additionally, the study of the active species by techniques such as EPR spectroscopy and MALDI-TOF spectrometry are presented. We will also demonstrate how, based on such techniques, highly stereospecific bis(amidinate) titanium complexes may be designed and applied in the polymerization of propylene.
The discovery of olefin polymerization mediated by heterogeneous catalytic systems by Ziegler1 and Natta2 made polyolefins one of the most important commodity synthetic polymers, represented mostly by polyethylene and polypropylene with a global production of more than 100 million tons per year.3,4 The development of homogeneous system analogues followed shortly thereafter, introducing metallocenes as precursors for olefin polymerization activated by alkylaluminium compounds.5,6 The metallocene systems perform in a single site catalysis nature, generating uniform polymeric materials in low yields. It was not until 1977 that methylaluminoxane (MAO) was introduced as a co-catalyst, capable of generating catalytic mixtures with outstanding activity.7–9
Since their first application as olefin polymerization catalysts, group 4 metallocenes have received considerable attention from the scientific community. Various metallocene systems have contributed valuable information regarding the key steps in the polymerization mechanism. For example, the empirical relationship between the pre-catalyst symmetry and the polymer structure was established.10 However, the search for more elaborate and easily accessible systems capable of producing polymers with various predicted stereostructures and precise molecular weights led to the development of a different class of compounds over recent decades. Numerous post-metallocene catalysts have been designed, possessing ancillary ligands containing donor motifs decorated with N, O and S atoms. For example, among the ligands containing nitrogen donors which have emerged, the catalysts containing amidinate ligands and their derivatives, such as guanidinates and aminopyridinates, have attracted considerable interest over the last 30 years.
The first neutral amidines were reported in 1868 in the pioneering work of Gerhardt,11 who obtained them as a product of heating aniline with benzonitrile. However, the field of amidine chemistry remained dormant for over a century until Sanger reported the synthesis of PhC(=NSiMe3)(N(SiMe3)2).12,13 Since then, amidinates have attracted the attention of numerous researchers and proved to be versatile ligands for the stabilization of various elements across the periodic table, and the application of these complexes has been demonstrated in the breadth of various chemical transformations.14–18
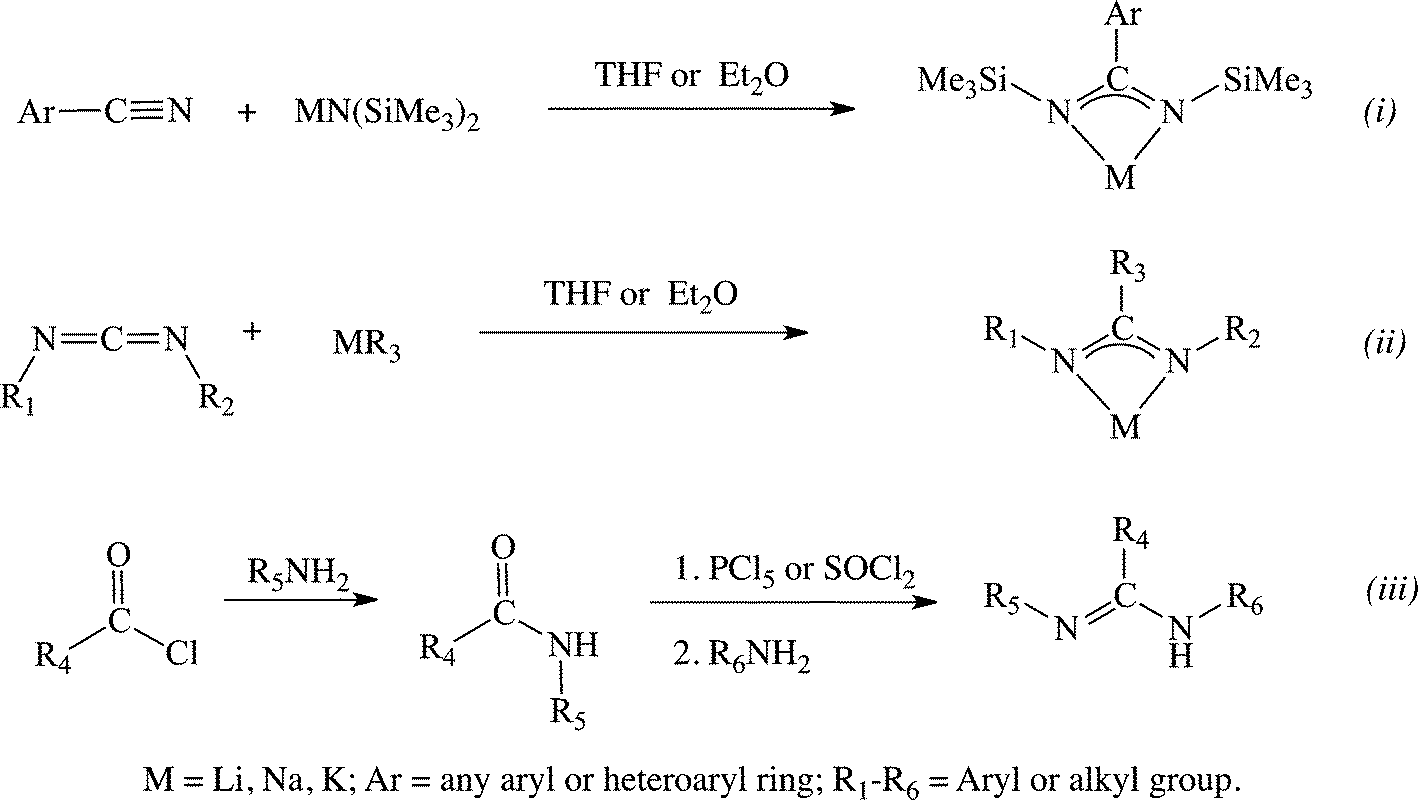
Anionic amidinates have emerged as an alternative to cyclopentadienyl (Cp) ligands, primarily due to their steric similarity.19,20 The main advantages of amidinates over Cp analogues are their ease of modification, allowing for fine tuning of the electronic and steric properties of the ligand, and tailoring of the specific properties of the resulting complexes, leading to a facile access to a large library of compounds. Similar to the carboxylate ligands, amidinates donate 4 electrons, which allows the resulting metal complexes to exhibit increased electrophilicity as compared to their metallocene equivalents. Amidines are most often synthesized following three major routes: i. a sigmatropic rearrangement during the reaction between nitriles and silyl amides12,21–23 (Scheme 1, (a), (a)), ii. via an insertion of a carbodiimide into a metal–carbon bond,24,25 (Scheme 1, (b)), and iii. through the condensation reaction between amines and acyl or imidoyl chlorides26–29 (Scheme 1, (c)).
 | ||
| Scheme 1 Synthetic routes for the synthesis of amidines and amidinates. | ||
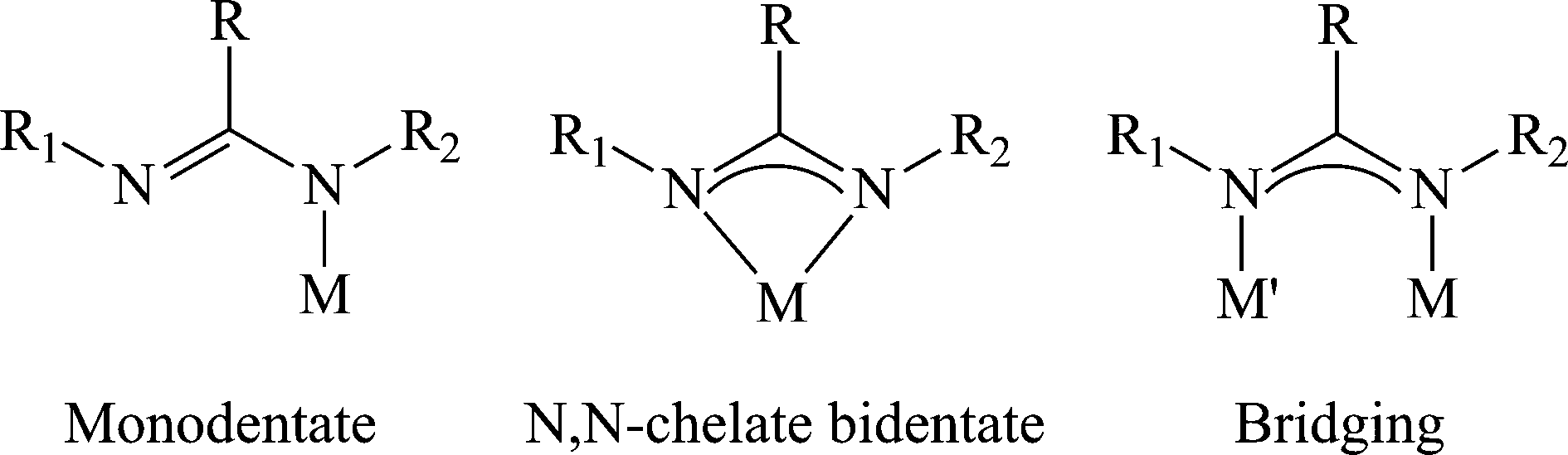
In addition to the variety of easily modified electronic and steric properties, the different connectivity modes of amidinates revealed a remarkable ability of this class of ligands to stabilize the metal center (Scheme 2), adding to the diversity and versatility of the amidinates as ancillary ligands.
 | ||
| Scheme 2 Connectivity modes of amidinate ligands with metal centers. | ||
Group 4 amidinates appeared in literature for the first time in 1988 (ref. 30) when PhC(N(SiMe3))2TiCl2 (1) was prepared,31 shortly followed by the synthesis of the zirconium analogue 2;32 the interest in amidinate ligands as ancillary ligands for achieving homogeneous post-metallocene olefin polymerization began to attract attention in 1995, when the [PhC(NTMS)2]ZrCl2 (2) and [p-Me-C6H4C(NTMS)2]ZrCl2 (3) complexes were used as precursors for the polymerization of ethylene after their activation with an excess of methylaluminoxane (MAO).33 The results revealed that the (bis)benzamidinate zirconium dichloride complex was more active than the corresponding mono(benzamidinate)cyclopentadienyl zirconium dichloride complex, (26 kg mol−1 atm−1 h−1 for 2 and 16 kg mol−1 atm−1 h−1 for 3), due to the increased electrophilic nature of the resulting complexes.33 However, the reactivity of the bis(benzamidinates) remained lower than that of the zirconium bis-metallocenes. Changing both of the N-amidinate substituents from SiMe3 to cyclohexyl (Cy) led to a similar reactivity of 16 kg mol−1 atm−1 h−1 for complex 4 (Scheme 3), under the same experimental conditions.34 The use of (PhC(NCy)2)2TiCl2 (5) resulted in a lower activity (7 kg mol−1 atm−1 h−1) compared with the analogous zirconium complex 4. As extensive studies on the polymerization of ethylene with group 4 amidinates and guanidinates have been previously reviewed,35 this perspective will concentrate on the application of group 4 amidinate complexes in the polymerization of propylene.
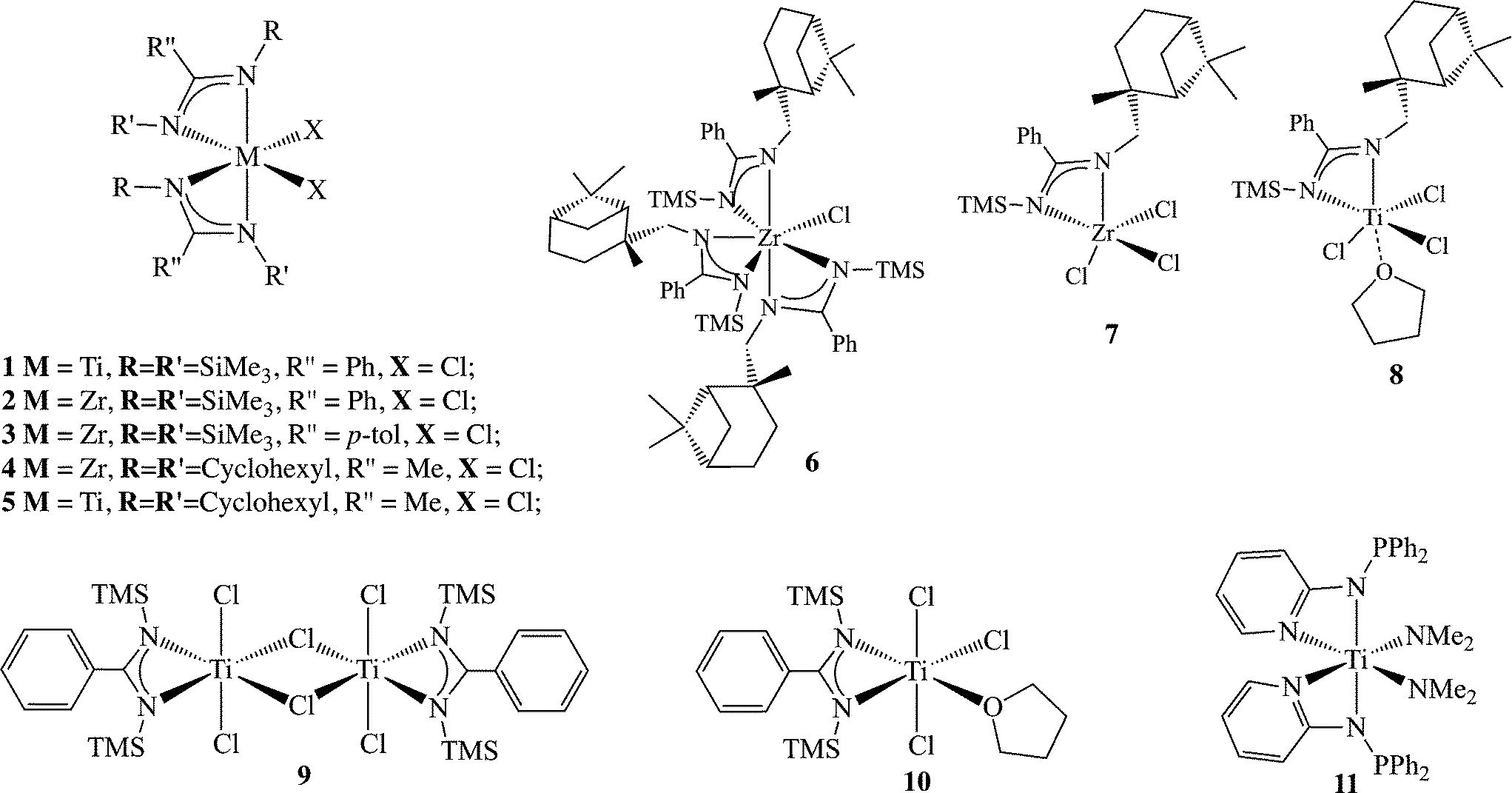 | ||
| Scheme 3 Complexes bearing various benzamidinates (1–10) and aminopyridinates (11) as ancillary ligands. | ||
After the activity of the bis(benzamidinate) group 4 complexes was established in the polymerization of ethylene, an immediate interest emerged in their application for the stereoregular polymerization of propylene. First, it has been found that the pressure of propylene in the reaction plays an important role in the stereochemistry of the resulting polymer, yielding an atactic elastomeric polypropylene or an isotactic solid material when the reaction proceeded at atmospheric or high pressure of monomer, respectively (Scheme 4). These results triggered significant interest, since the atactic polymer was formed despite the C2 symmetry of the pre-catalyst, which was expected to produce isospecific stereoselectivity.10,36 The deviation from Ewen empirical symmetry rules called for an investigation of the active species involved in the reaction.37
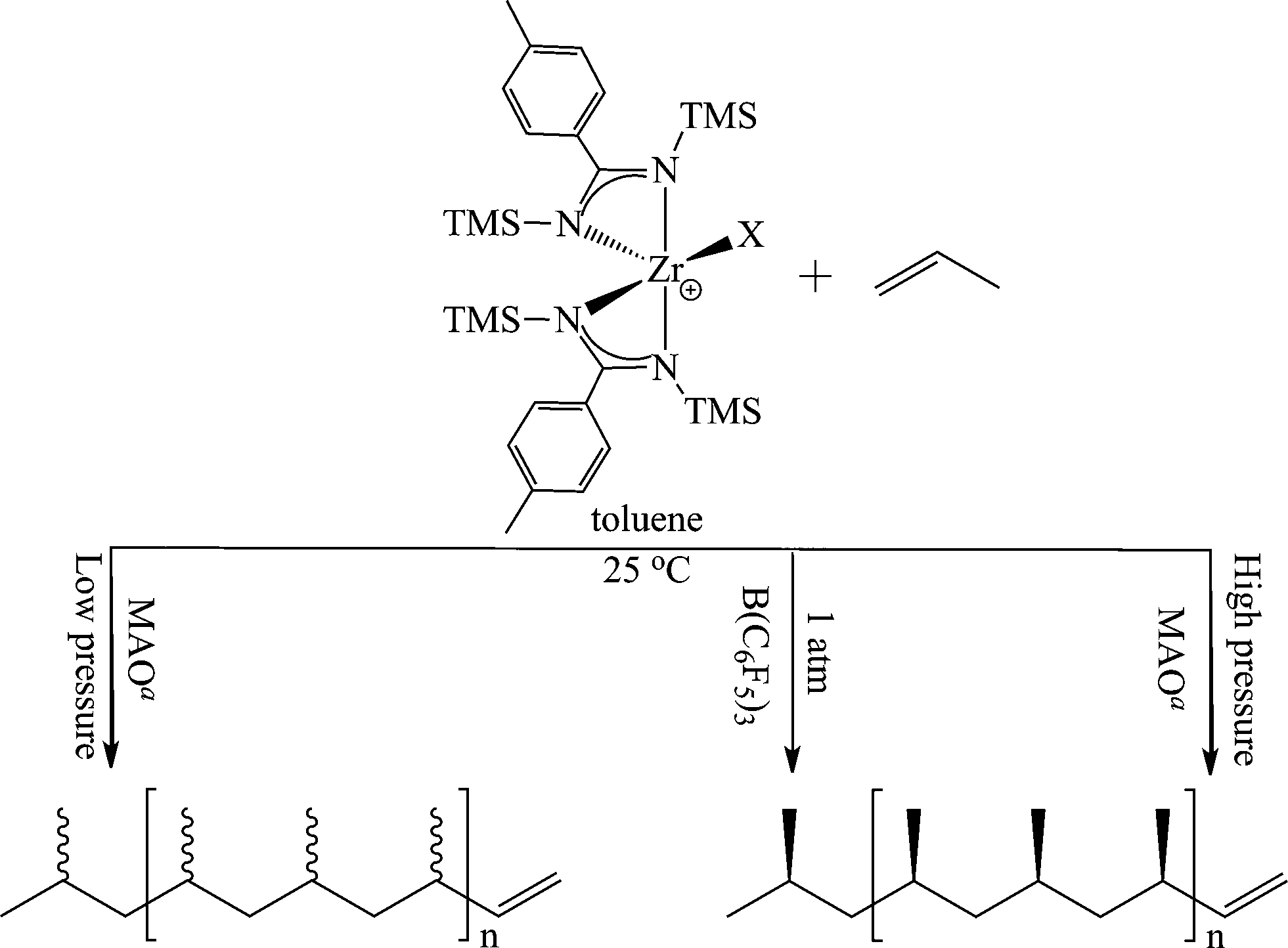 | ||
| Scheme 4 Reaction conditions affecting the stereoregularity of the polymers in the polymerization of propylene. aThe different MAO ratio allows the production of the isotactic material either as the major fraction or as a mixture with an elastomeric material. | ||
The formation of the elastomeric material has been proposed to originate due to a competing intermolecular epimerization of the last inserted monomer that exhibits higher reaction rates at low monomer concentrations.38,39 Additionally, the proposed mechanism has been corroborated by the isomerization of higher olefins such as 1-octene in the presence of the catalytically active mixture.40,41
In order to elucidate the source of the formation of the elastomeric material from the C2 symmetric precursors, deuterium labelled propylene was prepared and was followed by the formation of stereo-errors.42 The use of 2-D-propylene as a substrate revealed that the epimerization mechanism initially proposed by Busico43–45 and Resconi46,47 was not operative for these types of complexes, and a new type of intramolecular epimerization was discovered in which β-D elimination was followed by γ-H elimination, and then rapid reinsertion, resulting in the formation of CDH groups in the polymer (Scheme 5). The result has also been supported by the observation that conducting the reaction at a high pressure resulted in the formation of the stereoregular polypropylene, since at higher monomer concentrations, the rate of the stereospecific monomer insertion becomes faster than the epimerization rate.48
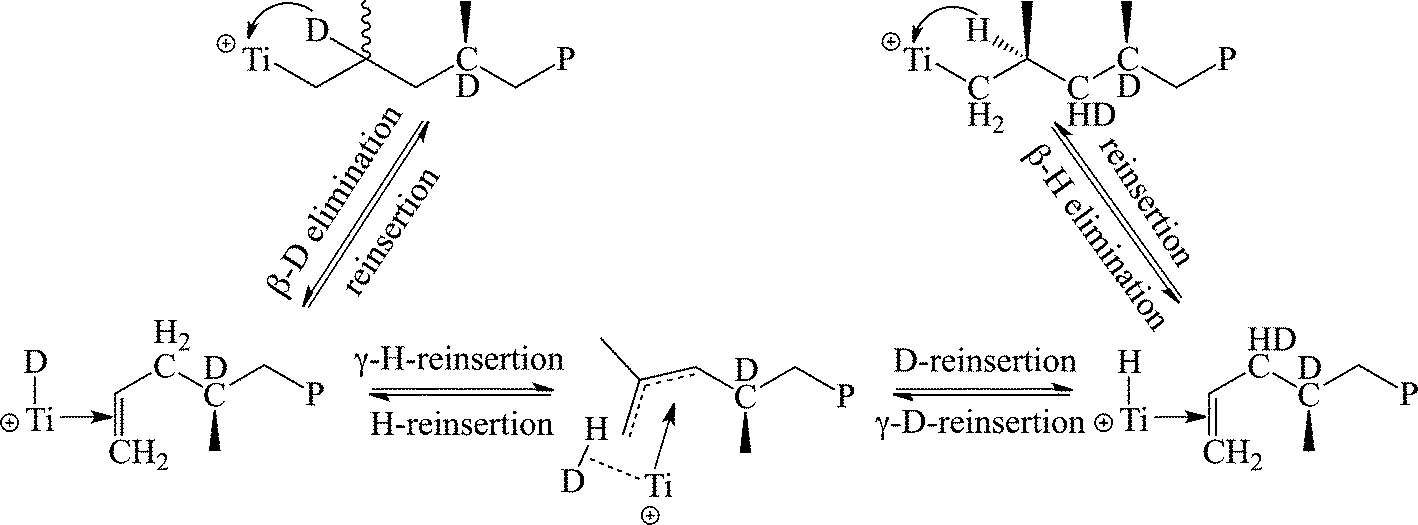 | ||
| Scheme 5 Mechanistic labelling studies of the stereo-error formation in polypropylene during the reaction catalyzed by bis(amidinate) complexes. | ||
The discovery that the bis(benzamidinate) complexes operate under a different catalytic mechanism from the metallocenes motivated the investigation of complexes with various symmetries bearing the benzamidinate ligand (complexes 1–10, Scheme 3). Complexes containing one, two, and three amidinates were prepared and tested for their catalytic activity in the polymerization of α-olefins. The chiral tris(amidinate) zirconium complex 6 and the bis(amidinate) zirconium complex 2 (C3 and C2 symmetry, respectively) generated a highly isotactic material under a high pressure of propylene (>92 mmmm%) as a mixture with an elastomeric material, while the mono(amidinate) zirconium complex 7, as well as the titanium mono(benzamidinate) analogue 8, generated atactic polypropylene. The similarity in the stereoregularity of the resulting polymers, when the C3 and C2 catalysts were used, suggested a similar active species. The dynamic behaviour of the benzamidinate ligand at the metal center caused the formation of a “C2 like” active site when the C3 symmetric precatalyst was used, leading to the formation of isotactic polypropylene. Similar ligand dynamics were found to be operative when C2 symmetric precatalysts were applied, resulting in the formation of a “C1 like” active site to yield an atactic polymer.34
The bis(benzamidinate) titanium dichloride complex 1 and the mono(benzamidinate) complexes (9 and 10) were tested in the polymerization of propylene.49,50 While bis(benzamidinate) titanium dichloride was prepared directly from the reaction of 2 equivalents of the lithiated ligand and 1 equivalent of TiCl4 in toluene, adjusting the ligand equivalents for the synthesis of mono(amidinate) titanium trichloride resulted in the formation of the dimeric complex 9. The mononuclear complex 10 may be easily generated by adding a coordinating solvent such as tetrahydrofuran to the binuclear complex 9.51 When activated with MAO and reacted with propylene, all three complexes yielded the same elastomeric polymers with similar kinetic behaviour, despite the difference in the symmetry of the precatalysts, suggesting that a similar active species is operative in the process.49 Interestingly, and in contrast to these findings, some titanium mono(amidinates) have demonstrated the ability to polymerize propylene with a predominantly syndiospecific stereochemistry at low temperatures (−60 °C, ~53 rrrr%). The elevation of the reaction temperature to 25 °C resulted, in some cases, in the formation of predominantly isotactic polypropylenes.52
To study the effect of the counter ion in the polymerization, the bis(benzamidinate) zirconium dichlorides were converted to the dimethyl analogues by reacting one equivalent of 2 and 3 with 2 equivalents of MeLi. The alkylated compounds were then activated with MAO or B(C6F5)3, and the nature of the co-catalyst was found to influence the stereoselectivity of the polymerization. When MAO was used as the co-catalyst activator, the resulting polymer was an atactic polymer; however, when B(C6F5)3 was utilized, a solid isotactic polypropylene was formed (Scheme 4).53 The reactivity of the active species generated by different co-catalysts has shown their influence not only on the stereoselectivity, but also on the reactivity of the species, demonstrating a lower activity when B(C6F5)3 was used as an activator, and establishing the importance of the counter ion near the cationic active species.
Surprisingly, when a mixture of both co-catalysts MAO and TTPB (trityl tetrakispentafluorophenylborate) (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, respectively) were used to activate the complex [PhC(NTMS)2]2ZrCl2 (2) or its dimethylated analogue, an extraordinary increase in the catalytic activity was observed (433 kg mol−1 h−1 as compared to the trace amounts of polypropylene obtained when activated with only 50 equiv. of MAO), yielding an elastomeric material. It was noteworthy that TTPB was found to react with the [PhC(NTMS)2]2ZrMe2 complex; however, the species formed was inactive in the polymerization of propylene. The methyl group is abstracted by the trityl cation (Ph3C+), and the borate moiety (B(C6F5)4−) of the TTPB serves as the counter ion. EPR experiments revealed that MAO is able to reduce, at this stage, the Zr(IV) to a Zr(III) species. The EPR signal corresponding to the reduced species disappears after the addition of propylene to the reaction mixture, indicating the oxidation of the metal to Zr(IV).54 C60 radical trapping indicated that a radical methyl moiety is abstracted from the metal center when reduced by MAO. Trapping of higher oligomeric chains was not observed by MALDI-TOF; however, when the Ti complex 1 was used, hexyl radicals were observed (see Scheme 6 for the proposed mechanism).
:1, respectively) were used to activate the complex [PhC(NTMS)2]2ZrCl2 (2) or its dimethylated analogue, an extraordinary increase in the catalytic activity was observed (433 kg mol−1 h−1 as compared to the trace amounts of polypropylene obtained when activated with only 50 equiv. of MAO), yielding an elastomeric material. It was noteworthy that TTPB was found to react with the [PhC(NTMS)2]2ZrMe2 complex; however, the species formed was inactive in the polymerization of propylene. The methyl group is abstracted by the trityl cation (Ph3C+), and the borate moiety (B(C6F5)4−) of the TTPB serves as the counter ion. EPR experiments revealed that MAO is able to reduce, at this stage, the Zr(IV) to a Zr(III) species. The EPR signal corresponding to the reduced species disappears after the addition of propylene to the reaction mixture, indicating the oxidation of the metal to Zr(IV).54 C60 radical trapping indicated that a radical methyl moiety is abstracted from the metal center when reduced by MAO. Trapping of higher oligomeric chains was not observed by MALDI-TOF; however, when the Ti complex 1 was used, hexyl radicals were observed (see Scheme 6 for the proposed mechanism).
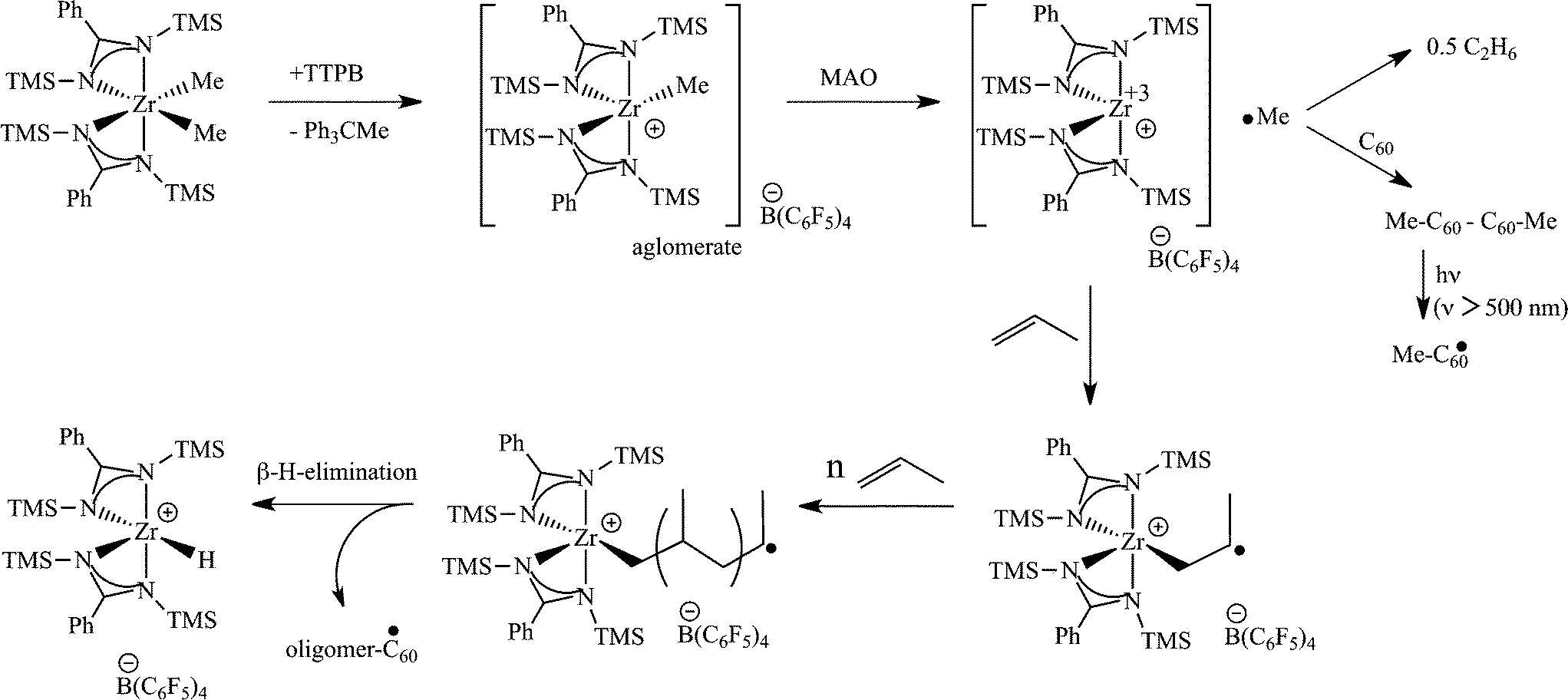 | ||
| Scheme 6 Proposed mechanism for the activation of [PhC(NTMS)2]2ZrMe2 by a mixture of MAO and TTPB. | ||
The importance of the solvent was investigated using complex 1, which revealed that an increase in the solvent polarity leads to a decrease in the molecular weight of the polymers; however, the stereoregularity of the resulting polymer remained around 8–12 mmmm%, regardless of the solvent.55 Complex 11, containing a slightly different aminopyridine ligand framework, was also studied in comparison to the amidine analogues.55,56 Additional aminopyridinate ligands have also been extensively investigated in parallel to the amidinate scaffolds, and comprehensive reviews summarizing the results have been previously published.57–65 However, while discussing the catalytic activity of such frameworks, the complexes were tested mostly in ethylene polymerization and in the co-polymerization of ethylene with propylene.
When complex 11 (Scheme 3) was used as a precatalyst for the polymerization of propylene, lower activities were observed as compared to the corresponding amidinate systems, similar to the behaviour seen in the previously published results.66 Interestingly, complex 11 followed the same trends in catalytic behaviour as its amidinate group 4 analogues.48
The presumed formation of a similar active species during the activation of the benzamidinate complexes (1, 9, 10) called for a deeper mechanistic investigation. The lack of stereospecificity during the polymerization with C2 precursors, and similar behaviour to the C1 precursors led to the conclusion that one of the amidinates dissociates from the metal center and migrates to the aluminium in the MAO (Scheme 7). The assumption was corroborated by NMR analysis of the reaction mixture of 1 with MAO by a systematic comparison to the mono(amidinate) group 4 complexes and respective mono(amidinate) aluminium dimethyl complex.50,67
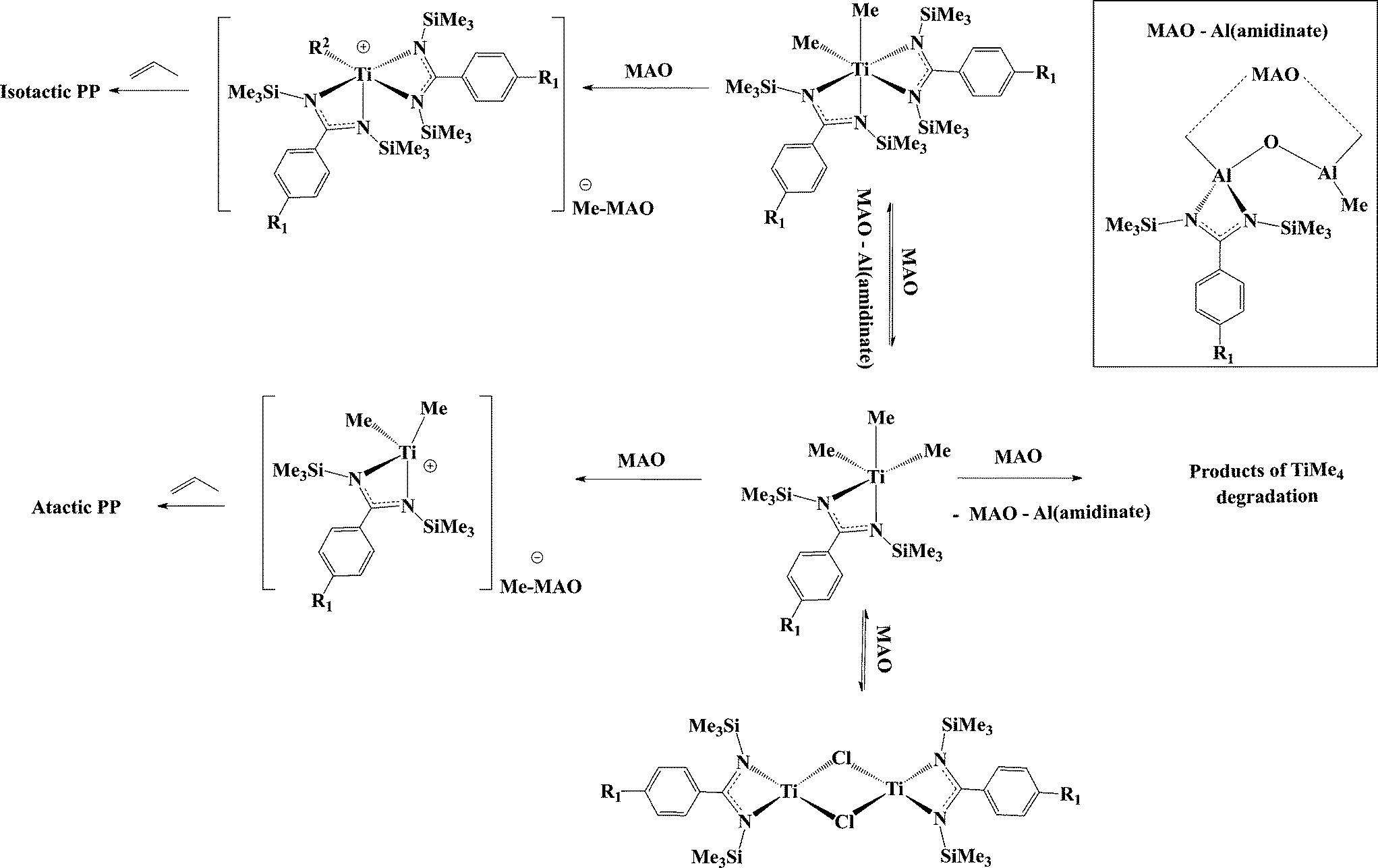 | ||
| Scheme 7 Generation of active species upon the activation of titanium bis(benzamidinates) with MAO, leading to the formation of two polymeric fractions. | ||
When the resulting polymer was fractionated with hot diethyl ether or hexane, two polymeric materials were obtained: a major, soluble atactic elastomer with similar structure to the elastomers described by Resconi,68 and a minor, insoluble isotactic fraction. These results revealed that the cationic bis(benzamidinate)titanium methyl complex is responsible for the formation of the isotactic fraction, whereas the corresponding cationic mono(benzamidinate) titanium species produces the elastomeric material.50
Contrary to the mono(amidinate) systems, some complexes bearing only one ancillary ligand demonstrated stereospecific behaviour towards propylene polymerization. High throughput screening methods have led to the discovery of the hafnium complexes, bearing pyridyl-amido ligands.69 The neutral ligands were reacted with the metal precursor, generating the mono substituted hafnium complex; however, a peculiar behaviour of the ligands has been observed during the reaction of the tris(dimethylamido) complexes to the methylated analogue by the reaction with AlMe3. Instead of the formation of the trimethyl derivative, the metallation of the aryl ring of the ligand was observed (Scheme 8).
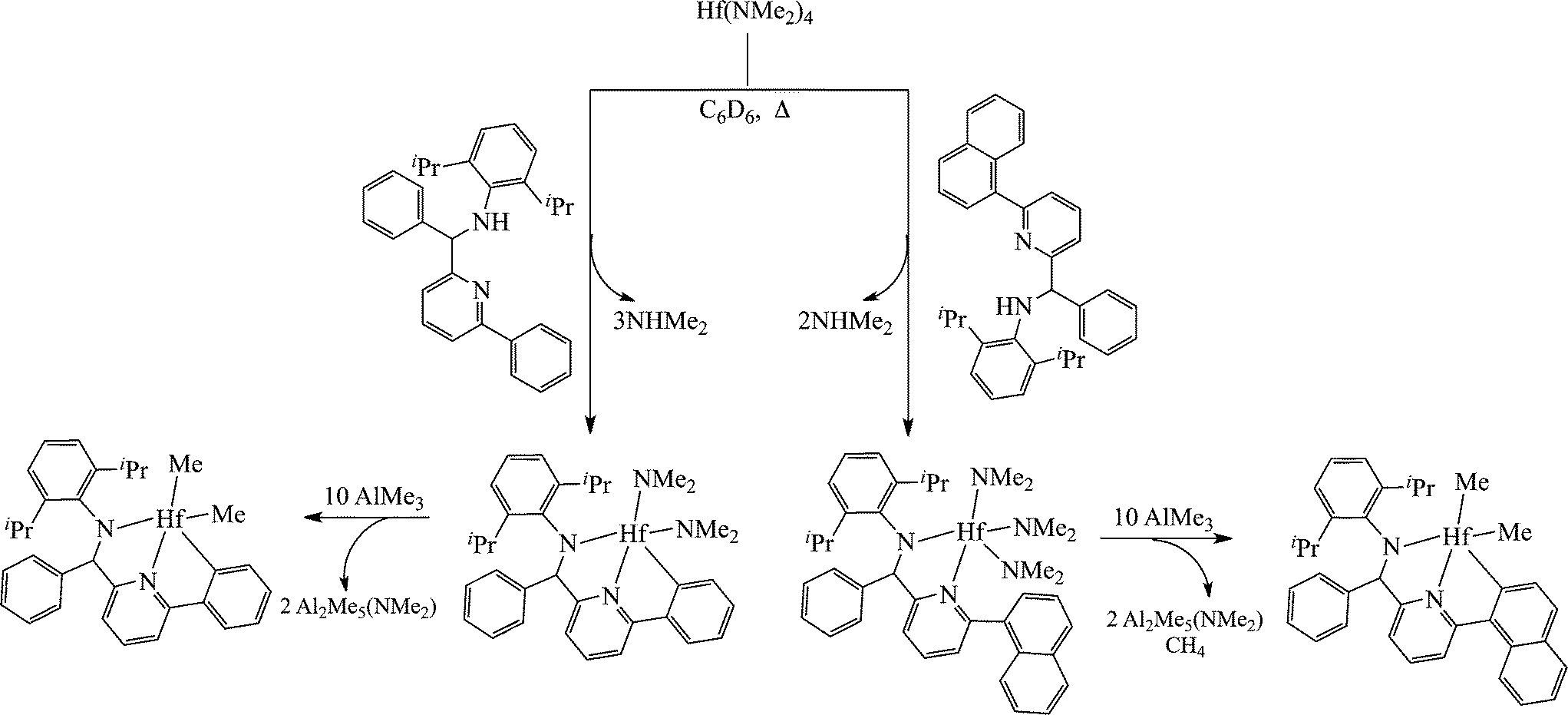 | ||
| Scheme 8 Formation of the mono-substituted hafnium pyridyl-amido complexes. | ||
The resulting mono pyridyl-amido complexes have shown the ability to generate isotactic polypropylene when activated by TTPB or with a combination of [Me2PhNH][B(C6F5)4] and Al(iBu)2H.70 The pyridyl-amido ligands are known to be prone to ortho-metallation of the aryl ring, and have been modified to allow the synthesis of numerous catalysts capable of producing the isotactic polypropylene, ranging between 56 and 94 mmmm%.70,71
Based on the mechanistic data available for benzamidinate complexes, various ligands were studied to find a correlation between the ligand structure and catalyst reactivity, as well as polymer properties. The first approach was to change the substituent on the ipso-carbon of the amidinate. The ligands were prepared following a sigmatropic rearrangement (vide supraScheme 1a), and the lithiated ligands were reacted with TiCl4 to generate complexes 12–21, and with ZrCl4, under the same reaction conditions, to generate complexes 22–27 (Scheme 9).
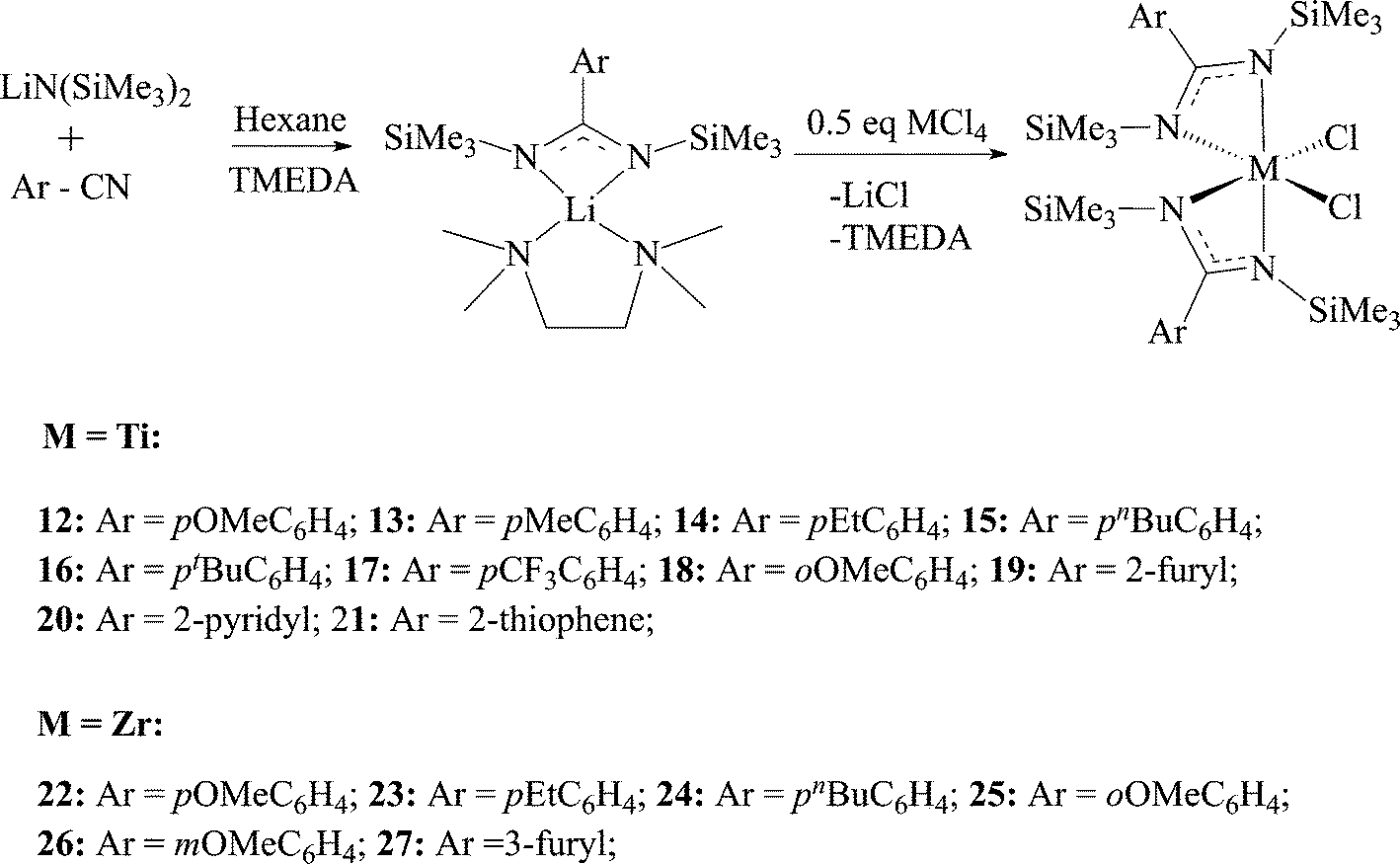 | ||
| Scheme 9 Synthesis of substituted bis(benzamidinate) titanium and zirconium dichloride complexes. | ||
When all the complexes were activated with MAO and reacted with propylene, two polymeric fractions were always generated, albeit in different ratios. For the quantitative comparison between the various complexes, the following equations for the rate of monomer insertion (Ri) (eqn (1)) and the rate of chain termination (Rt) (eqn (2)) were utilized for each complex on all fractions.67
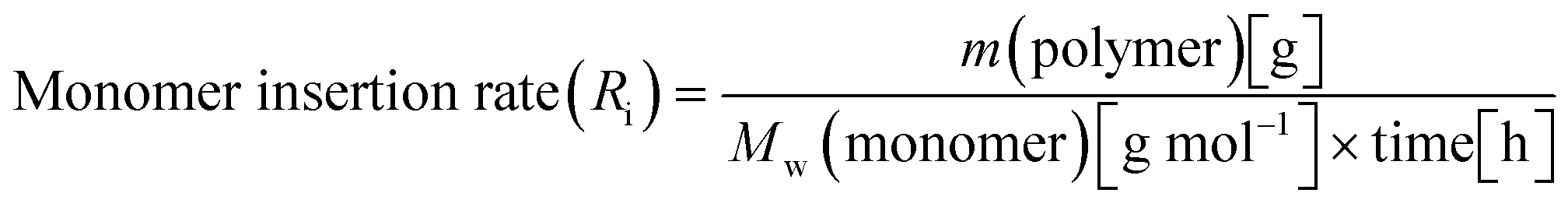 | (1) |
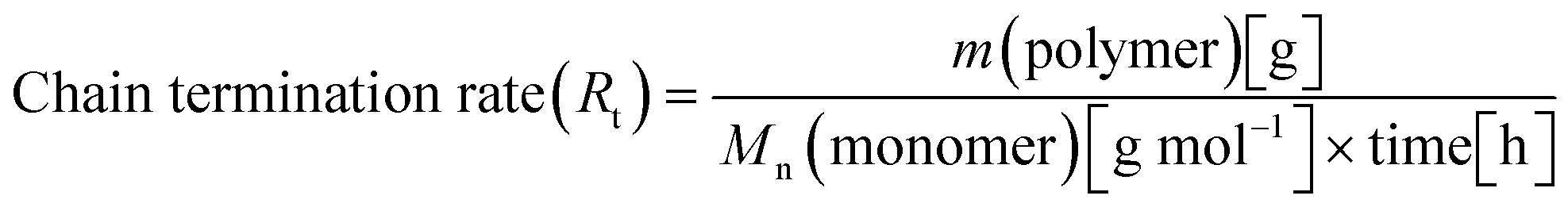 | (2) |
Interestingly, a linear relationship between the Rt of the elastomeric fraction and the steric parameter, Taft (Es), of the aryl substituent (Fig. 1a) at the para-position was revealed.67 These results indicated that the larger para-substituent induced a slower Rt and generates polymers with higher molecular weights. Mechanistic studies indicate that the benzamidinate ligand opens, rotates around the N–C amidinate bond, and disposes the aryl ring closer to the metal center, forming a CGC-type complex and placing the para aryl substituent in close proximity to the metal centre, impeding the chain β-H elimination and allowing for additional growth of the polymer chain. NOE experiments performed with complex 12 shows that the p-OMe substituent of the aromatic ring is located in close proximity to the TMS and the methyl group of MAO (a distance less than 5 Å), corroborating the proposed opening and rearrangement of the ligand (E-syn, Scheme 10).67,72
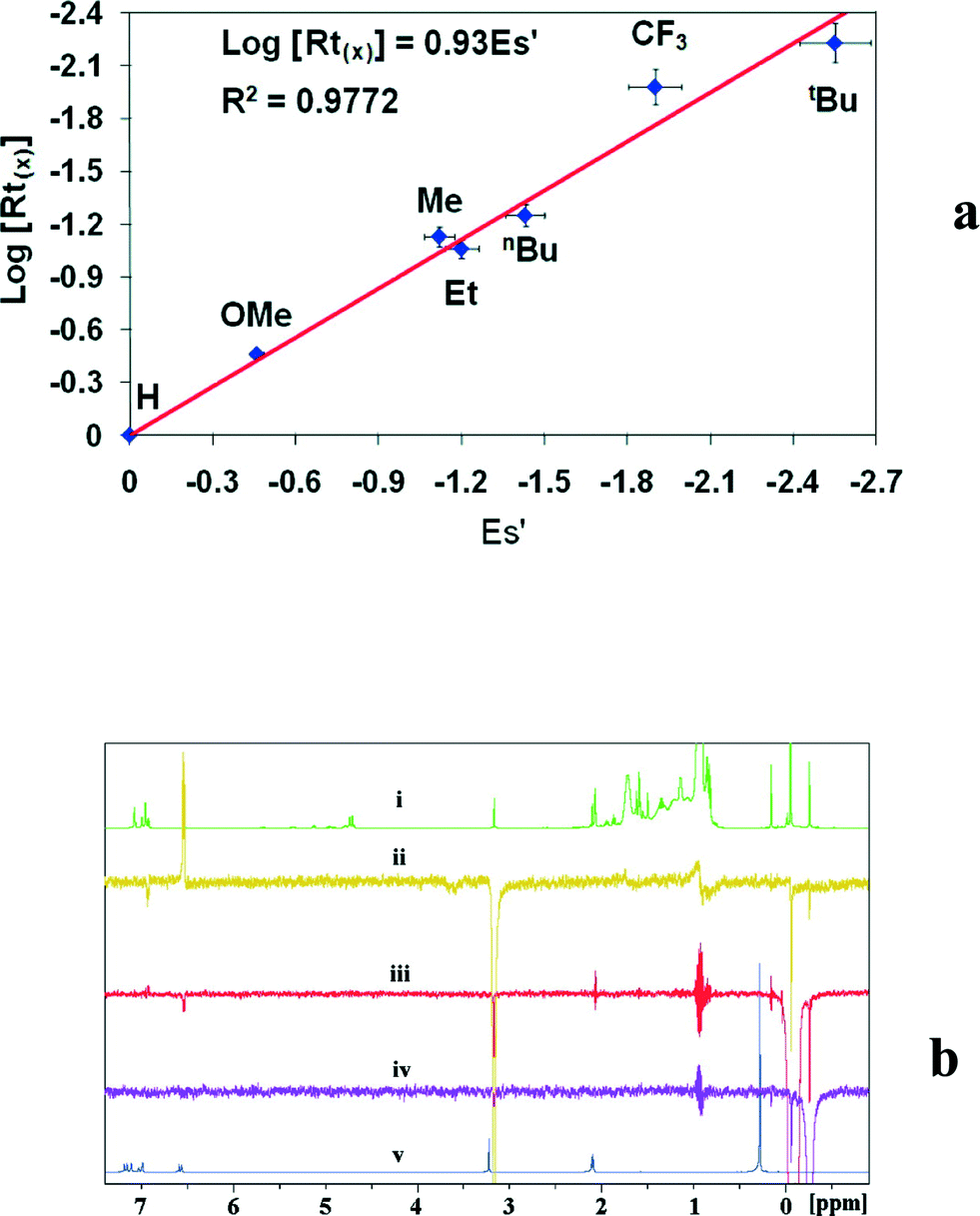 | ||
| Fig. 1 Mechanistic studies of the catalytically active species generated from the bis(benzamidinate) titanium dichloride complex 12: (a) Linear Free Energy Relationship (LFER) between the para substituent of the aryl of the benzamidinate ligand and the rate of the growing chain termination (log(Rt)); (b) 1H NMR monitoring of the catalytic mixture of complex 12 in C7D8: (i) 1H NMR spectrum; (ii–iv) 1H NMR spectra showing the NOE after irradiation of the OMe (3.19 ppm), TMS (0.04 ppm), and MAO (−0.24 ppm) Me signals, respectively; (v) 1H NMR spectrum of complex. “Reprinted with permission from S. Aharonovich, M. Botoshansky, Y. S. Balazs and M. S. Eisen, Organometallics, 2012, 31, 3435–3438. Copyright (2012) American Chemical Society”. | ||
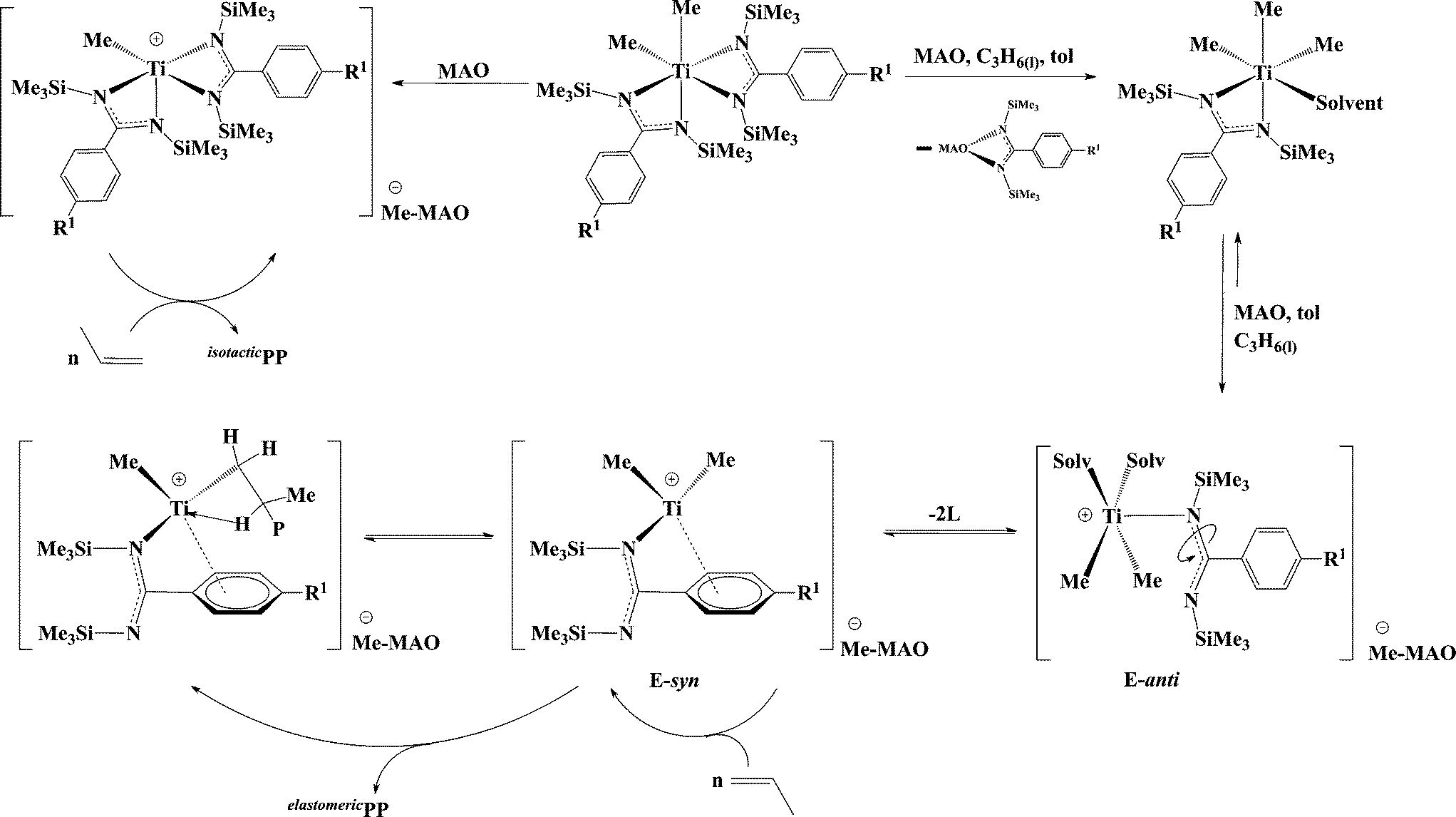 | ||
| Scheme 10 Generation of active species during the polymerization of propylene with bis(benzamidinate) titanium complexes. | ||
An unusual effect was observed when either a heterocyclic or an ortho-methoxy substituted amidinate was used. The opening and rearrangement of the amidinate induces the coordination of the heteroatom to the metal center, allowing the formation of larger metallacycles (5–6 membered rings) that are found to be highly active in the polymerization of propylene (complexes I–III) (Scheme 11).73,74
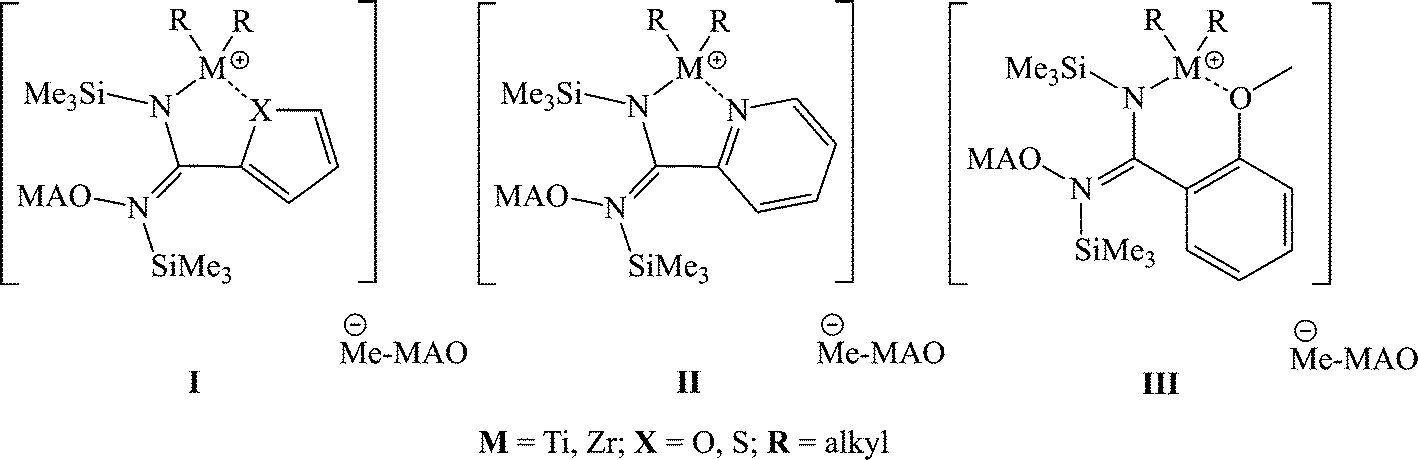 | ||
| Scheme 11 Plausible species formed as a result of the rearrangement of the remaining amidinate ligand in complexes 18–21 and 25. | ||
The results of the selected propylene polymerizations with bis(amidinate) group 4 complexes after activation with 1000 equivalents of MAO are presented in Table 1.
| Complex | Activity kg (PP) mol (M) h | Hexane soluble fraction | Hexane non-soluble fraction | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Weight% | M w × 103 | PD | mmmm% | Weight% | M w × 103 | PD | mmmm% | ||||||
| M | R1 | R2 | R3 | X | |||||||||
| Typical reaction conditions: 1000 equiv. of MAO, 6 mL of toluene, 30 mL of liquid propylene, room temperature.a (I)mmmm pentad by 13C-NMR, (S)rrrr pentad by 13C-NMR.b Not reported.c Active only at 60 °C. | |||||||||||||
| Ti67 | Ph | SiMe3 | Cl | 24.7 | 92.1 | 23 | 2.8 | 11.2 | 7.9 | 103 | 2.9 | 79.3 | |
| Ti67 | pMe-C6H4 | Cl | 15.8 | 86.7 | 122 | 2.9 | 15.5 | 13.3 | 59 | 3.2 | 73.4 | ||
| Ti67 | pEt-C6H4 | Cl | 10.2 | 88.2 | 149 | 3.7 | 12.7 | 11.8 | 66 | 3.2 | 74.5 | ||
| Ti67 | p n Bu-C6H4 | Cl | 9.9 | 83.5 | 188 | 3.6 | 11.2 | 16.5 | 58 | 2.9 | 76.4 | ||
| Ti67 | p t Bu-C6H4 | Cl | 24.0 | 91.8 | 22 | 5.1 | 8.9 | 8.2 | 55 | 2.9 | 73.4 | ||
| Ti67 | pOMe-C6H4 | Cl | 9.8 | 90.8 | 26 | 2.8 | 8.5 | 9.2 | 93 | 2.9 | 75.2 | ||
| Ti72 | pCF3-C6H4 | SiMe3 | Cl | 6.0 | 62.0 | 867 | 2.3 | 9.6 | 38.0 | 560 | 3.2 | 58.6 | |
| Ti72 | oOMe-C6H4 | Cl | 12.2 | 100 | 64 | 2.7 | 5.6 | Traces | — | — | — | ||
| Ti72 | pEt-C6H4 | Cl | 2.6 | 91.6 | 95 | 2.5 | 10.0 | 8.4 | 48 | 2.0 | 73.3 | ||
| Zr73 | Ph | SiMe3 | Cl | 3.4 | 44.1 | 47 | 6.8 | 19.5 | 55.9 | 75 | 6.2 | 63.5(I)a | |
| 48.4(S)a | |||||||||||||
| Zr73 | pOMe-C6H4 | Cl | 9.0 | 52.2 | 15 | 3.0 | 7.9 | 47.8 | 55 | 2.4 | 61.4(I)a | ||
| 53.8(S)a | |||||||||||||
| Zr73 | pEt-C6H4 | Cl | 2.6 | 53.8 | 16 | 2.7 | 15.7 | 46.2 | 108 | 7.7 | 62.0(I)a | ||
| 35.9(S)a | |||||||||||||
| Zr73 | p t Bu-C6H4 | Cl | 2.4 | 50.0 | 82 | 9.2 | 15.9 | 50.0 | 125 | 13.9 | 67.2(I)a | ||
| 49.2(S)a | |||||||||||||
| Zr73 | oOMe-C6H4 | Cl | 5.7 | 63.2 | 3793 | 2.3 | 15.9 | 36.8 | 117 | 14.7 | 71.0(I)a48.7(S)a | ||
| Zr73 | mOMe-C6H4 | Cl | 18 | 76.1 | 15 | 2.6 | 12.6 | 23.9 | 17 | 2.5 | 66.1(I)a | ||
| 40.0(S)a | |||||||||||||
| Zr73 | 3-Furyl | Cl | 12.8 | 72.6 | 51 | 5.12 | 21.6 | 27.4 | 91 | 8.2 | 84.2(I)a | ||
| 42.8(S)a | |||||||||||||
| Ti74 | 2-Furyl | Cl | 35.6 | 90.5 | 42 | 2.1 | 11.9 | 9.5 | 93.8 | 2.8 | 60.3 | ||
| Ti75 | 2-Thiophene | Cl | 32.8 | 80.0 | 303 | 19.4 | 10.2 | 20.0 | 784 | 2.4 | 61.0 | ||
| Ti74 | 2-Pyridyl | Cl | 230 | 96.6 | 226 | 2.6 | 7.7 | 3.4 | 71 | 3.1 | 55.8 | ||
| Ti76 | H | Cyclohexyl | Cyclohexyl | NMe2 | 25.3 | 80.4 | 32 | 2.5 | 10.0 | 19.6 | 196 | 40.0 | |
| Ti76 | H | o-OMe-C6H4 | o-OMe-C6H4 | NMe2 | 9.0 | — | — | — | — | 100 | 821 | 27.0 | |
| Ti76 | Me | o-OMe-C6H4 | o-OMe-C6H4 | NMe2 | 13.3 | 75.4 | 16 | 2.4 | 11.0 | 24.6 | 225 | 54.0 | |
| Ti76 | Et | o-OMe-C6H4 | o-OMe-C6H4 | NMe2 | 7.9 | 70.7 | 24 | 2.7 | 15.0 | 29.3 | 270 | 40.0 | |
| Ti77 | Ph | CH2CH2N(Me2)2 | NMe2 | 2.6 | nrb | nrb | nrb | nrb | 100.0 | 269 | 2.3 | 96.0 | |
| Ti77 | –CH2(2-pyridine) | NMe2 | 13.7 | nrb | nrb | nrb | nrb | 56.0 | 24 | 2.6 | 77.0 | ||
| Ti77 | –CH2(2-furyl) | NMe2 | 30.0 | nrb | nrb | nrb | nrb | 0.0 | 35 | 3.0 | 10.0 | ||
| Ti77 | Ph | C6F5 | –CH2CH2(2-pyridine) | NMe2 | 4.3 | nrb | nrb | nrb | nrb | 57 | 80 | 3.0 | 76.0 |
| Ti78 | iPr | C6F5 | NMe2 | 97.0 | 100.0 | 251 | 8.3 | — | — | — | — | — | |
| Ti78 | iPr | C6H5 | NMe2 | 33.5 | 100.0 | 55 | 3.9 | — | — | — | — | — | |
| Zr78 | iPr | C6F5 | NMe2 | 113.0c | 100.0 | 4.5 | 4.5 | — | — | — | — | — | |
Similar to the titanium complexes, a dynamic behaviour of the amidinate ligands has also been observed for the zirconium benzamidinate complexes. In general, zirconium bis(amidinates) are less reactive, but generate polymers with higher isotacticities as compared to the corresponding titanium analogues. The distribution between the elastomeric and stereospecific fractions for the generated polypropylenes by the zirconium complexes is also found to vary slightly, with increased mass percentages of the stereoregular fraction in the mixture. Surprisingly, analyses of the resulting polymer components have shown that the diethyl ether soluble fraction consisted of the expected atactic material; however, the insoluble fraction contained two products, an isotactic polypropylene formed by the site-control mechanism, and another moderately syndiotactic material formed by a chain control mechanism (Fig. 2).73
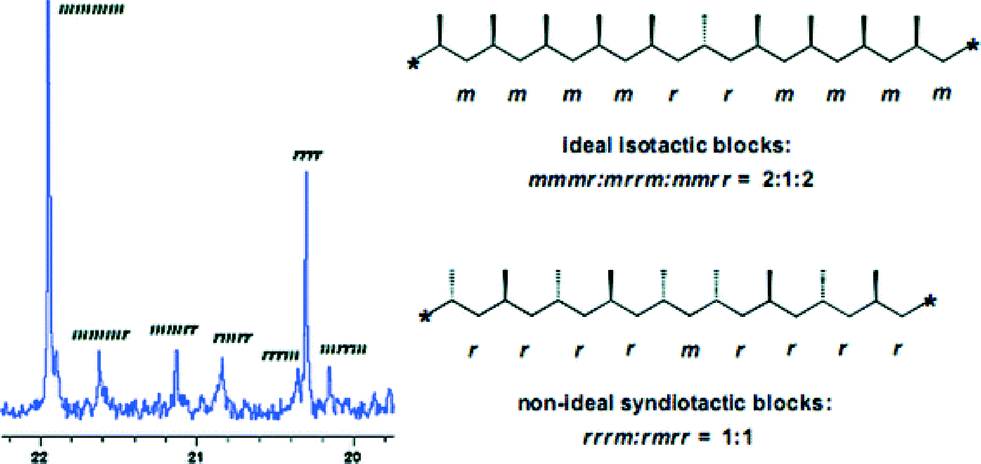 | ||
| Fig. 2 13C NMR spectrum of the methyl region of the hexane insoluble fraction of polypropylenes obtained by zirconium bis(amidinates). “Reproduced from ref. 73 with permission from The Royal Society of Chemistry”. | ||
The attempts to restrain the ligand around the metal center as a chelate were unsuccessful and resulted in polymeric materials as mixtures of the major elastomer and minor (between 10–15% w/w) stereoregular isotactic fraction. In order to eliminate the possibility of rearrangement into the CGC-type species, an aliphatic moiety (iPr) was introduced into the amidine ipso-carbon. The previous work by Fujita et al. with fluorinated FI-catalysts was shown to induce the living polymerization of ethylene, as well as the syndiospecific polymerization of propylene.79–81 These findings inspired the investigation into the use of fluorinated amidinates; these were prepared and introduced as ancillary ligands for group 4 olefin polymerization pre-catalysts. The ligands have been prepared through condensation between isobutyric acid and the appropriate anilines using (polyphosphoric trimethylsilyl ester) PPSE82 as the condensation agent. The ligands were added to Ti(NMe2)4 to generate the corresponding bis(amidinate) titanium bis(dimethylamido) complexes, which were used as polymerization precatalysts in combination with MAO as co-catalysts without any further chemical modification (Scheme 12).
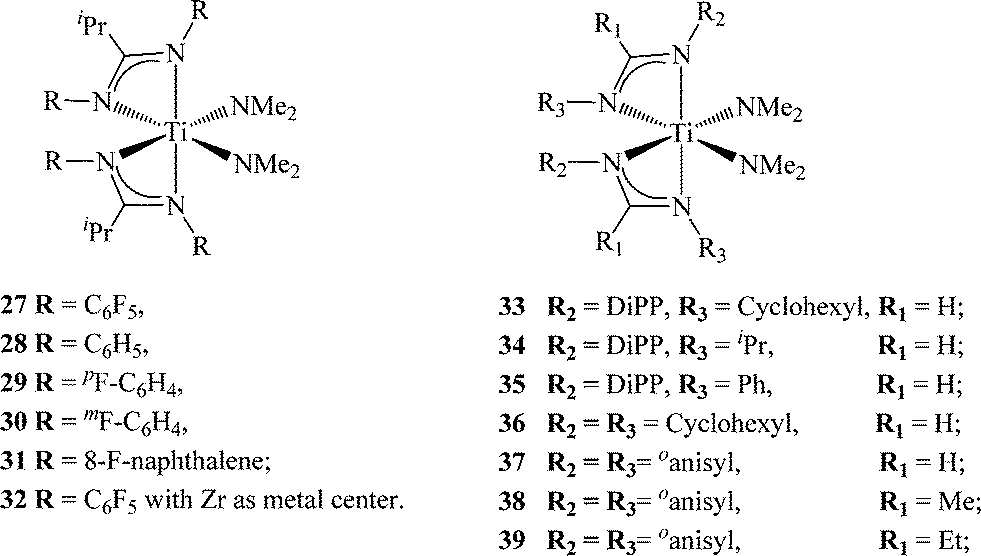 | ||
| Scheme 12 Group 4 amidinates containing electron-withdrawing fluorinated N-substituents, 27–32, and electron-donating N-substituents, 33–39. | ||
The introduction of electron-withdrawing fluorinated amidinates did not lead to similar trends as seen in the fluorinated FI catalysts (living polymerization of olefins,83,84 or the induction of unexpected stereospecificity in the resulting polymer84–86). The fluorinated bis(amidinates), however, demonstrated a different activity towards propylene as compared to other bis(benzamidinates) titanium complexes. The resulting polymer consisted of only one atactic elastomer, suggesting the absence of “C2 symmetry-like” species after activation with MAO; however, both amidinates remain coordinated to the metal center. It was shown that in the presence of electron-withdrawing ligands, the fluorinated bis(amidinate)titanium bis(dimethylamido) complexes exhibited a dynamic behaviour even in their neutral form. The NMR studies revealed that the ligands rearrange from a κ2 to κ1 mode of coordination, leading to an open catalytic site, impeding the stereo-control of the incoming monomer and resulting in the formation of a high molecular weight atactic elastomer. The positions of the fluorine atoms in the ligand were found to influence the Ri and Rt values as compared to the corresponding complex without fluorinated ligands (28). The ligand with only a para-F (complex 29) induces a faster Ri due to the increased electrophilicity of the metal center, whereas the complex containing a meta-F (complex 30) suppresses the Rtvia its interaction with the metal center. Interestingly, the fluorinated zirconium complex 32 showed no reactivity towards propylene at room temperature; however, at 60 °C, the complex exhibits an impressive activity of 113 kg mol−1 h−1 (Table 1), yielding a viscid, atactic, low molecular weight polymer.78
As the use of electron withdrawing groups (EWG) led to more active, but less stereoselective titanium and zirconium complexes compared to the unsubstituted amidinates, the study of electron donating groups (EDG) in the amidinate complexes represents a logical antithesis. It was expected that the introduction of EDGs into the amidinate framework would increase the electron density at the active site, thus decreasing ligand lability and inducing stereospecific polymerization. Cautious selection of the EDG was found prudent in order to minimally alter the steric effects of these new amidinates. For example, the introduction of bulky substituents, such as diisopropylphenyl (DiPP), at both nitrogen positions of the amidinate leads to a lack of reactivity of the ligands (RC(NDiPP)2− = R = Ph, Me, Et, iPr) with many titanium precursors at various ratios. The sterically demanding ligand PhC(NDiPP)2− has been previously reported to react with zirconium tetrachloride, forming the corresponding bis(benzamidinate)zirconium dichloride; however, the amidinates are unusually disposed in a trans fashion due to the steric influence.87 Introduction of two ligands containing the DiPP moiety was possible only when an asymmetric substitution design was utilized, keeping DiPP as the substituent of one nitrogen atom, while using a smaller group (cyclohexyl (33), iPr (34) or Ph (35)) at the second nitrogen.
The propylene polymerization activity of the asymmetric titanium complexes were found to be lower than those containing EWGs. In addition, similar trends in activity and polymer fractions were obtained as compared to the symmetric benzamidinates. Interestingly, the complexes bearing an N-alkyl substituent, such as isopropyl or cyclohexyl, were found to be more active than the corresponding N-aryl-substituted ligand.76 These results encouraged the preparation of the symmetric complex (33) containing two cyclohexyl or o-anisyl groups. Complex 36 was found to be most active among the complexes containing EDGs, allowing the formation of a stereospecific polymer (~20, 40 mmmm%). When the o-anisyl moiety was introduced into the amidinate framework (complexes 37–39), the polymerization behaviour was shown to also be influenced by the substituent at the ipso-carbon position, leading to isotactic polypropylene (up to 54 mmmm%) and high molecular weight polymers (up to ~821000 daltons) (Table 1). The remote location of the ipso-carbon substituent makes its impact on the catalytic behaviour an interesting mechanistic feature. It was shown for complex (39) that the anisyl group is unable to rotate freely, thus allowing the interaction of the methoxy moiety with the metal center, impeding the elimination of the growing chain and forming high molecular weight polymers.
As aforementioned, when the bis(benzamidinate) group 4 complexes are activated with MAO, the metal center undergoes reduction to the M(III) species after the dissociation of a methyl radical, which may be trapped by C60. The reduced metal is oxidized back to the active M(IV) species upon the addition of propylene (Scheme 6). Surprisingly, only the bis(amidinate) complexes bearing fluorinated N-substituents (27, and 29–32) were not reduced by MAO.
Since the immobilization of the ligand by the introduction of EDG was unsuccessful, because the methoxy groups were not coordinated to the metal center, an alkyl moiety was introduced which was connected to an electron donating pendant arm.88,89 The denticity of the ligands has been shown to vary as a function of the central metal atom size, thus increasing the coordination number as the size of the metal center increases.90,91 A number of bis(benzamidinate)titanium bis(dimethylamido) complexes have been prepared containing variable pendant arms (40–43) by reacting two equivalents of the neutral amidines with one equivalent of Ti(NMe2)4. From the X-ray crystallographic analysis of the molecular structures, it was evident that the arm did not coordinate to the metal centre in the neutral form. However, a strong relationship was seen between the nucleophilicity of the arm and the activity of the complexes, and the stereospecificity of the resulting polymers. Complex 40, containing the most nucleophilic moiety NMe2,92 promoted the formation of a highly isotactic polypropylene as a single fraction (Fig. 3) with the lowest yield. Only minor effects were exhibited by the length of the linker of the pendant arm. Pentad analysis of the resulting polymer revealed that the isotacticity is a result of a chain-end mechanism, which has not been previously observed at room temperature.77 When complex 43 was activated by 100 equivalents of MAO, the resulting polymer consisted of an isotactic polypropylene (5 kg mol−1 h−1); however, when the ratio of MAO was increased, the activity of the mixture became much higher, reaching a maximum of 48.3 kg mol−1 h−1 at a cat:MAO ratio of 1:400; however, the product consisted of two fractions: a major isotactic fraction and a minor elastomeric fraction. The use of 1000 equivalents of MAO slightly decreased the activity (43:MAO of 1:400 produced polypropylene with an activity of 48.3 kg mol−1 h−1, and 43:MAO of 1:1000 resulted in the formation of polymer with an activity of 30.0 kg mol−1 h−1), producing only an elastomeric material. The activation of complex 43 with MAO and TTPB (as shown above) resulted in an increased activity by an order of magnitude (230 kg mol−1 h−1); however, the resulting polymer was an atactic elastomer, thus indicating the importance of the counter-ion on the active site and its influence on the activity and stereoselectivity of polymerization (Scheme 13).
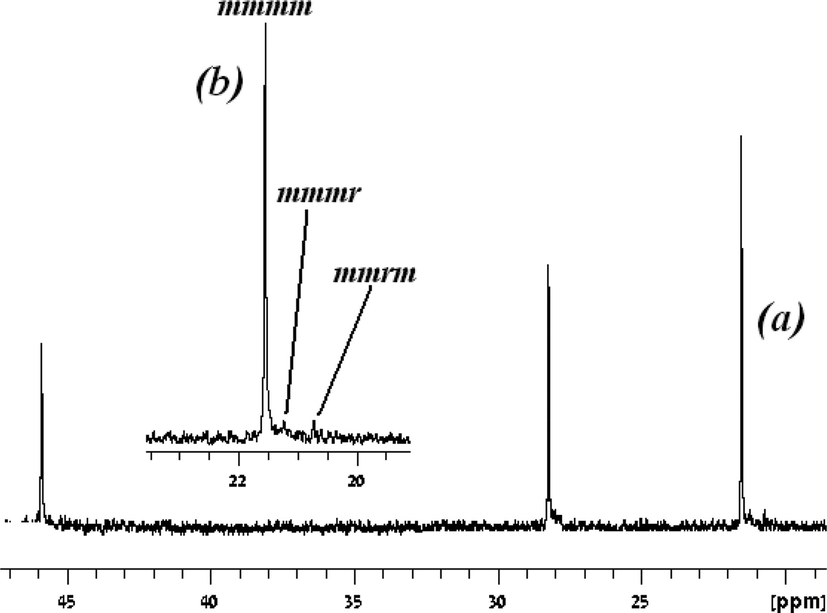 | ||
| Fig. 3
13C-NMR of polypropylene obtained from the catalytic mixture of 43:MAO = 1:1000: (a) full spectrum, and (b) enlarged methyl region. “Reprinted with permission from T. Elkin, M. Botoshansky, R. M. Waymouth, M. S. Eisen, Organometallics, 2014, 33, 840–843. Copyright (2014) American Chemical Society”. | ||
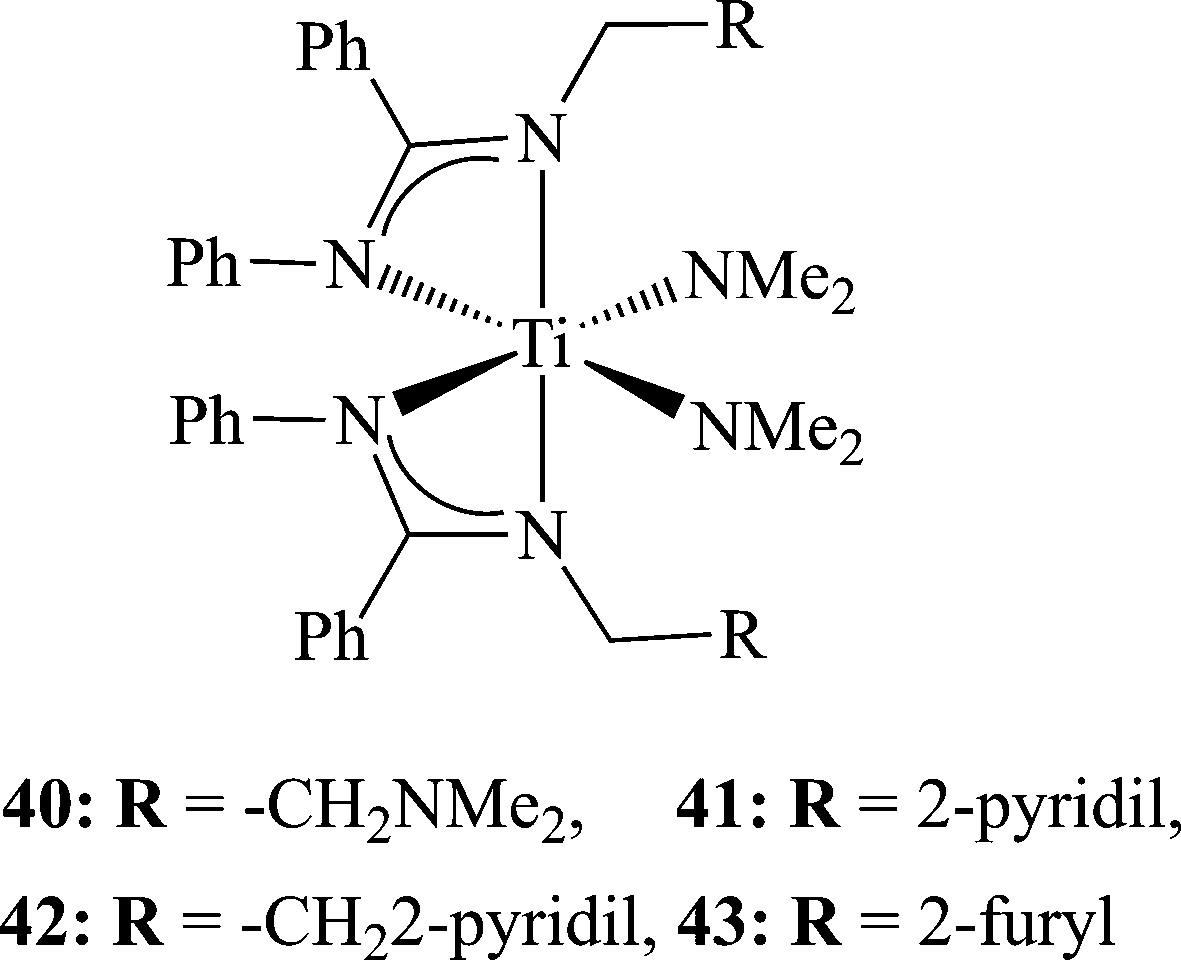 | ||
| Scheme 13 Bis(benzamidinate)titanium bis(dimethylamido) complexes containing electron donating pendant arms. | ||
Another approach for increasing the chelate effect in bis(amidinate) systems has been developed by bridging the two amidinates, analogous to the ansa-metallocenes. Such bridged systems may be approached using two main synthetic methods, sigmatropic rearrangement, leading to systems where amidinates are bridged with a silicon-containing chain, or an all carbon bridging chain formed by a condensation reaction. The first system has been shown to be synthetically challenging, and thus limiting the scope of the resulting complexes, allowing for the formation of only one mononuclear complex incorporating a silyl-bridged ligand around the metal centre and two working ligands (Scheme 14).93–98 Systems containing all carbon linkers have demonstrated more thermal and chemical robustness, and the complexes may be prepared enantiomerically pure.99,100 Catalytic studies with these new types of bridged complexes will be vital for understanding the structure and activity of the bis(amidinate) group 4 complexes as catalysts for olefin polymerization.
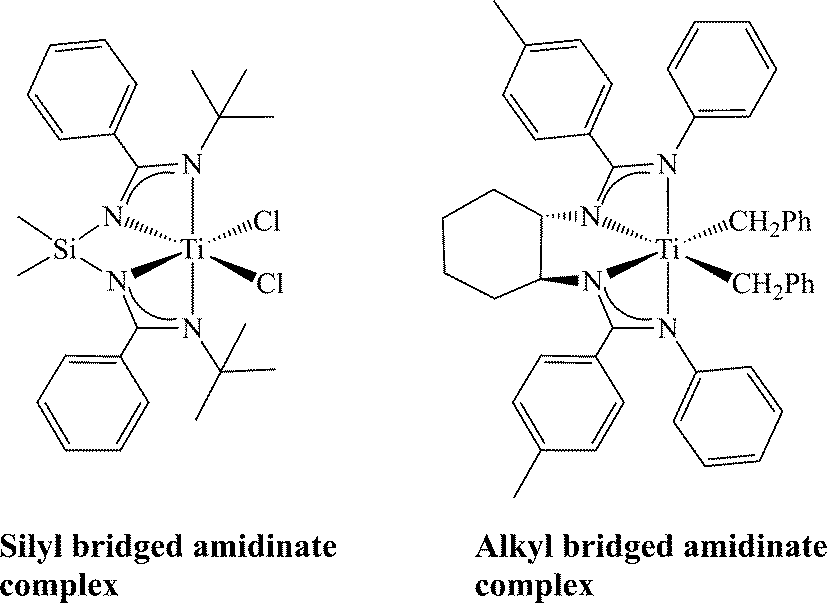 | ||
| Scheme 14 Examples of mononuclear linked bis(amidinate) titanium complexes. | ||
The versatility of the amidinate ligand increases even more when it is combined with other ancillary ligands, such as Cp ligands101–105 and imino-enamido ligands.106 These types of systems have been found to be useful in ethylene polymerization, as well as in the living polymerization of 1-hexene107–109 including co-polymer formations.110 For example, complex 43 demonstrated rather low activity in the polymerization of 1-hexene. After a slight modification of the ligand, complex 44 was shown to form a catalytically active species capable of generating highly isospecific polypropylene. Additionally, the same catalytic mixture containing 44:co-catalysts at 1:1 ratio, bearing amidinates and Cp ligands, serves as catalysts promoting living stereospecific polymerization of various monomers such as ethylene and 1-octene (Scheme 15).
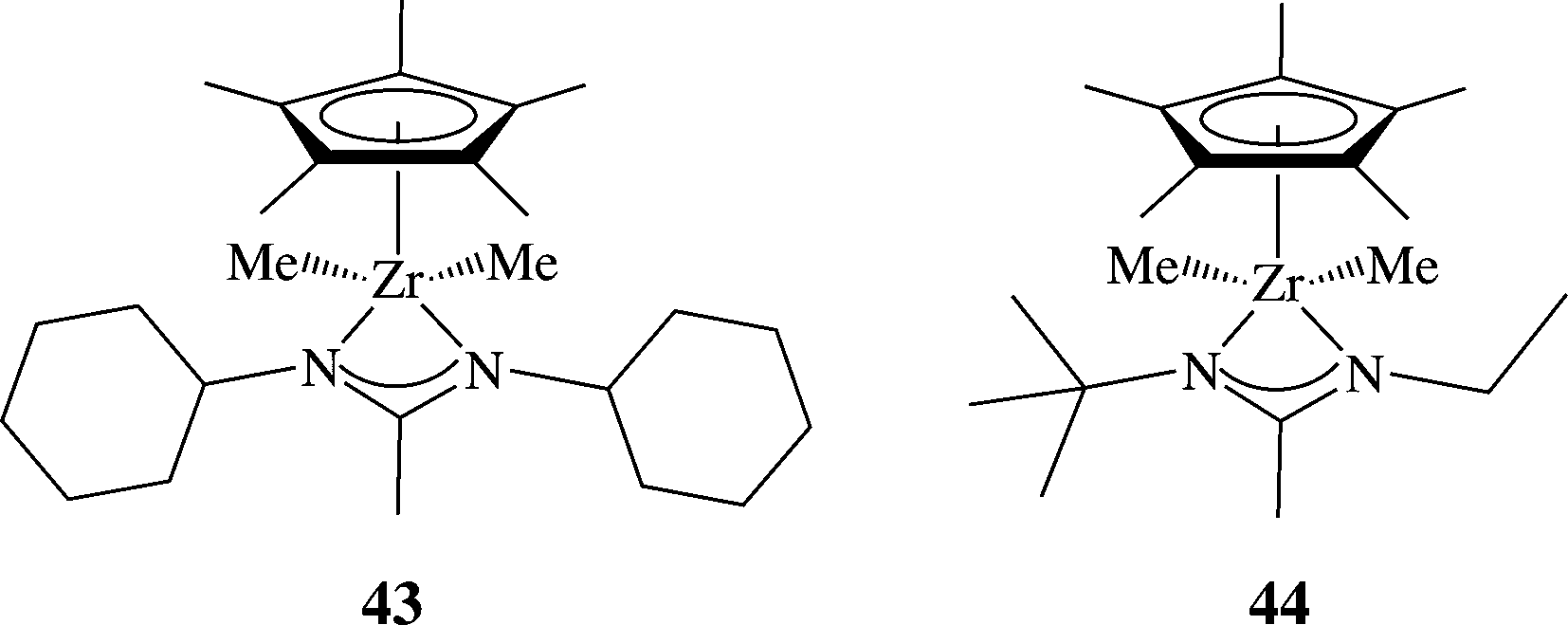 | ||
| Scheme 15 Structure of the mono(amidinate) mono Cp* complexes 43 and 44, promoting the living stereoselective polymerization of α-olefins and stereo-block polypropylene. | ||
When the activation of the mixture was generated by reacting one equivalent of 44 with only 0.5 equivalents of the co-catalyst [Me2PhNH][B(C6F5)4], the polymerization of the polymer resulted in the formation of atactic polypropylene under the conditions of the degenerative transfer mechanism. Due to this special feature, it became possible to produce isotactic-atactic stereo-block polymers with controlled lengths of each block and the whole polymer chain by simply adding more 44 or co-catalyst to the mixture at different times.111
In summary, the group 4 bis(amidinate) complexes and their derivatives, in our opinion, will continue to develop into versatile catalysts with numerous applications. Additionally, the study of the amidinates will inspire and contribute to the development of new ligand frameworks for olefin polymerization such as N,N-dialkylcarbamato complexes that have been recently applied to yield atactic polypropylene.112
Conclusions
Herein, we have presented recent advances in the application of amidinate group 4 complexes, thus revealing the effects of the ligands, metal centers, and various reaction conditions on the resulting polymerization. Simple amidinates demonstrate the ability to rearrange around the metal center and migrate to aluminium, leading to a loss of the stereospecificity of the product. The aromatic substitution on the ipso-carbon leads to the formation of a CGC-type species, inducing steric effects from the p-aryl substituent, and thus leading to an increased molecular weight of the obtained polymers and the reduced activity of the complexes for the larger substituent. The introduction of electron deficient substituents on the nitrogen sites of the ligand results in higher activity as compared to electron-donating moieties; however, the stereospecificity of the polymer for the former is completely lost. The immobility of the ligand was achieved when electron donating pendant arms were introduced into the ligand framework, resulting in the formation of isotactic polypropylene as a single product for the complexes bearing the most nucleophilic arm.Acknowledgements
This research was supported by the USA-Israel Binational Science Foundation under contract 2010109.Notes and references
- K. Ziegler, E. Holzkamp, H. Breil and H. Martin, Angew. Chem., 1955, 67, 541–547 CrossRef CAS.
- G. Natta, P. Pino, P. Corradini, F. Danusso, E. Mantica, G. Mazzanti and G. Moraglio, J. Am. Chem. Soc., 1955, 77, 1708–1710 CrossRef CAS.
- P. D. Hustad, Science, 2009, 325, 704–707 CrossRef CAS PubMed.
- R. C. Klet, C. N. Theriault, J. Klosin, J. A. Labinger and J. E. Bercaw, Macromolecules, 2014, 47, 3317–3324 CrossRef CAS.
- D. S. Breslow and N. R. Newburg, J. Am. Chem. Soc., 1957, 79, 5072–5073 CrossRef CAS.
- G. Natta, P. Pino, G. Mazzanti and U. Giannini, J. Am. Chem. Soc., 1957, 79, 2975–2976 CrossRef CAS.
- H. Sinn, W. Kaminsky, H.-J. Vollmer and R. Woldt, Angew. Chem., 1980, 92, 396–402 CrossRef CAS.
- W. Kaminsky, Macromol. Chem. Phys., 1996, 197, 3907–3945 CrossRef CAS.
- W. Kaminsky, Catal. Today, 2000, 62, 23–34 CrossRef CAS.
- J. A. Ewen, J. Mol. Catal. A: Chem., 1998, 128, 103–109 CrossRef CAS.
- C. Gerhardt, Liebigs Ann. Chem., 1858, 108, 214–223 CrossRef.
- A. R. Sanger, Inorg. Nucl. Chem. Lett., 1973, 9, 351–354 CrossRef CAS.
- R. T. Boeré, R. T. Oakley and R. W. Reed, J. Organomet. Chem., 1987, 331, 161–167 CrossRef.
- J. Barker and M. Kilner, Coord. Chem. Rev., 1994, 133, 219–300 CrossRef CAS.
- F. T. Edelmann, Coord. Chem. Rev., 1994, 137, 403–481 CrossRef.
- A. R. Katritzky, O. Meth-Cohn, C. J. Moody and C. W. Rees, Comprehensive Organic Functional Group Transformations: Synthesis: carbon with two attached heteroatoms with at least one carbon-to-heteroatom multiple link, Elsevier, 1995 Search PubMed.
- F. T. Edelmann, Chem. Soc. Rev., 2012, 41, 7657–7672 RSC.
- F. Edelmann, in Molecular Catalysis of Rare-Earth Elements, ed. P. W. Roesky, Springer, Berlin Heidelberg, 2010, vol. 137, pp. 109–163 Search PubMed.
- M. Wedler, F. Knösel, M. Noltemeyer, F. T. Edelmann and U. Behrens, J. Organomet. Chem., 1990, 388, 21–45 CrossRef CAS.
- M. Wedler, H. W. Roesky and F. Edelmann, J. Organomet. Chem., 1988, 345, C1–C3 CrossRef CAS.
- S. Aharonovich, M. Kapon, M. Botoshanski and M. S. Eisen, Organometallics, 2008, 27, 1869–1877 CrossRef CAS.
- G. Glatz, S. Demeshko, G. Motz and R. Kempe, Eur. J. Inorg. Chem., 2009, 1385–1392 CrossRef CAS.
- S. Aharonovich, M. Botoshanski, Z. Rabinovich, R. M. Waymouth and M. S. Eisen, Inorg. Chem., 2010, 49, 1220–1229 CrossRef CAS PubMed.
- M. Krasnopolski, R. W. Seidel, R. Goddard, J. Breidung, M. V. Winter, A. Devi and R. A. Fischer, J. Mol. Struct., 2013, 1031, 239–245 CrossRef CAS.
- J. Richter, J. Feiling, H.-G. Schmidt, M. Noltemeyer, W. Brüser and F. T. Edelmann, Z. Anorg. Allg. Chem., 2004, 630, 1269–1275 CrossRef CAS.
- C. E. Rodrigues-Santos, L. L. Leon, A. J. Bortoluzzi, M. M. Canto-Cavalheiro, G. C. Machado and A. Echevarria, Eur. J. Med. Chem., 2013, 67, 166–174 CrossRef CAS PubMed.
- M. M. A. C. C. F. J.-A. Gautier, The chemistry of the amidines and imidates, Wiley, London, 1975 Search PubMed.
- G. V. Boyd, in The chemistry of the amidines and imidates, ed. S. Patai and Z. Rappoport, Wiley, Chichester, 1991, ch. 7, vol. 2, pp. 339–366 Search PubMed.
- R. T. Boere, V. Klassen and G. Wolmershauser, J. Chem. Soc., Dalton Trans., 1998, 4147–4154 RSC.
- H. W. Roesky, B. Meller, M. Noltemeyer, H.-G. Schmidt, U. Scholz and G. M. Sheldrick, Chem. Ber., 1988, 121, 1403–1406 CrossRef CAS.
- D. G. Dick, J. J. H. Edema, R. Duchateau and S. Gambarotta, Inorg. Chem., 1993, 32, 1959–1962 CrossRef CAS.
- J. R. Hagadorn and J. Arnold, Organometallics, 1994, 13, 4670–4672 CrossRef CAS.
- D. Herskovics-Korine and M. S. Eisen, J. Organomet. Chem., 1995, 503, 307–314 CrossRef CAS.
- A. Littke, N. Sleiman, C. Bensimon, D. S. Richeson, G. P. A. Yap and S. J. Brown, Organometallics, 1998, 17, 446–451 CrossRef CAS.
- S. Collins, Coord. Chem. Rev., 2011, 255, 118–138 CrossRef CAS.
- G. W. Coates, Chem. Rev., 2000, 100, 1223–1252 CrossRef CAS PubMed.
- J. Richter, F. T. Edelmann, M. Noltemeyer, H.-G. Schmidt, M. Shmulinson and M. S. Eisen, J. Mol. Catal. A: Chem., 1998, 130, 149–162 CrossRef CAS.
- V. Volkis, M. Shmulinson, C. Averbuj, A. Lisovskii, F. T. Edelmann and M. S. Eisen, Organometallics, 1998, 17, 3155–3157 CrossRef CAS.
- M. Shmulinson, V. Volkis, A. Lisovskii, E. Nelkenbaum and M. S. Eisen, Polym. Adv. Technol., 2002, 13, 823–829 CrossRef CAS.
- C. Averbuj and M. S. Eisen, J. Am. Chem. Soc., 1999, 121, 8755–8759 CrossRef CAS.
- G. W. Coates and R. M. Waymouth, Science, 1995, 267, 217–219 CrossRef CAS PubMed.
- V. Volkis, M. Rodensky, A. Lisovskii, Y. Balazs and M. S. Eisen, Organometallics, 2006, 25, 4934–4937 CrossRef CAS.
- V. Busico and R. Cipullo, J. Am. Chem. Soc., 1994, 116, 9329–9330 CrossRef CAS.
- V. Busico, L. Caporaso, R. Cipullo, L. Landriani, G. Angelini, A. Margonelli and A. L. Segre, J. Am. Chem. Soc., 1996, 118, 2105–2106 CrossRef CAS.
- V. Busico, R. Cipullo, F. Cutillo and M. Vacatello, Macromolecules, 2001, 35, 349–354 CrossRef.
- L. Resconi, J. Mol. Catal. A: Chem., 1999, 146, 167–178 CrossRef CAS.
- G. Moscardi, L. Resconi and L. Cavallo, Organometallics, 2001, 20, 1918–1931 CrossRef CAS.
- E. Smolensky and M. S. Eisen, Dalton Trans., 2007, 5623–5650 RSC.
- C. Averbuj, E. Tish and M. S. Eisen, J. Am. Chem. Soc., 1998, 120, 8640–8646 CrossRef CAS.
- V. Volkis, A. Lisovskii, B. Tumanskii, M. Shuster and M. S. Eisen, Organometallics, 2006, 25, 2656–2666 CrossRef CAS.
- J. C. Flores, J. C. W. Chien and M. D. Rausch, Organometallics, 1995, 14, 1827–1833 CrossRef CAS.
- D. Liguori, F. Grisi, I. Sessa and A. Zambelli, Macromol. Chem. Phys., 2003, 204, 164–170 CrossRef CAS.
- V. Volkis, E. Nelkenbaum, A. Lisovskii, G. Hasson, R. Semiat, M. Kapon, M. Botoshansky, Y. Eishen and M. S. Eisen, J. Am. Chem. Soc., 2003, 125, 2179–2194 CrossRef CAS PubMed.
- V. Volkis, B. Tumanskii and M. S. Eisen, Organometallics, 2006, 25, 2722–2724 CrossRef CAS.
- V. Volkis, E. Smolensky, A. Lisovskii and M. S. Eisen, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4505–4516 CrossRef CAS.
- E. Smolensky, M. Kapon, J. D. Woollins and M. S. Eisen, Organometallics, 2005, 24, 3255–3265 CrossRef CAS.
- H. Fuhrmann, S. Brenner, P. Arndt and R. Kempe, Inorg. Chem., 1996, 35, 6742–6745 CrossRef CAS PubMed.
- R. Kempe and P. Arndt, Inorg. Chem., 1996, 35, 2644–2649 CrossRef CAS PubMed.
- R. Kempe, Eur. J. Inorg. Chem., 2003, 791–803 CrossRef CAS.
- W. P. Kretschmer, B. Hessen, A. Noor, N. M. Scott and R. Kempe, J. Organomet. Chem., 2007, 692, 4569–4579 CrossRef CAS.
- A. Noor, W. P. Kretschmer, G. Glatz, A. Meetsma and R. Kempe, Eur. J. Inorg. Chem., 2008, 5088–5098 CrossRef CAS.
- M. Hafeez, W. P. Kretschmer and R. Kempe, Eur. J. Inorg. Chem., 2011, 2011, 5512–5522 CrossRef CAS.
- A. Noor, W. P. Kretschmer, G. Glatz and R. Kempe, Inorg. Chem., 2011, 50, 4598–4606 CrossRef CAS PubMed.
- I. Haas, T. Dietel, K. Press, M. Kol and R. Kempe, Chem. – Eur. J., 2013, 19, 14254–14262 CrossRef CAS PubMed.
- X.-E. Duan, S.-F. Yuan, H.-B. Tong, S.-D. Bai, X.-H. Wei and D.-S. Liu, Dalton Trans., 2012, 41, 9460–9467 RSC.
- C. Morton, P. O'Shaughnessy and P. Scott, Chem. Commun., 2000, 2099–2100 RSC.
- S. Aharonovich, V. Volkis and M. S. Eisen, Macromol. Symp., 2007, 260, 165–171 CrossRef CAS.
- L. Resconi, R. L. Jones, A. L. Rheingold and G. P. A. Yap, Organometallics, 1996, 15, 998–1005 CrossRef CAS.
- T. R. Boussie, G. M. Diamond, C. Goh, K. A. Hall, A. M. LaPointe, M. K. Leclerc, V. Murphy, J. A. W. Shoemaker, H. Turner, R. K. Rosen, J. C. Stevens, F. Alfano, V. Busico, R. Cipullo and G. Talarico, Angew. Chem., Int. Ed., 2006, 45, 3278–3283 CrossRef CAS PubMed.
- G. M. Diamond, K. A. Hall, A. M. LaPointe, M. K. Leclerc, J. Longmire, J. A. W. Shoemaker and P. Sun, ACS Catal., 2011, 1, 887–900 CrossRef CAS.
- K. A. Frazier, R. D. Froese, Y. He, J. Klosin, C. N. Theriault, P. C. Vosejpka, Z. Zhou and K. A. Abboud, Organometallics, 2011, 30, 3318–3329 CrossRef CAS.
- S. Aharonovich, M. Botoshansky, Y. S. Balazs and M. S. Eisen, Organometallics, 2012, 31, 3435–3438 CrossRef CAS.
- S. Aharonovich, N. V. Kulkarni, J.-S. Zhang, M. Botoshansky, M. Kapon and M. S. Eisen, Dalton Trans., 2013, 42, 16762–16772 RSC.
- S. Aharonovich, Hetero-aza-allyl complexes of Li, Ti, Zr, and V: structure, reactivity, and catalytic propylene polymerization, Technion - Israel Institute of Technology, 2010 Search PubMed.
- E. Rabinovich, Organoactinide aryl-amidinate complexes, Technion - Israel Institute of Technology, 2010 Search PubMed.
- T. Elkin, N. V. Kulkarni, B. Tumanskii, M. Botoshansky, L. J. W. Shimon and M. S. Eisen, Organometallics, 2013, 32, 6337–6352 CrossRef CAS.
- T. Elkin, M. Botoshansky, R. M. Waymouth and M. S. Eisen, Organometallics, 2014, 33, 840–843 CrossRef CAS.
- T. Elkin, S. Aharonovich, M. Botoshansky and M. S. Eisen, Organometallics, 2012, 31, 7404–7414 CrossRef CAS.
- K. Kawai and T. Fujita, in Metal Catalysts in Olefin Polymerization, ed. Z. Guan, Springer, Berlin Heidelberg, 2009, vol. 26, pp. 3–46 Search PubMed.
- M. Mitani, J. Saito, S.-I. Ishii, Y. Nakayama, H. Makio, N. Matsukawa, S. Matsui, J.-I. Mohri, R. Furuyama, H. Terao, H. Bando, H. Tanaka and T. Fujita, Chem. Rec., 2004, 4, 137–158 CrossRef CAS PubMed.
- H. Makio and T. Fujita, Bull. Chem. Soc. Jpn., 2005, 78, 52–66 CrossRef CAS.
- S.-I. Ogata, A. Mochizuki, M.-A. Kakimoto and Y. Imai, Bull. Chem. Soc. Jpn., 1986, 59, 2171–2177 CrossRef CAS.
- S. Guo, S. Jie, H. Fan, J. Weng, W. Liu and B.-G. Li, J. Appl. Polym. Sci., 2013, 129, 1971–1977 CrossRef CAS.
- R. Furuyama, J. Saito, S. Ishii, H. Makio, M. Mitani, H. Tanaka and T. Fujita, J. Organomet. Chem., 2005, 690, 4398–4413 CrossRef CAS.
- H. Makio and T. Fujita, Acc. Chem. Res., 2009, 42, 1532–1544 CrossRef CAS PubMed.
- J. Saito, M. Mitani, M. Onda, J. Mohri, S. Ishii, Y. Yoshida, R. Furuyama, T. Nakano, N. Kashiwa and T. Fujita, in Studies in Surface Science and Catalysis, ed. M. O. Masakazu Anpo and Y. Hiromi, Elsevier, 2003, vol. 145, pp. 515–516 Search PubMed.
- E. Otten, P. Dijkstra, C. Visser, A. Meetsma and B. Hessen, Organometallics, 2005, 24, 4374–4386 CrossRef CAS.
- K.-M. Wu, C.-A. Huang, K.-F. Peng and C.-T. Chen, Tetrahedron, 2005, 61, 9679–9687 CrossRef CAS.
- C. L. Boyd, A. E. Guiducci, S. R. Dubberley, B. R. Tyrrell and P. Mountford, J. Chem. Soc., Dalton Trans., 2002, 4175–4184 RSC.
- K. Kincaid, C. P. Gerlach, G. R. Giesbrecht, J. R. Hagadorn, G. D. Whitener, A. Shafir and J. Arnold, Organometallics, 1999, 18, 5360–5366 CrossRef CAS.
- M. J. R. Brandsma, E. A. C. Brussee, A. Meetsma, B. Hessen and J. H. Teuben, Eur. J. Inorg. Chem., 1998, 1998, 1867–1870 CrossRef.
- G. Berionni, B. Maji, P. Knochel and H. Mayr, Chem. Sci., 2012, 3, 878–882 RSC.
- S.-D. Bai, H.-B. Tong, J.-P. Guo, M.-S. Zhou and D.-S. Liu, Inorg. Chim. Acta, 2009, 362, 1143–1148 CrossRef CAS.
- S.-D. Bai, H.-B. Tong, J.-P. Guo, M.-S. Zhou, D.-S. Liu and S.-F. Yuan, Polyhedron, 2010, 29, 262–269 CrossRef CAS.
- S. D. Bai, F. Guan, M. Hu, S. F. Yuan, J. P. Guo and D. S. Liu, Dalton Trans., 2011, 40, 7686–7688 RSC.
- S.-D. Bai, R.-Q. Liu, T. Wang, F. Guan, Y.-B. Wu, J.-B. Chao, H.-B. Tong and D.-S. Liu, Polyhedron, 2013, 65, 161–169 CrossRef CAS.
- T. Wang, J.-P. Zhao and S.-D. Bai, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, m654 CAS.
- S.-D. Bai, R.-Q. Liu, T. Wang, F. Guan, Y.-B. Wu, J.-B. Chao, H.-B. Tong and D.-S. Liu, Polyhedron, 2013, 65, 161–169 CrossRef CAS.
- T. S. Brunner, L. Hartenstein and P. W. Roesky, J. Organomet. Chem., 2013, 730, 32–36 CrossRef CAS.
- J. R. Hagadorn and J. Arnold, Angew. Chem., Int. Ed., 1998, 37, 1729–1731 CrossRef CAS.
- L. R. Sita and J. R. Babcock, Organometallics, 1998, 17, 5228–5230 CrossRef CAS.
- K. C. Jayaratne and L. R. Sita, J. Am. Chem. Soc., 2000, 122, 958–959 CrossRef CAS.
- K. C. Jayaratne and L. R. Sita, J. Am. Chem. Soc., 2001, 123, 10754–10755 CrossRef CAS PubMed.
- R. J. Keaton, L. A. Koterwas, J. C. Fettinger and L. R. Sita, J. Am. Chem. Soc., 2002, 124, 5932–5933 CrossRef CAS PubMed.
- D. A. Kissounko and L. R. Sita, J. Am. Chem. Soc., 2004, 126, 5946–5947 CrossRef CAS PubMed.
- J. Klosin, P. P. Fontaine, R. Figueroa, S. D. McCann and D. Mort, Organometallics, 2013, 32, 6488–6499 CrossRef CAS.
- D. A. Kissounko, J. C. Fettinger and L. R. Sita, Inorg. Chim. Acta, 2003, 345, 121–129 CrossRef CAS.
- Y. Zhang, E. K. Reeder, R. J. Keaton and L. R. Sita, Organometallics, 2004, 23, 3512–3520 CrossRef CAS.
- M. B. Harney, R. J. Keaton, J. C. Fettinger and L. R. Sita, J. Am. Chem. Soc., 2006, 128, 3420–3432 CrossRef CAS PubMed.
- Y. Zhang, R. J. Keaton and L. R. Sita, J. Am. Chem. Soc., 2003, 125, 9062–9069 CrossRef CAS PubMed.
- M. B. Harney, Y. Zhang and L. R. Sita, Angew. Chem., Int. Ed., 2006, 45, 2400–2404 CrossRef CAS PubMed.
- M. Hayatifar, C. Forte, G. Pampaloni, Y. V. Kissin, A. Maria Raspolli Galletti and S. Zacchini, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4095–4102 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |