
Novel iron(III) catalyst for the efficient and selective coupling of carbon dioxide and epoxides to form cyclic carbonates†
Antonio
Buonerba
ab,
Assunta
De Nisi
a,
Alfonso
Grassi
ab,
Stefano
Milione
ab,
Carmine
Capacchione
*ab,
Sergei
Vagin
c and
Bernhard
Rieger
c
aDipartimento di Chimica e Biologia, Università degli Studi di Salerno, via Giovanni Paolo II, 132-84084 Fisciano (SA), Italy. E-mail: ccapacchione@unisa.it
bNANOMATES, Research Centre for NANOMAterials and nanoTEchnology at Salerno University, 84084 Fisciano (SA), Italy
cWACKER Lehrstuhl für Makromolekulare Chemie, Technische Universität München, Lichtenbergstrasse 4, 85748 Garching, Germany
First published on 8th October 2014
Abstract
“Re-cycling carbon dioxide with iron”. The synthesis of cyclic organic carbonates in high yield, stereo- and chemo-selectivity was accomplished through the coupling of carbon dioxide and epoxides, catalysed by a novel air-stable and easy-to-handle thioether-triphenolate iron(III) complex.
The carbon dioxide produced by the combustion of fossil fuel is (alongside methane) considered to be the main cause of the so-called “greenhouse effect”. As a matter of fact the environment is unable to buffer the huge anthropogenic release of carbon dioxide and therefore this gas accumulates in the atmosphere, contributing to climate change.1 Efforts to reduce the CO2 production of the most advanced countries in recent years, should face the growing hunger for energy of the developing countries that primarily use coal as fuel in power plants. Indeed, coal has a higher emission of CO2 (950 g of CO2 per kWh) compared to natural gas and oil, and therefore such a situation resulted in a growing world production of CO2. Currently, despite carbon dioxide finding applications as a refrigerant, in fire extinguishers, as a supercritical solvent and as an extraction medium, for the recovery of oil and in the beverage industry, only 0.62% of the atmospheric CO2 is reutilized by humankind.1b The bottleneck for an intensive implementation of chemical processes based on CO2 lies in the thermodynamic stability of this molecule. Coupling with energy intensive starting materials or the supplying of external energy to the involved process are required. Among the possible applications, the synthesis of cyclic organic carbonates via cycloaddition of CO2 to epoxides represents a 100% atom-economical process.2 Cyclic organic carbonates are a class of value added products that, since their commercialization in the 1950s, have increased consistently in their importance as synthetic targets.3 Their high molecular dipole moments, dielectric constants and boiling temperatures yield them suitable as green, highly polar, aprotic solvents and as ion-carriers for lithium-based batteries. In addition, they find applications in the production of additives for lubricants and paints, as alkylating agents, in the synthesis of acyclic carbonates, and in the synthesis of polyurethanes and polycarbonates thorough ring opening polymerization. Industrially, cyclic organic carbonates are obtained from the condensation of phosgene and diols4 or from the coupling of carbon dioxide with diols,5 oxetane or epoxides.2
Typically, for the promotion of CO2–epoxide coupling binary catalytic systems, including a Lewis acid and a nucleophile, are employed. The Lewis acid activates the epoxide toward the ring opening resulting from the attack of the nucleophile to the less substituted carbon atom. This reaction is followed by a CO2 insertion by the nucleophile, leading to an alkyl emi-carbonato unit which can undergo ring closure, via a backbiting mechanism, producing the cyclic carbonate and regenerating the nucleophile (path A, Scheme 1). Concurrently the intermediate can participate in an alternate insertion of further molecules of epoxide and CO2, to afford a linear alternating polycarbonate (path B, Scheme 1).6 Moreover the consecutive insertion of epoxide molecules, leading to polyether linkages, is also possible (path C, Scheme 1). The selectivity and rate of the coupling are effected not only by the nature of the epoxide and catalytic system but also by the reaction conditions. Cyclic carbonates are thermodynamically favoured compared to polycarbonates, though a higher activation barrier generally has to be overcome to afford their formation.7 Quaternary ammonium or phosphonium (onium) salts,8 metal oxides and halides, ionic liquids9 and organocatalysts were found to be active for the synthesis of cyclic carbonates, though severe reaction conditions were generally required.2 In order to obtain a more active catalyst for the CO2–epoxide coupling under milder conditions various metal complexes combined with auxiliary nucleophiles, e.g. onium salts, have been intensively studied. Complexes of magnesium,10 aluminium,11 chromium,7b cobalt,12 zinc,13 niobium14 and other metals2 coupled mainly with porphyrins,10,15 phthalocyanines, salen,2f,7b,11c salphen13b–d or N-heterocyclic carbene16 ligands were found to be effective catalysts for this reaction. Although iron is widely available, cost effective and non-toxic, few examples of iron based catalysts for the CO2–epoxide coupling to provide cyclic carbonates have been reported to date.17 Indeed only recently a bimetallic macrocyclic-iron(III) catalyst has been shown by Williams and co-workers to be capable of affording both cyclic and poly-carbonates.17a Rieger and co-workers recently reported an iron(II)-Schiff base for propylene carbonate synthesis.17b More recently, Kleij and co-workers deeply investigated iron(III) chelated with aminotris(phenolates) as effective catalysts for the coupling of CO2 and epoxides, obtaining high activity and selectivity in sc-CO2.17c–f However, in many cases these catalysts require harsh reaction conditions or high catalyst loadings, which limit their broad applicability. Notably, in spite of the wide coordination chemistry of iron with sulfur containing enzymes and synthetic ligands18 such kinds of complexes have been barely investigated in catalysis.19 Indeed, the ligands used so far in the literature are based on the hard donors oxygen and nitrogen in combination with an iron(III) metal centre. The use of ligands containing soft donors such sulphur can lead to a more acidic iron(III) centre, resulting in an enhancement of the catalytic activity. With this target in mind we designed a new dithioether-triphenolate pro-ligand 3.
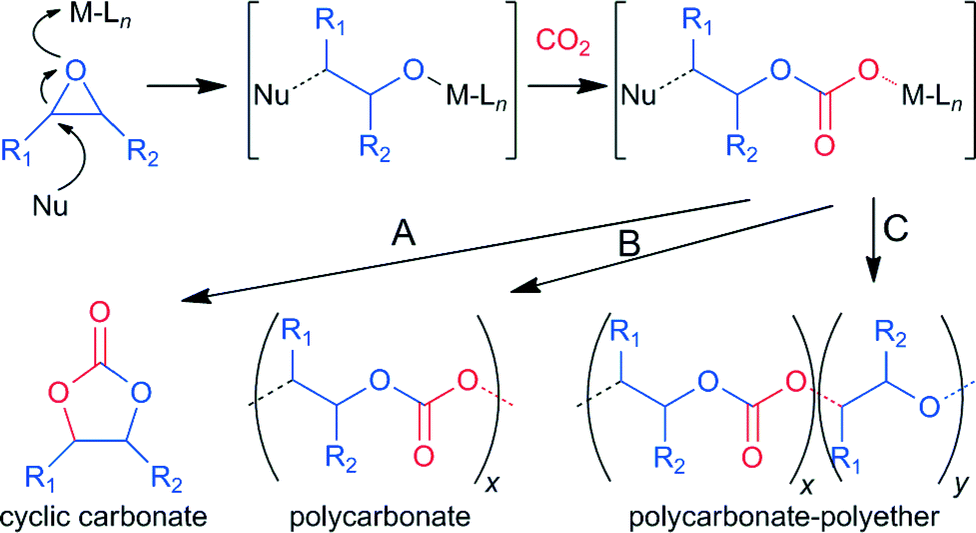 | ||
| Scheme 1 Mechanism for the formation of different products in CO2–epoxide coupling. A: backbiting; B: alternating epoxide/CO2 insertion; C: formation of polyether linkages. | ||
The synthetic strategy for affording the thioether-triphenolate ligand precursor 6,6′-(((5-(tert-butyl)-2-hydroxy-1,3-phenylene)bis(methylene))bis(sulfanediyl))bis(2,4-di-tert-butylphenol) (compound 3, Scheme 2) involves the coupling of 3,5-di-tert-butyl-2-hydroxythiophenol20 (1) and 2,6-di(bromomethyl)-4-(tert-butyl)phenol21 (2), via nucleophilic substitution (see Scheme 2). The novel ligand precursor 3 was recrystallized from acetone and fully characterized using 1H, 13C and two-dimensional NMR experiments, elemental analysis, MS, and FT-IR (see ESI†). The reaction of 3 with 3 equiv. of sodium hydride in THF and subsequent addition of the resulting sodium salt to FeCl3 affords the desired thioether-triphenolate iron(III) complex (4, Scheme 2). Complex 4 was recovered by recrystallization from THF, as small needle-shaped crystals that were unfortunately not suitable for structural resolution by means of single crystal X-ray diffraction. The formation of iron-ligand adduct 4 was initially confirmed by elemental analysis and FT-IR spectroscopy, (see ESI,† Fig. S9–S11). The ESI-mass spectrum of 4 shows a molecular ion peak at 1409.2 m/z, which is consistent with the dinuclear structure proposed (ESI,† Fig. S8). The Evans method22 was applied for determination of the magnetic moment in solution and a value of 7.34 μB resulted at 25 °C (the value is stable in the temperature range of 25–105 °C, see ESI,† Fig. S6). This value is higher than that predicted for a mononuclear high spin (HS) iron(III) complex and close to the value calculated for two isolated high spin (HS) iron(III) centres (8.37 μB),23 confirming the dimeric nature of the complex and indicating some degree of ferromagnetic coupling between the iron atoms, both being in the HS (s = 5/2) state. As expected for two HS iron(III) centres in an octahedral environment the UV-vis spectrum showed no d–d transitions, but only a strong ligand-to-metal charge transfer absorption at 620 nm (16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 129 cm−1; ε620 = 6508 L mol−1 cm−1). Furthermore, the octahedral coordination environment of the metal centre requires the coordination also of the sulfur atoms, which was in effect confirmed by a shift of the vibration bands in the infrared spectrum of the complex with respect to that of the free pro-ligand, in the spectral regions: 1440–1415 cm−1, 1270–1220 cm−1 and 700–600 cm−1, diagnostic of the alkyl-sulphide moiety (see Fig. S12–S14).24 In particular, the red-shift observed for the C–S stretching vibration accounts for a weakening of the bond as a result of the coordination of the sulphur atoms to the metal.
129 cm−1; ε620 = 6508 L mol−1 cm−1). Furthermore, the octahedral coordination environment of the metal centre requires the coordination also of the sulfur atoms, which was in effect confirmed by a shift of the vibration bands in the infrared spectrum of the complex with respect to that of the free pro-ligand, in the spectral regions: 1440–1415 cm−1, 1270–1220 cm−1 and 700–600 cm−1, diagnostic of the alkyl-sulphide moiety (see Fig. S12–S14).24 In particular, the red-shift observed for the C–S stretching vibration accounts for a weakening of the bond as a result of the coordination of the sulphur atoms to the metal.
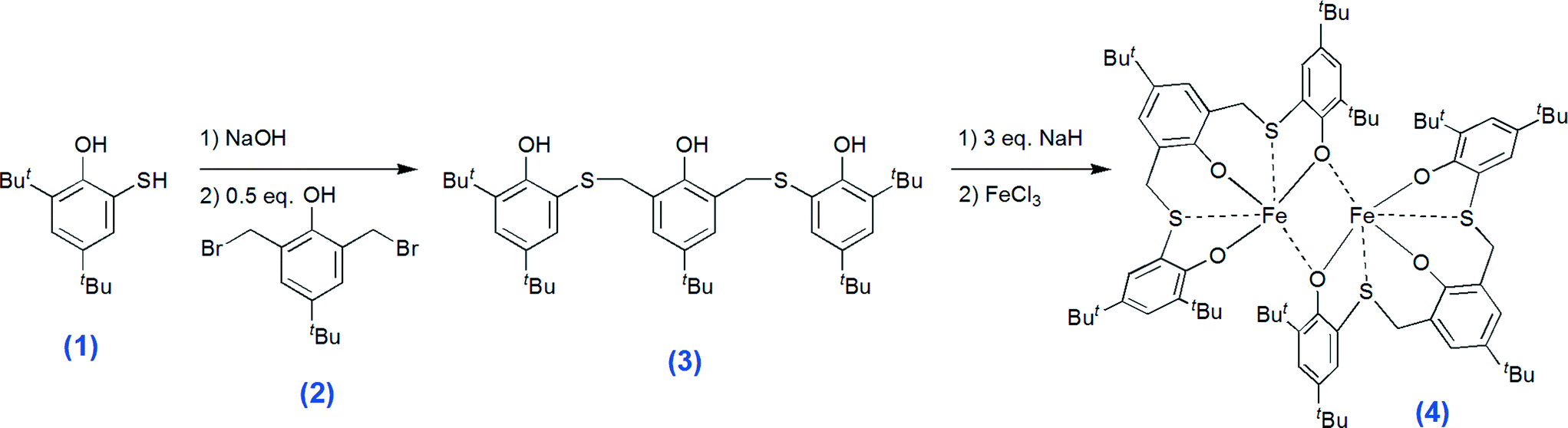 | ||
| Scheme 2 Synthesis of the iron(III) complex 4. | ||
Notably, the addition of pyridine or epichlorohydrin (ECH), for the evaluation of the stability of the complex,17d neither results in a change of the magnetic moment (evaluated in the presence of 50 equiv. of ECH with respect to the iron, in order to avoid a strong perturbation of the dielectric constant of the solvent media, see Fig. S6), nor in a change of the UV-vis spectrum (acquired in the presence of 3738 equiv. of ECH, see Fig. S17†), evidencing the stability of the dimeric state.
The iron(III) complex 4 was investigated as catalyst for the coupling of (±)-propylene oxide (PO) with carbon dioxide, in combination with several cocatalysts, for the selective synthesis of (±)-propylene carbonate (PC), under solvent free conditions. The main results are summarized in Table 1. A complete conversion of PO was observed within 6 h with a catalyst loading of 0.25 mol% and n-tetrabutylammonium bromide (TBAB) as the cocatalyst (2 MPa; 60 °C; TBAB:Fe molar ratio = 1, entry 1). Under the same conditions, a conversion of 68% of PO was achieved within 3 h, with a high turnover frequency (TOF) value of 90 h−1 (entry 2). As a consequence of these encouraging initial results, the catalyst and cocatalyst concentrations were both lowered by one order of magnitude; a conversion of 67% was reached in 24 h with a TOF value of 111 h−1 under these conditions (entry 3). The use of other common cocatalysts, such as n-tetrabutylammonium iodide (TBAI), bis(triphenylphosphine)iminium chloride (PPNCl) or 4-dimethylaminopyridine (DMAP), was less effective (entries 4–6). Iron(III) complex 4, in the absence of a cocatalyst, showed no reactivity (entry 7), whereas TBAB alone (entry 8) yields a PO conversion of only 14% within 24 h, thus highlighting a synergic effect between the catalyst and the cocatalyst (compare entries 8 and 3). Notably, by halving the cocatalyst loading (entry 9) a deterioration in the catalytic performance was observed; while a doubling of the cocatalyst loading improves the yield considerably, affording a conversion of 78% (entry 10). Further increasing the cocatalyst amount (entries 11–12) did not result in improved catalytic performances, and for this reason an optimal molar ratio of 2 was adopted for the subsequent catalytic tests (entries 13–19; Table 1). Remarkably, the temperature strongly affected the catalytic process. At 40 °C, a drop in the yield was observed (entry 13); while increasing the temperature (entries 14–16) resulted in a strong increase in the catalytic performance being observed. At 100 °C, a conversion of 87% after 6 h with the highest found TON and TOF values of 3480 and 580 h−1, respectively, was observed confirming additionally the thermal stability of the catalytic system. To the best of our knowledge, these catalytic performances lie among the highest reported for an iron based catalyst.17 The reason for the observed good activity of the complex 4 can be found in the weak coordination of the soft sulphur donor atoms to the hard Lewis acidic iron (III) metal centre. This interaction can facilitate the coordination of the epoxide to the metal centre, which is crucial for the subsequent attack of the nucleophile and the formation of the cyclic carbonate (see Scheme 1). In addition, the CO2 pressure seems to have no significant influence on the reaction course, in effect high TOF values were maintained at both low (entries 17–18) and high pressures (entry 19). This behaviour suggests a lesser impact on the energy barrier for the coupling with the epoxide from the CO2 insertion stage.
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entrya | Catalyst (mol%) | Cocatalyst (mol%) | Cocatalyst/Fe (molar ratio) | T (°C) | PCO2 (MPa) | t (h) | Conv.b,c (%) | TONd | TOFe (h−1) | |
| a Basic reaction conditions: 24.6 mg of complex 4 (1.75 × 10−5 mol), solventless. b Determined by NMR using mesitylene as an internal standard. c The selectivity for the formation of propylene carbonate was found to be >99%. d Overall turnover number (molPC molCat−1). e Overall turnover frequency (TON/reaction time). | ||||||||||
| 1 | 0.25 | TBAB | 0.5 | 1 | 60 | 2 | 6 | >99 | 394 | 66 |
| 2 | 0.25 | TBAB | 0.5 | 1 | 60 | 2 | 3 | 68 | 270 | 90 |
| 3 | 0.025 | TBAB | 0.05 | 1 | 60 | 2 | 24 | 67 | 2664 | 111 |
| 4 | 0.025 | TBAI | 0.05 | 1 | 60 | 2 | 24 | 31 | 1240 | 52 |
| 5 | 0.025 | PPNCl | 0.05 | 1 | 60 | 2 | 24 | 50 | 2000 | 83 |
| 6 | 0.025 | DMAP | 0.05 | 1 | 60 | 2 | 24 | 0 | — | — |
| 7 | 0.025 | — | — | 0 | 60 | 2 | 24 | 0 | — | — |
| 8 | — | TBAB | 0.05 | — | 60 | 2 | 24 | 14 | — | — |
| 9 | 0.025 | TBAB | 0.025 | 0.5 | 60 | 2 | 24 | 23 | 900 | 38 |
| 10 | 0.025 | TBAB | 0.1 | 2 | 60 | 2 | 24 | 78 | 3112 | 130 |
| 11 | 0.025 | TBAB | 0.25 | 5 | 60 | 2 | 24 | 56 | 2240 | 93 |
| 12 | 0.025 | TBAB | 0.35 | 7 | 60 | 2 | 24 | 64 | 2560 | 107 |
| 13 | 0.025 | TBAB | 0.1 | 2 | 40 | 2 | 24 | 6.4 | 256 | 11 |
| 14 | 0.025 | TBAB | 0.1 | 2 | 70 | 2 | 24 | 83 | 3320 | 138 |
| 15 | 0.025 | TBAB | 0.1 | 2 | 80 | 2 | 24 | 96 | 3840 | 160 |
| 16 | 0.025 | TBAB | 0.1 | 2 | 100 | 2 | 6 | 87 | 3480 | 580 |
| 17 | 0.025 | TBAB | 0.1 | 2 | 100 | 1 | 6 | 73 | 2920 | 487 |
| 18 | 0.025 | TBAB | 0.1 | 2 | 100 | 0.5 | 6 | 61 | 2440 | 407 |
| 19 | 0.025 | TBAB | 0.1 | 2 | 100 | 3 | 6 | 79 | 3160 | 526 |
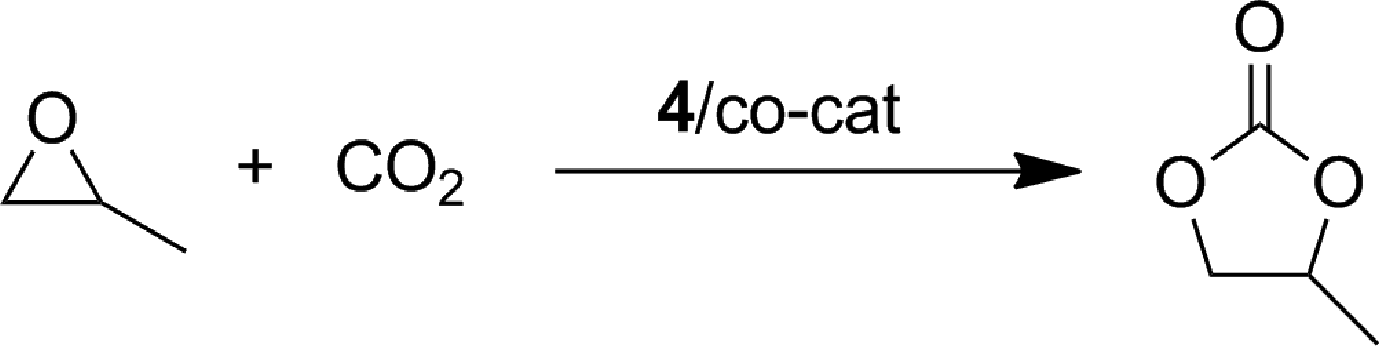
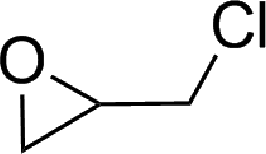
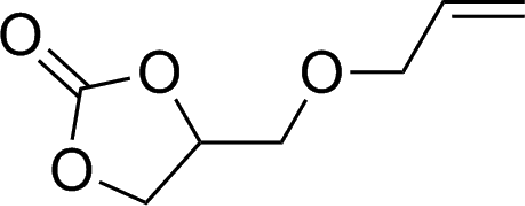
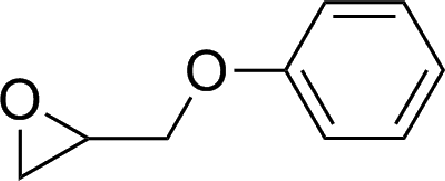
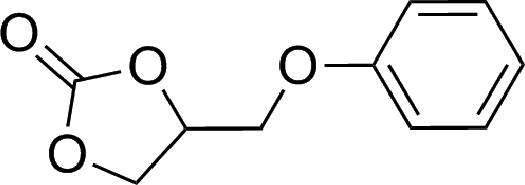
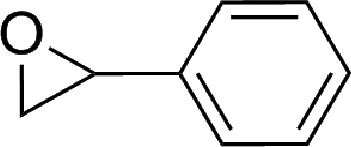
To expand the scope of the catalytic system, a series of differently substituted epoxides was screened as benchmark substrates (see Table 2). Monosubstituted epoxides containing functional groups, such as epichlorohydrin (78%, entry 20) or glycidol (95%, entry 21), gave conversions close to, or even superior to, that observed for CO2–PO coupling, showing that the presence of a functional group does not affect the catalyst performance. The increase of the size, in parallel with the reduction of the electron donating properties, of the substituents on the oxirane ring resulted in a slight decrease in the catalytic activity (entries 22–24) under the same reaction conditions. Enantiomerically enriched (R)-styrene oxide (ee = 94%) was also used in the coupling reaction with CO2, and showed a good retention of the stereochemistry (ee = 72%) giving (R)-styrene carbonate in 6 h, suggesting a good selectivity in the attack of the nucleophile. Finally, the cyclopentene and cyclohexene oxide, as expected for internal epoxides, were found to be less reactive toward the coupling with CO2 (entries 25–26). As a matter of fact, the corresponding cyclic carbonates consist, respectively, of a five- or six-membered aliphatic ring interconnected with a five-membered cycle comprising of the carbonate functionality, which results in an additional geometric strain. As a consequence, for these epoxides the formation of polycarbonates, even with a variable degree of polyether linkage, usually prevails over the cyclic carbonate formation, resulting in many cases in a mixture of products.7,17f,24 In our case, the cyclic carbonates were produced selectively with cis-stereochemistry, as pointed out by FT-IR analysis which revealed strong adsorption bands at 1800 and 1802 cm−1, previously assigned to the stretching of the carbonyl, respectively for the cis-cyclopentene carbonate25 and the cis-cyclohexene carbonate17a (see Fig. S15 and S16†). This unusual stereo control by iron catalysts was previously observed.17a
| Entrya | Substrate | Productb,c | Convb,c (%) | TONd | TOFe (h−1) |
|---|---|---|---|---|---|
| a Reaction conditions: substrate (7.0 × 10−2 mol), catalyst 4 (24.6 mg, 1.75 × 10−5 mol, 0.025 mol%), TBAB (7.0 × 10−5 mol, 0.1 mol%), CO2 (2 MPa); 100 °C, 6 h. b Determined by NMR (using mesitylene as an internal standard). c The selectivity for the formation of the cyclic carbonate was found to be >99%. d Overall turnover number (molPC molCat−1). e Overall turnover frequency (TON/reaction time). f Gave (R)-styrene carbonate in 86% selectivity and 72% ee). g A selectivity >99% for the formation of the cis isomer of the carbonate was observed. | |||||
| 20 |
|
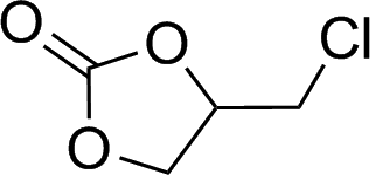
|
78 | 3120 | 520 |
| 21 |
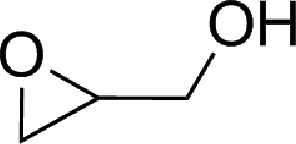
|
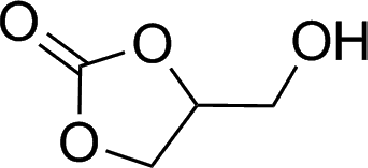
|
95 | 9800 | 633 |
| 22 |
|
|
55 | 2200 | 367 |
| 23 |
|
|
43 | 1720 | 287 |
| 24f |
|
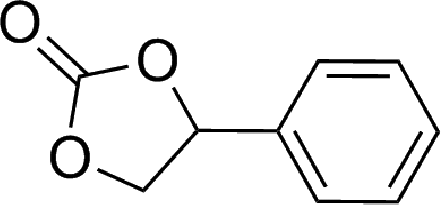
|
39 | 1560 | 260 |
| 25g |
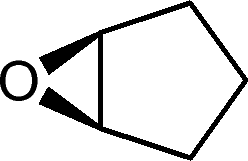
|
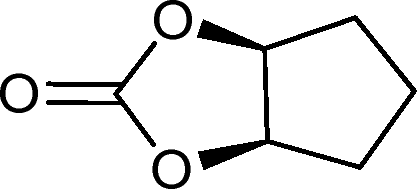
|
27 | 1077 | 180 |
| 26g |
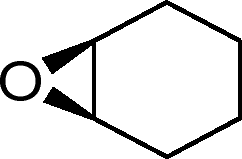
|
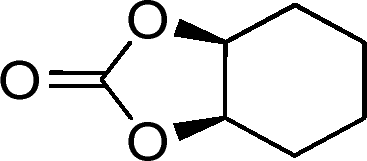
|
13 | 520 | 87 |
Conclusions
In summary, herein we reported on the efficient synthesis of cyclic carbonates by means of a new air stable and easy-to-handle iron(III) thioether-triphenolate complex. Experimental evidences indicated a dimeric nature for this novel complex. In the presence of tetrabutylammonium bromide, the title iron(III) complex 4 was very effective in the coupling of CO2 with epoxides, giving excellent results in terms of activity, chemo- and stereo-selectivity, under solvent-free conditions. To the best of our knowledge, this catalyst showed the highest TOF for a solventless cycloaddition of CO2 to propylene oxide under moderate reaction conditions. The catalyst was found to be thermally stable and tolerant of some functional groups (e.g. hydroxyl and vinyl groups). The dependence of the catalytic performance on the co-catalyst concentration and the steric and electron-donating properties of the substituents of the epoxide, as well as the low dependence of the activity on CO2 pressure, preliminarily indicates a greater influence of the stage of the ring-opening of the epoxide on the activation barrier for this reaction. In addition, this catalytic system showed good stereo control for the cycloaddition of CO2 to enantiomerically enriched (R)-styrene oxide or cyclic epoxides yielding, respectively, the (R)-styrene carbonate and the corresponding cis-cyclic carbonate isomers with good selectivity. These results clearly show that iron(III) based catalysts are a promising alternative to more toxic and/or expensive metals for the formation of cyclic carbonates from carbon dioxide and epoxides. Further work is currently in progress in order to establish a more refined catalyst structure–reactivity relationship.Acknowledgements
Financial support is acknowledged from the Ministero dell'Istruzione dell'Università e della Ricerca (MIUR, Roma, Italy for FARB-2013), the Regione Campania (POR FSE, project: “MAteriali e STrutture Intelligenti”, MASTRI, Code 4-17-3, CUPB25B09000010007) and the Centro di Tecnologie Integrate per la Salute (Project PONa3_00138) for the 600 MHz NMR instrument time. The Alexander von Humboldt Foundation is deeply acknowledged for a renewal fellowship (C.C.). The authors are also grateful to Dr. Carmen Talotta, Dr. Patrizia Oliva and Dr. Patrizia Iannece from Università degli Studi di Salerno for technical assistance.Notes and references
- (a) M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975–2992 RSC; (b) M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2013, 114, 1709–1742 CrossRef PubMed; (c) D. J. Darensbourg, Inorg. Chem., 2010, 49, 10765–10780 CrossRef CAS PubMed; (d) I. Omae, Coord. Chem. Rev., 2012, 256, 1384–1405 CrossRef CAS PubMed; (e) E. A. Quadrelli, G. Centi, J.-L. Duplan and S. Perathoner, ChemSusChem, 2011, 4, 1194–1215 CrossRef CAS PubMed; (f) Activation of Carbon Dioxide, ed. S. L. Suib, Elsevier, Amsterdam, 2013 Search PubMed.
- For some comprehensive books and reviews on CO2–epoxide couplings to synthesise cyclic carbonates see: (a) M. North, in New and Future Developments in Catalysis, ed. S. L. Suib, Elsevier, Amsterdam, 2013, pp. 379–413 Search PubMed; (b) M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS PubMed; (c) R. Li, X. Tong, X. Li and C. Hu, Pure Appl. Chem., 2012, 84, 621–636 CrossRef CAS; (d) N. Kielland, C. J. Whiteoak and A. W. Kleij, Adv. Synth. Catal., 2013, 355, 2115–2138 CrossRef CAS; (e) M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC; (f) A. Decortes, A. M. Castilla and A. W. Kleij, Angew. Chem., Int. Ed., 2010, 49, 9822–9837 CrossRef CAS PubMed.
- (a) B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CrossRef PubMed; (b) A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951–976 CrossRef CAS PubMed.
- A.-H. Liu, Y.-N. Li and L.-N. He, Pure Appl. Chem., 2012, 84, 581–602 CrossRef CAS.
- M. Tamura, M. Honda, Y. Nakagawa and K. Tomishige, J. Chem. Technol. Biotechnol., 2014, 89, 19–33 CrossRef CAS.
- (a) X.-B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 RSC; (b) P. P. Pescarmona and M. Taherimehr, Catal. Sci. Technol., 2012, 2, 2169–2187 RSC; (c) D. J. Darensbourg and M. W. Holtcamp, Coord. Chem. Rev., 1996, 153, 155–174 CrossRef CAS; (d) C. J. Whiteoak and A. W. Kleij, Synlett, 2013, 24, 1748–1756 CrossRef CAS PubMed.
- (a) D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS PubMed; (b) D. J. Darensbourg, J. C. Yarbrough, C. Ortiz and C. C. Fang, J. Am. Chem. Soc., 2003, 125, 7586–7591 CrossRef CAS PubMed.
- (a) V. Caló, A. Nacci, A. Monopoli and A. Fanizzi, Org. Lett., 2002, 4, 2561–2563 CrossRef PubMed; (b) N. Aoyagi, Y. Furusho and T. Endo, Tetrahedron Lett., 2013, 54, 7031–7034 CrossRef CAS PubMed.
- A.-L. Girard, N. Simon, M. Zanatta, S. Marmitt, P. Goncalves and J. Dupont, Green Chem., 2014, 16, 2815–2825 RSC.
- T. Ema, Y. Miyazaki, S. Koyama, Y. Yano and T. Sakai, Chem. Commun., 2012, 48, 4489–4491 RSC.
- (a) C.-Y. Li, D.-C. Liu and B.-T. Ko, Dalton Trans., 2013, 42, 11488–11496 RSC; (b) M. A. Fuchs, C. Altesleben, T. A. Zevaco and E. Dinjus, Eur. J. Inorg. Chem., 2013, 2013, 4541–4545 CrossRef CAS; (c) R. Luo, X. Zhou, S. Chen, Y. Li, L. Zhou and H. Ji, Green Chem., 2014, 16, 1496–1506 RSC; (d) C. J. Whiteoak, N. Kielland, V. Laserna, E. C. Escudero-Adán, E. Martin and A. W. Kleij, J. Am. Chem. Soc., 2013, 135, 1228–1231 CrossRef CAS PubMed; (e) C. J. Whiteoak, N. Kielland, V. Laserna, F. Castro-Gómez, E. Martin, E. C. Escudero-Adán, C. Bo and A. W. Kleij, Chem. – Eur. J., 2014, 20, 2264–2275 CrossRef CAS PubMed.
- P. Yan and H. Jing, Adv. Synth. Catal., 2009, 351, 1325–1332 CrossRef CAS.
- (a) M. A. Fuchs, S. Staudt, C. Altesleben, O. Walter, T. A. Zevaco and E. Dinjus, Dalton Trans., 2014, 43, 2344–2347 RSC; (b) M. V. Escárcega-Bobadilla, M. Martínez Belmonte, E. Martin, E. C. Escudero-Adán and A. W. Kleij, Chem. – Eur. J., 2013, 19, 2641–2648 CrossRef PubMed; (c) M. Taherimehr, A. Decortes, S. M. Al-Amsyar, W. Lueangchaichaweng, C. J. Whiteoak, E. C. Escudero-Adan, A. W. Kleij and P. P. Pescarmona, Catal. Sci. Technol., 2012, 2, 2231–2237 RSC; (d) A. Decortes, M. Martinez Belmonte, J. Benet-Buchholz and A. W. Kleij, Chem. Commun., 2010, 46, 4580–4582 RSC.
- A. Monassier, V. D'Elia, M. Cokoja, H. Dong, J. D. A. Pelletier, J.-M. Basset and F. E. Kühn, ChemCatChem, 2013, 5, 1321–13243 CrossRef CAS.
- T. Ema, Y. Miyazaki, T. Taniguchi and J. Takada, Green Chem., 2013, 15, 2485–2492 RSC.
- L. Yang and H. Wang, ChemSusChem, 2014, 7, 962–998 CrossRef CAS PubMed.
- (a) A. Buchard, M. R. Kember, K. G. Sandeman and C. K. Williams, Chem. Commun., 2011, 47, 212–214 RSC; (b) J. E. Dengler, M. W. Lehenmeier, S. Klaus, C. E. Anderson, E. Herdtweck and B. Rieger, Eur. J. Inorg. Chem., 2011, 2011, 336–343 CrossRef; (c) C. J. Whiteoak, E. Martin, M. M. Belmonte, J. Benet-Buchholz and A. W. Kleij, Adv. Synth. Catal., 2012, 354, 469–476 CrossRef CAS; (d) C. J. Whiteoak, B. Gjoka, E. Martin, M. M. Belmonte, E. C. Escudero-Adán, C. Zonta, G. Licini and A. W. Kleij, Inorg. Chem., 2012, 51, 10639–10649 CrossRef CAS PubMed; (e) C. J. Whiteoak, E. Martin, E. Escudero-Adán and A. W. Kleij, Adv. Synth. Catal., 2013, 355, 2233–2239 CrossRef CAS; (f) M. Taherimehr, S. M. Al-Amsyar, C. J. Whiteoak, A. W. Kleij and P. P. Pescarmona, Green Chem., 2013, 15, 3083–3090 RSC; (g) M. A. Fuchs, T. A. Zevaco, E. Ember, O. Walter, I. Held, E. Dinjus and M. Doring, Dalton Trans., 2013, 42, 5322–5329 RSC; (h) M. Adolph, T. A. Zevaco, C. Altesleben, O. Walter and E. Dinjus, Dalton Trans., 2014, 43, 3285–3296 RSC.
- (a) S. Sproules and K. Wieghardt, Coord. Chem. Rev., 2010, 254, 1358–1382 CrossRef CAS PubMed; (b) A. Volbeda and J. C. Fontecilla-Camps, Coord. Chem. Rev., 2005, 249, 1609–1619 CrossRef CAS PubMed; (c) A. K. Justice, R. C. Linck, T. B. Rauchfuss and S. R. Wilson, J. Am. Chem. Soc., 2004, 126, 13214–13215 CrossRef CAS PubMed; (d) Z. Li, Y. Ohki and K. Tatsumi, J. Am. Chem. Soc., 2005, 127, 8950–8951 CrossRef CAS PubMed.
- (a) Iron Catalysis Fundamentals and Applications, ed. B. Plietker, Springer, 2011 Search PubMed; (b) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2010, 111, 1293–1314 CrossRef PubMed; (c) C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217–6254 CrossRef CAS PubMed.
- S. D. Pastor and D. Z. Denney, J. Heterocycl. Chem., 1988, 25, 681–683 CrossRef CAS.
- M. V. Baker, M. J. Bosnich, D. H. Brown, L. T. Byrne, V. J. Hesler, B. W. Skelton, A. H. White and C. C. Williams, J. Org. Chem., 2004, 69, 7640–7652 CrossRef CAS PubMed.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- F. A. Cotton, G. Wilkinson, C. A. Murillo and E. M. Bochmann, Advanced Inorganic Chemistry, Wiley, 6th edn, 1999 Search PubMed.
- (a) The Sadtler handbook of infrared spectra, ed. W. W. Simons, Sadtler Research Laboratories, Philadelphia, 1978 Search PubMed; (b) Interpreting Infrared, Raman, and Nuclear Magnetic Resonance Spectra, ed. R. A. Nyquist, Academic Press, San Diego, 2001, pp. 65–83 Search PubMed; (c) D. J. Darensbourg, S. J. Lewis, J. L. Rodgers and J. C. Yarbrough, Inorg. Chem., 2003, 42, 581–589 CrossRef CAS.
- D. J. Darensbourg, S.-H. Wei, A. D. Yeung and W. C. Ellis, Macromolecules, 2013, 46, 5850–5855 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and further NMR, MS, FT-IR, UV-vis spectroscopic characterizations. See DOI: 10.1039/c4cy01187b |
| This journal is © The Royal Society of Chemistry 2015 |