Fe-containing polyimide-based high-performance ORR catalysts in acidic medium: a kinetic approach to study the durability of catalysts
Received
2nd August 2014
, Accepted 5th September 2014
First published on 5th September 2014
Abstract
The ORR activity and durability of Fe-containing non-precious N-doped carbon catalysts in acidic medium were studied using a rotating ring-disk electrode voltammetry and XPS technique. The catalysts (Fe/PI) were synthesised from the pyrolysis of the Fe(acac)3 and polyimide nanoparticle (PI) mixture. The catalytic activity and durability of Fe/PI are superior to that of the conventional phthalocyanine-based catalyst. The onset potential of ORR was 0.915 V vs. RHE in 0.5 M H2SO4 which is very close to that of a commercially available Pt/C catalyst. The Fe/PI catalyst sustains its activity and stability even after 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 110 repeating potential steps between 0.6 and 1.0 V vs. RHE in O2-saturated 0.5 M H2SO4 and 1110 potential cycles between 1.0 and 1.5 V vs. RHE in argon-saturated 0.5 M H2SO4, respectively. The kinetic and mechanistic analyses of the ORR on this catalyst indicate that the ORR, as a whole, follows a 4-electron (parallel) pathway. The N 1s XPS spectra before and after the durability test indicate that pyridinic and graphite-like nitrogens take part in improving the ORR activity, whereas nitrogen oxides have a negative role in the ORR. The loss of ORR activity was observed after the acid washing of the catalysts, which suggests the role of Fe in the overall 4-electron reduction of O2.
110 repeating potential steps between 0.6 and 1.0 V vs. RHE in O2-saturated 0.5 M H2SO4 and 1110 potential cycles between 1.0 and 1.5 V vs. RHE in argon-saturated 0.5 M H2SO4, respectively. The kinetic and mechanistic analyses of the ORR on this catalyst indicate that the ORR, as a whole, follows a 4-electron (parallel) pathway. The N 1s XPS spectra before and after the durability test indicate that pyridinic and graphite-like nitrogens take part in improving the ORR activity, whereas nitrogen oxides have a negative role in the ORR. The loss of ORR activity was observed after the acid washing of the catalysts, which suggests the role of Fe in the overall 4-electron reduction of O2.
1. Introduction
Recently, many researchers have focused on developing non-precious metal catalysts for oxygen reduction reaction (ORR) in polymer electrolyte membrane fuel cells in order to overcome the cost and scarcity of platinum and its alloy catalysts. Non-precious metal nitrogen-doped carbon catalysts have been proposed as promising catalysts for this purpose.1–18 The ORR activity of these catalysts is sometimes found to be better than that of platinum-based catalysts in alkaline media but is inferior to that of platinum-based catalysts in acidic media. Moreover, their metal-free catalysts are active only in alkaline media.17,19–24 Early studies were focused on heat-treated transition metal macrocyclic complexes,25–28 and later, nitrogen-containing organic polymers, especially polyaniline (PANI), have been used as precursors29–32 for preparing the effective ORR catalysts. Recently, many researchers have confined their attention to the heat treatment of small nitrogen-containing organic molecules33–41 and also to N-doped carbon nanotubes (NCNT)4,10,42–46 and N-doped graphene (NGr)11,46–61 for obtaining better ORR activity in acidic as well as in alkaline media. In addition, our research group has recently reported a very active non-precious metal catalyst prepared from polyimide nanoparticles,62 which is one of the catalysts exhibiting the best fuel cell performance in the world and can be prepared from ordinary chemicals, such as pyromellitic acid dianhydride (PMDA) and 4,4′-oxydianiline (ODA), which are the most common polyimide precursors.
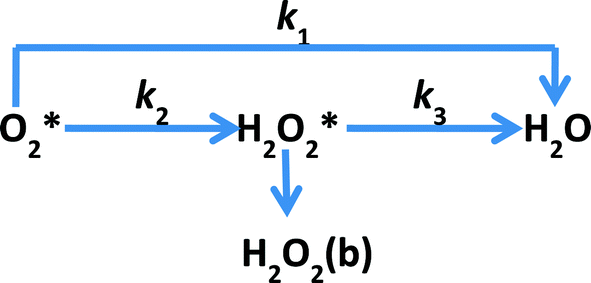
Studying the kinetics of such highly active catalysts is very useful in understanding the mechanism of ORR and further improving the catalytic performance. Rotating ring-disc electrode voltammetry is often used to study the kinetics of the ORR. In general, the mechanism of ORR is considered to follow one of the two different reduction pathways or both of them during the reduction of O2 to water as proposed by Damjanovic et al.63 (Scheme 1). The two pathways of ORR are the direct reduction involving 4-electron transfer (direct pathway) and the stepwise reduction via a H2O2 intermediate involving 2-electron transfer in each step (sequential pathway). This model was proposed with three assumptions: (a) no catalytic decomposition of H2O2, (b) adsorption–desorption of H2O2 is fast and in equilibrium and (c) re-oxidation of H2O2 is negligible.
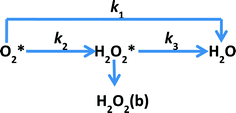 |
| | Scheme 1 Proposed model for the electrochemical reduction of O2 by Damjanovic et al.63 Indices b and * designate the bulk species and the species in the vicinity of the disk electrode, respectively. | |
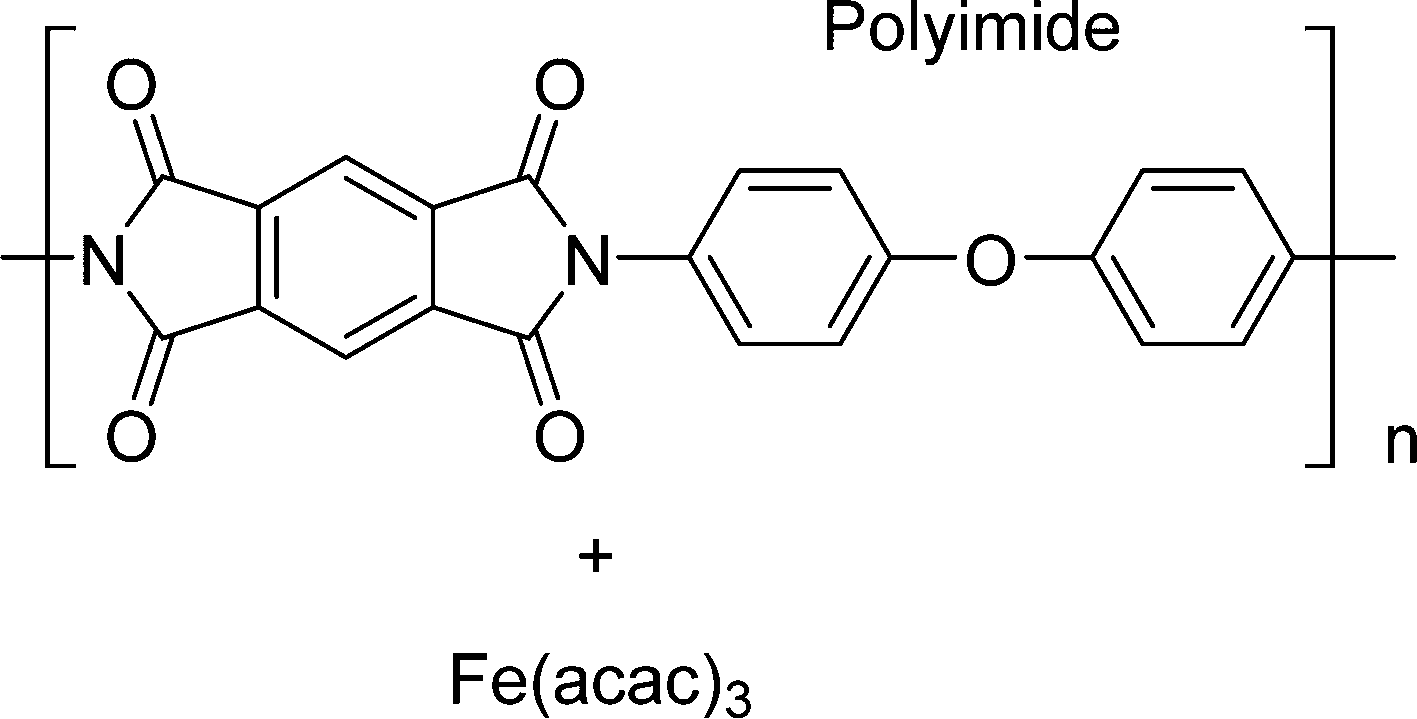
In this work, using rotating ring-disk electrode voltammetry, we have studied the ORR activity of the Fe/PI and acid-washed Fe/PI (hereafter referred to as Fe/PI-aw) catalysts. Fig. 1 shows the precursors for the Fe/PI catalyst: polyimide and Fe(acac)3 as carbon–nitrogen and Fe sources, respectively. The effect of acid washing of the Fe/PI catalyst on its durability and activity in ORR is examined. The durability of the catalysts is studied in two different modes of potential application with reference to those used in the durability test of Pt/C catalysts.64,65 Based on the individual rate constants of ORR (k1, k2 and k3) estimated according to the Damjanovic model63 before and after the durability test, the ORR performance and durability of Fe/PI and Fe/PI-aw catalysts are discussed. In addition, the possibility of the disproportionation reaction of H2O2 catalysed by the porous Fe/PI and Fe/PI-aw catalysts is also discussed.
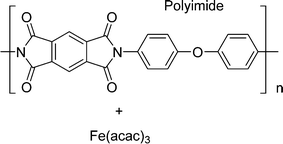 |
| | Fig. 1 The chemical structures of the Fe and N sources for Fe/PI and Fe/PI-aw. | |
2. Experimental
2.1 Preparation of Fe-containing polyimide catalyst (Fe/PI)
Fe/PI was prepared according to the recently reported procedure.62 A solution of pyromellitic acid dianhydride (PMDA) in acetone was added to a solution of 4,4′-oxydianiline (ODA) and tris(acetylacetonato) iron(III) (Fe(acac)3, >98.0%, Dojindo) in acetone (>99.0%, Wako). The amount of Fe species was 2 wt% relative to the resulting polyimide. The mixture was stirred at 0 °C for 30 min and the solvent was removed with a rotary evaporator to collect poly(amic acid). The curing reaction was conducted by heating the poly(amic acid) at 200 °C under vacuum to obtain fine particles of polyimide. The thus-obtained composite was converted into Fe/PI by multistep pyrolysis.66 The precursor was heated at 600 °C under nitrogen atmosphere for 5 h and washed with concentrated HCl. Then, the product was again heated at 800 °C under ammonia atmosphere for 1 hour and washed with concentrated HCl. Finally, it was further heated at 1000 °C under ammonia atmosphere for 1 hour to obtain Fe/PI. Fe/PI-aw was prepared by washing Fe/PI with concentrated HCl.
2.2 Preparation of Fe/PI-modified GC electrodes
The composition and coating of the Fe/PI ink play an important role in preparing a uniform thin layer of Fe/PI on the GC electrode surface. 5 mg of each Fe/PI was placed into a 1 ml bottle followed by the addition of 150 μl of ethanol, 150 μl of Milli-Q water (18.2 MΩ) and 50 μl of 5 wt% Nafion. This mixture was sonicated in an ice-cold water bath for 1 hour. 4 μl of the uniformly dispersed ink was taken using a micropipette and transferred onto a clean, polished GC disk electrode, and the whole surface of the GC disk electrode was covered with the ink in uniform thickness. (Care must be taken that the ink should cover the GC disk of the rotating ring-disk electrode completely and it should not flow into the Teflon gap between the ring and disk electrodes.) The electrode was then dried under low flow of argon environment until it dried completely. The thus-prepared Fe/PI-modified GC electrodes contained 0.2 mg of Fe/PI per cm2 with 0.1 mg of the Nafion binder per cm2.
2.3 Electrochemical measurements
All the measurements were carried out in 0.5 M H2SO4 using a CHI bi-potentiostat (model 700D) in a custom-built electrochemical cell. A rotating ring-disk electrode (RRDE) with a GC disk (6 mm in diameter, 0.283 cm2) and a Pt ring (0.252 cm2), a carbon plate (~7 cm2) and Ag/AgCl (sat. KCl) were used as a working electrode, a counter electrode and a reference electrode, respectively. The working electrode was polished with 1 μm and 0.06 μm alumina powder to mirror finish and sonicated to remove the alumina particles before each experiment. After the disk electrode was modified with each Fe/PI, its surface was completely wetted with 0.5 M H2SO4 without any air droplets on the surface. The pretreatment of the Fe/PI-modified GC disk electrode was carried out by repeating potential scan at 0.05 V s−1 between 1.1 and 0.05 V in argon-saturated 0.5 M H2SO4 until stable voltammograms were obtained. Similarly, the Pt ring electrode was also treated by potential scanning at 0.05 V s−1 between the onset potential of hydrogen evolution (ca. 0 V) and 1.2 V until a voltammogram characteristic of a clean Pt electrode was obtained. The thus-pretreated electrodes were used to study the ORR by scanning the disk electrode potential from 1.1 to 0.05 V in O2-saturated 0.5 M H2SO4 at a scan rate of 5 mV s−1 at various electrode rotation speeds of 400 to 3600 rpm. At the same time, the Pt ring electrode was polarized at a constant potential of 1.2 V to measure the amount of H2O2 formed during the ORR at the disk electrode (the collection efficiency is 0.37). The blank response was also measured in argon-saturated 0.5 M H2SO4 under the same experimental conditions used above. Electrode potential was finally corrected to a reversible hydrogen electrode (RHE) by adding 0.227 V, estimated from the pH of the 0.5 M H2SO4, to the electrode potential measured against Ag/AgCl (sat. KCl). Unless otherwise mentioned, the potentials in this article are referred to as a RHE.
The durability or stability test of Fe/PI was carried out in two different modes of potential application: the stability of the “carbon support” in Fe/PI was tested using the protocol based on the potential cycling between (typically using 10110 and 1110 cycles) 1.0 and 1.5 V at a scan rate of 0.5 V s−1 in argon-saturated 0.5 M H2SO4. On the other hand, the stability in the Fe/PI′s ORR activity was tested by repeating the potential step between the ORR-off region (1.0 V for 3 s) and the ORR-on region (0.6 V for 3 s) for 11110 times (max). Hereafter, the former and latter durability tests will be called start–stop durability (SSD) test and loading durability (LD) test, respectively.64,65
2.4 XPS measurements
For XPS measurements, the Fe/PI samples, which were prepared by a coating procedure similar to that used in the preparation of Fe/PI-modified GC disk electrodes for their electrochemical measurements, were placed under ultra-high vacuum conditions (10−6 Pa) where an ESCA3400 electron spectrometer (SHIMADZU) with an unmonochromatized X-ray source [Mg Kα (hν = 1253.6 eV) anode, emission current of 20 mA and acceleration voltage of 10 kV] was employed, operating at 200 W. The photoelectron take-off angle was set to 75°, the analyzer pass energy was set to 75 eV, and the XPS spectra were corrected to the internal reference spectra of C 1s at 284.5 eV, being the binding energy of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of the Fe/PI in order to compensate for the electrostatic charging. Then, the core level spectra were carefully deconvoluted to investigate the different nitrogen species, i.e., pyridinic nitrogen, pyrrolic nitrogen, graphite-like nitrogen and nitrogen oxide on the Fe/PI and Fe/PI-aw surfaces. The XPS spectra of the N core levels were fitted with partial Gaussian functions (usually around 50–80% and the rest being Lorentzian).
C bond of the Fe/PI in order to compensate for the electrostatic charging. Then, the core level spectra were carefully deconvoluted to investigate the different nitrogen species, i.e., pyridinic nitrogen, pyrrolic nitrogen, graphite-like nitrogen and nitrogen oxide on the Fe/PI and Fe/PI-aw surfaces. The XPS spectra of the N core levels were fitted with partial Gaussian functions (usually around 50–80% and the rest being Lorentzian).
3. Results and discussion
3.1 XPS analysis
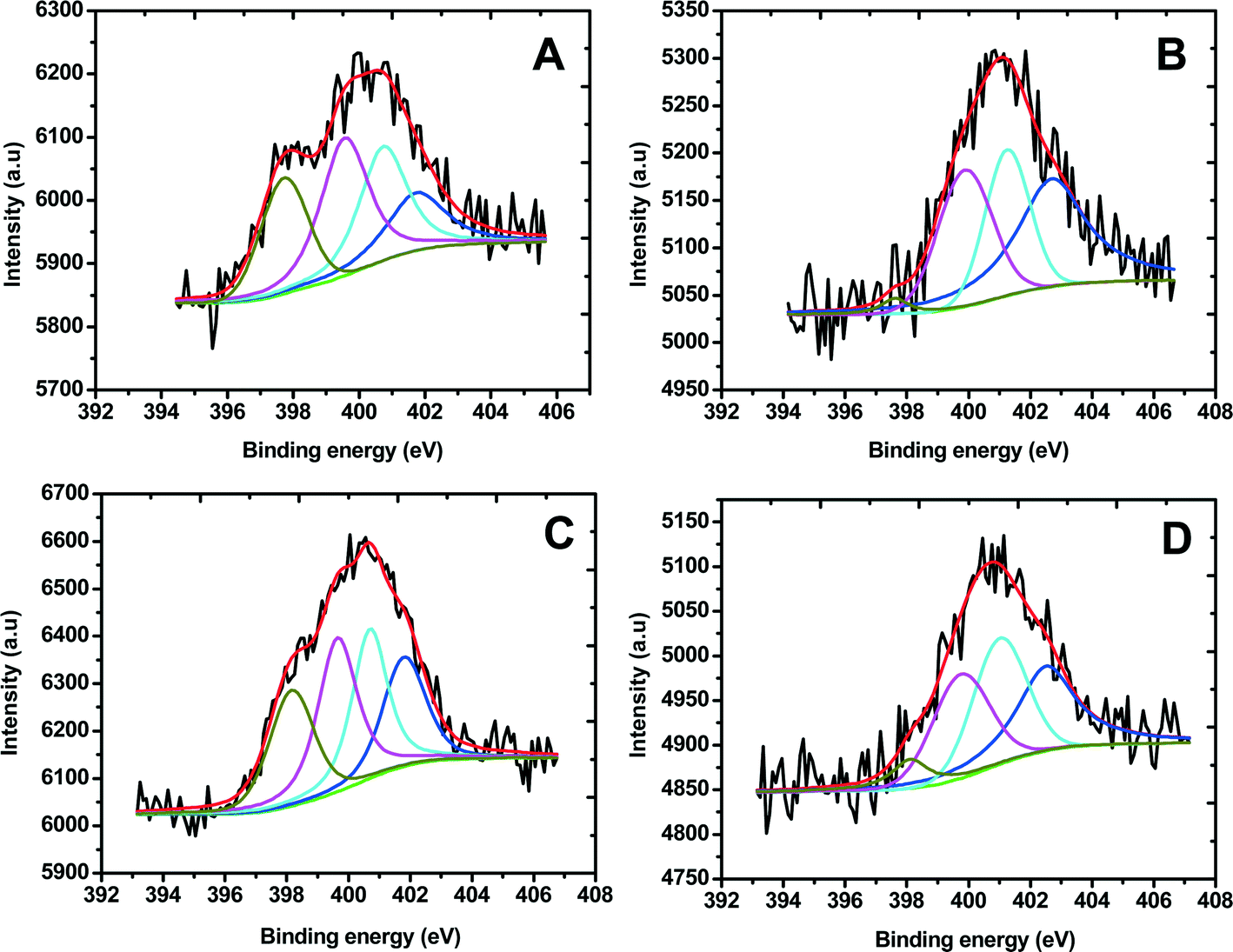
Fig. 2 shows the N 1s XPS spectra of Fe/PI and Fe/PI-aw before and after the LD test (11110 potential steps). The N 1s spectra were carefully deconvoluted to investigate the chemical state of nitrogen in these catalysts. The four common nitrogen groups observed in nitrogen-containing carbonaceous materials are pyridinic nitrogen (Py-N), pyrrolic nitrogen (Pr-N), graphite-like nitrogen (Q-N) and nitrogen oxide (N-O), the peaks of which were observed in the regions of 398.2–398.7, 400.1–400.3, 401.5–401.2 and 402.2–403.1 eV, respectively.67 The percentage of each type of nitrogen can be calculated from the corresponding peak area before and after the LD test (Table 1). Previous studies suggest that Py-N and Q-N improve the ORR activity,68–70 whereas N-O shows a negative effect on the ORR. In this study, we have found that the amount of Py-N and Q-N is 22% and 27%, respectively, for the Fe/PI catalyst, whereas in the Fe/PI-aw catalyst, the quantity of Py-N and Q-N decreases to 0% and 21%, respectively. This observation indicates that the acid-washed catalyst did not render high ORR activity compared with the non-washed one. Moreover, the amount of N-O is abnormally high in the Fe/PI-aw catalyst (48%), also supporting a poor activity of the acid-washed catalyst towards the ORR. However, the situation changes after the durability test of the Fe/PI-aw catalyst, that is, after the 11110 durability cycles the relative amount of Py-N and Q-N increases, while the N-O amount decreases considerably, being in agreement with the increasing activity of the Fe/PI-aw catalyst towards the ORR after the durability test (see the inset in Fig. 5B). Each quantity of the nitrogen species before and after the durability test of 11110 potential steps remains almost unchanged for the Fe/PI catalyst, reflecting a high stability towards the ORR. The relative quantity of Pr-N in both catalysts decreases by ca. 2% after the durability test. The XPS peak found at 711 eV for Fe/PI corresponds to Fe-2p3/2, but this peak was not observed for Fe/PI-aw. This confirmed the removal of iron from the catalyst by acid washing.
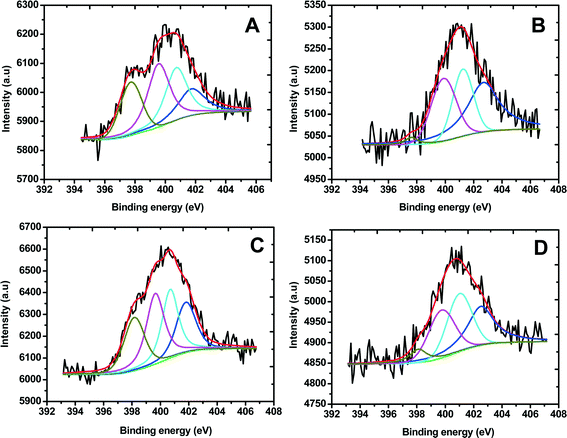 |
| | Fig. 2 Core level N-1s XPS spectra obtained for (A) Fe/PI and (B) Fe/PI-aw. The deconvoluted peaks correspond to pyridinic nitrogen (dark yellow), pyrrolic nitrogen (magenta), graphite-like nitrogen (cyan) and nitrogen oxide (blue), and the overall fitting in each spectrum is shown by a red line. C and D correspond to the XPS spectra of Fe/PI and Fe/PI-aw after 11110 cycles of the LD test. | |
Table 1 Percentages of each kind of nitrogen surface groups in Fe/PI and Fe/PI-aw before and after the LD test (11110 potential steps)
| Types of nitrogen groups |
Fe/PI |
Fe/PI-aw |
| Before |
After |
Before |
After |
| Pyridinic |
22 |
23 |
0 |
9 |
| Pyrrolic |
33 |
30 |
31 |
28 |
| Graphite-like |
27 |
26 |
21 |
31 |
| Nitrogen oxide |
18 |
21 |
48 |
32 |
Table 2 Tafel slopes and exchange current densities for the ORR at Fe/PI, Fe/PI-aw and Pt/C
| Catalysts |
Tafel slope (mV dec−1) |
Exchange current density (A cm−2) |
|
Lower current density region.
Higher current density region.
|
| Fe/PI |
−66 |
1.1 × 10−10 |
| Fe/PI-aw |
−65 |
4.6 × 10−11 |
| Pt/C |
−77a, 167b |
3.9 × 10−8,a, 3.9 × 10−5,b |
3.2 Rotating ring-disk electrode voltammetry
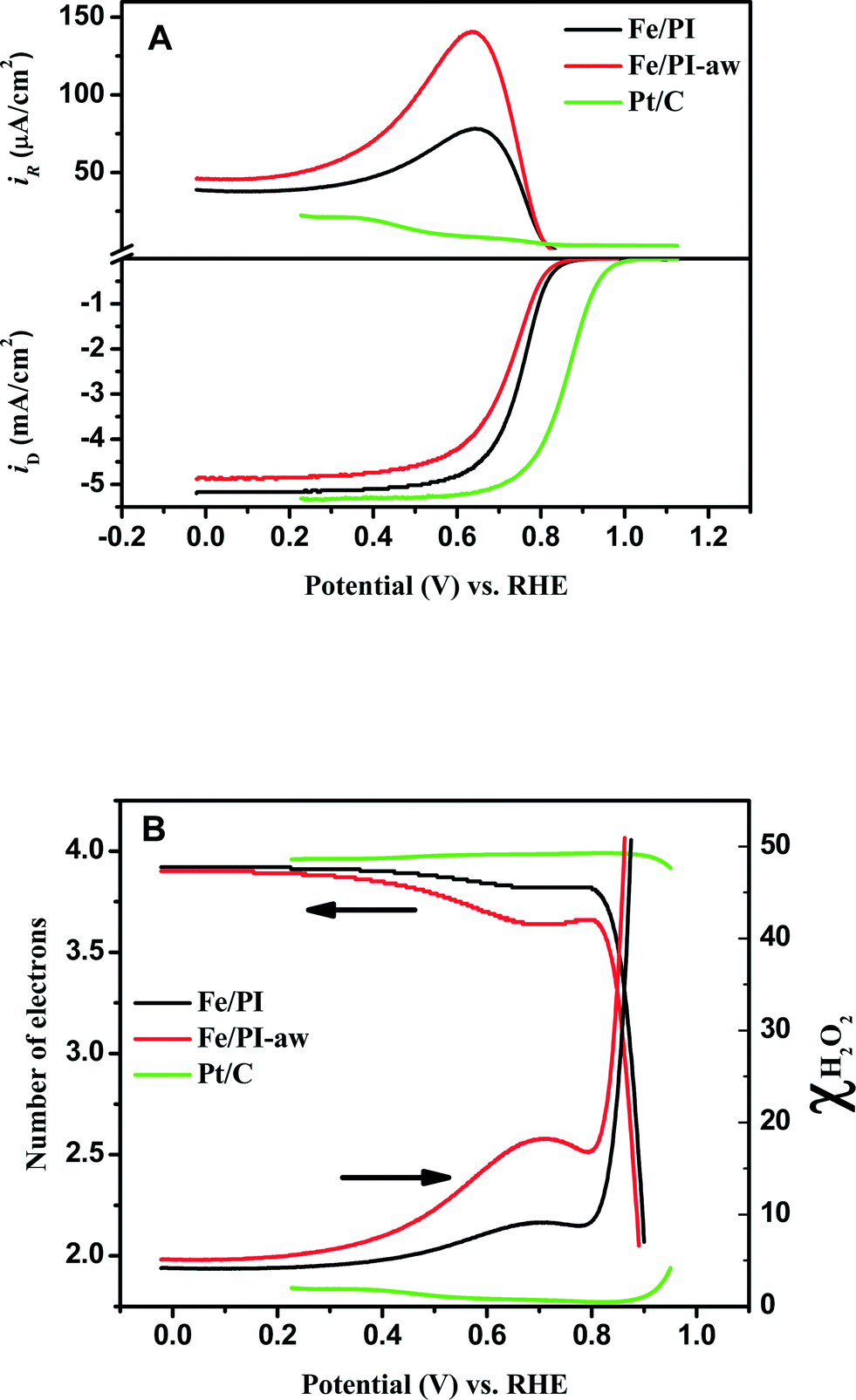
The RRDE voltammograms obtained for Fe/PI and Fe/PI-aw catalysts (before the durability test) at 1600 rpm in O2-saturated 0.5 M H2SO4 are shown in Fig. 3A. The onset potential (Eonset) of the ORR was measured as a potential at which the disk current reaches 10 μA cm−2. The Fe/PI catalyst exhibits a fairly positive Eonset (0.915 V) which is ~100 mV more negative than that of the commercially available Pt/C catalyst (TEC 10E50E (46 wt% Pt, purchased from Tanaka Kikinzoku Kogyo (TKK) Corp., Japan)) with a loading density of 55.8 μgPt cm−2, and Fe/PI-aw shows a slight negative shift in Eonset (0.902 V). The half-wave potential (E1/2) obtained at Fe/PI-aw (0.724 V) was found to be more negative than that at Fe/PI (0.751). The limiting current density (iDL) obtained at Fe/PI is close to that obtained at the Pt/C. A slight decrease in the iDL was observed for the acid-washed catalyst.
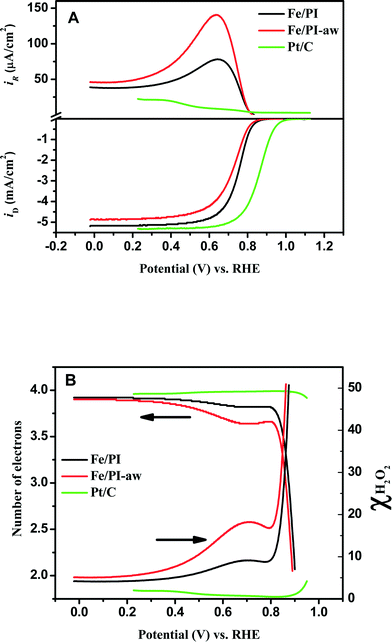 |
| | Fig. 3 (A) Steady-state voltammograms for the ORR obtained at the Fe/PI and Fe/PI-aw catalyst-modified GC disk–Pt ring electrode in O2-saturated 0.5 M H2SO4 solution. The lower panel shows the disk currents, and the corresponding Pt ring currents are shown in the upper panel corresponding to the oxidation of H2O2 produced at the relevant disk electrode. Potential scan rate: 5 mV s−1. Electrode rotation speed: 1600 rpm. The Pt ring electrode was potentiostated at 1.2 V. The voltammograms indicated by green lines were obtained at the Pt/C-modified GC disk–Pt ring electrode (Pt loading of Pt/C: 55.8 μg cm−2) under the same conditions as mentioned above. (B) Variation of the number of electrons (n) and χH2O2 with the disk electrode potential during the ORR at the Fe/PI, Fe/PI-aw and Pt/C catalysts. | |
3.3 Number of electrons and percentage of hydrogen peroxide
The background-subtracted RRDE voltammograms were used for the estimation of potential-dependent parameters of ORR such as the number of electrons (n), the percentage of hydrogen peroxide produced (χH2O2) and the rate constants. The estimation of n in the kinetically controlled region helps us to identify the reaction pathway for the reduction of oxygen to water as described in Scheme 1. The value of n can be estimated using the ring and disc currents as follows:71–74| | 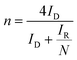 | (1) |
where ID and IR represent the disk and ring currents, respectively. N is the collection efficiency (in this case, 0.37). Similarly, χH2O2 is also estimated using the following equation:71–74| | 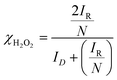 | (2) |
Potential-dependent variation in n and χH2O2 is shown in Fig. 3B. n increases as the electrode potential becomes more negative, and at the limiting current region, its values are 3.8 to 3.95 (depending on the catalysts), which are very close to that expected for 4-electron reduction of oxygen to water. The percentage of H2O2 increases as the potential becomes more positive and the value is ~20% in the ORR onset region, whereas at the limiting current region, it is less than 5%, indicating that the ORR actually follows both a direct pathway of the 4-electron reduction of O2 to H2O and a sequential pathway, where O2 is first reduced to H2O2 followed by the reduction of H2O2 to H2O, rather than only either of both, i.e., a parallel pathway (Scheme 1).
3.4 Stability tests
3.4.1 Stability of the carbon support.
The stability test of Fe/PI as “carbon support” was carried out at the potential region where carbon corrosion may occur using the protocol (i.e., SSD test) described in the experimental section. E1/2 and IDL are the two important parameters reflecting the stability of Fe/PI. E1/2 is shifted to the negative direction of the potential after the stability test (the degree in E1/2 shift is noted by the difference in E1/2 (ΔE1/2) before and after the stability test), and the ΔE1/2 values for the Fe/PI and Fe/PI-aw catalysts are 40 and 60 mV, respectively. The decreased IDL after the SSD test is attributed to the leaching of the ORR active sites from the electrode surface, probably resulting from the carbon corrosion of Fe/PI. The difference in iDL before and after the stability test was found to be within 5%.
3.4.2 Stability of the Fe/PI catalyst in its ORR activity.
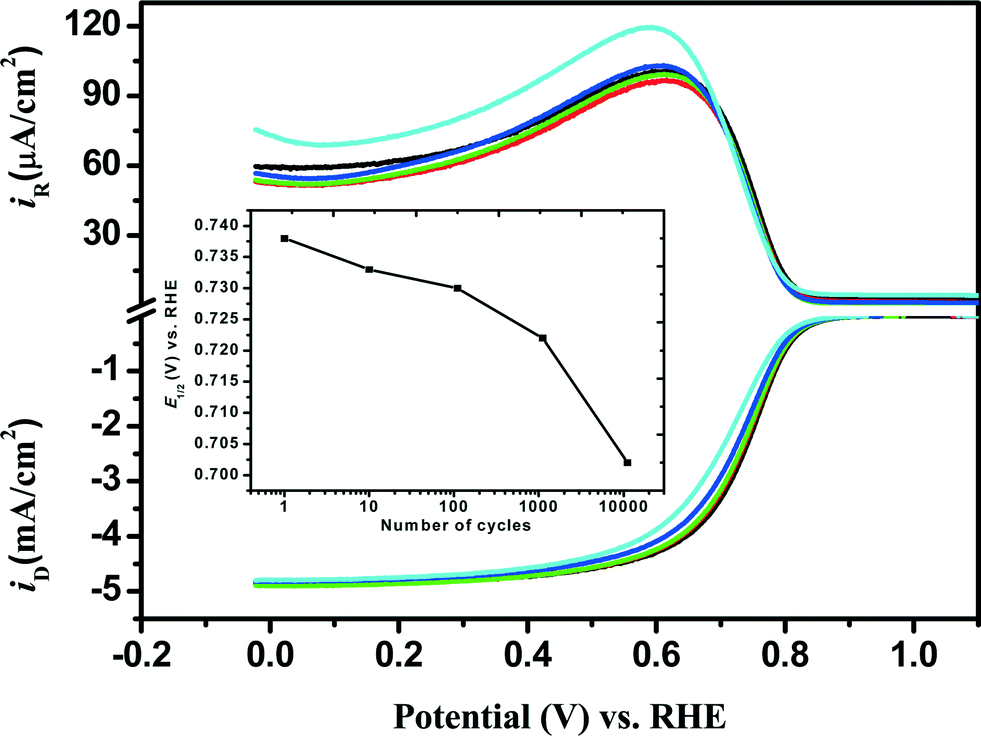
The stability in the Fe/PI′ ORR activity was also examined by applying the potential-step protocol (i.e., LD test) as mentioned in the experimental section. In this study, the potential step was repeated 11110 times (max.) and the ORR performance of the individual catalysts was checked at the 10th, 110th, 1110th and 11110th steps using the RRDE voltammetry and the results are shown in Fig. 4. The negative shift of E1/2 was observed after the LD test as in the case of the SSD test. This shift increased with increasing number of potential steps and was 36 mV after 11110 potential steps. No appreciable changes were observed in Eonset and IDL. However, n and χH2O2 decreased and increased, respectively, with increasing number of potential steps. The values of n and χH2O2 (%) before the LD test were found to be, for example, approximately 3.9 and 6% (at 0 V), respectively, whereas they did not change so much after 11110 LD potential steps. In the case of Fe/PI-aw, the values of n and χH2O2 (%) before and after the LD test are found to be approximately 3.8 and 11%, respectively, at 0 V.
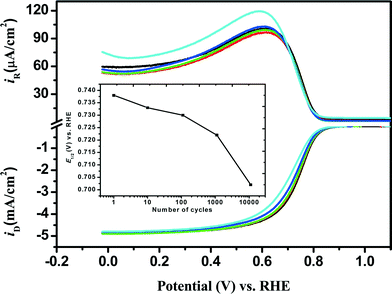 |
| | Fig. 4 Comparison of RRDE voltammograms obtained for the ORR at the Fe/PI/GC electrode after each stage of the LD test: before LD test (black) and after 10 (red), 110 (green), 1110 (blue) and 11110 (cyan) potential steps. The inset shows the variation of E1/2 with the number of the potential steps. | |
3.5 Kinetics and mechanism of ORR
To study the kinetics and mechanism of the ORR, the analytical procedure based on a RRDE voltammetry on the ORR (Scheme 1), which was originally given by Damjanovic et al.63 and later developed by Hsueh et al.,75 was used in this study. The individual rate constants (k1, k2 and k3) were estimated from the slopes and intercepts of the ID/IRvs. ω−1/2 and IDL/(IDL − ID) vs. ω−1/2 plots, where IDL and ω are the limiting disk current and rotational speed, respectively. The plots of IDL/(IDL − ID) vs. ω−1/2 at various potentials indicated a linear behaviour with an intercept equal to unity, suggesting that the decomposition (i.e., disproportionation) of the intermediate H2O2 formed during the ORR is negligible. The catalytic decomposition of H2O2 on the surface of the catalysts has also been studied separately as shown later. Following the procedure of Hsueh et al., the individual rate constants are calculated using the following equations:75| | 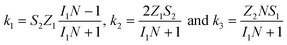 | (3) |
where S1 and I1 are the slope and the intercept of the ID/IRvs. ω−1/2 plot, respectively, and S2 indicates the slope of the IDL/(IDL − ID) vs. ω−1/2 plot. 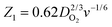 and
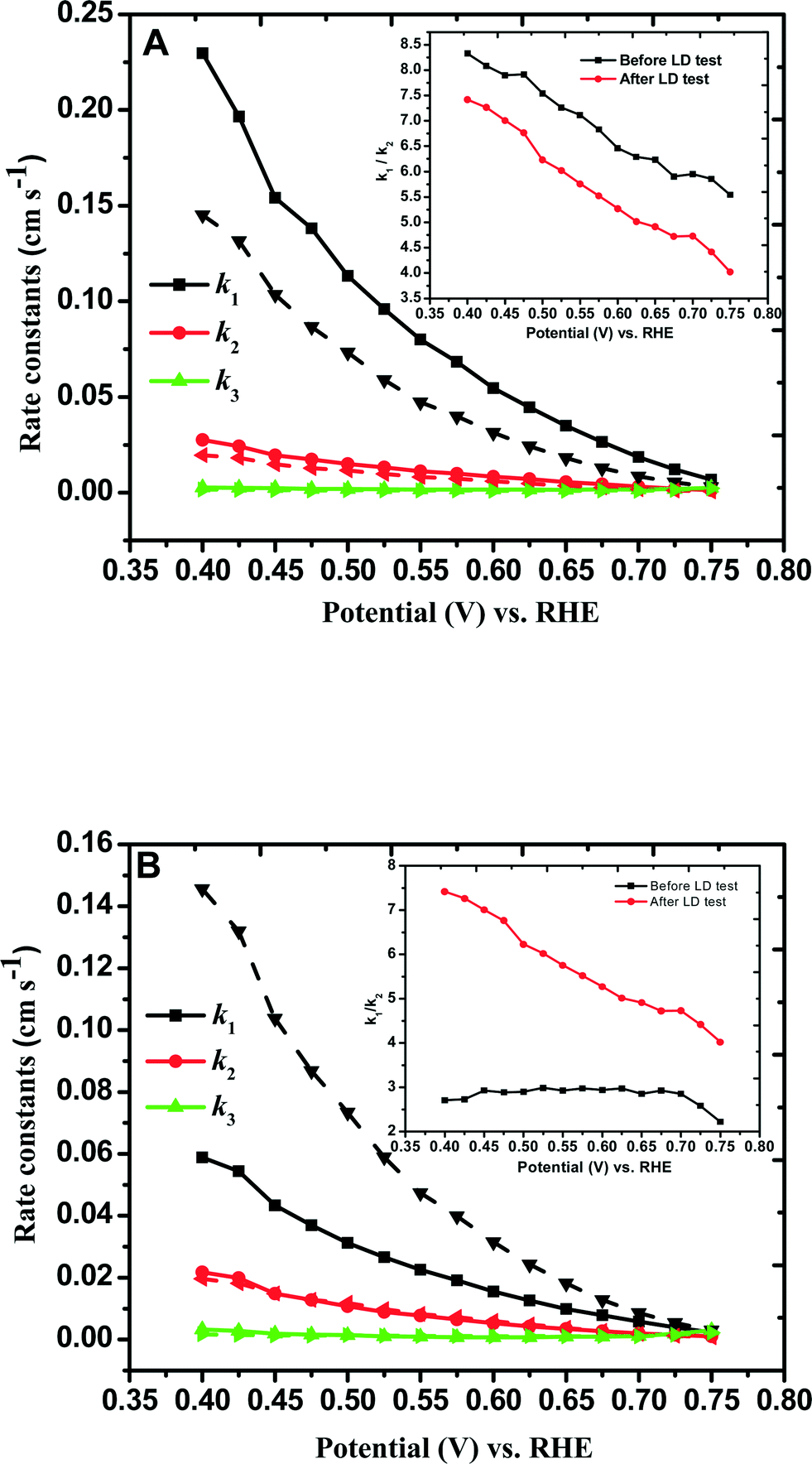
and 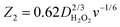 with the values of DO2 = 1.4 × 10−5 cm2 s−1, DH2O2 = 6.8 × 10−6 cm2 s−1 and ν = 0.01 cm2 s−1.76 The estimated rate constants in the potential range from 0.75 to 0.4 V are shown typically in Fig. 5A and B for the Fe/PI and Fe/PI-aw catalysts, respectively. The rate constants k1 and k2 are both potential dependent, while k3 is almost potential independent. The ratios of k1/k2 are much larger than unity in the examined range of potential for the Fe/PI catalyst (0.4–0.7 V), i.e., 5.5–8.3 and 4–7.5 before and after the LD test, respectively (the inset in Fig. 5A), indicating that O2 is mainly reduced to H2O via the 4-electron reduction path and only trace amounts of O2 are reduced via the sequential path which involves H2O2 as an intermediate. Similarly, Fe/PI-aw shows that the ratios of k1/k2 are larger than unity, but after the durability test the ratios are increased from ca. 2–3 to ca. 4–7.5, i.e., the 4-electron reduction is facilitated (cf. the inset in Fig. 5B). The k1/k2 ratios are almost linearly dependent on potential and the values are smaller at more positive potential. The rate constant k3 is smaller than k2. This indicates that the intermediate H2O2 formed in the sequential path is reduced to H2O at a relatively slow rate. A similar behaviour has also been reported for the Mo–Ru–Se-based electrocatalyst for ORR.76
with the values of DO2 = 1.4 × 10−5 cm2 s−1, DH2O2 = 6.8 × 10−6 cm2 s−1 and ν = 0.01 cm2 s−1.76 The estimated rate constants in the potential range from 0.75 to 0.4 V are shown typically in Fig. 5A and B for the Fe/PI and Fe/PI-aw catalysts, respectively. The rate constants k1 and k2 are both potential dependent, while k3 is almost potential independent. The ratios of k1/k2 are much larger than unity in the examined range of potential for the Fe/PI catalyst (0.4–0.7 V), i.e., 5.5–8.3 and 4–7.5 before and after the LD test, respectively (the inset in Fig. 5A), indicating that O2 is mainly reduced to H2O via the 4-electron reduction path and only trace amounts of O2 are reduced via the sequential path which involves H2O2 as an intermediate. Similarly, Fe/PI-aw shows that the ratios of k1/k2 are larger than unity, but after the durability test the ratios are increased from ca. 2–3 to ca. 4–7.5, i.e., the 4-electron reduction is facilitated (cf. the inset in Fig. 5B). The k1/k2 ratios are almost linearly dependent on potential and the values are smaller at more positive potential. The rate constant k3 is smaller than k2. This indicates that the intermediate H2O2 formed in the sequential path is reduced to H2O at a relatively slow rate. A similar behaviour has also been reported for the Mo–Ru–Se-based electrocatalyst for ORR.76
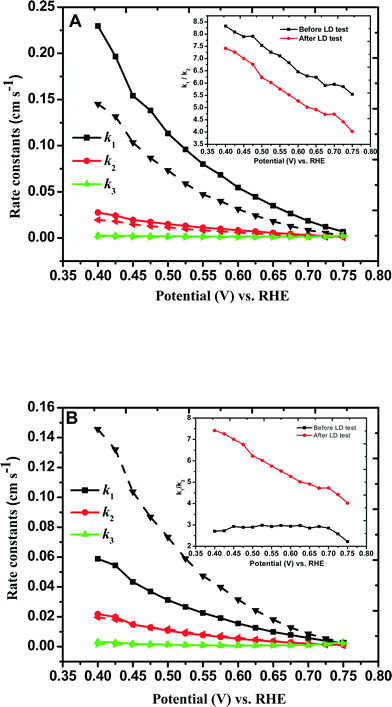 |
| | Fig. 5 Comparison of individual rate constants (k1, k2 and k3) of the ORR at (A) Fe/PI/GC and (B) Fe/PI-aw/GC electrodes before (solid lines) and after (dashed lines) the LD test. The insets indicate the potential-dependent k1/k2 values before and after the durability. | |
As discussed in section 3.4.2, a negative shift of E1/2, a decrease in n and an increase in H2O2 generation were observed after the durability test. These preliminary observations indicate that the electrode kinetics of the ORR becomes more sluggish after the durability test. An appreciable decrease in k1 was observed for Fe/PI, typically 42%. A similar trend was observed for k2 and k3, but the extent of decrease was not as high as that of k1. On the other hand, interestingly, the rate constants were found to increase after the durability test for Fe/PI-aw, although this remains to be clarified. Furthermore, Fe/PI-aw showed less stability compared with Fe/PI.
3.6 Disproportionation of H2O2
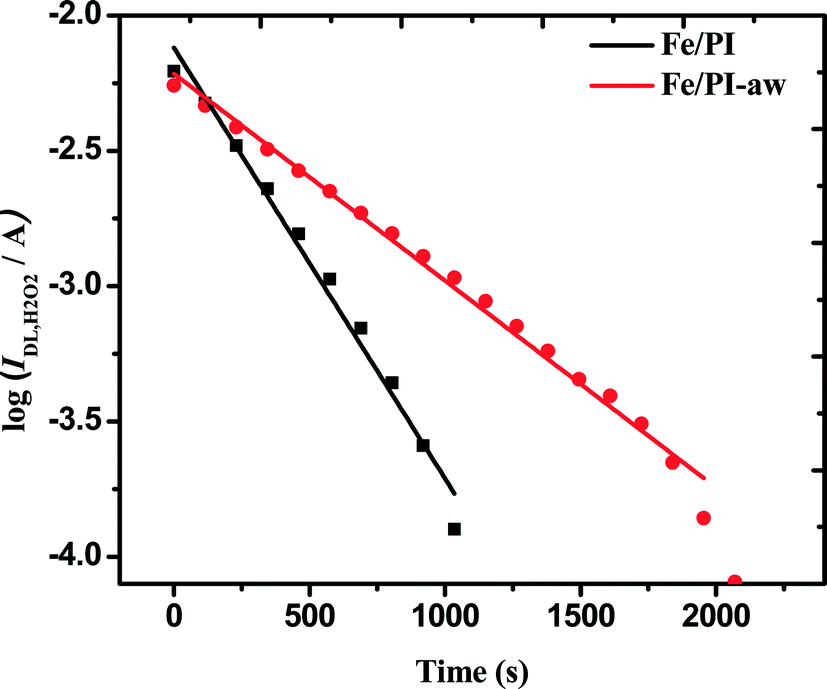
It is important in clarifying the overall ORR mechanism to know the catalytic decomposition of the H2O2 intermediate formed during the ORR. In the above-mentioned kinetic analysis, based on the fact that IDL/(IDL − ID) vs. ω−1/2 plots are linear with an intercept of unity, we have considered that the disproportionation of H2O2 on the catalyst surface is negligible. This was actually confirmed by estimating the disproportionation rate constants in the presence of Fe/PI or Fe/PI-aw according to the procedure proposed by Jaouen and Dodelet.77 A known amount (2 mg) of each of the catalysts was added to the known volume (50 mL) of O2-saturated 0.1 M HClO4 and the solution was stirred with a uniform distribution of the catalyst. 51.1 μL of 30% H2O2 was added to the solution, resulting in an initial H2O2 concentration of 10 mM. Immediately after addition of H2O2, the RRDE voltammetric diffusion-limited current (IDL,t) was measured at constant intervals with the Pt/C electrode (the loading density of the Pt/C electrode is 37.2 μgPt cm−2) at 1600 rpm rotational speed. The RRDE voltammogram was also measured before the addition of H2O2 in the O2-saturated solution and the obtained time-independent current limited by O2 diffusion is given as IDL,O2. Finally, the time-dependent H2O2 diffusion-limited current (IDL,H2O2) was calculated as follows:| | | IDL,H2O2 = IDL,t − IDL,O2 | (4) |
The rate constants (k4) of the homogeneous disproportionation reaction of H2O2 can be calculated from log(IDL,H2O2) vs. time plots as shown in Fig. 6. The slopes for both catalysts gave the values of k4.
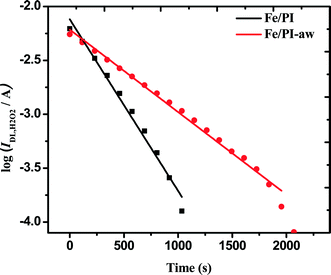 |
| | Fig. 6 Variation of the H2O2 diffusion-limited current at 0.2 V vs. Ag/AgCl (sat. KCl) with time elapsed since 10 mM H2O2 was introduced in O2-saturated 0.1 M HClO4 solution containing 2 mg of each catalyst. | |
To compare the thus-estimated disproportionation rate constants with the ORR rate constants, the unit (cm s−1) of k1, k2 and k3 needs to be converted to the same unit of the homogenous rate constant (k4) by taking into account the thickness (l) of the catalyst coated on the electrode. The ORR rate constants divided by the thickness of the film leads to the unit of s−1 as shown in Appendix A. The thickness of the film coated on the GC surface was estimated to be 23 ± 3 μm using a 3D laser scanning microscope (Keyence Co., Japan). Finally, k1/l, k2/l and k3/l are calculated as 104.3, 12.0 and 1.2 s−1, respectively, for the Fe/PI catalyst and 26.1, 9.4 and 1.4 s−1, respectively, for the Fe/PI-aw catalyst. k4 is normalized to the amount of the catalyst coated on the electrode for comparison and the estimated values are 10.4 × 10−5 and 5.0 × 10−5 s−1 for the Fe/PI and Fe/PI-aw catalysts, respectively. These values are very low compared with k1/l, k2/l and k3/l, and hence, the disproportionation of H2O2 is negligible. From the mechanistic point of view, it is expected that k4 might have an influence on k3 but not on k1 and k2.75 As mentioned above, k4 is too small compared to k3. Hence, the catalytic disproportionation reaction of H2O2 on these catalysts is considered to be very slow and negligible.
3.7 Tafel analysis
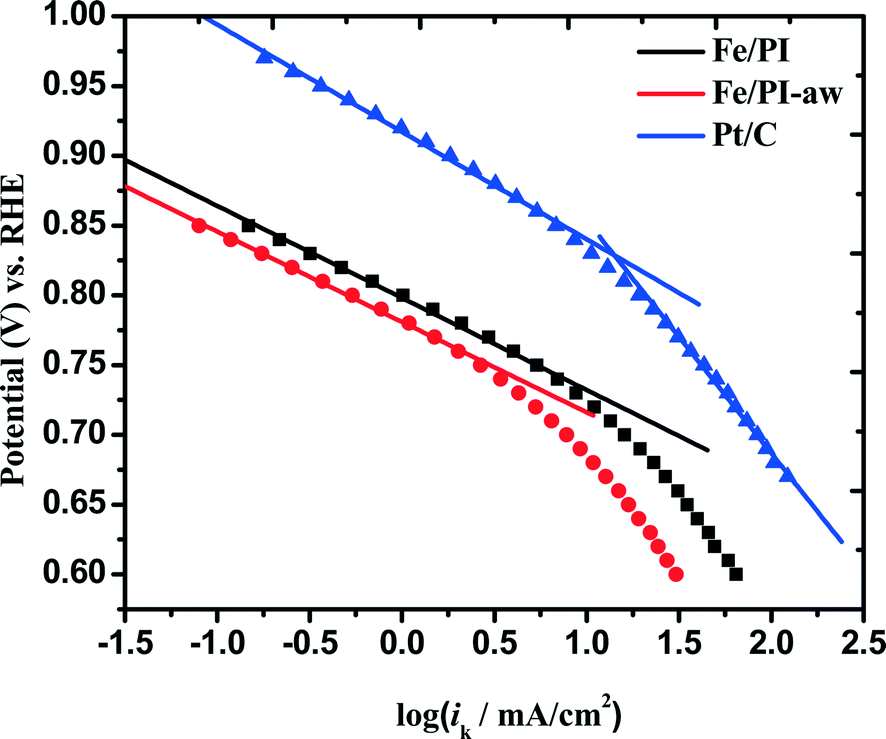
Tafel plots are very useful in examining the mechanism of ORR on a particular catalyst. For estimating the kinetic current to obtain the mass transfer-corrected Tafel plot, the modified Koutecky–Levich equation was used as shown below:| | 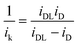 | (5) |
where ik and iDL are the kinetic and limiting current densities, respectively. The estimated ik values are plotted against potential in Fig. 7. For both Fe/PI and Fe/PI-aw catalysts, the good linear plots were obtained, and thus, the Tafel slopes were estimated to be −66 and −65 mV dec−1 for Fe/PI and Fe/PI-aw catalysts, respectively (Table 2). It is well recognized for Pt/C catalysts73,78–80 that the slope of −60 mV dec−1 is expected for the ORR at a low current density (lcd) region, and it is attributed to the adsorbed oxygen species with the fast initial electron transfer step followed by the rate-determining chemical step or adsorption under the Temkin adsorption conditions and larger values of Tafel slopes (from ca. −120 to −160 mV dec−1) are usually obtained at a high current density (hcd) region in agreement with the expectation that the initial electron transfer is the rate-determining step under the Langmuir adsorption conditions. The exchange current density (Iex) can be estimated from the intercept of a linear Tafel plot and it is a key parameter to judge the catalytic efficiency. For Pt/C, these values were estimated to be 3.9 × 10−8 and 3.9 × 10−5 A cm−2 for lcd and hcd regions, respectively. The Iex values obtained for Fe/PI and Fe/PI-aw are 1.1 × 10−10 and 4.6 × 10−11 A cm−2, respectively, which are by two to three orders of magnitude smaller than that at lcd for the Pt/C. The Fe/PI catalyst gives a larger Iex compared with Fe/PI-aw.
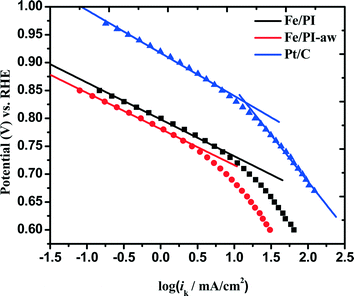 |
| | Fig. 7 Mass transfer-corrected Tafel plots for the ORR at Fe/PI, Fe/PI-aw and Pt/C catalysts. | |
4. Conclusions
In conclusion, we have successfully demonstrated the high performance of Fe-containing N-doped carbon catalyst (Fe/PI) for the ORR in acidic medium. Fe/PI was prepared from the high-temperature pyrolysis of the polyimide precursor with the Fe(acac)3. The durability of Fe/PI, which was estimated based on the SSD and LD tests, is relatively good in acidic medium compared with other catalysts reported in the literature.16,81–83 The XPS and RRDE results suggested that the catalyst enriched with Py-N and Q-N shows a high activity towards the ORR, while that enriched with N-O shows less activity. The ORR rate constants (k1, k2 and k3) were estimated by a RRD voltammetry, indicating that the 4-electron reduction of oxygen is relatively predominant compared to the 2-electron reduction. The rate constant of the disproportionation reaction of H2O2 was estimated; it is by about 4 to 6 orders of magnitude smaller than the ORR rate constants, and hence, in the present case, the disproportionation reaction is actually negligible. The role of iron was also studied by comparing the activity and durability in ORR of the Fe/PI and Fe/PI-aw catalysts. Its role in the catalytic performance is not known exactly, but it seems true that Fe plays a vital role in the overall 4-electron reduction of O2.
Appendix A
According to the definition of the Damjanovic model, the current corresponding to the reduction of H2O2 to water (I3) can be represented aswhere SD, F and C*2 represent the surface area of the disk electrode, the Faraday constant and the concentration of H2O2, respectively. The rate of the reduction of H2O2 is also defined as| | 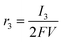 | (A2) |
Substituting eqn (A1) with eqn (A2),
| | 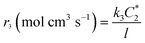 | (A3) |
where
l is the thickness of the electrode evolved from the ratio of the surface area to the volume of the electrode. The rate of the disproportionation reaction of H
2O
2 (
r4) can be written as
| | 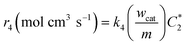 | (A4) |
where
k4 is the homogeneous disproportionation rate constant (s
−1), and
wcat and
m are the weight of the catalyst on the electrode surface in the ORR study and the weight of the catalyst used for the disproportionation study (2 mg), respectively. By taking
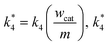
can be compared with
k3/
l. Similarly,
k1/
l and
k2/
l are also compared with
k*4.
Acknowledgements
This work was financially supported by the New Energy and Industrial Technology Development Organization (NEDO), Japan. The authors thank Dr. T. Fuchigami and Dr. D. Zhang for helpful discussions.
References
- S. Gupta, D. Tryk, I. Bae, W. Aldred and E. Yeager, J. Appl. Electrochem., 1989, 19, 19–27 CrossRef CAS.
- F. Jaouen, S. Marcotte, J.-P. Dodelet and G. Lindbergh, J. Phys. Chem. B, 2003, 107, 1376–1386 CrossRef CAS.
- M. Lefevre, E. Proietti, F. Jaouen and J.-P. Dodelet, Science, 2009, 324, 71–74 CrossRef CAS PubMed.
- S. Kundu, T. C. Nagaiah, W. Xia, Y. Wang, S. Van Dommele, J. H. Bitter, M. Santa, G. Grundmeier, M. Bron, W. Schuhmann and M. Muhler, J. Phys. Chem. C, 2009, 113, 14302–14310 CAS.
- T. C. Nagaiah, S. Kundu, M. Bron, M. Muhler and W. Schuhmann, Electrochem. Commun., 2010, 12, 338–341 CrossRef CAS PubMed.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed.
- G. Wu, K. L. More, P. Xu, H.-L. Wang, M. Ferrandon, A. J. Kropf, D. J. Myers, S. Ma, C. M. Johnston and P. Zelenay, Chem. Commun., 2013, 49, 3291–3293 RSC.
- Z. Chen, D. Higgins and Z.-W. Chen, Electrochim. Acta, 2010, 55, 4799–4804 CrossRef CAS PubMed.
- Z. Chen, D. Higgins, H. Tao, R. S. Hsu and Z. Chen, J. Phys. Chem. C, 2009, 113, 21008–21013 CAS.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- L. Qu, Y. Liu, J.-B. Baek and L. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed.
- M. Chokai, M. Taniguchi, S. Moriya, K. Matsubayashi, T. Shinoda, Y. Nabae, S. Kuroki, T. Hayakawa, M.-A. Kakimoto, J.-I. Ozaki and S. Miyata, J. Power Sources, 2010, 195, 5947–5951 CrossRef CAS PubMed.
- J.-I. Ozaki, S.-I. Tanifuji, A. Furuichi and K. Yabutsuka, Electrochim. Acta, 2010, 55, 1864–1871 CrossRef CAS PubMed.
- G. Ma, R. Jia, J. Zhao, Z. Wang, C. Song, S. Jia and Z. Zhu, J. Phys. Chem. C, 2011, 115, 25148–25154 CAS.
- M. H. Robson, A. Serov, K. Artyushkova and P. Atanassov, Electrochim. Acta, 2013, 90, 656–665 CrossRef CAS PubMed.
- H. Peng, Z. Mo, S. Liao, H. Liang, L. Yang, F. Luo, H. Song, Y. Zhong and B. Zhang, Sci. Rep., 2013, 3, 1765 Search PubMed.
- L. Qu, Y. Liu, J.-B. Baek and L. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed.
- H. Niwa, M. Saito, M. Kobayashi, Y. Harada, M. Oshima, S. Moriya, K. Matsubayashi, Y. Nabae, S. Kuroki, T. Ikeda, K. Terakura, J.-I. Ozaki and S. Miyata, J. Power Sources, 2013, 223, 30–35 CrossRef CAS PubMed.
- C. Han, J. Wang, Y. Gong, X. Xu, H. Li and Y. Wang, J. Mater. Chem. A, 2014, 2, 605–609 CAS.
- S.-Y. Wang, D.-S. Yu, L.-M. Dai, D.-W. Chang and J.-B. Baek, ACS Nano, 2011, 5, 6202–6209 CrossRef CAS PubMed.
- X. Zhou, J. Qiao, L. Yang and J. Zhang, Adv. Energy Mater., 2014, 4, 1301523 Search PubMed.
- Z.-W. Liu, F. Peng, H.-J. Wang, H. Yu, W.-X. Zheng and J. Yang, Angew. Chem., Int. Ed., 2011, 50, 3257–3261 CrossRef PubMed.
- Y. Li, Y. Zhao, H. Cheng, Y. Hu, G. Shi, L. Dai and L. Qu, J. Am. Chem. Soc., 2012, 134, 15–18 CrossRef CAS PubMed.
- X. Bo and L. Guo, Phys. Chem. Chem. Phys., 2013, 15, 2459–2465 RSC.
- K. Wiesener, D. Ohms, V. Neumann and R. Franke, Mater. Chem. Phys., 1989, 22, 457–475 CrossRef CAS.
- K. A. Radyushkina and M. R. Tarasevich, Elektrokhimiya, 1986, 22, 1155–1170 CAS.
- H. Meng, N. Larouche, M. Lefevre, F. Jaouen, B. Stansfield and J.-P. Dodelet, Electrochim. Acta, 2010, 55, 6450–6461 CrossRef CAS PubMed.
- J. A. R. Van Veen, J. F. Van Baar and K. J. Kroese, J. Chem. Soc., Faraday Trans. 1, 1981, 77, 2827–2843 RSC.
- G. Wu, K. Artyushkova, M. Ferrandon, A. J. Kropf, D. Myers and P. Zelenay, ECS Trans., 2009, 25, 1299–1311 CAS.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed.
- N. Gavrilov, M. Vujkovic, I. A. Pasti, G. Ciric-Marjanovic and S. V. Mentus, Electrochim. Acta, 2011, 56, 9197–9202 CAS.
- J. Chlistunoff, J. Phys. Chem. C, 2011, 115, 6496–6507 CAS.
- S. Maldonado and K. J. Stevenson, J. Phys. Chem. B, 2005, 109, 4707–4716 CrossRef CAS PubMed.
- P. H. Matter, E. Wang, M. Arias, E. J. Biddinger and U. S. Ozkan, J. Phys. Chem. B, 2006, 110, 18374–18384 CrossRef CAS PubMed.
- S. Shanmugam and T. Osaka, Chem. Commun., 2011, 47, 4463–4465 RSC.
- J. Tian, L. Birry, F. Jaouen and J. P. Dodelet, Electrochim. Acta, 2011, 56, 3276–3285 CrossRef CAS PubMed.
- W. Yang, T.-P. Fellinger and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 206–209 CrossRef CAS PubMed.
- H. Jiang, Y. Zhu, Q. Feng, Y. Su, X. Yang and C. Li, Chem. – Eur. J., 2014, 20, 3106–3112 CrossRef CAS PubMed.
- G. Nam, J. Park, S. T. Kim, D.-B. Shin, N. Park, Y. Kim, J.-S. Lee and J. Cho, Nano Lett., 2014, 14, 1870–1876 CrossRef CAS PubMed.
- W. Wei, H. Liang, K. Parvez, X. Zhuang, X. Feng and K. Muellen, Angew. Chem., Int. Ed., 2014, 53, 1570–1574 CrossRef CAS PubMed.
- R. Zheng, Z. Mo, S. Liao, H. Song, Z. Fu and P. Huang, Carbon, 2014, 69, 132–141 CrossRef CAS PubMed.
- Z. Chen, D. Higgins and Z. Chen, Carbon, 2010, 48, 3057–3065 CrossRef CAS PubMed.
- D. Yu, Q. Zhang and L. Dai, J. Am. Chem. Soc., 2010, 132, 15127–15129 CrossRef CAS PubMed.
- D. Yu, Y. Xue and L. Dai, J. Phys. Chem. Lett., 2012, 3, 2863–2870 CrossRef CAS.
- H. T. Chung, J. H. Won and P. Zelenay, Nat. Commun., 2013, 4, 1922 CrossRef PubMed.
- S. Ratso, I. Kruusenberg, M. Vikkisk, U. Joost, E. Shulga, I. Kink, T. Kallio and K. Tammeveski, Carbon, 2014, 73, 361–370 CrossRef CAS PubMed.
- I.-Y. Jeon, D. Yu, S.-Y. Bae, H.-J. Choi, D. W. Chang, L. Dai and J.-B. Baek, Chem. Mater., 2011, 23, 3987–3992 CrossRef CAS.
- P. Xu, D. Wu, L. Wan, P. Hu and R. Liu, J. Colloid Interface Sci., 2014, 421, 160–164 CrossRef CAS PubMed.
- M. Vikkisk, I. Kruusenberg, U. Joost, E. Shulga, I. Kink and K. Tammeveski, Appl. Catal., B, 2014, 147, 369–376 CrossRef CAS PubMed.
- C. He, J. J. Zhang and P. K. Shen, J. Mater. Chem. A, 2014, 2, 3231–3236 CAS.
- X. Bo, C. Han, Y. Zhang and L. Guo, ACS Appl. Mater. Interfaces, 2014, 6, 3013–3020 Search PubMed.
- Y. Jiang, Y. Lu, X. Lv, D. Han, Q. Zhang, L. Niu and W. Chen, ACS Catal., 2013, 3, 1263–1271 CrossRef CAS.
- J. Bai, Q. Zhu, Z. Lv, H. Dong, J. Yu and L. Dong, Int. J. Hydrogen Energy, 2013, 38, 1413–1418 CrossRef CAS PubMed.
- Z. Yang, Z. Yao, G. Li, G. Fang, H. Nie, Z. Liu, X. Zhou, X. A. Chen and S. Huang, ACS Nano, 2012, 6, 205–211 CrossRef CAS PubMed.
- L.-J. Yang, S.-J. Jiang, Y. Zhao, L. Zhu, S. Chen, X.-Z. Wang, Q. Wu, J. Ma, Y.-W. Ma and Z. Hu, Angew. Chem., Int. Ed., 2011, 50, 7132–7135 CrossRef CAS PubMed.
- G. Wu, N. H. Mack, W. Gao, S. Ma, R. Zhong, J. Han, J. K. Baldwin and P. Zelenay, ACS Nano, 2012, 6, 9764–9776 CrossRef CAS PubMed.
- K. Parvez, S. Yang, Y. Hernandez, A. Winter, A. Turchanin, X. Feng and K. Müllen, ACS Nano, 2012, 6, 9541–9550 CrossRef CAS PubMed.
- G. Wu, H. T. Chung, M. Nelson, K. Artyushkova, K. L. More, C. M. Johnston and P. Zelenay, ECS Trans., 2011, 41, 1709–1717 CAS.
- Z. Luo, S. Lim, Z. Tian, J. Shang, L. Lai, B. MacDonald, C. Fu, Z. Shen, T. Yu and J. Lin, J. Mater. Chem., 2011, 21, 8038–8044 RSC.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- Y. Li, Y. Zhao, H. Cheng, Y. Hu, G. Shi, L. Dai and L. Qu, J. Am. Chem. Soc., 2011, 134, 15–18 CrossRef PubMed.
- Y. Nabae, Y. Kuang, M. Chokai, T. Ichihara, A. Isoda, T. Hayakawa and T. Aoki, J. Mater. Chem. A, 2014, 2, 11561–11564 CAS.
- A. Damjanovic, M. A. Genshaw and J. O. M. Bockris, J. Chem. Phys., 1966, 45, 4057–4059 CrossRef CAS PubMed.
- A. Ohma, K. Shinohara, A. Iiyama, T. Yoshida and A. Daimaru, ECS Trans., 2011, 41, 775–784 CAS.
- H. Yano, T. Akiyama, P. Bele, H. Uchida and M. Watanabe, Phys. Chem. Chem. Phys., 2010, 12, 3806–3814 RSC.
- Y. Nabae, M. Sonoda, C. Yamauchi, Y. Hosaka, A. Isoda and T. Aoki, Catal. Sci. Technol., 2014, 4, 1400–1406 CAS.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS.
- C. V. Rao, C. R. Cabrera and Y. Ishikawa, J. Phys. Chem. Lett., 2010, 1, 2622–2627 CrossRef CAS.
- N. P. Subramanian, X. Li, V. Nallathambi, S. P. Kumaraguru, H. Colon-Mercado, G. Wu, J.-W. Lee and B. N. Popov, J. Power Sources, 2009, 188, 38–44 CrossRef CAS PubMed.
- G. Liu, X. Li, P. Ganesan and B. N. Popov, Electrochim. Acta, 2010, 55, 2853–2858 CrossRef CAS PubMed.
- M. Lefevre and J.-P. Dodelet, Electrochim. Acta, 2003, 48, 2749–2760 CrossRef CAS.
- S. L. Gojković, S. Gupta and R. F. Savinell, Electrochim. Acta, 1999, 45, 889–897 CrossRef.
- U. A. Paulus, T. J. Schmidt, H. A. Gasteiger and R. J. Behm, J. Electroanal. Chem., 2001, 495, 134–145 CrossRef CAS.
- E. Claude, T. Addou, J. M. Latour and P. Aldebert, J. Appl. Electrochem., 1998, 28, 57–64 CrossRef CAS.
- K. L. Hsueh, D. T. Chin and S. Srinivasan, J. Electroanal. Chem. Interfacial Electrochem., 1983, 153, 79–95 CrossRef CAS.
- N. Alonso-Vante, H. Tributsch and O. Solorza-Feria, Electrochim. Acta, 1995, 40, 567–576 CrossRef CAS.
- F. Jaouen and J.-P. Dodelet, J. Phys. Chem. C, 2009, 113, 15422–15432 CAS.
- N. M. Marković, R. R. Adžić, B. D. Cahan and E. B. Yeager, J. Electroanal. Chem., 1994, 377, 249–259 CrossRef.
- V. Stamenković, T. J. Schmidt, P. N. Ross and N. M. Marković, J. Electroanal. Chem., 2003, 554–555, 191–199 CrossRef.
- N. Wakabayashi, M. Takeichi, M. Itagaki, H. Uchida and M. Watanabe, J. Electroanal. Chem., 2005, 574, 339–346 CrossRef CAS PubMed.
- Y. Li, W. Zhou, H. Wang, L. Xie, Y. Liang, F. Wei, J.-C. Idrobo, S. J. Pennycook and H. Dai, Nat. Nanotechnol., 2012, 7, 394–400 CrossRef CAS PubMed.
- X. Li, G. Liu and B. N. Popov, J. Power Sources, 2010, 195, 6373–6378 CrossRef CAS PubMed.
- D. Geng, Y. Chen, Y. Chen, Y. Li, R. Li, X. Sun, S. Ye and S. Knights, Energy Environ. Sci., 2011, 4, 760–764 CAS.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
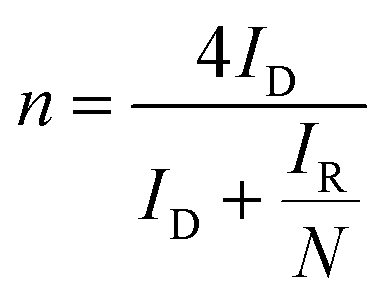
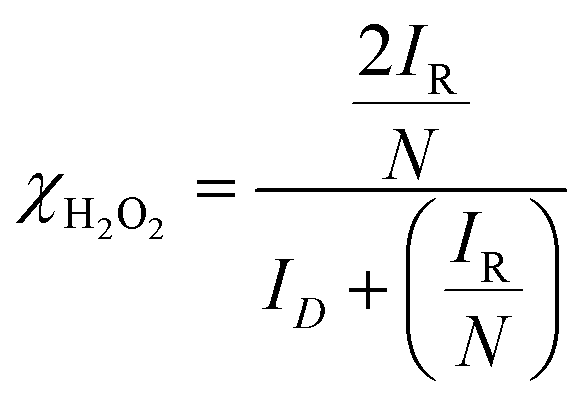
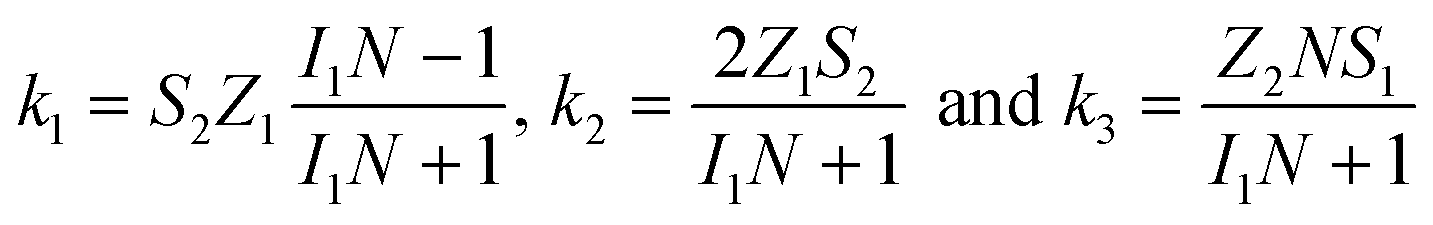
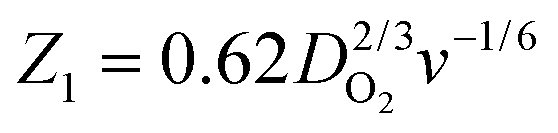 and
and 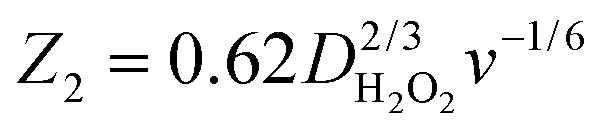 with the values of DO2 = 1.4 × 10−5 cm2 s−1, DH2O2 = 6.8 × 10−6 cm2 s−1 and ν = 0.01 cm2 s−1.76 The estimated rate constants in the potential range from 0.75 to 0.4 V are shown typically in Fig. 5A and B for the Fe/PI and Fe/PI-aw catalysts, respectively. The rate constants k1 and k2 are both potential dependent, while k3 is almost potential independent. The ratios of k1/k2 are much larger than unity in the examined range of potential for the Fe/PI catalyst (0.4–0.7 V), i.e., 5.5–8.3 and 4–7.5 before and after the LD test, respectively (the inset in Fig. 5A), indicating that O2 is mainly reduced to H2O via the 4-electron reduction path and only trace amounts of O2 are reduced via the sequential path which involves H2O2 as an intermediate. Similarly, Fe/PI-aw shows that the ratios of k1/k2 are larger than unity, but after the durability test the ratios are increased from ca. 2–3 to ca. 4–7.5, i.e., the 4-electron reduction is facilitated (cf. the inset in Fig. 5B). The k1/k2 ratios are almost linearly dependent on potential and the values are smaller at more positive potential. The rate constant k3 is smaller than k2. This indicates that the intermediate H2O2 formed in the sequential path is reduced to H2O at a relatively slow rate. A similar behaviour has also been reported for the Mo–Ru–Se-based electrocatalyst for ORR.76
with the values of DO2 = 1.4 × 10−5 cm2 s−1, DH2O2 = 6.8 × 10−6 cm2 s−1 and ν = 0.01 cm2 s−1.76 The estimated rate constants in the potential range from 0.75 to 0.4 V are shown typically in Fig. 5A and B for the Fe/PI and Fe/PI-aw catalysts, respectively. The rate constants k1 and k2 are both potential dependent, while k3 is almost potential independent. The ratios of k1/k2 are much larger than unity in the examined range of potential for the Fe/PI catalyst (0.4–0.7 V), i.e., 5.5–8.3 and 4–7.5 before and after the LD test, respectively (the inset in Fig. 5A), indicating that O2 is mainly reduced to H2O via the 4-electron reduction path and only trace amounts of O2 are reduced via the sequential path which involves H2O2 as an intermediate. Similarly, Fe/PI-aw shows that the ratios of k1/k2 are larger than unity, but after the durability test the ratios are increased from ca. 2–3 to ca. 4–7.5, i.e., the 4-electron reduction is facilitated (cf. the inset in Fig. 5B). The k1/k2 ratios are almost linearly dependent on potential and the values are smaller at more positive potential. The rate constant k3 is smaller than k2. This indicates that the intermediate H2O2 formed in the sequential path is reduced to H2O at a relatively slow rate. A similar behaviour has also been reported for the Mo–Ru–Se-based electrocatalyst for ORR.76
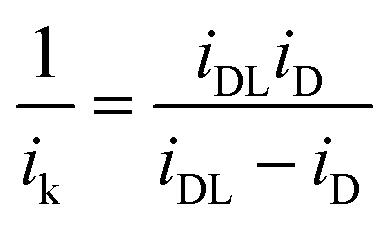
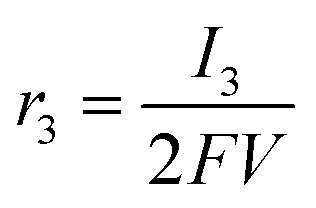
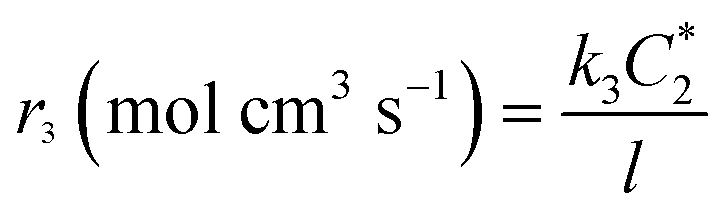
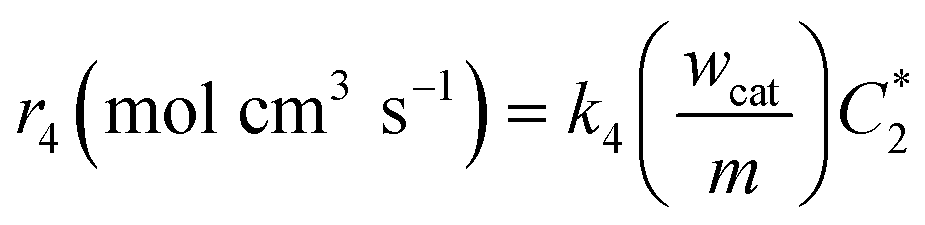
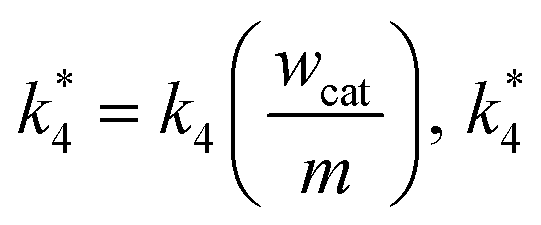 can be compared with k3/l. Similarly, k1/l and k2/l are also compared with k*4.
can be compared with k3/l. Similarly, k1/l and k2/l are also compared with k*4.