
Perspective on CO oxidation over Pd-based catalysts
You
Zhou
,
Zongyuan
Wang
and
Changjun
Liu
*
Collaborative Innovation Center of Chemical Science and Engineering, School of Chemical Engineering and Technology, Tianjin University, Tianjin 300072, China. E-mail: ughg_cjl@yahoo.com; Fax: +86 22 27406490; Tel: +86 22 27406490
First published on 29th September 2014
Abstract
CO oxidation is one of the most extensively investigated reactions in the field of heterogeneous catalysis because of its importance in both environmental protection and fundamental studies. CO oxidation over Pd catalysts has attracted significant attention not only because of its high activity. It is an excellent model reaction for the study of the structure, metal–support interaction and other issues of various Pd-based catalysts with many important applications beyond CO oxidation. It is simple and can be easily operated with low cost. Density functional theoretical studies can be directly and easily combined with experimental investigations. Carbon monoxide itself is an excellent probe molecule for catalyst characterization. In this perspective, we summarize the progress in the study of CO oxidation over Pd-based catalysts. Various influencing factors will be discussed. Future challenges and opportunities will be addressed.
 You Zhou | Mr. You Zhou was born in Hubei, China in 1986. He received his bachelor's degree in 2009, master's degree in 2011 and Ph.D degree in 2014, all at Tianjin University. He has published two papers in Chem. Commun. and Ind. Eng. Chem. Res. on CO oxidation over Pd catalysts. He is now working in Wanhua, Yantai, China. |
 Zongyuan Wang | Mr. Zongyuan Wang was born in Henan, China in 1988. He received his bachelor's degree from Tianjin University in 2012. He is now a graduate student in the same university. He has published one paper in Scientific Reports on the 3D-printed polymer composites. |
 Changjun Liu | Dr. Chang-jun Liu, a Chang Jiang Distinguished Professor and a Fellow of the Royal Society of Chemistry, received his Ph.D degree from Tianjin University in 1993. His research interests include catalysis, CO2 utilization and plasma nanoscience. He has published more than 200 papers that have attracted more than 3000 citations. He is now an editorial board member of Appl. Catal., B, J. CO2Utilization and J. Energy Chem. He is now an advisory board member of Greenhouse Gases: Science & Technology. He served as the 2010 Program Chair of ACS Fuel Chemistry Division and the Chair of the 10th International Conference on CO2 Utilization. |
Introduction
CO oxidation is one of the most extensively investigated reactions in the field of heterogeneous catalysis because of its importance in both environmental protection and fundamental studies.1–8 According to the Web of Science (SCIE), 5498 papers have been published since 1995 (as of Sept. 10, 2014) with the title ‘CO oxidation’. These papers have attracted 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 837 citations (19.87 citations per article). This is definitely a hot topic on such a simple molecule.9–13
837 citations (19.87 citations per article). This is definitely a hot topic on such a simple molecule.9–13
Carbon monoxide or CO is a colourless, odourless but poisonous and even deadly gas. It is produced from the incomplete burning or partial oxidation of various fuels like coal, gasoline and natural gas. Some specific cases like CO2 lasers also generate CO. It is very necessary to remove CO from enclosed atmospheres and various residue gases. In addition, even trace CO is harmful to Pt catalysts with applications such as in fuel cells. The purification of feed gases for these applications is of course mandatory.
From the aspect of fundamental studies, CO is exactly a frequently used probe molecule for studying the catalyst structure, adsorption/desorption and reaction mechanism. With the potential competitive adsorption and activation of oxygen, CO oxidation is an excellent reaction for the evaluation of catalysts with easy operation and low cost.
Catalysts for CO oxidation can be grouped into two main categories: non-noble metal catalysts and supported noble metal catalysts. Non-noble metal catalysts mainly include copper-, cobalt-, and manganese-based catalysts and their mixtures. The commercial Hopcalite catalyst, in which CuO and MnO2 are principal components, is a representative of these catalysts.14 In addition, the supported Cu catalyst has also been widely studied.15,16 CuO can be highly dispersed on CeO2, ZnO and Al2O3 with intense interaction. For example, Pillai et al.17 prepared a 60% CuO–40% ZnO catalyst by co-precipitation and aging treatment, which can complete the oxidation of CO into CO2 at room temperature. They found that CuO can be diluted with ZnO, which can change the surface morphology of CuO, resulting in an amorphous phase. It promotes the formation of more weakly adsorbed CO on the surface of the catalyst and the evolution of active copper oxide species (CuOx). Although non-noble metal catalysts are cheap, they have some disadvantages that cannot be ignored. Their activity is still not high in general. They are normally sensitive to H2O.
Supported noble metal catalysts, especially Pt-, Pd-, Au- and Ru-based samples (and their mixtures such as bi-metal or tri-metal catalysts), have been extensively studied for CO oxidation.18–21 Among them, Au catalysts always exhibit excellent activity at lower temperatures, while Pd and Pt catalysts have higher turnover frequencies at relatively high temperatures (not less than 100 °C). Normally, the adsorption of CO on Pd and Pt sites is very strong and almost exclusive at low temperature, leaving O2 with no active sites for activation. As the temperature rises, O2 adsorption and activation occur and the reaction rate of CO oxidation increases. Therefore, over Pd and Pt catalysts, CO oxidation always proceeds in a competitive Langmuir–Hinshelwood mechanism with high apparent activation energies (Ea). Considering the low activity of Pt catalysts, Pd-based catalysts have drawn much more attention due to their high activity and application for the interpretation of the metal–support interaction.
The properties of Pd catalysts are influenced by many factors, such as promoters, preparation methods, supports, Pd structure, and so on. In this perspective, we attempt to summarize the studies on the factors influencing CO oxidation over Pd-based catalysts. The future development will be discussed at the end of the article.
Effects of additives
As a promoter, ceria can improve the dispersion of the noble metal and the ability of oxygen storage and release. It can thereby enhance the activity of CO oxidation. As for Pd catalysts, CeO2 is mostly applied as a support for the specific metal–support interaction that allows synergistic oxidation/reduction of both the metal and CeO2, which favours the catalytic performance.22–25 Apart from ceria, many other species can also be used as additives to promote the performance of Pd catalysts in CO oxidation.The addition of Zn to Pd catalysts can lower the binding energy of CO, which will be beneficial to CO oxidation. Even submonolayer quantities of Zn deposited onto Pd catalysts will change the CO binding energy, and the increased coverage brings more pronounced influence. Johnson et al.26 reported PdZn bimetallic nanoparticles that are homogeneous in both phase and composition. The aerosol-derived bimetallic samples were used to investigate the effect of the reduced binding energy of CO on Zn-modified Pd catalysts for CO oxidation. The activities of the Pd metal (fcc structure), α-PdZn (Zn solid solution in fcc Pd) and β-PdZn (50:50 intermetallic PdZn) powder samples were compared, as shown in Fig. 1. It was found that the initial CO oxidation reactivity (per Pd surface atom) is as much as 10 times higher on the α-PdZn surface and about 5 times higher on the β-PdZn surface compared to that on metallic Pd. The weakening of the CO bond and the easier binding of oxygen to Pd sites modified by Zn were considered as the reasons for the higher reactivity. However, most of the initial CO oxidation enhancement is lost once the steady state is achieved, which is presumably caused by the disruption of the Zn–Pd interaction after the oxidation of Zn. Obviously, the addition of oxophilic metals such as Zn can modify the reactivity of Pd catalysts. It is important to prevent the oxidation of the oxophilic component in order to achieve stable high activities.
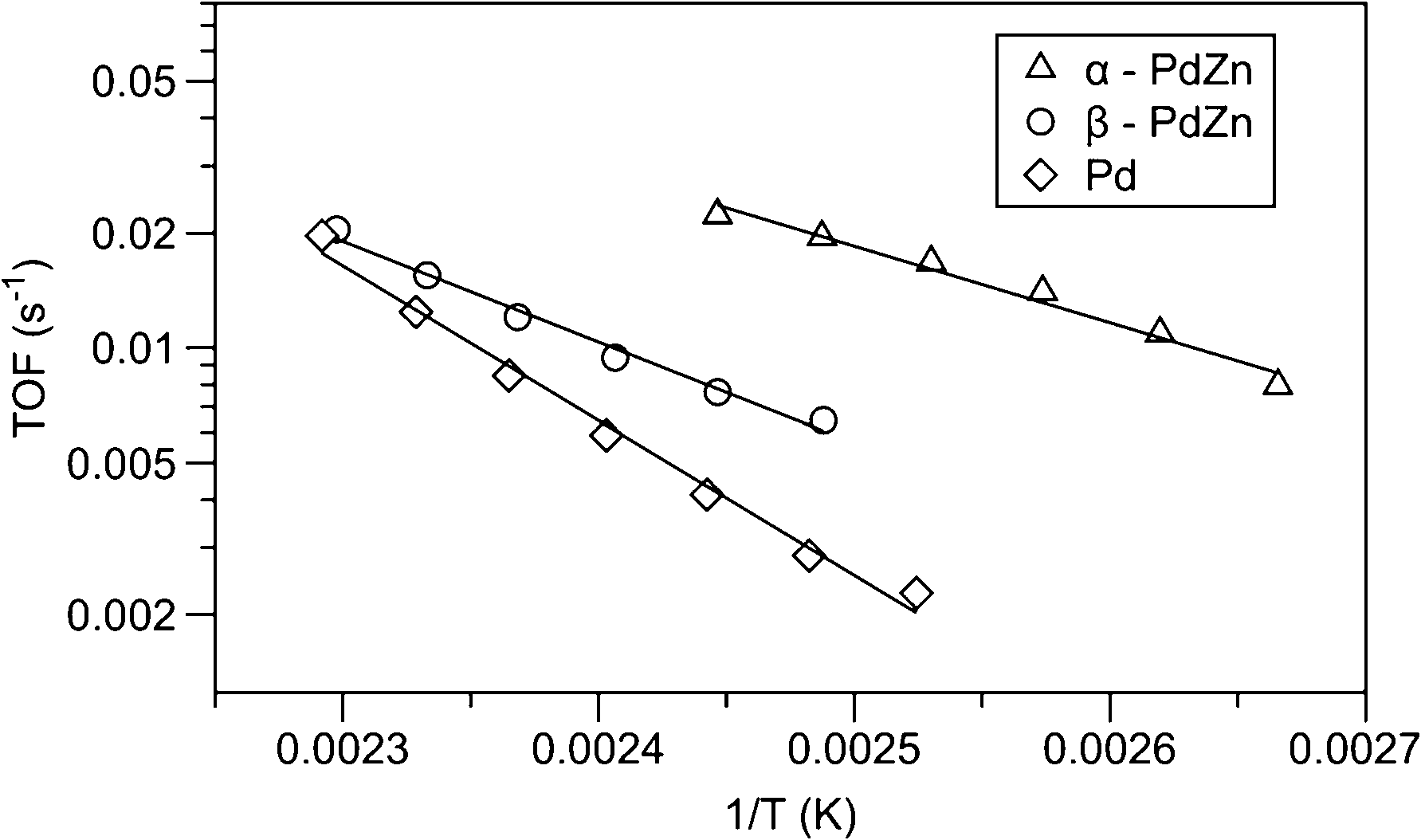 | ||
| Fig. 1 Arrhenius plots for CO oxidation over Pd, α-PdZn and β-PdZn catalysts.26 | ||
Alkaline earth metals are also common additives for improving the activity of Pd-based catalysts. Tanikawa and Egawa prepared barium fixed alumina (Pd/Ba/Al) and ceria–zirconia mixed oxide (Pd/Ba/CZ) supported Pd catalysts and investigated the effect of barium on CO oxidation over these two catalysts.27 As shown in Fig. 2, the properties of the support are significantly influenced by the addition of barium. For the Pd/Ba/Al catalysts, the increasing barium loading gradually improves the CO oxidation activity. For the Pd/Ba/CZ catalysts, however, the presence of barium reduces the activity for CO oxidation. The higher barium loading causes lower activity. It was proposed that the CZ support could enhance the activation of oxygen at surface anion vacancies that are important for CO oxidation. When barium is added, the contact between the Pd particles and the CZ support may be prevented, and the oxygen supply from CZ to Pd would be inhibited, which results in the reducing activity of Pd/Ba/CZ for CO oxidation. Considering the results of XPS and DRIFT spectra, they concluded that the adsorption strength of CO on palladium could be weakened by barium in Pd/Ba/Al, while in Pd/Ba/CZ the oxygen mobility from CZ to palladium is inhibited by barium.
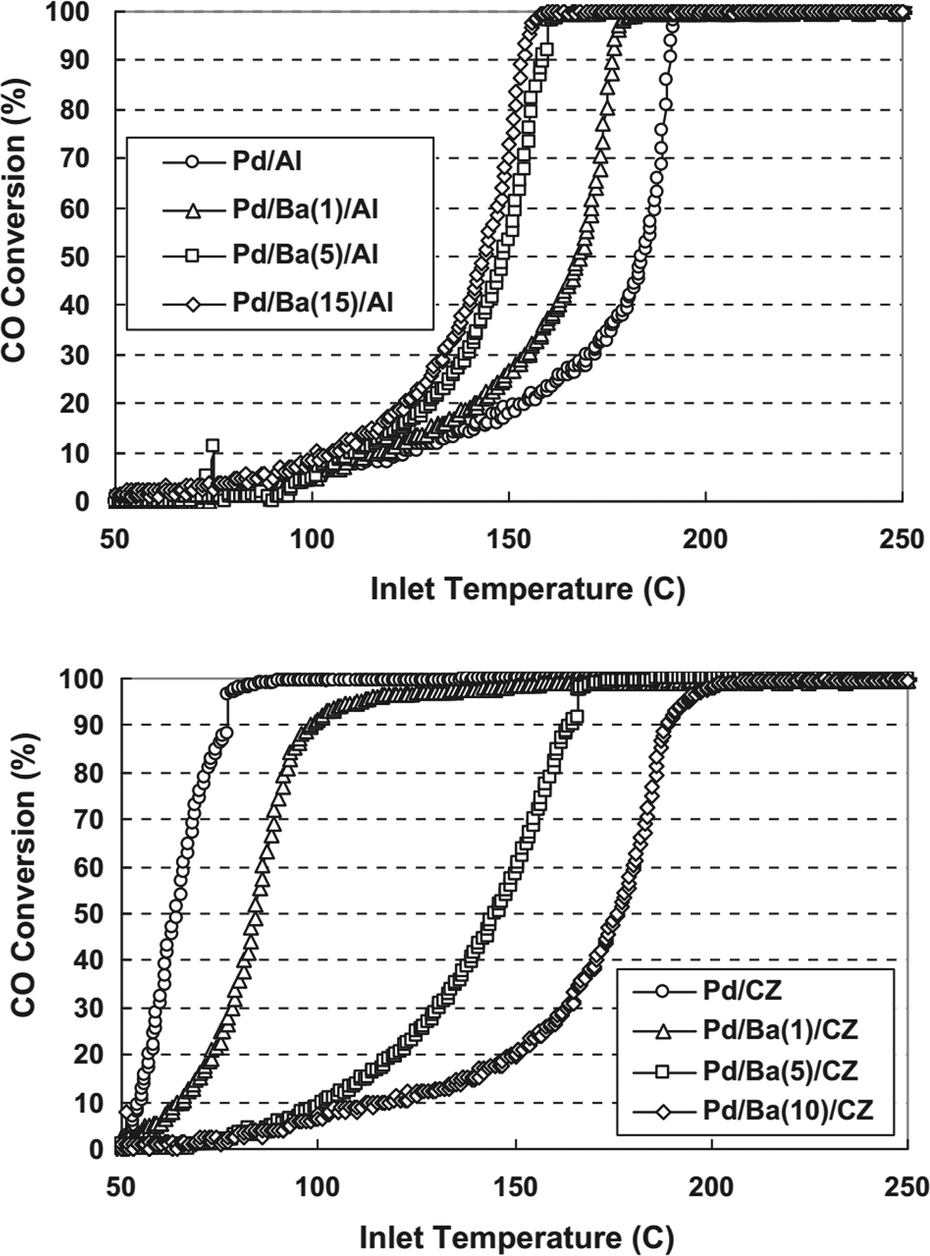 | ||
| Fig. 2 CO conversion in CO oxidation as a function of the amount of Ba loading for (up) Pd/Ba/Al catalysts and (down) Pd/Ba/CZ catalysts. Reproduced with permission from ref. 27. Copyright 2011, Elsevier. | ||
Additives in the support can also lead to better performance of the supported Pd catalysts. In Gaudet's work, they found that the La-stabilized alumina improves the dispersion of Pd particles.28 To investigate the role of the La-stabilized alumina support in CO oxidation, they prepared 2.5% Pd/Al2O3 and 2.5% Pd/La–Al2O3 and found that the La-containing catalyst has higher CO conversion and lower onset temperature for CO oxidation. As shown in Fig. 3(a), the 2.5% Pd/La–Al2O3 sample is much more active for CO oxidation. Calculated from the data in Fig. 3(b), the apparent activation energies (EA) are 16 kcal mol−1 for Pd/Al2O3 and 15 kcal mol−1 for Pd/La–Al2O3. What is more, the results of STEM and EXAFS analyses indicate that the addition of La increases not only the Pd dispersion but also the TOF, indicating that the factors can increase the CO oxidation rate and improve dispersion. A further study shows that the presence of La is helpful for increasing the preference of oxygen adsorption. As shown in Scheme 1, for Pd/La–Al2O3, O could displace CO from 3-fold hollow sites, resulting in improved O coverage and higher TOF. Meanwhile, a bulk oxide or a passivation layer, which might deactivate the catalyst, would not form. The authors believe that the La species help to modify the nucleation of Pd. The resulting smaller Pd clusters exhibit facile redox behaviour at low temperatures, which are less susceptible to poisoning by CO.
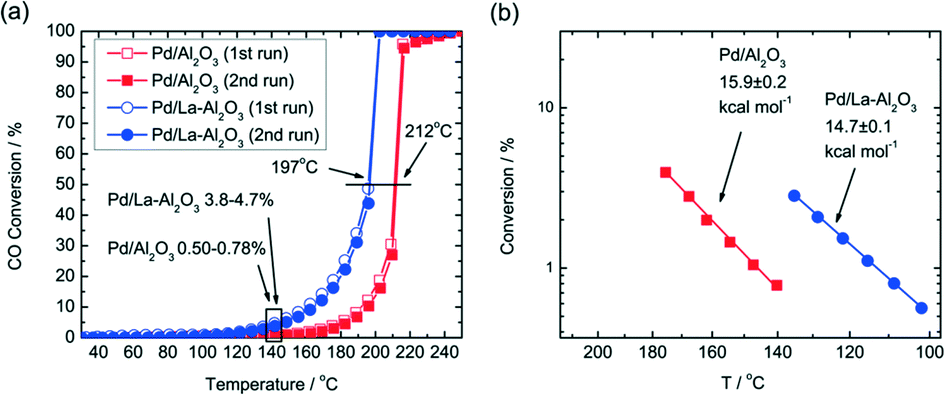 | ||
| Fig. 3 (a) Light-off traces of 2.5% Pd/La–Al2O3 and 2.5% Pd/Al2O3 and (b) the associated Arrhenius plot. Reproduced with permission from ref. 28. Copyright 2013, American Chemical Society. | ||
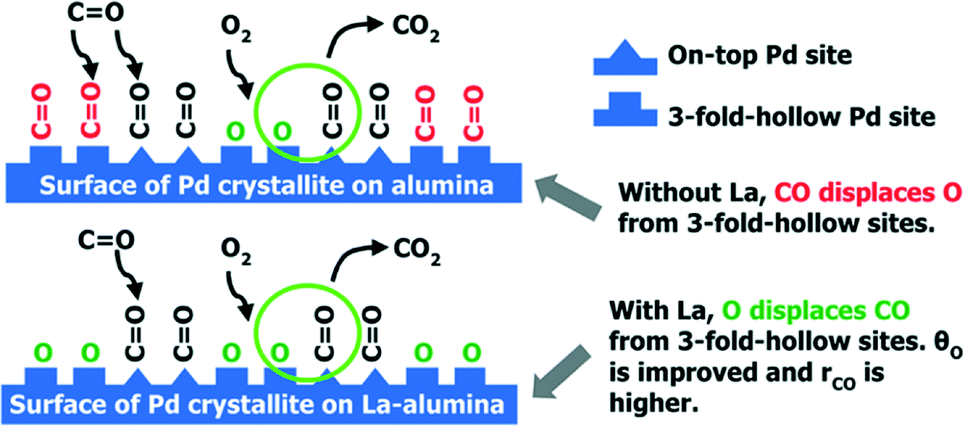 | ||
| Scheme 1 CO oxidation over Pd/Al2O3 and Pd/La–Al2O3. Reproduced with permission from ref. 28. Copyright 2013, American Chemical Society. | ||
In diesel applications, the Pd–Pt bi-metal catalyst is more active than either alone. The synergistic effect between Pd and Pt is useful to weaken the adsorption of CO, which is helpful for improving the activity in low-temperature CO oxidation. We prepared Pd/MIL-101 and Pt–Pd/MIL-101 catalysts by the impregnation method and electron reduction. For the bi-metal sample, Pd was firstly loaded and reduced then Pt was added subsequently. As shown in Fig. 4, the Pt–Pd/MIL-101 sample exhibits much higher activity. From the results of DRIFT in Fig. 5, we found that the CO molecules are strongly absorbed on the Pd sites in the Pd/MIL-101 sample. Meanwhile, CO2 and formate species also keep strong interaction with the sample. It could be concluded that Pd/MIL-101 could strongly adsorb the carbon species, leading to the low efficiency of the active sites. As for Pt–Pd/MIL-101, the addition of Pt significantly changes the adsorption properties of the sample. Compared with those for Pd/MIL-101, the bands of the carbon species for the Pt–Pd/MIL-101 sample are much weaker. It indicates that the interaction between Pt and Pd could weaken the adsorption capacity of the metal particles for carbon species. To further investigate the reason for the better CO oxidation activity of Pt–Pd/MIL-101, we carried out CO-TPD on the two samples. As shown in Fig. 6, there are two distinct CO desorption peaks in the CO-TPD profiles for both the two samples. Peak α, which is centred at 76 °C for Pd/MIL-101 and at 85 °C for Pt–Pd/MIL-101, is attributed to the weakly adsorbed CO species. Although these CO species are not active for CO oxidation, they compete with O2 to adsorb on the active sites in the low-temperature region, resulting in poor reactive oxygen species as well as low activity of the samples. Peak β, centred at 262 °C for both samples, is assigned to the moderately adsorbed CO species on the samples, which plays the key role in CO oxidation. Obviously, peak β of Pt–Pd/MIL-101 has stronger intensity and larger area than that of Pd/MIL-101, which indicates that there are more active CO species adsorbed on the Pt–Pd/MIL-101 sample. The desorption of CO species in the peak β part of Pt–Pd/MIL-101 starts at a much lower temperature than that of Pd/MIL-101, which might account for the better activity of the bimetallic catalyst.
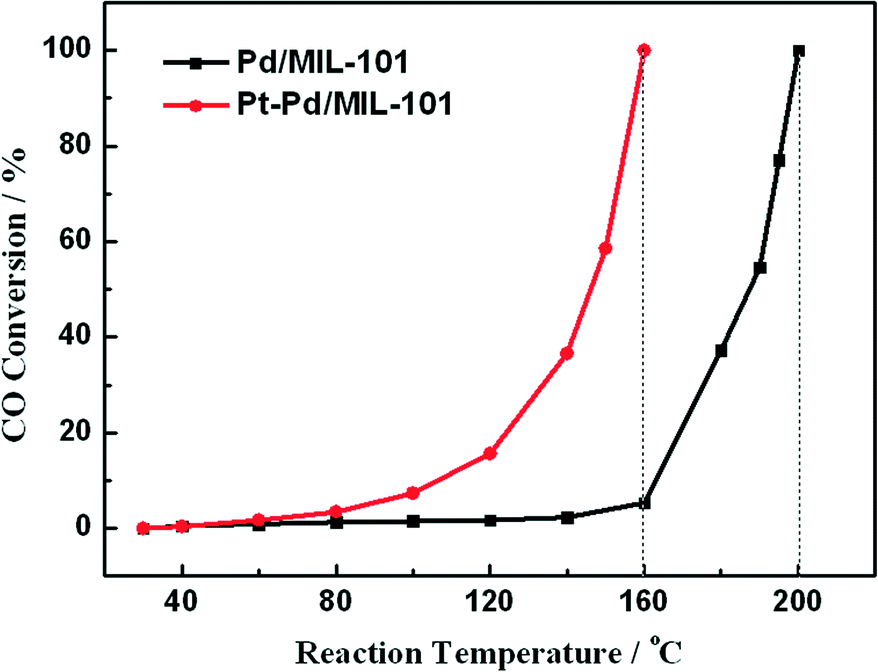 | ||
| Fig. 4 Results of CO oxidation over Pd/MIL-101 (Pd 5%, 10 mg) and Pt–Pd/MIL-101 (Pd 1.8%, Pt 3.2%, 10 mg). | ||
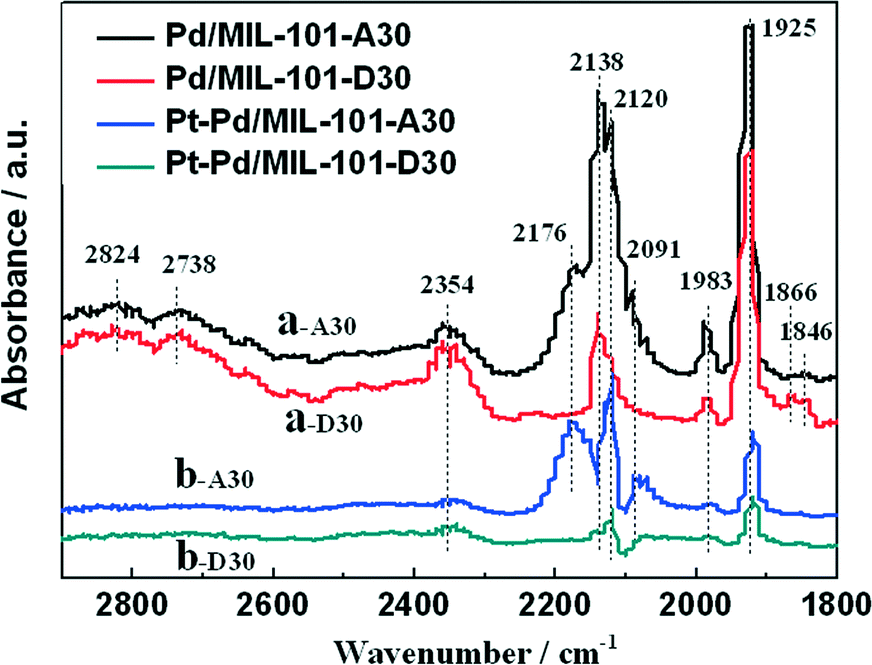 | ||
| Fig. 5 DRIFT spectra of CO absorbed on (a) Pd/MIL-101 and (b) Pt–Pd/MIL-101 at 25 °C (A30: absorbing for 30 min; D30: desorbing for 30 min). | ||
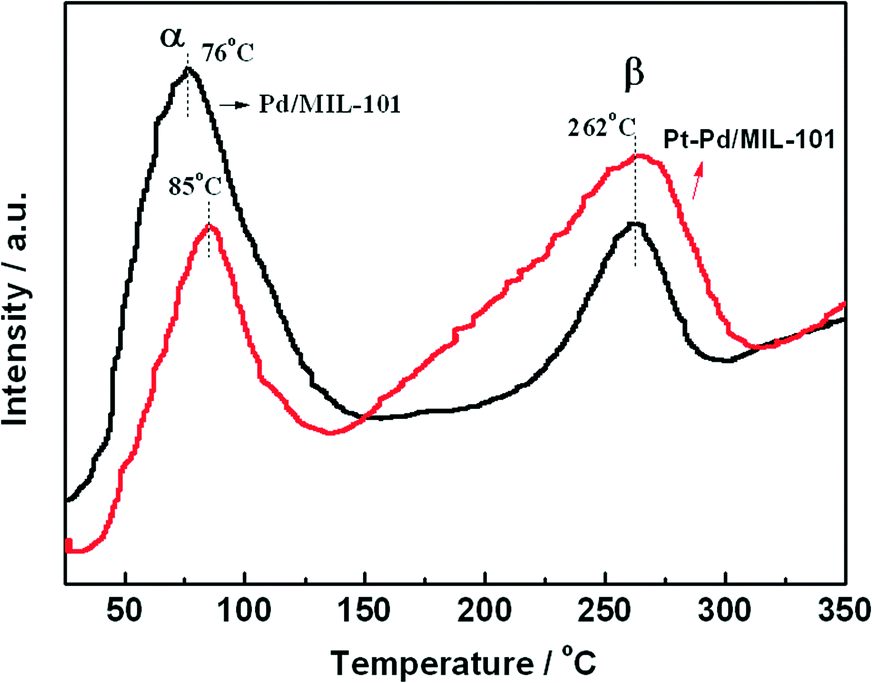 | ||
| Fig. 6 CO-TPD profiles of Pd/MIL-101 and Pt–Pd/MIL-101. Experimental conditions: flow rate = 30 mL min−1 (He), heating rate = 5 °C min−1. | ||
Effect of alloyed catalysts
Regarding the bimetallic catalysts, the effect of alloyed catalysts should not be ignored.29–31 A higher activity and enhanced stability of Pd-alloy-based catalysts are always expected. Unfortunately, as indicated by Singh and Xu,30 only a few reported studies can be found in the literature on CO oxidation over alloyed Pd catalysts. However, these reported studies indeed show improved activity with enhanced stability. Morfin et al.32 found that the IrPd catalysts revealed a marked superiority over their pure metal counterparts and an Ir + Pd mechanical mixture in terms of CO conversion and selectivity. Shan et al.33 found that PdNi nanoalloy catalysts exhibited remarkable tunability in terms of the phase state, bimetallic composition, and atomic-scale structure, which were responsible for the origin of the structural synergy for CO oxidation reaction over the nanoalloy catalysts. There is also pronounced synergistic enhancement in supported bimetallic palladium–gold catalysts. The Pd–Au alloys are superior to pure palladium for CO oxidation.34,35 Goodman and his co-workers postulated that Au atoms tune Pd–Pd bonds in model bimetallic compounds via an “ensemble effect”, which enhances catalytic performance. However, the surface structure of gold-modified palladium remains conjectural. Furthermore, the precise mechanistic role of gold in accelerating low-temperature reactions remains ambiguous.Ham et al.36 presented a theoretical explanation on how PdAu alloyed catalysts can enhance the oxidation of CO molecules based on density functional theory (DFT) studies of CO adsorption and oxidation on AuPd/Pd(111) surfaces. They suggested that the enhanced activity is principally attributed to the possible existence of “partially poisoned” Pd ensembles that accommodate fewer CO molecules than Pd atoms. AuPd alloys resist CO poisoning. Kim et al.37 also conducted a DFT study on CO-adsorption-induced Pd surface segregation in Au/Pd bimetallic surfaces, dynamics of Pd–Au swapping, the effect of defects on the swapping rate, and the reaction mechanism of CO oxidation. They found that Pd clusters consisting of at least four Pd atoms prefer to bind O2 rather than CO. These clusters facilitate the rapid dissociation of O2 and supply reactive oxygen species for CO oxidation. Their findings suggest that geometric, electronic, and dynamic effects should be considered in the function of bimetallic alloys or nanoparticles whose components asymmetrically interact with reacting molecules.
Xu et al.38 synthesized a series of nanocrystalline biphasic Pd–Au nanoalloy catalysts with different compositions, supported on silica fume, in order to investigate the activity for CO oxidation as a function of bulk composition and to understand the key role of modified surface chemistries in mixed metal catalysts. They found that, except for the pure metals, these materials invariably showed biphasic separation into palladium- and gold-rich components. Optimal performance is achieved for a catalyst of bulk composition Pd4Au1, a mixture of Pd90Au10 (72.5 at.%) and Pd31Au69 (27.5 at.%), which was remarkably active at 300 K and more stable than a pure Au catalyst. CO oxidation reaction was conducted on all catalysts. The activities from highest to lowest for all catalyst pairs are in the sequence: Au > Pd:Au = 4:1 > 8:1 > 16:1 > Pd > 2:1 > 1:1.6 > 1:3.4. However, Pd4Au1 exhibits superior activity and stability as compared to pure gold, with the latter showing a 50% loss of activity after 10 h at 353 K as compared to 5% for Pd4Au1 in an ideal exhaust gas of 5.0 kPa CO, 5.0 kPa O2, and Ar balance. In addition, in the simulated exhaust gas (5.0 kPa CO, 5.0 kPa O2, 10.0 kPa H2O, 20.0 kPa CO2, and Ar balance), there was a substantial loss of activity for pure Au, while only a mild deterioration was observed for Pd4Au1. The catalyst characterization indicated that, at Pd-rich compositions, hollow spheres or annular morphologies are formed, while Au-rich crystals are often twinned. Pd atoms in the surface might be surrounded or separated by Au. Surface defects, ascribed as metal vacancies, are directly observed and contribute to enhance low-temperature CO oxidation. Pd–Au alloy catalysts showed significant activity and stability toward low-temperature CO oxidation even at 300 K for bulk compositions from Pd to Pd4Au1, but showed degraded performance at higher Au contents due to higher activation that was accompanied by greater surface coverage of Au. These results demonstrate that gold-containing biphasic Pd nanoalloys are excellent catalysts for CO oxidation.
Influence of the preparation method
To obtain supported noble metal catalysts with a small particle size and high distribution, as well as excellent performance in reaction, the preparation method plays a crucial role.Santos et al.39 investigated the activity of noble metal catalysts (Pd, Pt, Ir, Rh and Au) supported on TiO2 for the CO oxidation reaction. Two methods, liquid-phase reduction deposition (LPRD) and incipient wetness impregnation (IMP), were used to prepare the catalysts for comparison. It was found that the dispersion of the metallic phase was significantly affected by the preparation method. For example, the average size of Pd particles prepared by IMP was 8.0 nm, while that prepared by LPRD was 3.0 nm. Meanwhile, the LPRD-prepared Pd sample has a dispersion of 28%, much higher than that of the IMP Pd sample (16%). Subsequently, the performance of the catalysts towards CO oxidation is different. LPRD shows better results, as shown in Fig. 7.
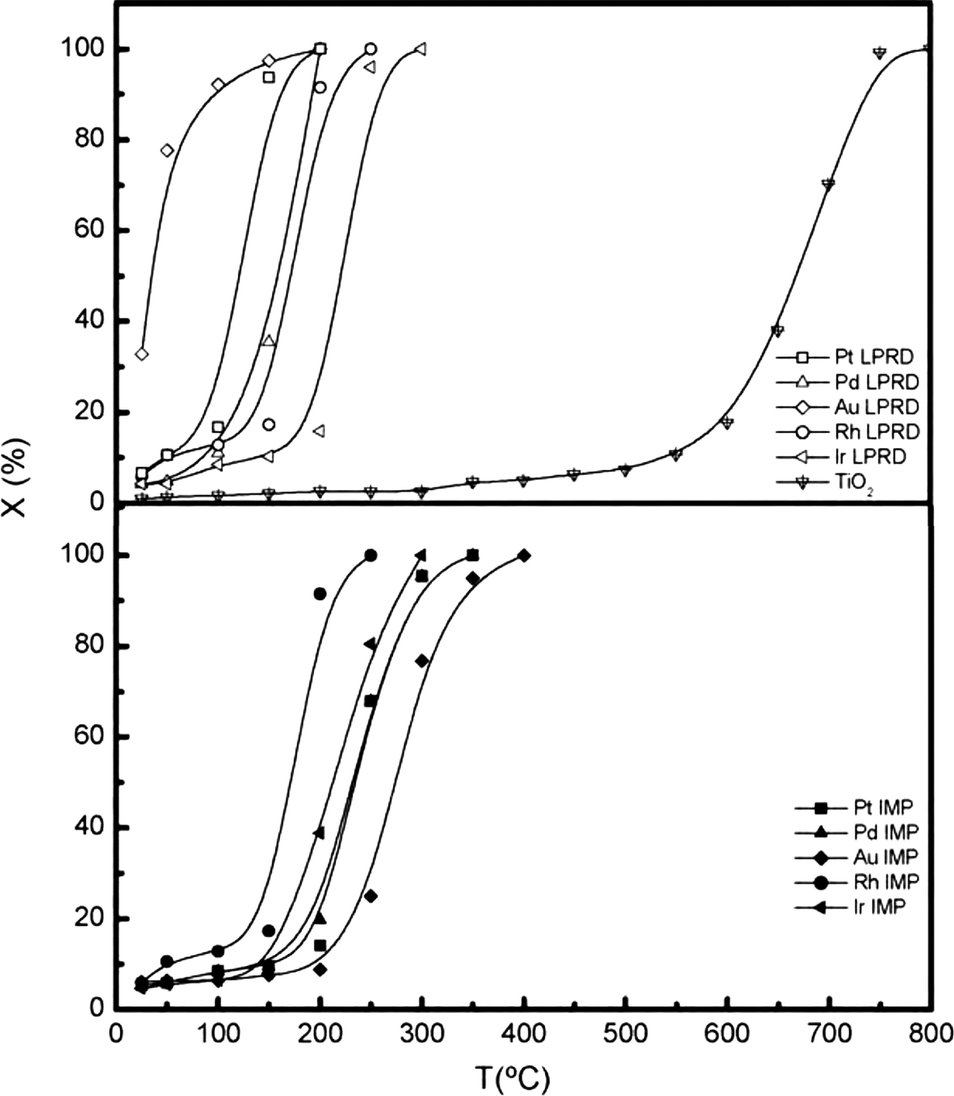 | ||
| Fig. 7 Light-off curves for CO oxidation over supported noble metal catalysts prepared by different methods. Reproduced with permission from ref. 39. Copyright 2010, Elsevier. | ||
During the normal impregnation method, the use of different solvents would lead to distinguishing particle distribution and thereby different catalytic properties. We have found that the solvent applied in the impregnation method has a significant effect on CO oxidation over the covalent organic polymer-4 (COP-4, a new porous organic polymer) supported palladium catalyst.40N,N-Dimethylformamide (DMF) and distilled water were used as the solvents for novel electron reduction to convert Pd ion into metallic species. As shown in Fig. 8, when DMF was used as the solvent, Pd particles are distributed in the pores of COP-4 with a uniform size of 1.1 nm. When water was used, the average size of Pd particles, obviously outside the pores, becomes 2.4 ± 0.1 nm. In our experiment, DMF can easily infiltrate into the pores of COP-4, while water is kept outside the pores. Therefore, we can say that the Pd particles are confined in the pores after the metal ions in DMF are reduced, while the Pd ions in water are transferred to Pd particles outside the pores without confinement of the pores. The DRIFT spectra of CO at room temperature show that there are different active sites on the two samples, which show different activities for CO oxidation.
 | ||
| Fig. 8 TEM images of (a) COP-4, (b and c) Pd/COP-4-DE, and (d–f) Pd/COP-4-WE. (D = DMF, W = water, E = electron reduction).40 | ||
We further prepared two more Pd/COP-4 samples by hydrogen reduction,41 compared with the samples reduced by electron reduction, to check the effect of the reduction method. The relatively high temperature (300 °C) of hydrogen reduction leads to the formation of much larger Pd particles outside the pores of COP-4. The average size of the particles in Pd/COP-4-DH and Pd/COP-4-WH (H = hydrogen reduction) is 7.7 ± 1.8 nm and 9.9 nm, respectively. However, the samples prepared by hydrogen reduction have better activity for CO oxidation than those prepared by electron reduction, as shown in Fig. 9. Comparing the DRIFT spectra of CO of the four Pd/COP-4 samples, we found that the Pd/COP-4 samples prepared by electron reduction have much stronger interaction with the CO molecules and mainly show the Pd(111) lattice plane, which should be the reason for their relatively low activity for CO oxidation. The results indicated that not only the solvents but also the reduction methods will influence the properties of the catalysts. What is more, the activities of the Pd/COP-4 samples prepared by hydrogen reduction are compared with that of a MOF-5 supported Pd catalyst, as shown in Table 1. The TOF values of Pd/COP-4 samples are much higher than that of Pd/MOF-5, indicating the superiority of the COP-4 material with large amounts of nitrogen atoms.
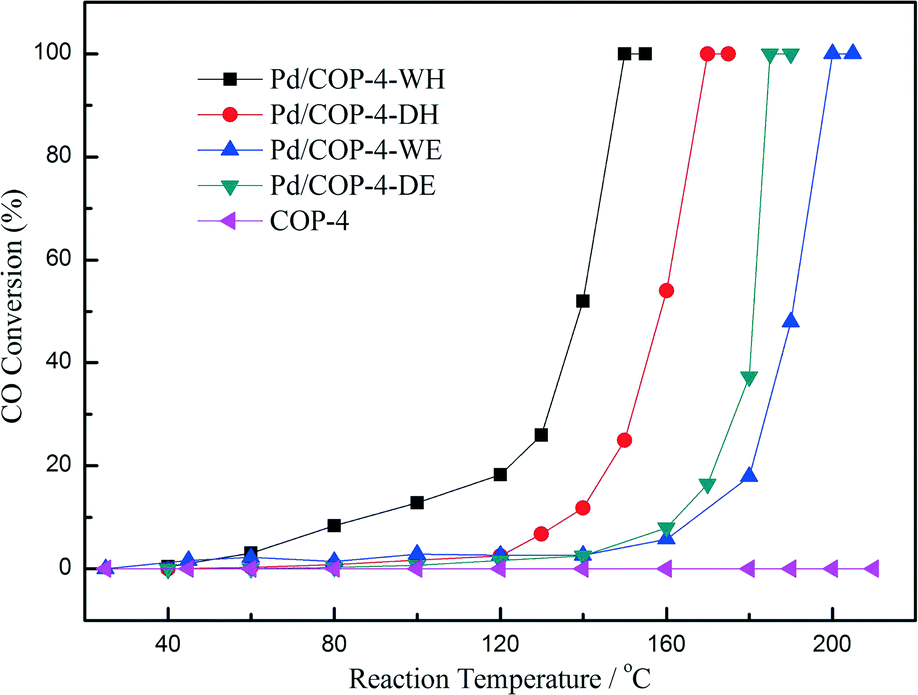 | ||
| Fig. 9 Light-off curves for CO oxidation over COP-4 supported Pd catalysts. Reproduced with permission from ref. 41. Copyright 2014, American Chemical Society. | ||
| Dispersion | TOF130°C × 103 s−1 | TOF140°C × 103 s−1 | |
|---|---|---|---|
| Pd/COP-4-DH | 0.14 | 14.9 | 25.8 |
| Pd/COP-4-WH | 0.11 | 72.9 | 145.8 |
| 1%Pd/MOF-5-DH | 0.45 | 1.8 | 3.2 |
Wang et al.42 reported the effect of preparation conditions on the SBA-15 supported Pd catalysts for CO oxidation. Using electron reduction, they synthesized highly dispersed Pd nanoparticles within the uniform channel of SBA-15. The obtained Pd/SBA-15 catalyst shows good activity for CO oxidation. Then the electron reduced Pd/SBA-15 (P–Pd/SBA-15) is calcined under air and further reduced thermally with hydrogen. Apart from a slight increase in the particle size, the hydrogen reduced Pd/SBA-15 (H–Pd/SBA-15) has higher activity for CO oxidation compared with P–Pd/SBA-15, as shown in Fig. 10. It indicates that the preparation conditions exhibit a significant influence on the catalyst activity. This study suggests that CO oxidation over Pd/SBA-15 is a structure-sensitive reaction. The structure change induced by the thermal treatment is the reason for the improvement in the activity, which is implied by the DRIFT studies. Normally, linear CO is preferentially formed on low-coordinated Pd atoms, while smaller particles always expose a larger fraction of low-coordinated atoms than larger ones. Therefore, the structure change can be induced by the preferential adsorption of CO at bridge sites. This is an advantage for reaction between CO and O adatoms. It changes the reaction rate, resulting in a higher activity for CO oxidation over the H–Pd/SBA-15 sample.
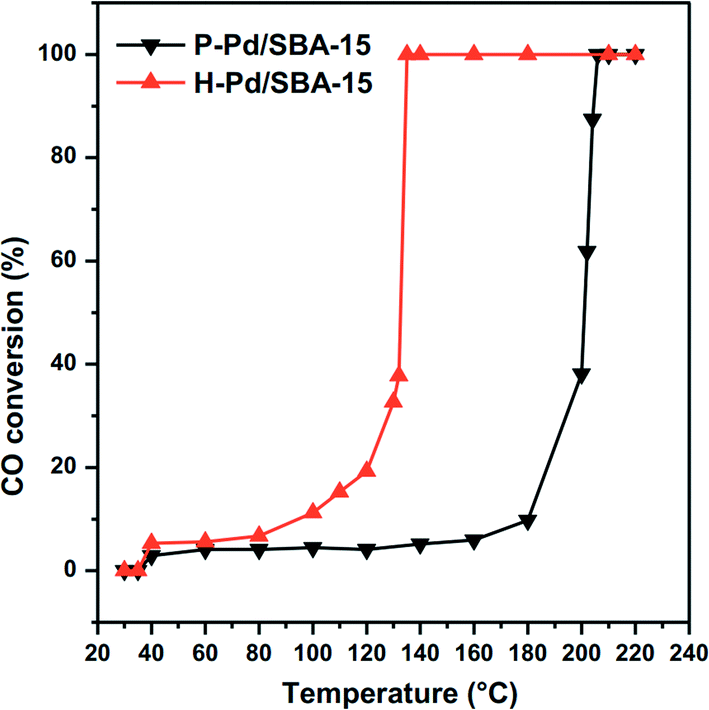 | ||
| Fig. 10 Light-off curves for CO oxidation over Pd/SBA-15 samples. Reproduced with permission from ref. 42. Copyright 2011, Elsevier. | ||
Ivanova et al.43 used γ-Al2O3 as a support to prepare Pd catalysts and investigated the effect of the calcination temperature. The properties of the catalysts, denoted as Pd/Al2O3(X)-Y (X—the calcination temperature of the support, °C; Y—the calcination temperature of the catalyst, °C), were studied. Palladium particles in the Pd/Al2O3 (550)-450 catalyst are almost completely decorated with a thin layer of an aluminate phase, which cannot be reduced by hydrogen in the temperature range of −15 to 450 °C. In Pd/Al2O3(800)-T catalysts, decorated metallic particles with a core–shell structure are formed. When the calcination temperature of the catalyst changes from 450 °C to 1000 °C gradually, the morphological form of the active component converts from the core–shell structure to the combination of two phases, Pd0 and PdO, with the gradually decreasing Pd0/PdO ratio. Meanwhile, the interaction between the Pd phases and the support is weakened, and the palladium particles grow larger. After calcination of the Pd/Al2O3 catalyst at 1200 °C, the coarse PdO particles are reduced into Pd0. However, the palladium particles lose interaction with the support, gathering into large agglomerates and exhibiting poor catalytic performance for CO oxidation. The light-off curves for CO oxidation over the Pd/Al2O3 catalysts are shown in Fig. 11. It could be found that the efficiency of the palladium catalysts for CO oxidation depends on the support calcination temperature. The increase of the support calcination temperature from 550 to 800 °C makes the catalyst more active (for the catalysts calcined at 450 °C). When the calcination temperature is not more than 800 °C, the characteristic of CO oxidation has no big difference. If the calcination temperature is further increased, the CO conversion shows a sharp decrease.
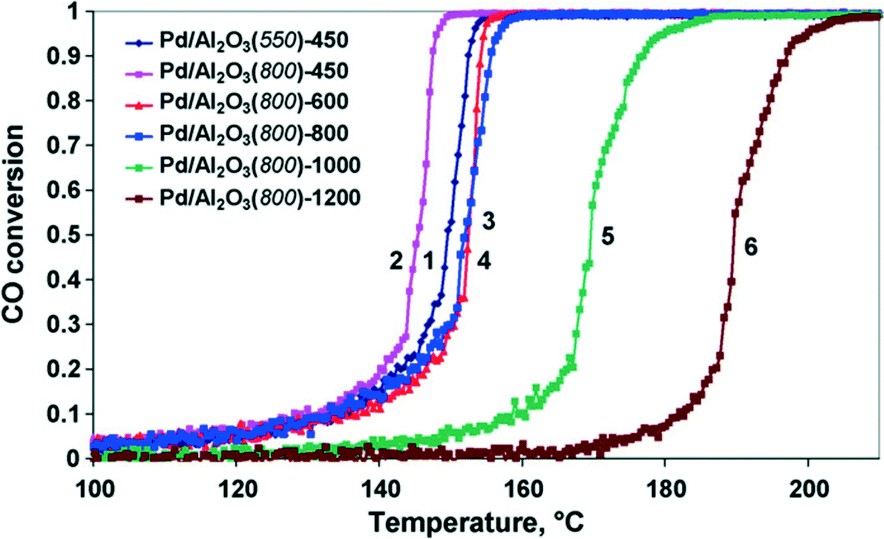 | ||
| Fig. 11 Temperature dependence of the CO conversion for catalysts: Pd/Al2O3(550)-450 (1), Pd/Al2O3(800)-450 (2), Pd/Al2O3(800)-600 (3), Pd/Al2O3(800)-800 (4), Pd/Al2O3(800)-1000 (5), and Pd/Al2O3(800)-1200 (6). Reproduced with permission from ref. 43. Copyright 2010, Elsevier. | ||
For supported metal catalysts, high-temperature aging always results in sintering or re-dispersion, which is mastered by metal–support interaction and gas atmosphere. Hinokuma et al.44 found that the Pd/CeO2 catalyst after thermal aging at 900 °C in air has obviously increased activity for ambient-temperature CO oxidation. Fig. 12a exhibits the temperature dependence of the conversion of CO to CO2 before and after thermal aging at 900 °C, while Fig. 12b shows CO conversion measured at 50 °C versus the aging temperature. The activation is demonstrated to be driven by metal–support interactions followed by phase transformation. Details are discussed in the following mechanism shown in Fig. 13. Firstly, the bonding of Pd–O–Ce is formed at the PdO/CeO2 interface, which allows high dispersion of Pd oxide into the surface structure of sintered CeO2 crystallites. However, the surface Pd oxide species themselves could not act as active sites for low-temperature CO oxidation. When the aging temperature reaches 800 °C or higher, the Pd–O–Ce moiety becomes unstable. Finally, the PdO/Pd phase equilibration at 900 °C will cause the thermodynamic dissociation of Pd oxide species to yield metallic Pd nanoparticles, which can activate CO. Compared with other supported catalysts such as Pd/γ-Al2O3 and Pt/CeO2, which are deactivated by sintering, the Pd/CeO2 catalyst is an exceptional case; that is, significant thermal sintering in the presence of O2 converts it to a very active catalyst for ambient-temperature CO oxidation.
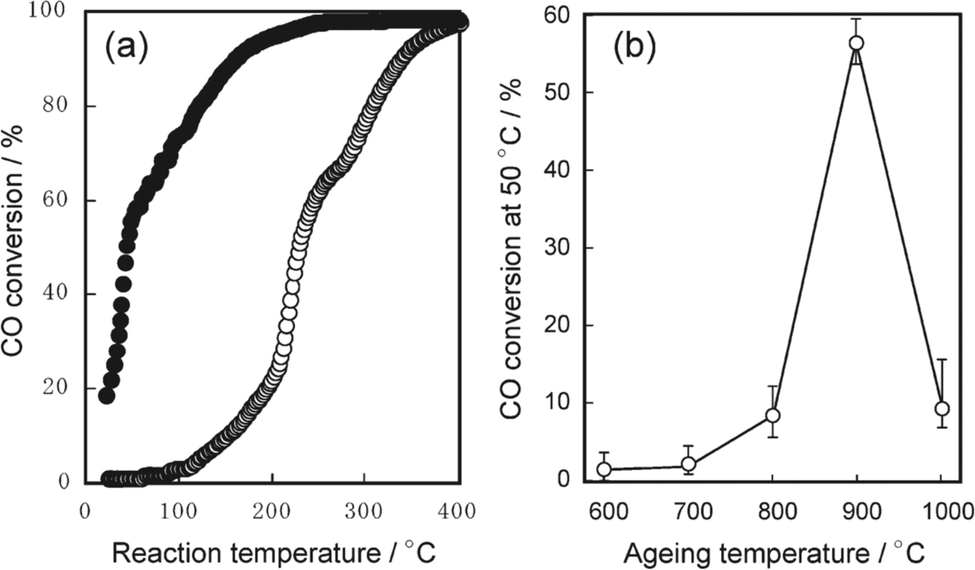 | ||
| Fig. 12 (a) Light-off curve of CO oxidation over Pd/CeO2 before (○) and after (●) aging at 900 °C; (b) conversion of CO to CO2 at 50 °C as a function of aging temperature. Reproduced with permission from ref. 44. Copyright 2010, American Chemical Society. | ||
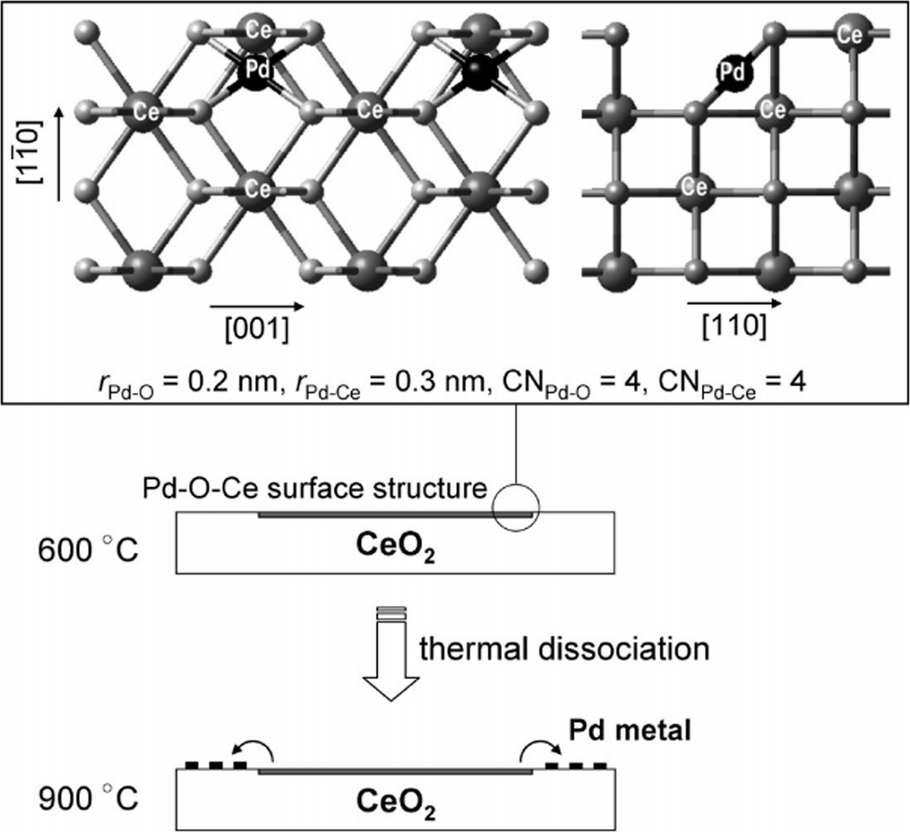 | ||
| Fig. 13 Possible mechanism for sintering-induced activation of Pd/CeO2. Reproduced with permission from ref. 44. Copyright 2010, American Chemical Society. | ||
Effect of the Pd structure
The shape or morphology of the nanoparticles is also an important factor which influences the catalytic performance of metal catalysts.45 When the crystalline shape is tuned, the exposed facets on the surface of metallic nanocrystals will alter, and then the catalytic properties of metal nanocrystals will be different.Wang et al.46 synthesized palladium nanocrystals with cubic, octahedral, and spherical morphologies and dispersed them onto the inert silica support. Carbon monoxide oxidation was chosen to investigate the activity of these catalysts. The results shown in Fig. 14 indicate that the catalytic activities of the Pd catalysts are influenced by the shape of Pd nanocrystals. The apparent activation energies of CO oxidation are calculated to be 76.5, 52.9 and 42.6 kJ mol−1 for the cubic, octahedral and spherical Pd catalysts, respectively. The spherical Pd/SiO2 catalyst possesses the lowest activation energy and shows the best catalytic performance, while the cubic Pd/SiO2 sample has the highest activation energy with the poorest activity. Their further investigations indicate that the octahedral and spherical Pd nanocrystals with mainly exposed Pd(111) planes are more active for CO oxidation, compared with the cubic Pd nanocrystals which are enclosed by the Pd(100) plane. It means that the exposed facets of Pd nanocrystals influence the adsorption and desorption properties of the CO molecule and consequently the catalytic activity for CO oxidation. The results of CO-TPD and DRIFT indicate that the interaction between CO species and Pd(100) is weak, while the CO molecule has much stronger interaction with Pd(111) under ambient conditions. It is inferred that the appropriate adsorption strength of the CO molecule on Pd(111) planes is beneficial to the enhancement of the catalytic activity.
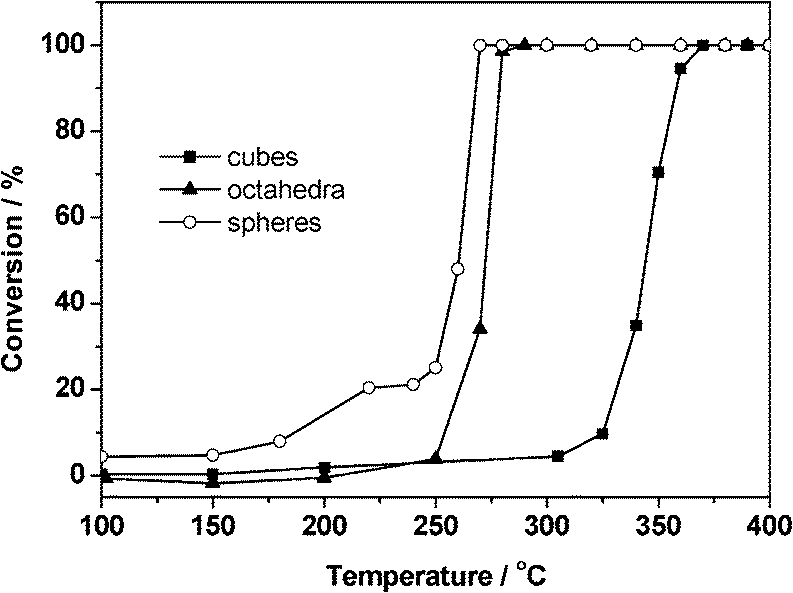 | ||
| Fig. 14 CO oxidation activity of Pd/SiO2 catalysts with different particle shapes.46 | ||
In addition to the facets exposed on the surface, the size of Pd nanocrystals is also important for the catalytic activity. As for CO oxidation, many studies have demonstrated that the Pd(100) plane of Pd single crystals is superior in activity to the Pd(110) and Pd(111) planes. By manipulating the rate of reduction of the precursor, Jin et al.47 successfully synthesized Pd nanocrystals enclosed by (100) facets with sizes controlled in the range of 6–18 nm. When the concentrations of Br− and Cl− ions added to the reaction mixture are adjusted, the rate of reduction of Na2PdCl4 can be controlled. They immobilized the Pd nanocrystals on ZnO nanowires and investigated their activity for CO oxidation. As shown in Fig. 15, the nanocrystal size shows great influence on the catalytic activity for CO oxidation. The 6 nm Pd cubes/bars have much higher CO conversion than the 10 and 18 nm Pd cubes/bars at the same reaction temperature. The activity can be remarkably enhanced by decreasing the size of the Pd cubes/bars. The complete conversion temperature and the intrinsic turnover frequency (TOF) are also affected by the size of the Pd cubes/bars in the same way. When the size is reduced from 18 nm to 6 nm, the complete conversion temperature is lowered by ~80 °C. The TOF at 140 °C of the 6 nm Pd cubes/bars is approximately 3 and 10 times higher, respectively, than those of the 10 and 18 nm Pd cubes/bars. It indicates that the size of the Pd nanocrystals determines their activity for CO oxidation.
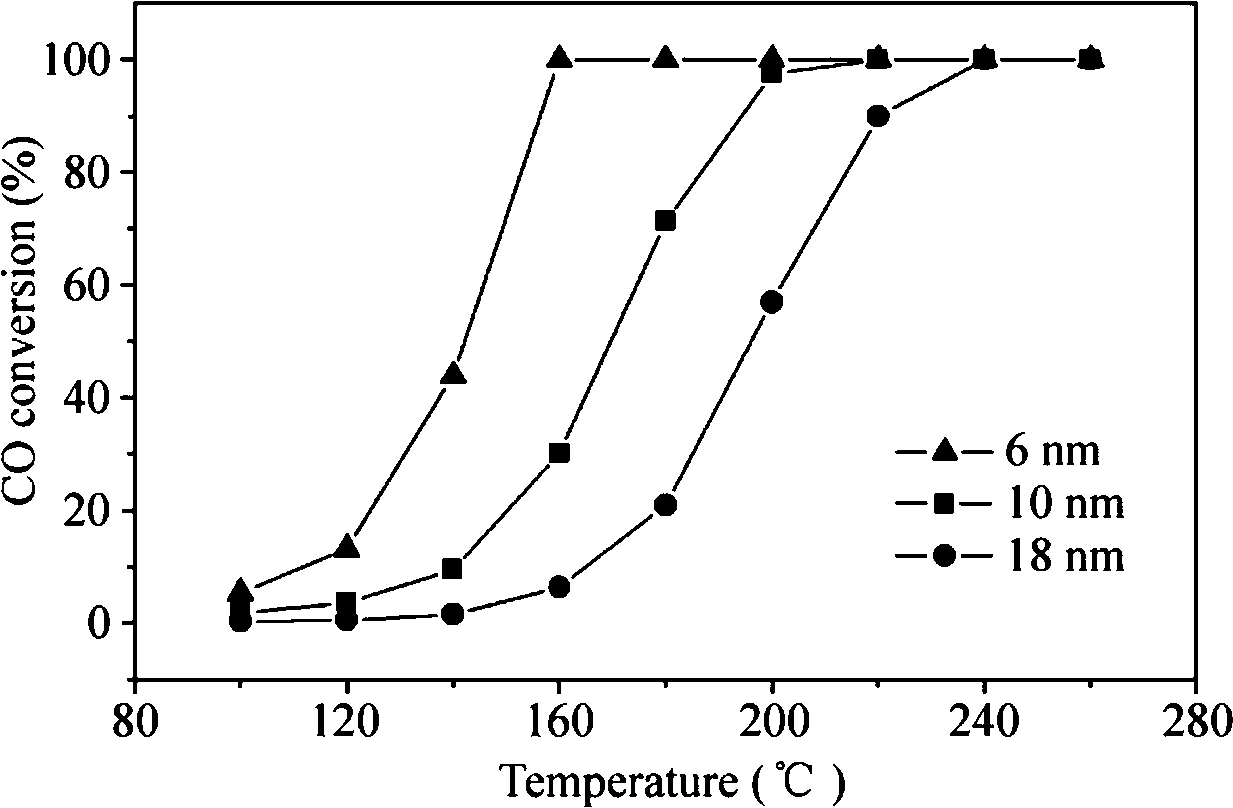 | ||
| Fig. 15 CO conversion as a function of temperature for Pd cubes/bars with different sizes supported on ZnO nanowires. Reproduced with permission from ref. 47. Copyright 2011, Springer. | ||
Study of the active sites
Pavlova et al.48 reported that, at saturation coverages of carbon monoxide on supported palladium particles, the most reactive weakly bound forms of CO (both linear and bridge-bonded) appear to be mainly located at defect centres generated by reconstruction of the open (100) and (110) faces. There are some disputes on the oxidation state of active Pd species for CO oxidation. A common assertion exists stating that PdO is a poor CO oxidation catalyst. In addition, the case states that lower dispersion of Pd is more preferable for higher activity to some degree. Highly dispersed Pd particles tend to be oxidized to PdO or ionic Pd2+, which are less active than metallic Pd. It is believed that the metallic state of Pd is more important than Pd dispersion for improving the catalytic activity.Satsuma et al.49 prepared different oxides (CeO2, TiO2, Al2O3, ZrO2, and SiO2) supported Pd catalysts in order to investigate the controlling factors on CO oxidation. As shown in Fig. 16, the Pd/CeO2 sample shows the highest activity. Comparing the temperature at 50% conversion of CO, the light-off temperature follows the order of Pd/CeO2 < Pd/TiO2 < Pd/Al2O3 < Pd/ZrO2 ≤ Pd/SiO2. As shown in Table 2, Pd/TiO2, with the lowest Pd dispersion, has higher catalytic activity than Pd/Al2O3 at lower temperatures, indicating that Pd dispersion is not the most preferential activity-controlling factor of supported Pd catalysts. Further CO-TPR analysis indicates that the order of catalytic activity is fairly in agreement with the reduction temperature of supported Pd to metallic species. It suggests that the reduction of the Pd ion to the metallic state is the most important controlling factor for CO oxidation. By using the series of Pd/SiO2 as model catalysts, metallic Pd can be ensured to be the active sites for low-temperature CO oxidation, and the reaction follows the typical Langmuir–Hinshelwood mechanism. Further research on contribution of supports shows that the oxygen storage property of the samples and their catalytic activity for CO oxidation agree well, which suggests that the oxygen storage property of the support is another key factor for CO oxidation. Both CeO2 and TiO2 have outstanding oxygen release and storage capacities, thus the activities of Pd/CeO2 and Pd/TiO2 are better. In conclusion, there are two controlling factors for CO oxidation over supported Pd catalysts. One is the reducibility of PdO to Pd, and the other is the oxygen storage property of the support.
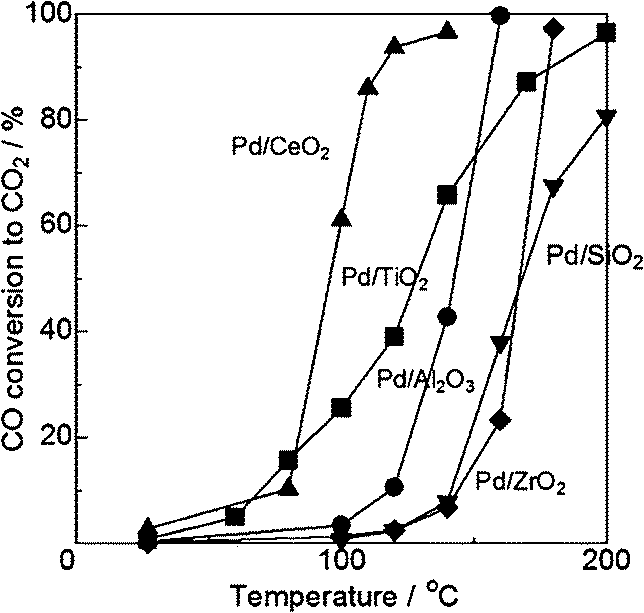 | ||
| Fig. 16 CO conversion as a function of temperature for Pd catalysts. Reproduced with permission from ref. 49. Copyright 2013, Elsevier. | ||
| Catalyst | Surface area (m2 g−1) | Pd dispersion (%) |
|---|---|---|
| Pd/CeO2 | 26 | 25 |
| Pd/TiO2 | 39 | 8.9 |
| Pd/Al2O3 | 111 | 54 |
| Pd/ZrO2 | 11 | 10 |
| Pd/SiO2 | 101 | 16 |
For its prominent redox property, the reducible support CeO2 plays an important role in the catalytic oxidation of CO. Generally, the metal–support interface is believed to be where the reaction takes place. However, no final conclusion is made about the nature of the active sites/phase. As for the Pd/CeO2 supported catalysts, the PdO species are considered as the active sites for the oxidation of CO.50–52 The PdO–CeO2 system may contain complex PdO species, such as free surface PdO species with different particle sizes and Pd2+ ions in the CeO2 matrix. Therefore, it is also difficult to identify the active sites in the PdO–CeO2 catalysts. Meng et al.53 prepared the catalyst PdO/Ce1−xPdxO2−δ, which contains both free surface PdO species and PdO species in Ce1−xPdxO2−δ solid solution, and the PdO/CeO2 catalyst, which contains only free surface PdO species. After the removal of the free surface PdO species by nitric acid, the catalyst Ce1−xPdxO2−δ which contains only Pd2+ species in the CeO2 matrix can also be obtained. Then, the catalytic activities of the samples for CO oxidation were examined, with the results shown in Fig. 17. Obviously, the PdO/Ce1−xPdxO2−δ catalyst has the highest activity, in which the complete CO conversion is realized at 160 °C. The Ce1−xPdxO2−δ and PdO/CeO2 catalysts have similar activity, with a complete CO conversion at 220 °C. What is more, the results indicate that, as for the activity of the individual PdO species, the free PdO species on the surface of the PdO/Ce1−xPdxO2−δ catalyst has the best performance with a reaction rate of 969.3 μmolCO gPd−1 s−1, and that on the surface of the PdO/CeO2 catalyst has a much lower reaction rate of 109.0 μmolCO gPd−1 s−1, while Pd2+ in the CeO2 lattice shows the lowest activity with a reaction rate of 13.2 μmolCO gPd−1 s−1. As a simplified reaction model, Scheme 2 is used to demonstrate the CO oxidation pathways on the samples. For the catalytic cycle, CO chemisorbed on the surface of PdO firstly, and then the chemisorbed CO migrated to the interface of PdO and the support. After the activation of O2 on the oxygen vacancies and the formation of active oxygen, the reaction between the chemisorbed CO at interface and the nearby active oxygen occurs. At last, the refilling of oxygen vacancies would be finished by gas-phase O2.
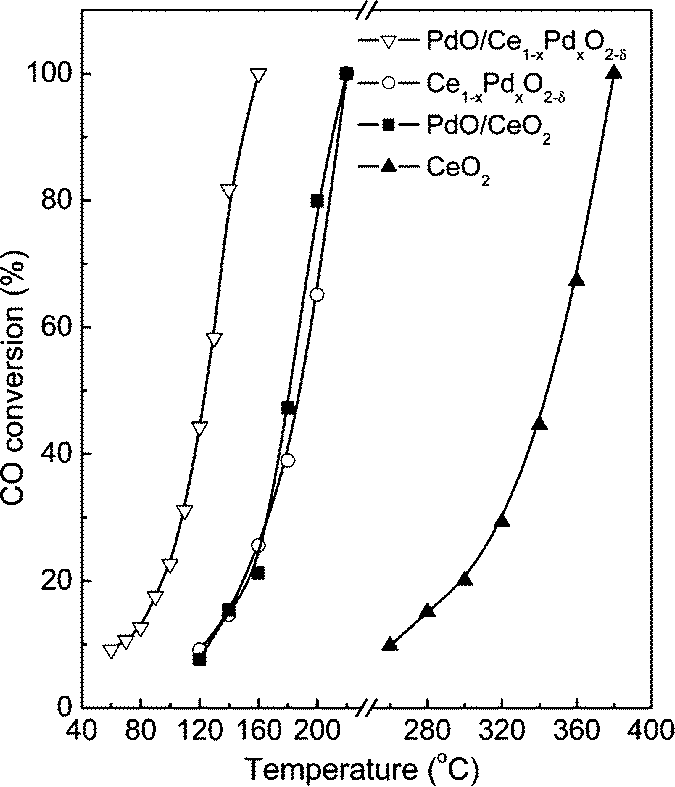 | ||
| Fig. 17 Catalytic activities of the samples for CO oxidation. Reproduced with permission from ref. 53. Copyright 2011, American Chemical Society. | ||
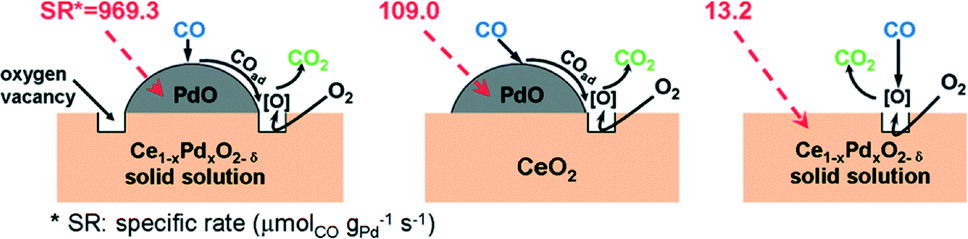 | ||
| Scheme 2 Reaction model of CO oxidation over catalysts. Reproduced with permission from ref. 53. Copyright 2011, American Chemical Society. | ||
Effect of the support
CO oxidation is a reaction that follows bi-functional reactions at the metal and support interface. The properties of the support have a significant influence on the activity of supported Pd catalysts. For CO oxidation, the reported Pd catalysts are usually supported on metal oxides, like SiO2, Al2O3, CeO2, TiO2, CoOx and MnOx. The metal oxides can be simply divided into two classes, active and inert. Known as oxygen storage materials, CeO2 and TiO2 possess redox properties. The Co–O bond in Co3O4 is relatively weak. The lattice oxygen makes Co3O4 show high reactivity for CO oxidation even at low temperature. As for Mn2O3, it contains a large amount of labile oxygen that is useful for CO oxidation. In comparison, SiO2 and Al2O3 are always considered as inert supporting materials. In addition, some new materials have been employed as supports, such as graphene or graphene oxide, in which oxygen-containing groups are rich. This would be helpful for CO oxidation. Apart from reactive oxygen, the basic property of the support is an important factor too.Kochubey et al.54 prepared SiO2, TiO2 and Al2O3 supported Pd clusters and investigated the influence of the support on the low-temperature activity of Pd. They found that the morphology and the spatial homogeneity of the Pd species on the support are different with different supports. Pd on SiO2 appears to have a fairly perfect structure, while the structure of Pd clusters on TiO2 and γ-Al2O3 is strongly disturbed by the interaction with the support. For Pd/TiO2, flat clusters of Pd are probably stabilized due to a structural matching with this support. For Pd/Al2O3, the absence of any preferential shape for Pd clusters on alumina could indicate a strong interaction of Pd with defect centres on the surface of a disordered spinel support. Furthermore, at saturation coverages, weakly bound CO (both linear and bridge-bonded) are the most reactive. Obviously, this depends upon the type of the support.
Li et al.55 prepared graphene supported palladium catalysts by the impregnation and hydrogen reduction methods. Pd nanoparticles are highly dispersed on the support. Through the density functional theory (DFT) study and the catalyst characterization using Raman and X-ray photoelectron spectroscopy, they found that the oxygen-containing groups in graphene are useful for stabilizing Pd clusters. It was concluded that the Pd clusters tended to take on a multilayer structure, in which the first layer mainly presented as the PdOx form, and the dominant component of other layers is the Pd metal. The resulting catalysts show excellent activity and high stability for CO oxidation, following the Langmuir–Hinshelwood mechanism. The Arrhenius plots of graphene supported palladium catalysts (Pd-GE) shown in Fig. 18 are basically consistent with those of the other Pd supported catalysts in the literature.56
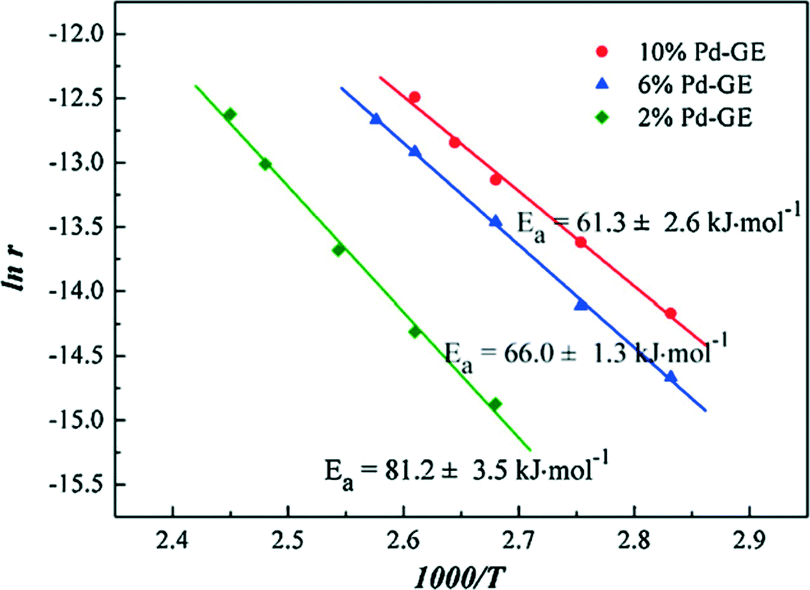 | ||
| Fig. 18 Arrhenius plots for CO oxidation over Pd-GE catalysts. Reproduced with permission from ref. 55. Copyright 2012, Elsevier. | ||
Liu et al.57 prepared a series of FeOx and Al2O3 supported Pd catalysts that exhibit high CO oxidation activity. They found that Pd catalysts on FeOx and Al2O3 show different Ea. The values are 34 kJ mol−1 and 71 kJ mol−1 for FeOx and Al2O3 supported Pd catalysts, respectively. Al2O3 can hardly activate O2 for CO oxidation, while adsorption and activation of molecular oxygen occur on the FeOx support. Furthermore, they found that the Fe2O3 supported Pd catalysts, calcined at high temperature, exhibit similar Ea to that of Pd/FeOx but far inferior catalytic activity. It means that different CO oxidation mechanisms or pathways might happen over FeOx and Fe2O3 supported Pd catalysts. The characterization indicates that Pd on Fe(OH)x can facilitate Fe3+ reduction and hydroxyl loss at relatively low temperatures, resulting in the production of large numbers of oxygen vacancies. The partially reduced FeOx support acts as an oxygen supply in CO oxidation. Then for CO oxidation over Pd/FeOx, Pd plays the role of active sites for CO activation and FeOx acts as active oxygen supply, which results in the low activation energy and high activity.
For CO oxidation, the basic property of the support may induce the adjustment in the electronic structure of the metallic NPs and directly alter the adsorption intensity of surface CO molecules. This will further change the catalytic activity of the supported catalysts. Liu et al.58 prepared SiO2, TiO2 and MgO supported Pd nanocube catalysts and evaluated the effect of the basic property of the support. As shown in Fig. 19, the Pd/MgO sample has the best catalytic activity for CO oxidation. To elucidate the origin of the difference, they investigated the surface acid–base properties of the supports and the CO adsorption behaviours of the catalysts. The results of the “temperature-dependent” and the “time-dependent” DRIFT spectra indicate that CO adsorption on the Pd/MgO catalyst is the strongest with the largest adsorption amount. Among the three supports, MgO is the most basic, which can enhance the amount and stability of CO adsorption on (100) planes of the Pd nanocubes. In CO oxidation, a “volcano-shaped correlation” curve exists between the CO adsorption behaviour and the catalytic activity. For Pd/MgO, the CO adsorption approaches the top of the “volcano-shaped” curve and the CO oxidation activity is better than those of the other two samples.
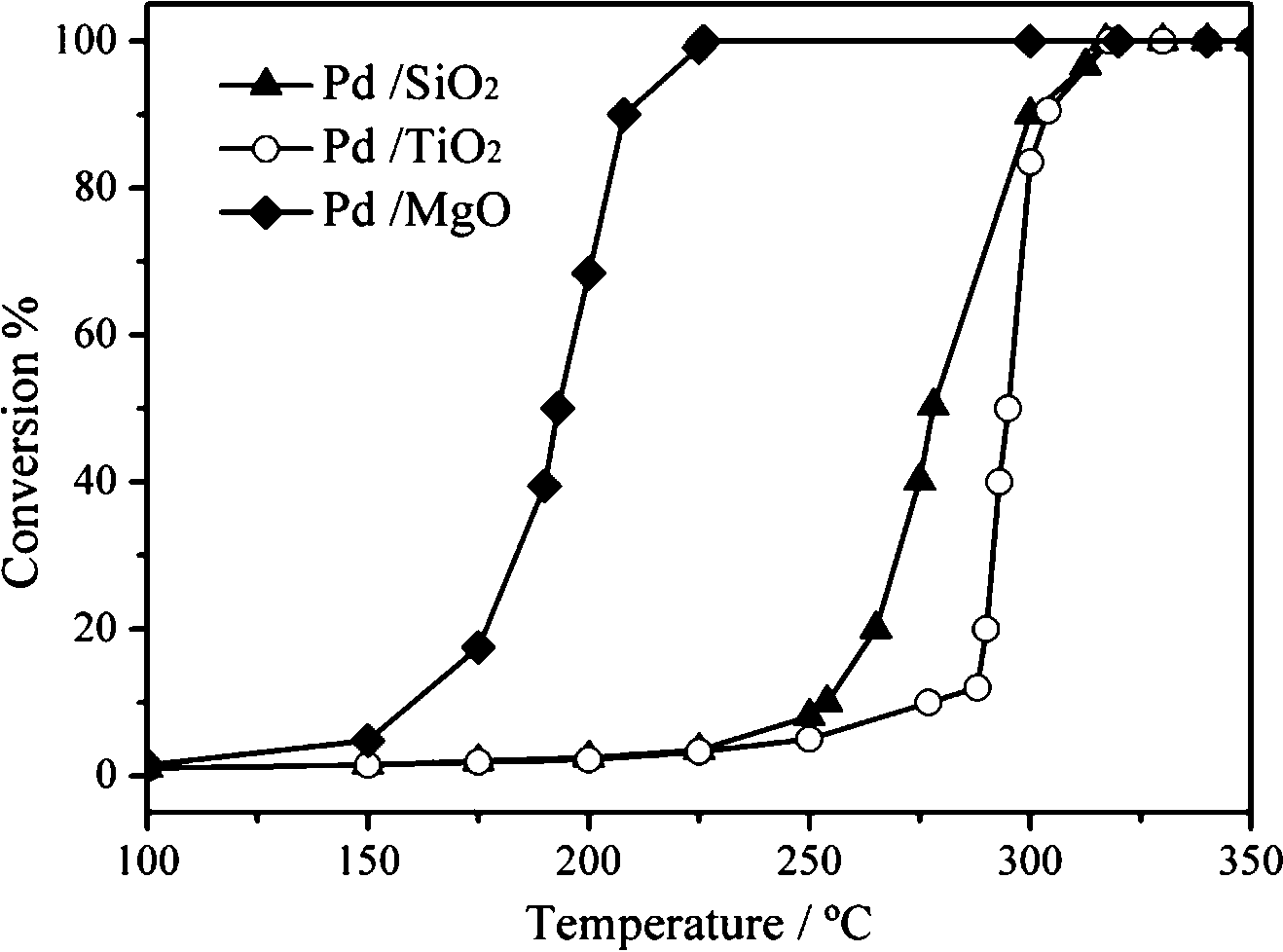 | ||
| Fig. 19 CO oxidation activity over the supported Pd catalysts. Reproduced with permission from ref. 58. Copyright 2014, Elsevier. | ||
Jin et al.59 synthesized a series of highly ordered mesoporous metal oxides (meso-MOx) by a nanoreplication method, such as CeO2, Co3O4, Mn2O3, SnO2 and TiO2, and investigated the CO oxidation activity of meso-MOx supported Pd catalysts (Pd/meso-MOx). As shown in Fig. 20, meso-Co3O4 and meso-Mn2O3 exhibit much higher catalytic activities for CO oxidation than meso-CeO2, meso-SnO2 and meso-TiO2. The temperature at which CO is converted to CO2 completely follows the order of Co3O4 > Mn2O3 > CeO2 > SnO2 > TiO2. After the loading of Pd, the light-off curves for CO oxidation are shown in Fig. 21(a). Compared with the corresponding mesoporous metal oxides, the CO oxidation activities of the Pd/meso-MOx samples all increased, with Pd/meso-Co3O4 showing the highest activity. Meanwhile, resulting from the well-defined pore size and higher surface areas of meso-MOx, the Pd/meso-MOx samples also exhibit higher activities than the corresponding Pd/bulk-MOx samples. It indicates that the properties of the support would affect the activity of the catalyst. However, the activity amplifications of Pd/meso-Co3O4 and Pd/meso-Mn2O3 are slight, while those of Pd/meso-CeO2, Pd/meso-SnO2 and Pd/meso-TiO2 are significant. The results of XPS indicate that a strong interaction exists between Pd species and meso-CeO2/meso-SnO2/meso-TiO2, while a weak interaction between Pd species and meso-Co3O4 and meso-Mn2O3 is observed. It suggests that there is a synergistic effect between the Pd species and the supports. The nature of the support strongly affects the catalytic activities of the samples. Furthermore, the activities of the supported Pd catalysts can be improved by a pre-reduction treatment. As shown in Fig. 21(b), after H2 pre-treatment, the catalytic activities of the samples for CO oxidation all increased. Among the samples, Pd/meso-CeO2 exhibits the highest activity after the promotion via H2 pre-treatment. It was thought that the pre-treatment can enhance the metal–support interaction and facilitate the formation of oxygen vacancies and hydroxyl groups, which will influence the electronic state of surface active sites. From the above results, the authors estimated that the activity of supported Pd catalyst is dependent on the type of support and the interaction between the metal and the support, and H2 pre-treatment might enhance the metal–support interaction.
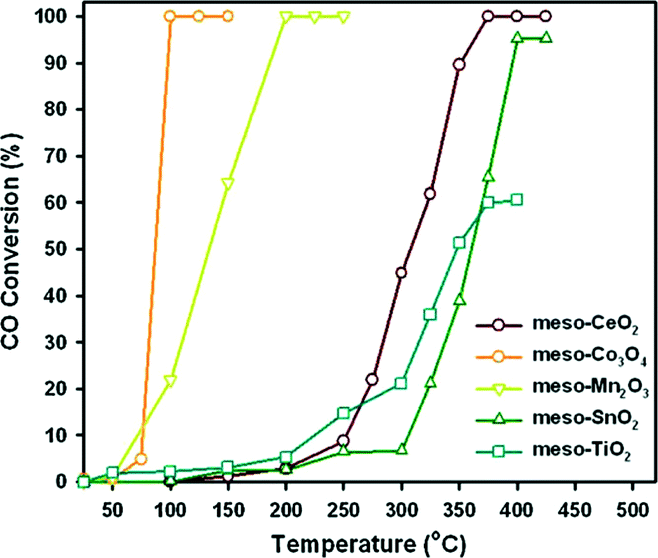 | ||
| Fig. 20 CO conversion over meso-MOx catalysts. Reproduced with permission from ref. 59. Copyright 2012, Elsevier. | ||
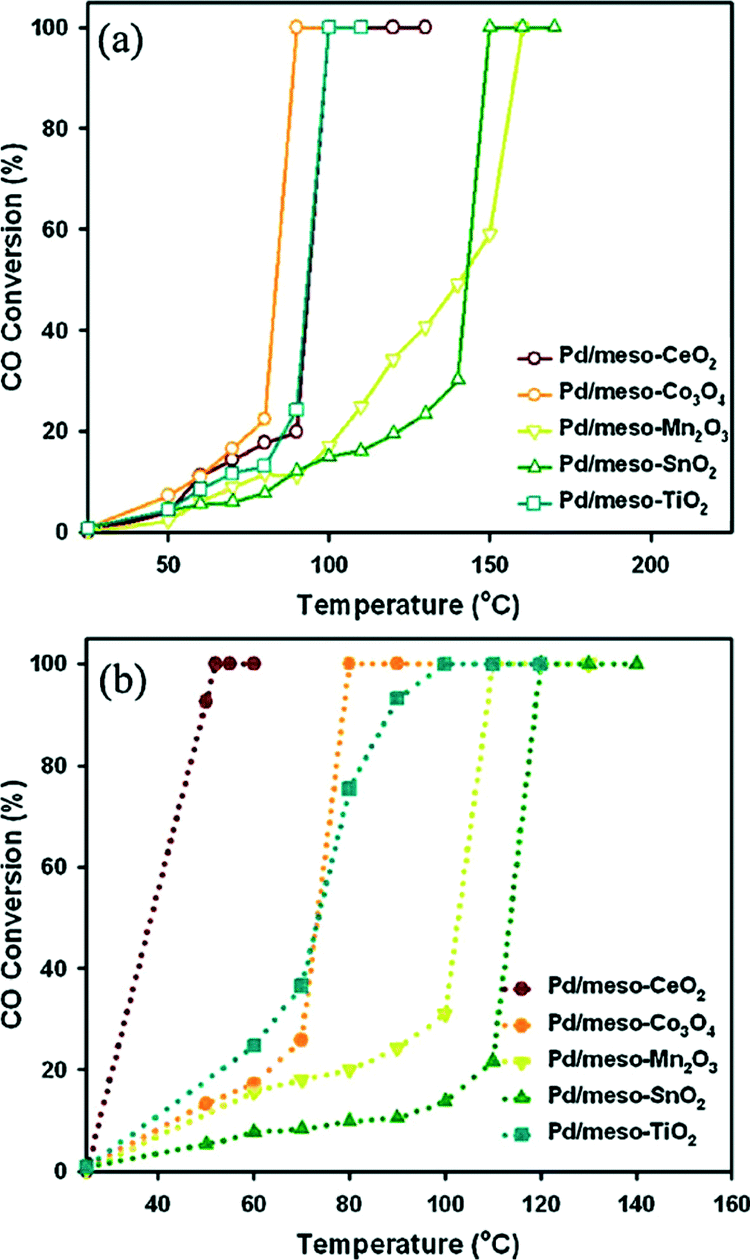 | ||
| Fig. 21 Light-off curves for CO oxidation over Pd/meso-MOx catalysts (a) without H2 pre-treatment and (b) with H2 pre-treatment. Reproduced with permission from ref. 59. Copyright 2012, Elsevier. | ||
Conclusions and outlook
In this perspective, the factors influencing the activity of supported Pd catalysts for CO oxidation are discussed. Additives, such as ceria, zinc, alkaline earth metals, and so on, can be used as promoters to enhance the activity of catalyst for CO oxidation. They may lower the binding energy of CO, enhance the thermal durability of the support, or improve the dispersion of Pd species. The preparation method also plays a crucial role. Different preparation methods will change the distribution of Pd particles, as well as the interaction between the Pd species and the support. The Pd structure decides the exposed facets on the surface, which strongly influences the performance in CO oxidation. The properties of the support are another key role in deciding the activity of the catalyst. The basic property and oxygen storage capacity both have important influence.Pd-based catalysts are very active and important not only for CO oxidation but also for many other reactions.60–64 Pd-based catalysts have been extensively applied in industrial productions. CO oxidation is an excellent model reaction for studying the structure, metal–support interaction and other issues of various Pd-based catalysts. It is simple and can be easily operated with low cost. Carbon monoxide itself is an excellent probe molecule for catalyst characterization. Density functional theoretical studies have been a convenient tool for the study.64,65 We can expect further increase in publications and applications of Pd-based catalysts.
However, even for simple CO oxidation, many challenges regarding the use of Pd catalysts remain. The low-temperature activity of Pd catalysts is not impressive. The role of the metal–support interface is still not clear. The defect control is not easy. The geometrical and electronic roles of alloyed Pd catalysts need to be further studied. It is still very difficult to in situ characterize the reaction. A significant effort is being made towards the size and structure control of Pd catalysts but it still remains as a big challenge to chemists worldwide. High activity of single atomic Pd catalysts or Pd cluster catalysts has been expected but we need to find a way to achieve it. The catalysts with one-dimensional or two-dimensional Pd nanoparticles have attracted much attention but their progress is very slow. Innovation in catalyst preparation is immediately needed.
Acknowledgements
The support from the National Natural Science Foundation of China (#91334206) is appreciated. The authors are thankful for the help of Prof. Qiang Xu from AIST, Japan.Notes and references
- Y. X. Shen, Y. Guo, L. Wang, Y. Q. Wang, Y. L. Guo, X. Q. Gong and G. Z. Lu, Catal. Sci. Technol., 2011, 1, 1202 CAS.
- L. Q. Liu, F. Zhou, L. G. Wang, X. J. Qi, F. Shi and Y. Q. Deng, J. Catal., 2010, 274, 1 CrossRef CAS PubMed.
- Y. Zhao, C. L. Zhong and C.-J. Liu, Catal. Commun., 2013, 38, 74 CrossRef CAS PubMed.
- Q. X. Cai, X. D. Wang and J. G. Wang, J. Phys. Chem. C, 2013, 117, 21331 CAS.
- H. B. Na, T. Zhu and Z. M. Liu, Catal. Sci. Technol., 2014, 7, 2051 Search PubMed.
- B. T. Qiao, A. Q. Wang, X. F. Yang, L. F. Allard, Z. Jiang, Y. T. Cui, J. Y. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634 CrossRef CAS PubMed.
- J. J. Wang, Z. Y. Wang and C.-J. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 12860 CAS.
- R. V. Gulyaev, E. M. Slavinskaya, S. A. Novopashin, D. V. Smovzh, A. V. Zaikovskii, D. Yu. Osadchii, O. A. Bulavchenko, S. V. Korenev and A. I. Boronin, Appl. Catal., B, 2014, 147, 132 CrossRef CAS PubMed.
- K. Shimizu, Y. Kamiya, K. Osaki, H. Yoshidad and A. Satsuma, Catal. Sci. Technol., 2012, 2, 767 CAS.
- N. Li, Q. Y. Chen, L. F. Luo, W. X. Huang, M. F. Luo, G. S. Hu and J. Q. Lu, Appl. Catal., B, 2013, 142–143, 523 CrossRef CAS PubMed.
- Y. X. Shen, Y. Guo, L. Wang, Y. Q. Wang, Y. L. Guo, X. Q. Gong and G. Z. Lu, Catal. Sci. Technol., 2011, 1, 1202 CAS.
- M. Kurnatowska, L. Kepinski and W. Mista, Appl. Catal., B, 2012, 117–118, 135 CrossRef CAS PubMed.
- S. T. Chen, R. Si, E. Taylor, J. Janzen and J. Y. Chen, J. Phys. Chem. C, 2012, 116, 12969 CAS.
- H. Chen, X. L. Tong and Y. D. Li, Appl. Catal., A, 2009, 370, 59 CrossRef CAS PubMed.
- C. J. Tang, J. F. Sun, X. J. Yao, Y. Cao, L. C. Liu, C. Y. Ge, F. Gao and L. Dong, Appl. Catal., B, 2014, 146, 201 CrossRef CAS PubMed.
- J. Q. Lu, C. X. Sun, N. Li, A. P. Jia and M. F. Luo, Appl. Surf. Sci., 2013, 287, 124 CrossRef CAS PubMed.
- U. R. Pillai and S. Deevi, Appl. Catal., B, 2006, 65, 110 CrossRef CAS PubMed.
- E. M. C. Alayon, J. Singh, M. Nachtegaal, M. Harfouche and J. A. van Bokhoven, J. Catal., 2009, 263, 228 CrossRef CAS PubMed.
- P. J. Berlowitz, C. H. F. Peden and D. W. Goodman, J. Phys. Chem., 1988, 92, 5213 CrossRef CAS.
- M. S. Chen, Y. Cai, Z. Yan, K. K. Gath, S. Axnanda and D. Wayne Goodman, Surf. Sci., 2007, 601, 5326 CrossRef CAS PubMed.
- E. D. Park and J. S. Lee, J. Catal., 1999, 186, 1 CrossRef CAS.
- A. I. Boronin, E. M. Slavinskaya, I. G. Danilova, R. V. Gulyaev, Y. I. Amosov, P. A. Kuznetsov, I. A. Polukhina, S. V. Koscheev, V. I. Zaikovskii and A. S. Noskov, Catal. Today, 2009, 144, 201 CrossRef CAS PubMed.
- E. L. Wilson, Q. Chen, W. A. Brown and G. J. Thornton, J. Phys. Chem. C, 2007, 111, 14215 CAS.
- H. Q. Zhu, Z. F. Qin, W. J. Shan, W. J. Shen and J. G. Wang, J. Catal., 2004, 225, 267 CrossRef CAS PubMed.
- R. J. Farrauto and R. M. Heck, Catal. Today, 1999, 51, 351 CrossRef CAS.
- R. S. Johnson, A. DeLaRiva, V. Ashbacher, B. Halevi, C. J. Villanueva, G. K. Smith, S. Lin, A. K. Datye and H. Guo, Phys. Chem. Chem. Phys., 2013, 15, 7768 RSC.
- K. Tanikawa and C. Egawa, Appl. Catal., A, 2011, 403, 12 CrossRef CAS PubMed.
- J. R. Gaudet, A. de la Riva, E. J. Peterson, T. Bolin and A. K. Datye, ACS Catal., 2013, 3, 846 CrossRef CAS.
- S. Y. Shan, J. Luo, L. F. Yang and C. J. Zhong, Catal. Sci. Technol., 2014, 4, 3570 CAS.
- A. K. Singh and Q. Xu, ChemCatChem, 2013, 5, 652 CrossRef CAS.
- F. Gao and D. W. Goodman, Chem. Soc. Rev., 2012, 41, 8009 RSC.
- F. Morfin, S. Nassreddine, J. L. Rousset and L. Piccolo, ACS Catal., 2012, 2, 2161 CrossRef CAS.
- S. Y. Shan, V. Petkov, L. F. Yang, J. Luo, P. Joseph, D. Mayzel, B. Prasai, L. Y. Wang, M. Engelhard and C. J. Zhong, J. Am. Chem. Soc., 2014, 136, 7140 CrossRef CAS PubMed.
- R. W. Scott, C. Sivadinarayana, O. M. Wilson, Z. Yan, D. W. Goodman and R. M. Crooks, J. Am. Chem. Soc., 2005, 127, 1380 CrossRef CAS PubMed.
- A. M. Venezia, L. F. Liotta, G. Pantaleo, V. La Parola, G. Deganello, A. Beck, Z. Koppany, K. Frey, D. Horvath and L. Guczi, Appl. Catal., A, 2003, 251, 359 CrossRef CAS.
- H. C. Ham, J. A. Stephens, G. S. Hwang, J. Han, S. W. Nam and T. H. Lim, J. Phys. Chem. Lett., 2012, 3, 566 CrossRef CAS.
- H. Y. Kim and G. Henkelman, ACS Catal., 2013, 3, 2541 CrossRef CAS.
- J. Xu, T. White, P. Li, C. H. He, J. G. Yu, W. K. Yuan and Y. F. Han, J. Am. Chem. Soc., 2010, 132, 10398 CrossRef CAS PubMed.
- V. P. Santos, S. A. C. Carabineiro, P. B. Tavares, M. F. R. Pereira, J. J. M. Órfão and J. L. Figueiredo, Appl. Catal., B, 2010, 99, 198 CrossRef CAS PubMed.
- Y. Zhou, Z. H. Xiang, D. P. Cao and C. J. Liu, Chem. Commun., 2013, 49, 5633 RSC.
- Y. Zhou, Z. H. Xiang, D. P. Cao and C. J. Liu, Ind. Eng. Chem. Res., 2014, 53, 1359 CrossRef CAS.
- H. P. Wang and C. J. Liu, Appl. Catal., B, 2011, 106, 672 CrossRef CAS PubMed.
- A. S. Ivanova, E. M. Slavinskaya, R. V. Gulyaev, V. I. Zaikovskii, O. A. Stonkus, I. G. Danilova, L. M. Plyasova, I. A. Polukhina and A. I. Boronin, Appl. Catal., B, 2010, 97, 57 CrossRef CAS PubMed.
- S. Hinokuma, H. Fujii, M. Okamoto, K. Ikeue and M. Machida, Chem. Mater., 2010, 22, 6183 CrossRef CAS.
- W. E. Kaden, W. A. Kunkel, M. D. Kane, F. S. Roberts and S. L. Anderson, J. Am. Chem. Soc., 2010, 132, 13097 CrossRef CAS PubMed.
- R. Wang, H. He, L. C. Liu, H. X. Dai and Z. Zhao, Catal. Sci. Technol., 2012, 2, 575 CAS.
- M. S. Jin, H. Y. Liu, H. Zhang, Z. X. Xie, J. Y. Liu and Y. N. Xia, Nano Res., 2011, 4, 83 CrossRef CAS PubMed.
- S. N. Pavlova, V. A. Sadykov, V. A. Razdobarov and E. A. Paukshtis, J. Catal., 1996, 161, 507 CrossRef CAS.
- A. Satsuma, K. Osaki, M. Yanagihara, J. Ohyama and K. Shimizu, Appl. Catal., B, 2013, 132–133, 511 CrossRef CAS PubMed.
- A. Hadi and I. I. Yaacob, Catal. Today, 2004, 96, 165 CrossRef CAS PubMed.
- S. H. Oh and G. B. Hoflund, J. Phys. Chem. A, 2006, 110, 7609 CrossRef CAS PubMed.
- M. F. Luo, Z. Y. Pu, M. He, J. Jin and L. Y. Jin, J. Mol. Catal. A: Chem., 2006, 260, 152 CrossRef CAS PubMed.
- L. Meng, A. P. Jia, J. Q. Lu, L. F. Luo, W. X. Huang and M. F. Luo, J. Phys. Chem. C, 2011, 115, 19789 CAS.
- D. I. Kochubey, S. N. Pavlova, B. N. Novgorodov, G. N. Kryukova and V. A. Sadykov, J. Catal., 1996, 161, 500 CrossRef CAS.
- Y. Z. Li, Y. Yu, J. G. Wang, J. Song, Q. Li, M. D. Dong and C. J. Liu, Appl. Catal., B, 2012, 125, 189 CrossRef CAS PubMed.
- L. Liu, F. Zhou, L. Wang, X. Qi, F. Shi and Y. Deng, J. Catal., 2010, 274, 1 CrossRef CAS PubMed.
- L. Q. Liu, F. Zhou, L. G. Wang, X. J. Qi, F. Shi and Y. Q. Deng, J. Catal., 2010, 274, 1 CrossRef CAS PubMed.
- X. J. Liu, R. Wang, L. Y. Song, H. He, G. Z. Zhang, X. H. Zi and W. G. Qiu, Catal. Commun., 2014, 46, 213 CrossRef CAS PubMed.
- M. S. Jin, J. N. Park, J. K. Shon, J. H. Kim, Z. H. Li, Y. K. Park and J. M. Kim, Catal. Today, 2012, 185, 183 CrossRef CAS PubMed.
- D. B. Pacardo and MR Knecht, Catal. Sci. Technol., 2013, 3, 745 CAS.
- H. C. Ma, W. Cao, Z. K. Bao and Z. Q. Lei, Catal. Sci. Technol., 2012, 2, 2291 CAS.
- F. F. Lu, D. H. Sun, J. L. Huang, M. M. Du, F. Yang, H. M. Chen, Y. L. Hong and Q. B. Li, ACS Sustainable Chem. Eng., 2014, 2, 1212 CrossRef CAS.
- C. J. Liu, Y. Zhao, Y. Z. Li, D. S. Zhang, Z. Chang and X. H. Bu, ACS Sustainable Chem. Eng., 2014, 2, 3 CrossRef CAS.
- J. Y. Ye, C. J. Liu and Q. F. Ge, Phys. Chem. Chem. Phys., 2012, 14, 16660 RSC.
- D. Vogel, C. Spiel, Y. Suchorski, A. Trinchero, R. Schlogl, H. Gronbeck and G. Rupprechter, Angew. Chem., Int. Ed., 2012, 51, 10041 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |