
Ruthenium(II)-catalyzed switchable C3-alkylation versus alkenylation with acrylates of 2-pyridylbenzofurans via C–H bond activation†
Yadagiri
Kommagalla
,
Venkanna Babu
Mullapudi
,
Fredi
Francis
and
Chepuri V.
Ramana
*
Division of Organic Chemistry, CSIR-National Chemical Laboratory, Dr. Homi Bhabha Road, Pune-411008, India. E-mail: vr.chepuri@ncl.res.in; Fax: +91 20 25902629; Tel: +91 20 2590 2577
First published on 1st October 2014
Abstract
We documented an interesting observation of ruthenium(II)-catalyzed benzofuran C–H activation and subsequent functionalization with acrylates that reveals that a simple base can switch the process from alkylation to alkenylation.
The transition metal-catalyzed carbon–carbon and carbon–heteroatom bond formation via the activation of sp2 and sp3 C–H bonds is recognized as a powerful alternative to classical cross-coupling reactions involving a preformed organometallic partner.1 Since the discovery of Ru(0)-catalyzed chelation-assisted C–H bond functionalization and subsequent alkylation via insertion of olefin into the (C–Ru–H) Ru–H bond and reductive elimination by Murai and co-workers in 1993,2 this reaction has been extensively investigated by employing various transition metal complexes.3 However, olefins with conjugation, such as α,β-unsaturated acceptors, were found not to perform this alkylation.4 Interestingly, a few reports on the utilization of conjugated olefins such as acrylates in these directed C–H alkylation reactions have appeared only recently with ruthenium(II)-complexes,5 favoured by strongly chelating directing groups,5b whereas there have been several examples since 2011 of alkenylation with acrylates employing the Ru(II)/Cu(II) catalytic system.6 Thus, it is a challenging task to design efficient catalytic systems for tuning the alkylation over alkenylation and vice versa.
Benzofuran is one of the commonly encountered structural units in natural products and pharmaceutically important compounds.7 The functionalization of benzofurans via C–H activation is attractive because this enables an efficient approach to various analogues from simple precursors.8 2-(2-Pyridyl)benzofuran derivatives have recently been identified as promising scaffolds in the PET imaging of β-amyloid (Aβ) plaques and thus for the diagnosis of and the discovery of effective therapeutic agents for Alzheimer's disease.9 However, as the reports on either the synthesis or the functionalization of 2-(2-pyridyl)benzofurans are few, the C–H activation and selective functionalization of 2-(2-pyridyl)benzofurans will provide simple tactics and promote further advances in this class of compounds.10
Recently, we have showed that an acyl-directing group in 2-aroylbenzofuran could allow the insertion of an acrylate into the C3–H bond of furan, but more importantly, the nature of the catalyst could selectively lead to the branched insertion product (Ru(PPh3)3Cl2) or to the linear insertion product {[Ru(p-cymene)Cl2]2}.5a Of course, alkylation took place due to the preference of the RCO directing group. Thus, it was of interest to understand how different directing groups could selectively orchestrate the insertion leading to alkylation or favour β-elimination and thus alkenylation.11 We now wish to report our initial findings and to show for the first time how acrylates can, via ruthenium(II) C–H bond activation, simply lead to alkylation or alkenylation of heterocyclic C–H bonds [Fig. 1(a) and (b), respectively]. We reasoned that due to the pyridine directing group being a strong electron donor, the Ru–C bond in the intermediate ruthenacycle is expected to be more polarized and thus favour the coordination and insertion of the olefin based upon the electronic preferences, i.e. the electrophilic β-carbon of the acrylate binding to nucleophilic carbon providing the linear alkylation product. This was indeed observed, but in addition, the conditions for selective alkylation or alkenylation were discovered.
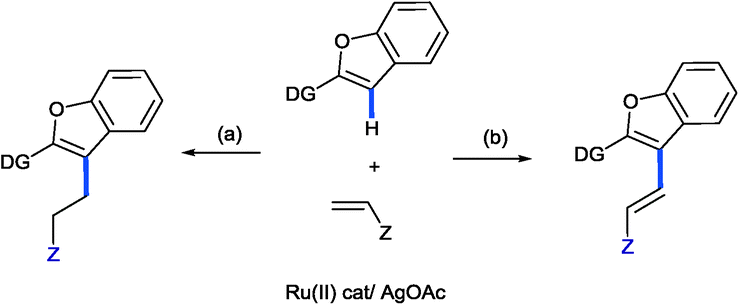 | ||
| Fig. 1 Proposal for the selective alkylation or alkenylation with acrylates. | ||
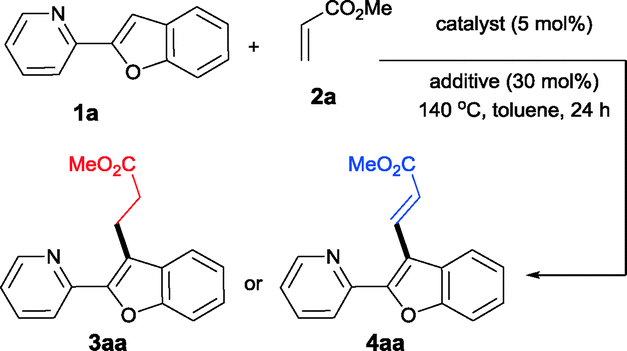
Our investigations in this regard started with the reaction of 2-pyridylbenzofuran and methyl acrylate in the presence of Ru(PPh3)3Cl2, AgOAc and in the presence of K2CO3 in toluene at 140 °C for 24 h. Surprisingly, the formation of the alkenylated product 4aa with complete linear selectivity has been observed. Linear selectivity was anticipated; however, the observed complete cross-dehydrogenative coupling without any alkylation product was surprising, since no oxidant was employed, such as the Cu(II) salt.5a However, considering the fact that the base was employed in excess and that in Heck-like couplings it is known that the base facilitates the final reductive elimination step, the same reaction was attempted in the absence of base.12 Interestingly, when the reaction was conducted under identical conditions except for the absence of base, the linear alkylation product 3aa was obtained exclusively. This important observation revealed that with this Ru(II) complex, the alkylation reactions proceed through the ruthenacycle formation followed by the commonly proposed coordinative insertion mechanism and that the involvement of a Ru–H intermediate is not necessary.
Next, the compatibility of other bases has been studied. As summarized in Table 1, with many other bases employed the reactions are incomplete and the starting 1a was recovered in substantial amounts. When Cu(OAc)2 was used as an additive in place of AgOAc, the reaction in the presence of base provided 4aa in comparable yields. However, when there was no base, unlike with AgOAc, where the alkylation reaction was exclusive, with Cu(OAc)2 the products resulting from both the alkylation and the alkenylation were obtained in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 ratio. Next, [Ru(p-cymene)Cl2]2 was examined as a catalyst for these transformations. The reactions were conducted under similar conditions and the results are comparable. In the presence of base, 4aa was obtained in 69% yield, whereas in the absence of base over the [Ru(p-cymene)Cl2]2 catalyst, the linear alkylation product 3aa was obtained exclusively. This is quite important, as it reveals that with both the ruthenium complexes, the course of the reactions seems to be similar.
:4 ratio. Next, [Ru(p-cymene)Cl2]2 was examined as a catalyst for these transformations. The reactions were conducted under similar conditions and the results are comparable. In the presence of base, 4aa was obtained in 69% yield, whereas in the absence of base over the [Ru(p-cymene)Cl2]2 catalyst, the linear alkylation product 3aa was obtained exclusively. This is quite important, as it reveals that with both the ruthenium complexes, the course of the reactions seems to be similar.
|
|
|||||
|---|---|---|---|---|---|
| S. No | Catalyst | Base | Additive | 3aa | 4aa |
| a Reagents and conditions: benzofuran (1 eq.), methyl acrylate (3 eq.), catalyst (5 mol%), base (3 eq. unless otherwise mentioned), additive (30 mol%), 140 °C, toluene, 24 h. b Isolated yield after column chromatographic purification. c 25% of 1a was recovered. d 63% of 1a was recovered. e 61% of 1a was recovered. f 41% of 1a was recovered. | |||||
| 1 | Ru(PPh3)3Cl2 | K2CO3 | AgOAc | No | 77 |
| 2 | Ru(PPh3)3Cl2 | Absent | AgOAc | 75 | No |
| 3 | Ru(PPh3)3Cl2 | Na2CO3 | AgOAc | No | 74c |
| 4 | Ru(PPh3)3Cl2 | Cs2CO3 | AgOAc | No | 66d |
| 5 | Ru(PPh3)3Cl2 | NaHCO3 | AgOAc | No | 63e |
| 6 | Ru(PPh3)3Cl2 | NaOAc | AgOAc | No | 42f |
| 7 | Ru(PPh3)3Cl2 | K2CO3 | Cu(OAc)2 | No | 67 |
| 8 | [Ru(p-cymene)Cl2]2 | K2CO3 | AgOAc | No | 69 |
| 9 | [Ru(p-cymene)Cl2]2 | Absent | AgOAc | 66 | No |
| 10 | Ru(PPh3)3Cl2 | Absent | Cu(OAc)2 | 31 | 43 |
Having discovered two complementary conditions for alkylation or alkenylation, we next explored the generality of these reactions by employing a wide range of olefins. Table 2 summarizes the results obtained with the alkylation and alkenylation reactions. Various acrylates such as ethyl- and tert-butyl- (2b and 2c) gave the corresponding alkylation or alkenylation products in moderate to good isolated yields. However, the reactions with N-isopropylacrylamide (2d) were found to be sluggish and the products were obtained in poor yields. With styrene (2e), the linear alkylated product was obtained exclusively even in the presence of K2CO3. As shown in Table 2, the presence of an electron-withdrawing substituent on the benzofuran ring has, at most, only a nominal influence on the selectivity. Interestingly, when a methyl group is present in place of chlorine, the yields were found to decrease slightly. These observations indicate that the C–H bond strength of the C3 carbon of benzofuran and the steric environment around the olefin have some influence on the yield of the reaction.
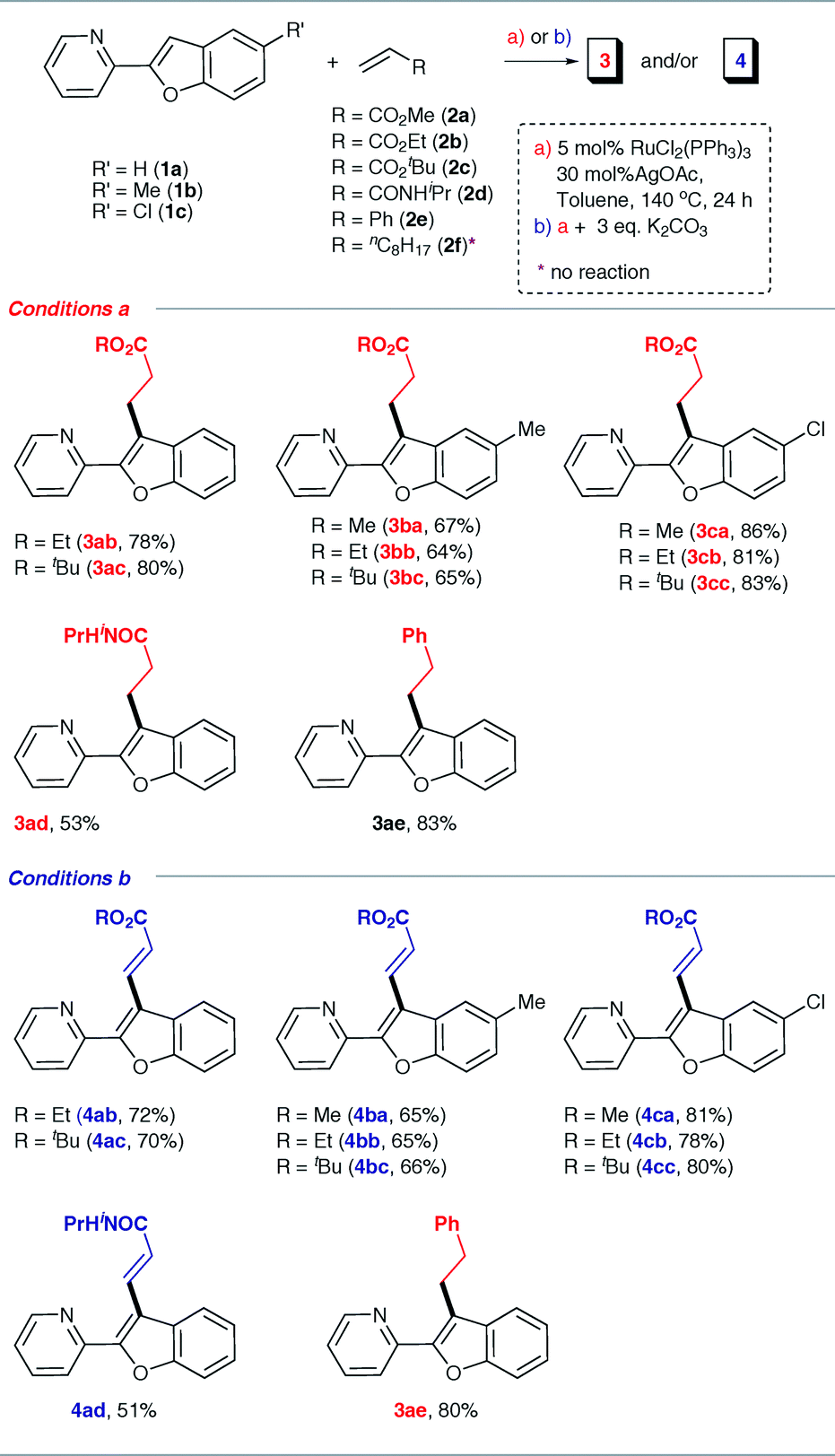
|
Deuterium labelling experiments have been carried out in order to understand the course of the deprotonation and also the possibility of hydrometalation or carbometalation followed by proto-demetalation. The reactions were conducted in the presence of D2O and under conditions a) and b), excluding the olefin. Deuterium incorporation was observed at the C3 position of benzofuran. The C3-deuterated benzofuran 5 reacted with acrylate under the optimized conditions. As shown in Scheme 1, only a nominal incorporation of deuterium at the α-position of the alkylated product was observed.
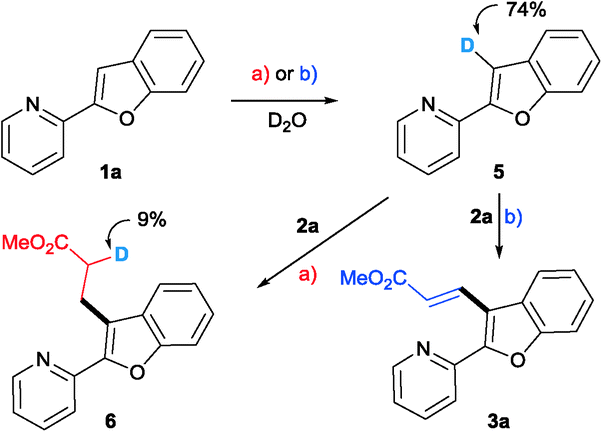 | ||
| Scheme 1 Deuterium labelling experiments. | ||
Although the mechanism cannot be demonstrated yet, we extend the following proposal to explain these two reactions. The reaction of the Ru(PPh3)3Cl2 complex with 2 eq. of AgOAc generates Ru(OAc)2(PPh3)3.13,14 The coordination of the pyridyl nitrogen and the subsequent acetate-mediated deprotonation lead to the intermediate ruthenacycle I and release AcOH. The easy dissociation of the AcO–Ru(II) bond from complex I, especially in the presence of AcOH, favours the coordination of the olefin to the cationic intermediate II. Due to the polar Ru–C bond present in complex II, the olefin insertion takes place with addition of the nucleophilic Ru–C carbon atom to the acrylate electrophilic β-carbon to give the linear insertion product precursor III. When there is no base present, this intermediate, upon proto-demetalation by the released AcOH, affords the linear alkylation product 3 and thus regenerates the ruthenium acetate complex. When the base is present, since β-elimination (commonly encountered in Heck coupling) is not possible (as it requires the M–Cα and Cβ–H bonds to align in a syn-coplanar arrangement), we propose that the base abstracts the β-proton at the anti-position of the Ru atom to form the alkenylation product 4 (Scheme 2).15
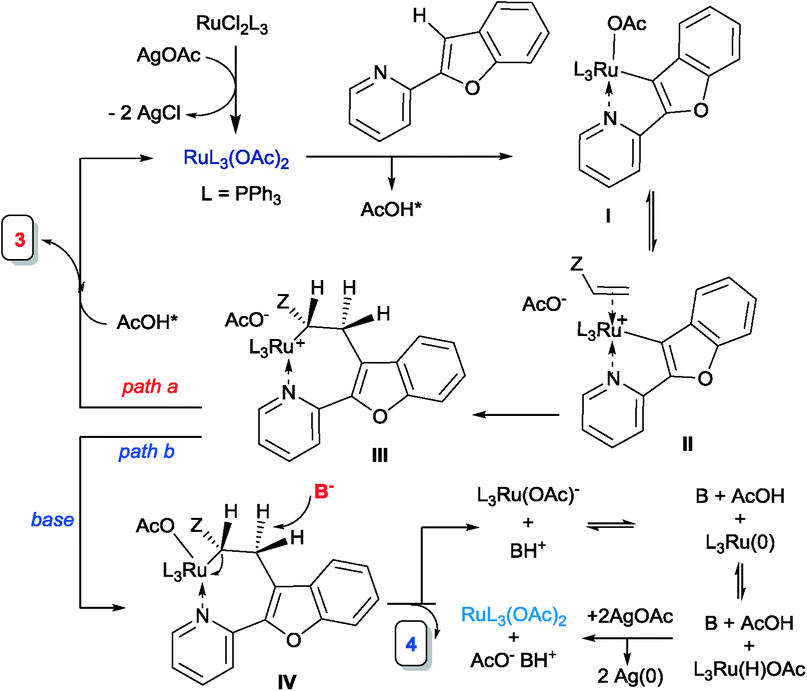 | ||
| Scheme 2 Proposed mechanism for the alkylation vs. alkenylation. | ||
Conclusions
In conclusion, our current investigations reveal two important observations in the directed C–H activation and functionalization. First, the donating ability of the chelating group, inter alia, the electronic factors override the steric influence when the Ru–C bond is polar. Secondly, we show for the first time that the presence of base avoids alkylation and favours, in the absence of an oxidant (CuII), dehydrogenative cross coupling processes via C–H activation. We believe that these findings will provide fresh impetus for further efforts to understanding how the nature of the substrate, catalyst and base will be fine-tuned in order to achieve alkylation or alkenylation with the desired regioselectivity.Acknowledgements
We thank the CSIR (India) for funding this project under the CSIR-NCL NICE program (CSC0109) and a research fellowship to YK.Notes and references
- (a) D. A. Culkin and J. F. Hartwig, Acc. Chem. Res., 2003, 36, 234–245 CrossRef CAS PubMed; (b) F. Bellina and R. Rossi, Chem. Rev., 2010, 110, 1082–1146 CrossRef CAS PubMed; (c) S. Y. Zhang, F. M. Zhang and Y. Q. Tu, Chem. Soc. Rev., 2011, 40, 1937–1949 RSC; (d) J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS PubMed; (e) V. Ritleng, C. Sirlin and M. Pfeffer, Chem. Rev., 2002, 102, 1731–1769 CrossRef CAS PubMed.
- S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529–531 CrossRef CAS.
- (a) X. Chen, K. M. Engle, D. H. Wang and J. Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed; (b) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed; (c) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814–825 CrossRef CAS PubMed; (d) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed; (e) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef CAS PubMed; (f) J. Wencel-Delord, T. Droge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740–4761 RSC.
- F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda, N. Chatani and S. Murai, Bull. Chem. Soc. Jpn., 1995, 68, 62–83 CrossRef CAS.
- (a) Y. Kommagalla, K. Srinivas and C. V. Ramana, Chem. – Eur. J., 2014, 20, 7884–7889 CrossRef CAS PubMed; (b) G. Rouquet and N. Chatani, Chem. Sci., 2013, 4, 2201–2208 RSC.
- (a) T. Ueyama, S. Mochida, T. Fukutani, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2011, 13, 706–708 CrossRef CAS PubMed; (b) L. Ackermann and J. Pospech, Org. Lett., 2011, 13, 4153–4155 CrossRef CAS PubMed; (c) P. B. Arockiam, C. Fischmeister, C. Bruneau and P. H. Dixneuf, Green Chem., 2011, 13, 3075–3078 RSC; (d) K. Padala and M. Jeganmohan, Org. Lett., 2011, 13, 6144–6147 CrossRef CAS PubMed; (e) V. Lanke and K. R. Prabhu, Org. Lett., 2013, 15, 2818–2821 CrossRef CAS PubMed.
- (a) L. D. Quin and J. A. Tyrell, Fundamentals of Heterocyclic Chemistry: Importance in Nature and in the Synthesis of Pharmaceuticals, Wiley, New Jersey, 2007, p. 196 Search PubMed; (b) G. Zeni and R. C. Larock, Chem. Rev., 2006, 106, 4644–4680 CrossRef CAS PubMed; (c) H. Stuppner, et al. , J. Nat. Prod., 2011, 74, 1779–1786 CrossRef PubMed; (d) K. M. Zareba, Drugs Today, 2006, 42, 75–86 CrossRef CAS PubMed.
- (a) R. Rossi, F. Bellina, M. Lessi and C. Manzini, Adv. Synth. Catal., 2014, 356, 17–117 CrossRef CAS; (b) S. Anwar, W.-Y. Huang, C.-H. Chen, Y.-S. Cheng and K. Chen, Chem. – Eur. J., 2013, 19, 4344–4351 CrossRef CAS PubMed; (c) A. Carrer, D. Brinet, J.-C. Forent, P. Rousselle and E. Bertounesque, J. Org. Chem., 2012, 77, 1316–1327 CrossRef CAS PubMed.
- (a) M. Ono, Y. Cheng, H. Kimura, M. Cui, S. Kagawa, R. Nishii and H. Saji, J. Med. Chem., 2011, 54, 2971–2979 CrossRef CAS PubMed; (b) B.-M. Swahn, D. Wensbo, J. Sandell, D. Sohn, C. Slivo, D. Pyring, J. Malmstrom, E. Arzel, M. Vallin, M. Bergh, F. Jeppsson, A. E. Johnson, A. Jureus, J. Neelissen and S. Svensson, Bioorg. Med. Chem. Lett., 2010, 20, 1976–1980 CrossRef CAS PubMed; (c) E. Arzel, B. Swahn, D. Wensbo, B. M. Svahn and D. Winsbor, Int. PCT Appl., WO2008108730, 2008.
- J. Zhao, H. Fang, C. Xie, J. Han, G. Li and Y. Pan, Asian J. Org. Chem., 2013, 2, 1044–1047 CrossRef CAS.
- (a) S.-Y. Tang, J. Zhang and Y. Fu, Comput. Theor. Chem., 2013, 1007, 31–40 CrossRef CAS PubMed; (b) V. F. Kuznetsov, K. Abdur-Rashid, A. J. Lough and D. G. Gusev, J. Am. Chem. Soc., 2006, 128, 14388–14396 CrossRef CAS PubMed.
- (a) M. Beller, A. Zapf and T. H. Riermeier, in Transition Metals for Organic Synthesis, ed. M. Beller and B. Carsten, Wiley-VCH, Weinheim, 2004, vol. 1, p. 271 Search PubMed; (b) R. F. Heck and J. P. Nolley Jr, J. Org. Chem., 1972, 37, 2320–2322 CrossRef CAS; (c) J. P. Knowles and A. Whiting, Org. Biomol. Chem., 2007, 5, 31–44 RSC.
- (a) F. Pozgan and P. H. Dixneuf, Adv. Synth. Catal., 2009, 351, 1737–1743 CrossRef CAS; (b) P. B. Arockiam, V. Poirier, C. Fischmeister, C. Bruneau and P. H. Dixneuf, Green Chem., 2009, 11, 1871–1875 RSC; (c) B. Li, K. Devaraj, C. Darcel and P. H. Dixneuf, Tetrahedron, 2012, 68, 5179–5184 CrossRef CAS PubMed; (d) B. Li, T. Roisnel, C. Darcel and P. H. Dixneuf, Dalton Trans., 2012, 41, 10934–10937 RSC.
- E. F. Flegeau, C. Bruneau, P. H. Dixneuf and A. Jutand, J. Am. Chem. Soc., 2011, 133, 10161–10170 CrossRef CAS PubMed.
- The exclusive alkylation that resulted in the case of styrene (even in the presence of base) indicates the possible synergistic assistance of the carboxylate group either in stabilizing the alkylruthenium intermediate resulting from the coordinative insertion or the deprotonation of the syn-hydrogen.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data and spectra of all new compounds. See DOI: 10.1039/c4cy01268b |
| This journal is © The Royal Society of Chemistry 2015 |