
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Beyond iron: non-classical biological functions of bacterial siderophores
Timothy C.
Johnstone
and
Elizabeth M.
Nolan
*
Department of Chemistry, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: lnolan@mit.edu; Tel: +617-452-2495
First published on 18th February 2015
Abstract
Bacteria secrete small molecules known as siderophores to acquire iron from their surroundings. For over 60 years, investigations into the bioinorganic chemistry of these molecules, including fundamental coordination chemistry studies, have provided insight into the crucial role that siderophores play in bacterial iron homeostasis. The importance of understanding the fundamental chemistry underlying bacterial life has been highlighted evermore in recent years because of the emergence of antibiotic-resistant bacteria and the need to prevent the global rise of these superbugs. Increasing reports of siderophores functioning in capacities other than iron transport have appeared recently, but reports of “non-classical” siderophore functions have long paralleled those of iron transport. One particular non-classical function of these iron chelators, namely antibiotic activity, was documented before the role of siderophores in iron transport was established. In this Perspective, we present an exposition of past and current work into non-classical functions of siderophores and highlight the directions in which we anticipate that this research is headed. Examples include the ability of siderophores to function as zincophores, chalkophores, and metallophores for a variety of other metals, sequester heavy metal toxins, transport boron, act as signalling molecules, regulate oxidative stress, and provide antibacterial activity.
 Timothy C. Johnstone | Timothy Johnstone completed his B.Sc. (Hons) studies at McGill University in 2009, where he worked under the guidance of Professors Bruce Arndtsen, Scott Bohle, Jay Nadeau, and Derek Gray. He subsequently carried out his doctoral research under the supervision of Professor Stephen Lippard at the Massachusetts Institute of Technology. Following completion of his thesis on the chemistry and nanodelivery of platinum anticancer agents in 2014, he joined the laboratory of Professor Elizabeth Nolan at MIT. There, he is working on siderophore-antibiotic conjugates for antibacterial applications. |
 Elizabeth M. Nolan | Elizabeth Nolan is an Associate Professor of Chemistry at the Massachusetts Institute of Technology. She received her Ph.D. under the guidance of Professor Stephen J. Lippard at MIT in 2006 and was a post-doctoral fellow with Professor Christopher T. Walsh at Harvard Medical School. Her independent research focuses on the roles of transition metal ions in the host/microbe interaction with particular emphasis on metal-chelating human host-defense proteins as well as bacterial siderophores. |
Introduction
Iron is an essential nutrient for nearly all known life forms. The caveat is included in the preceding statement because certain lactobacilli and the causative agent of Lyme disease, Borrelia burgdorferi, do not require iron for growth.1,2 Iron is commonly used in biological systems most likely because of its Earth abundance and the breadth of the chemistry that it can undergo.3 This element exists in two readily inter-convertible oxidation states: ferrous and ferric, or Fe(II) and Fe(III), respectively. The ability to convert between these two states allows iron to play a pivotal role in numerous electron transfer processes.4 The precisely tuned steric and electronic environments within enzyme active sites permit access to more highly oxidized, Fe(IV) and Fe(V), or reduced, Fe(I), states that are essential intermediates in catalytic biochemical transformations that range from methane oxidation to proton reduction.5,6 Moreover, iron centres can access different spin states, which allows chemical reactions to occur at this metal centre that are not as readily feasible in purely organic systems.7,8 Despite the ubiquity of iron in biology, the very nature of its aqueous chemistry dictates that, under aerobic conditions and at neutral pH, the concentration of dissolved iron is lower than the concentration needed to sustain bacterial life.9 In this regard, and in the discussions below, dissolved or soluble iron refers to solvated aquo/hydroxo complexes of the metal.The solubility of Fe(III) in water at neutral pH is typically quoted to be 10−18 M,10,11 a value derived from the solubility product of Fe(OH)3, Ksp ≈ 10−39,12 and the concentration of hydroxide at neutral pH, [OH] = 10−7 M. An alternative soluble Fe(III) concentration of 1.4 nM has been proposed on theoretical grounds following re-evaluation of the importance of Fe(OH)2+ (aq.) in the speciation of this element.13,14 Regardless of this correction, the concentration of Fe(III) in solution falls far below the level to which iron is concentrated within bacteria. For instance, spectrochemical analysis revealed the dry weight of Escherichia coli to be 0.021% Fe.15 A cell volume of 10−15 L16,17 and a dry mass of 10−13 g17 affords a whole cell concentration of Fe that is approximately 10−3 M. This concentration deviates from the value previously quoted (10−6 M);11 however, a comparison of either value to the solubility of Fe(III) highlights the extent to which bacteria concentrate Fe obtained from the environment. For commensal and pathogenic bacteria that colonize humans, iron concentrations are even more limited. The low levels of Fe(III) that exist freely in solution are toxic to mammals and are accordingly suppressed by a number of mechanisms. Most notably, Fe(III) levels in human serum are maintained at approximately 10−24 M by transferrin,18 an abundant iron transport protein. We note that this concentration of Fe(III) in serum is obtained by using the transferrin log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K1 = 22.7, logK2 = 22.1, and the estimation that at any given time about 30% of the binding sites of transferrin in blood are empty.18
K1 = 22.7, logK2 = 22.1, and the estimation that at any given time about 30% of the binding sites of transferrin in blood are empty.18
In order to acquire iron under these conditions, bacteria employ a number of transport mechanisms.10 One of the most striking is the secretion of siderophores, small molecules that exhibit high binding affinity for iron.19 A similar strategy is employed by fungi20 and plants21 but the focus of this Perspective will be on bacterial siderophores. These chelators exhibit proton-independent stability constants (β) for iron complexation ranging from approximately 1010 to 1049.22,23 The latter is the β110 value for the complex of Fe(III) and enterobactin6−, where the βMLH notation indicates the formation constant for a given stoichiometry of metal (M), fully deprotonated ligand (L), and protons (H). Although the β values of different siderophores may not be directly comparable because of differences in ligand pKa values and metal:ligand binding ratios, enterobactin is widely credited as the siderophore with the highest known affinity for iron.24 This statement is borne out by metrics, such as pFe(III),† which more accurately compare the stabilities of Fe(III) complexes under physiologically relevant conditions.24 The enormous range in stability constants reflects the diversity of chemical composition across the >260 siderophores of known chemical structure.25,26 The chemical structures of the siderophores discussed in this paper are collected in Charts 1–9. The chemical structures and stereochemical assignments are in general agreement with those given in a recent comprehensive review of the siderophore literature,26 and when differences occur they are noted in the Chart captions. Table 1 lists all of the siderophores discussed in this Perspective in alphabetical order, provides the locations of the corresponding chemical structures, and summarizes the non-classical functions of each.
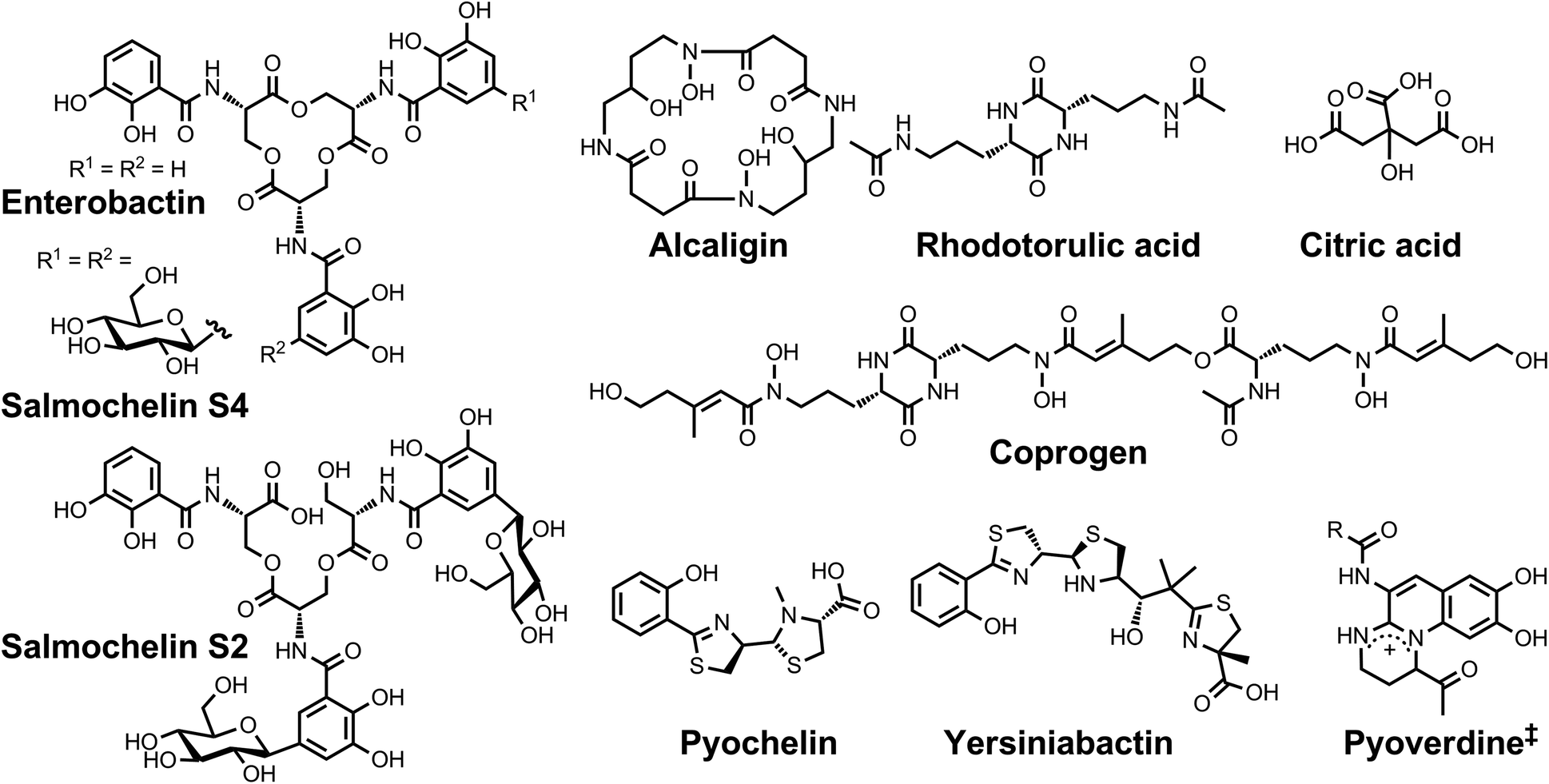 | ||
| Chart 1 Chemical structures of the siderophores discussed in the introduction. The stereochemistry of yersiniabactin was taken from ref. 31. The structure of salmochelin S2 is that reported in ref. 32. An alternative structure of a linearized salmochelin S4 in which a glucosyl unit is attached at the carboxylic acid end of the linearized trimer instead of the alcohol end has also been reported.33 The enantiomer of the form of pyochelin shown, enantio-pyochelin, is a known siderophore. ‡Many pyoverdines exist and are distinguished by the nature of the peptide chain attached at the R position. | ||
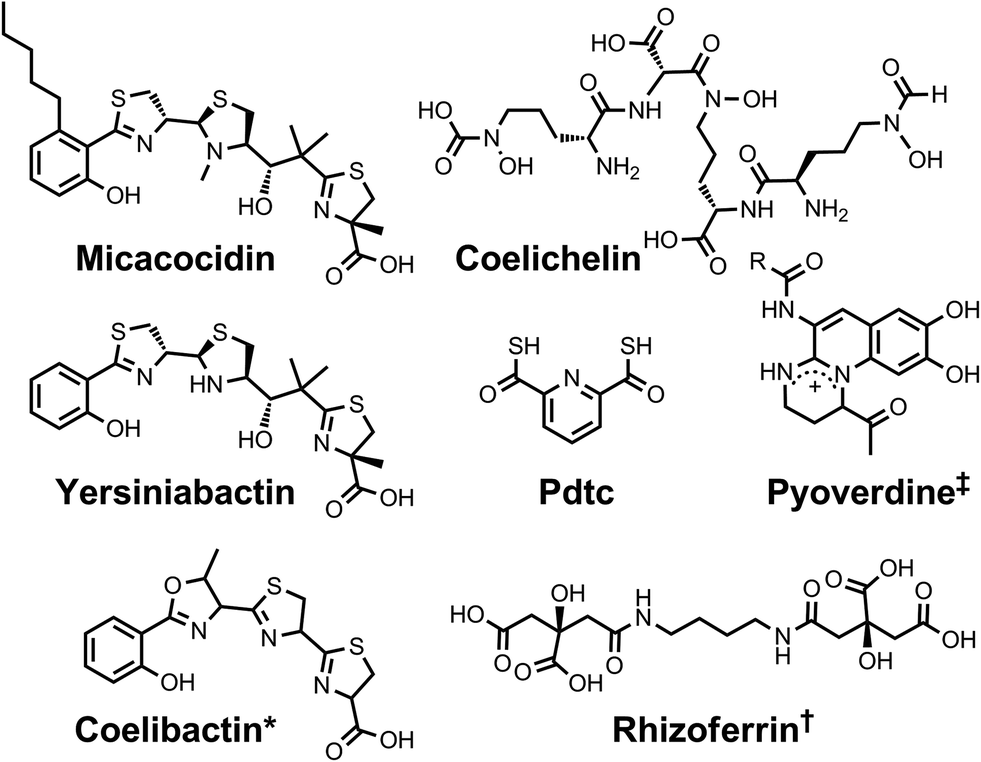 | ||
| Chart 2 Chemical structures of the siderophores discussed in Siderophores as Zincophores. The stereochemistry of yersiniabactin was taken from ref. 31. *The structure of coelibactin is tentative and based on bioinformatics analyses; stereochemistry has yet to be established.56†Rhizoferrin can occur as shown (S,S) or in the enantiomeric (R,R) form. ‡Many pyoverdines exist and are distinguished by the nature of the peptide chain attached at the R position. Pdtc is pyridine-2,6-dithiocarboxylic acid. | ||
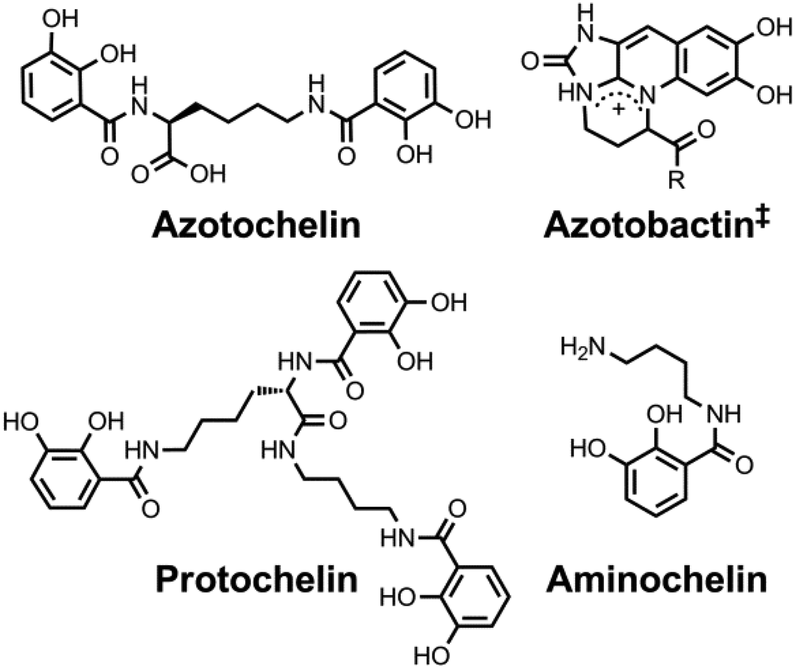 | ||
| Chart 3 Chemical structures of the siderophores discussed in Fuelling Nitrogenase: Molybdenum and Vanadium. ‡Azotobactin has a peptide chain as the R substituent. | ||
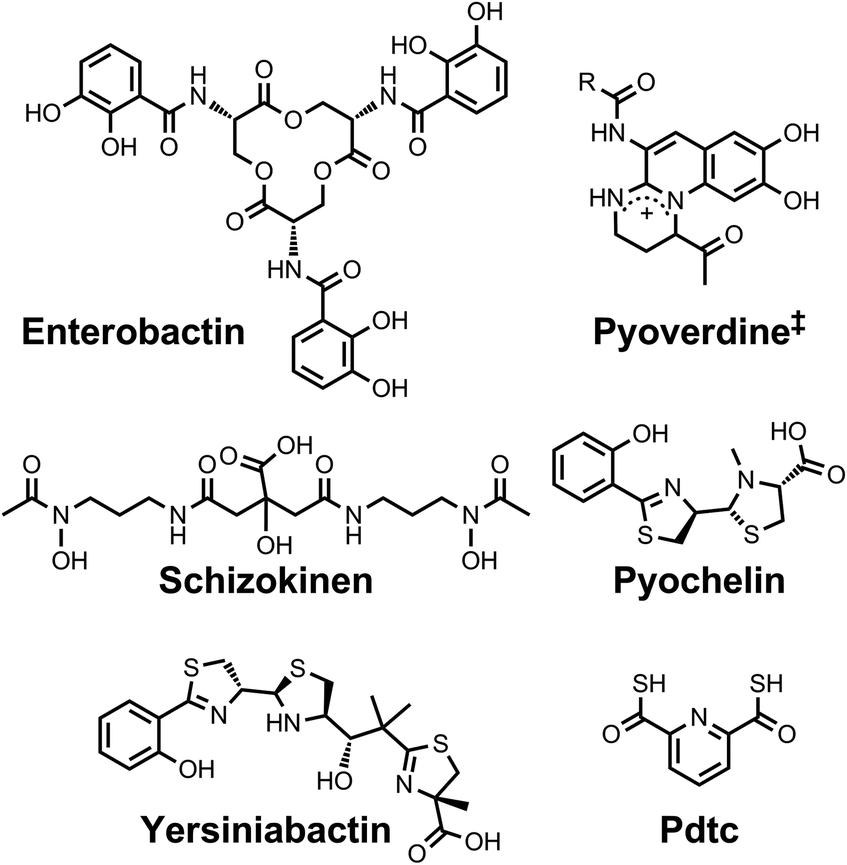 | ||
| Chart 4 Chemical structures of the siderophores discussed in The Interaction of Siderophores with Copper. The stereochemistry of yersiniabactin was taken from ref. 31. The enantiomer of the form of pyochelin shown, enantio-pyochelin, is a known siderophore. ‡Many pyoverdines exist and are distinguished by the nature of the peptide chain attached at the R position. Pdtc is pyridine-2,6-dithiocarboxylic acid. | ||
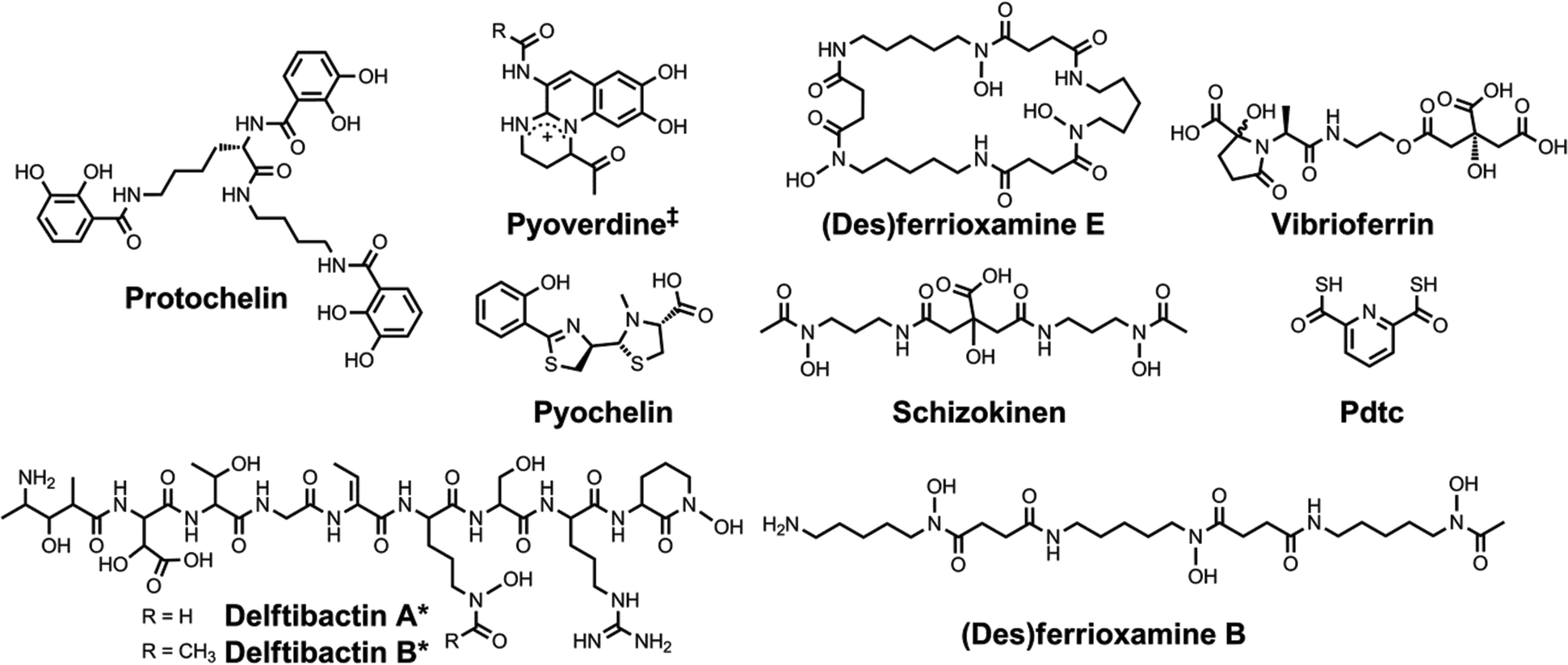 | ||
| Chart 5 Chemical structures of the siderophores discussed in Other Metals: Transport and Sequestration and Marine Siderophores and Boron. The enantiomer of the form of pyochelin shown, enantio-pyochelin, is a known siderophore. ‡Many pyoverdines exist and are distinguished by the nature of the peptide chain attached at the R position. *The stereochemistry of the delftibactins has not been reported.142,143 The chemical structure of vibrioferrin was obtained from ref. 144. Pdtc is pyridine-2,6-dithiocarboxylic acid. | ||
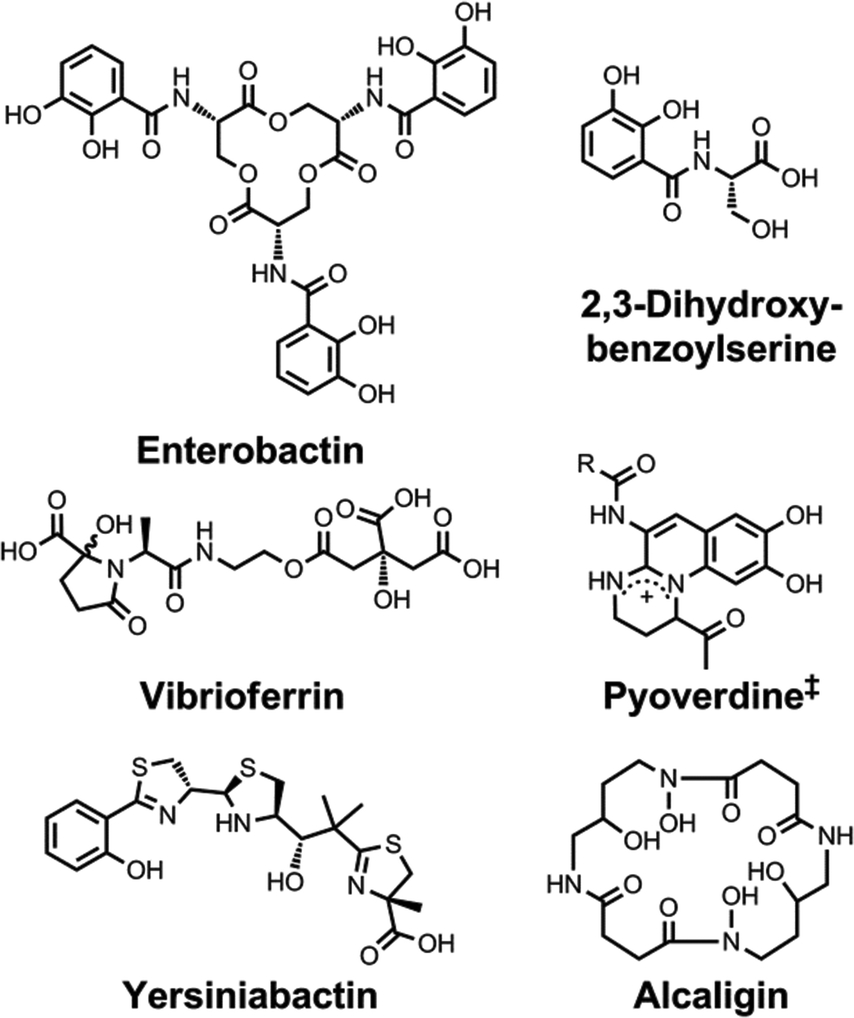 | ||
| Chart 6 Chemical structures of the siderophores discussed in Siderophores as Signalling Molecules. The stereochemistry of yersiniabactin was taken from ref. 31. ‡Many pyoverdines exist and are distinguished by the nature of the peptide chain attached at the R position. The chemical structure of vibrioferrin was obtained from ref. 144. | ||
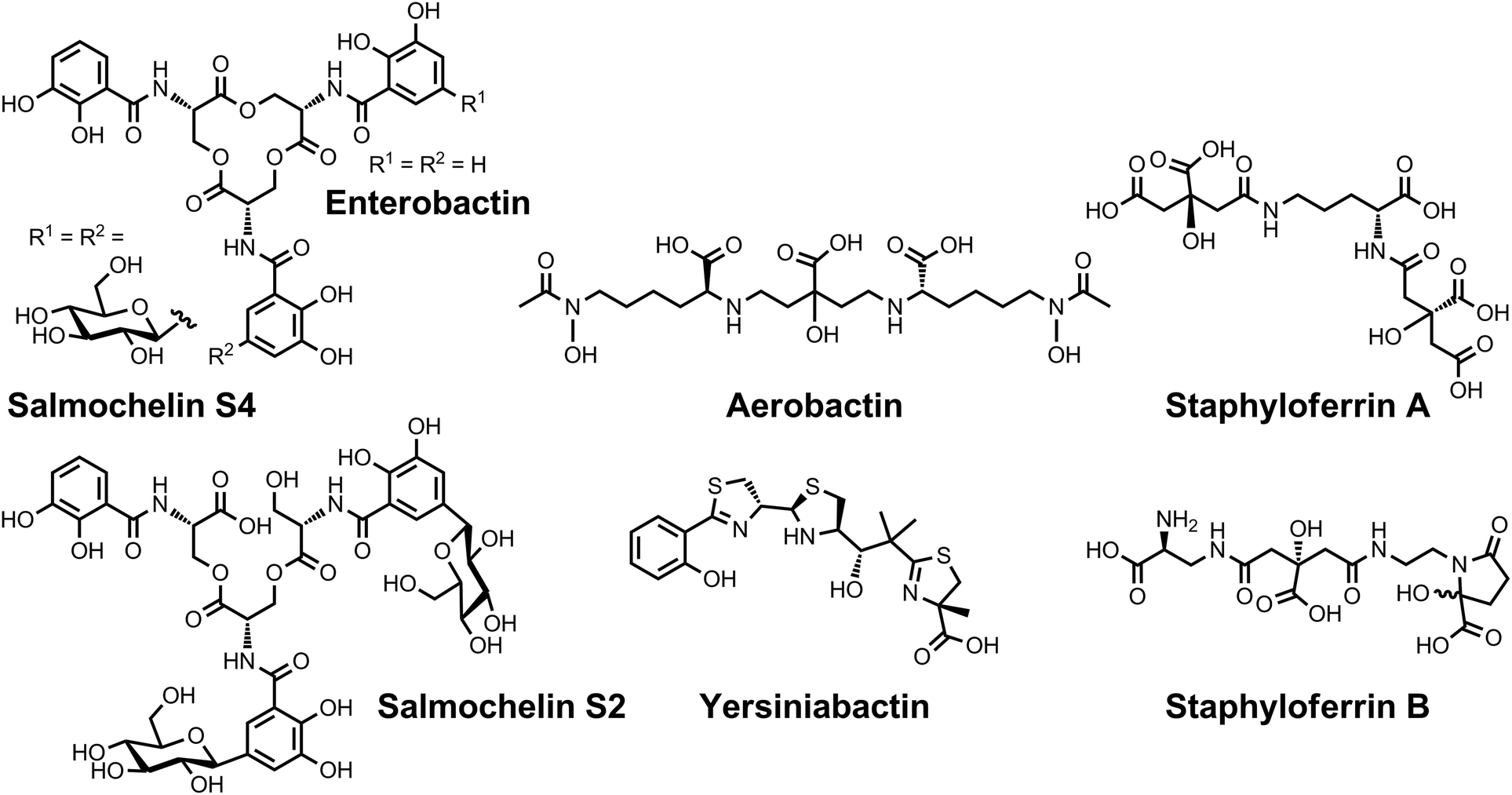 | ||
| Chart 7 Chemical structures of the siderophores discussed in Protection from Oxidative Stress. The stereochemistry of yersiniabactin was taken from ref. 31. The chemical structures of staphyloferrin A and staphyloferrin B were obtained from ref. 205 and 206. The structure of salmochelin S2 is that reported in ref. 32. An alternative structure of a linearized salmochelin S4 in which a glucosyl unit is attached at the carboxylic acid end of the linearized trimer instead of the alcohol end has also been reported.33 The stereochemistry of aerobactin is drawn so as to be consistent with the observation that only L-lysine is released upon degradation of this compound.245 | ||
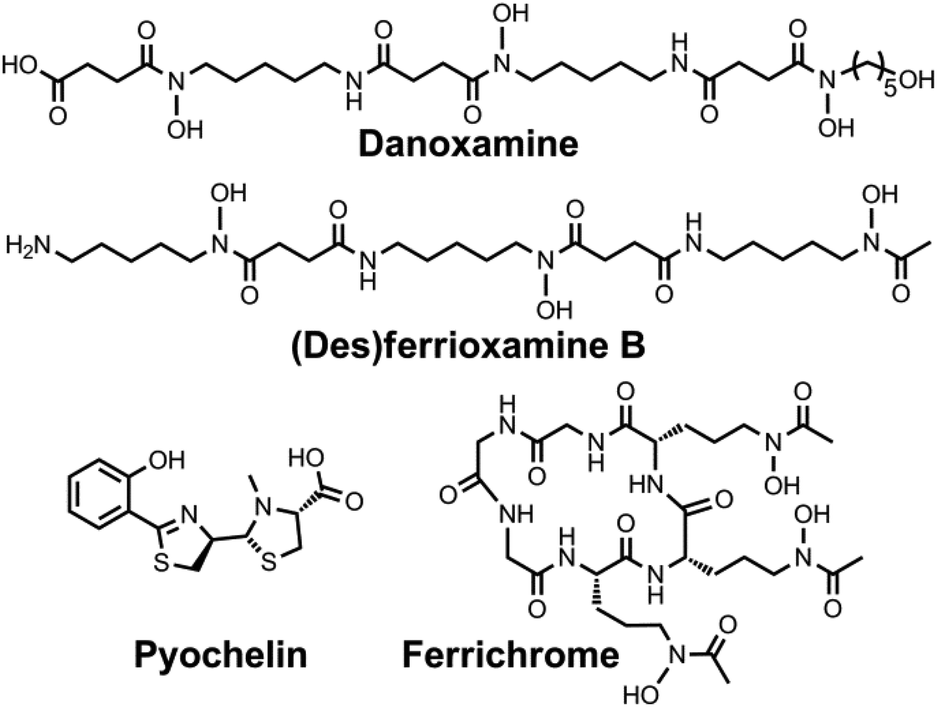 | ||
| Chart 8 Chemical structures of the siderophores discussed in Sideromycins: Siderophores as Antibiotics. The enantiomer of the form of pyochelin shown, enantio-pyochelin, is a known siderophore. | ||
 | ||
| Chart 9 Natural sideromycins of known structure. The iron-binding siderophore unit is black, the antibiotic warhead is shown in red, and if a linker is present it is shown in blue. *It is also possible that water is added such that this position bears a gem-diol. | ||
| Siderophore | Non-classical function(s) | Chart(s) containing structure |
|---|---|---|
| Aerobactin | — | 6 |
| Alcaligin | Cell signalling | 1, 6 |
| Aminochelin | Mo/V transport | 3 |
| Azotobactin | Mo/V transport | 3 |
| Azotochelin | Mo/V transport | 3 |
| Citrate | — | 1 |
| Coelibactin | Zn binding | 2 |
| Coelichelin | Zn binding | 2 |
| Coprogen | — | 1 |
| Danoxamine | Antibiotic component | 7 |
| Delftibactins (A, B) | Other non-iron metallophore | 5 |
| (Des)ferrioxamine B | Other non-iron metallophore | 5 |
| (Des)ferrioxamine E | Other non-iron metallophore | 5 |
| 2,3-Dihydroxybenzoylserine | Cell signalling | 6 |
| Enterobactin | Cell signalling | 1, 4, 6, 7 |
| Oxidative stress response | ||
| Ferrichrome | Antibiotic component | 7 |
| Micacocidin | Zn binding | 2 |
| Protochelin | Mo/V transport | 3 |
| Other non-iron metallophore | ||
| Pyochelin | Cu binding | 1, 4 |
| Other non-iron metallophore | ||
| Antibiotic activity | ||
| Pyoverdines | Zn binding | 1, 2, 4, 6, 7 |
| Cu binding | ||
| Other non-iron metallophore | ||
| Cell signalling | ||
| Pyridine-2,6-dithiocarboxylate (pdtc) | Zn binding | 2, 4 |
| Cu binding | ||
| Other non-iron metallophore | ||
| Rhizoferrin | Zn binding | 2 |
| Rhodotorulic acid | — | 1 |
| Salmochelins (S2, S4) | Oxidative stress response | 1, 7 |
| Schizokinen | Cu binding | 4 |
| Other non-iron metallophore | ||
| Staphyloferrin A | Oxidative stress response | 7 |
| Staphyloferrin B | Oxidative stress response | 7 |
| Vibrioferrin | Boron transport | 5, 6 |
| Cell signalling | ||
| Yersiniabactin | Zn binding | 1, 2, 4, 6 |
| Cu binding | ||
| Cell signalling | ||
| Oxidative stress response |
The typical metal-binding motifs – catecholates, hydroxamates, and α-hydroxycarboxylic acids – select for Fe(III) in accordance with the hard-soft acid–base theory. These hard oxygen-atom donors are well-matched to the hard ferric ion. Many siderophores present six coordinating atoms to the metal centre in a pseudooctahedral geometry and a number of these ligands exhibit a metal-binding pocket that is preorganized, typically via macrocyclization. For example, the linear dihydroxybenzoylserine trimer, a product of the hydrolysis of enterobactin, exhibits a proton-independent formation constant of 1043, as compared to the cyclized parent compound, which exhibited a corresponding β value of 1049.27 Additionally, the macrocyclic dihydroxamate siderophore alcaligin binds Fe(III) 32 times more strongly than the linear dihydroxamate rhodotorulic acid, as determined by comparing the proton-independent stability constants of the 1:1 Fe–siderophore complexes.28 In addition to macrocyclization, another general strategy employed in siderophore biosynthesis is heterocyclization, exemplified by the enzyme-catalyzed cyclodehydration of X-cysteine and X-serine dipeptide motifs to form thiazoline and oxazoline rings, respectively.29 These heterocycles can undergo further modifications, including dehydration and aromatization to afford thiazoles and oxazoles, or reduction to afford thiazolidines and oxazolidines. Such chemistry, used in the biosynthesis of siderophores such as yersiniabactin and pyochelin, rigidifies the peptide backbone and increases the basicity of the formerly peptidic nitrogen atom, allowing it to better coordinate iron.30
Many bacteria produce, or are capable of utilizing, multiple siderophores. For instance, the laboratory strain E. coli K-12 expresses machinery for the uptake of ferric complexes of enterobactin, linearized enterobactin, citrate, ferrichrome, rhodotorulate, and coprogen.24E. coli K-12 cannot produce the latter three siderophores, however.34 Siderophores used by a non-producer organism are called xenosiderophores. The FoxA-mediated uptake of ferrioxamine by Yersinia enterocolitica and the uptake of enterobactin via Pseudomonas aeruginosa PfeA and PirA are further examples of xenosiderophore utilization.35–37 The ability of organisms to use redundant siderophore-meditated iron uptake pathways has prompted the question as to why such overlap exists.
One answer to this question is that different siderophores can better extract iron from different sources or under different environmental conditions.38–40 Additionally, certain siderophores permit bacterial pathogens to better evade the host immune system. For example, Salmonella enterica produce enterobactin to satisfy their nutritional requirements, but humans have evolved a defence mechanism whereby the secreted host-defence protein lipocalin-2 binds and sequesters ferric enterobactin. In response, Salmonella produce salmochelins,41 enterobactin derivatives that are glucosylated at the C5 position of one or more catechol rings to prevent capture by lipocalin-2.42 Certain pathogenic E. coli also produce salmochelins to overcome the host response to infection.41P. aeruginosa strains typically produce at least two siderophores, pyoverdine and pyochelin.43 Pyoverdines coordinate Fe(III) with high affinity (for pyoverdine PaA, Ka = 1030.8 and pFe(III) = 27),44 whereas pyochelin exhibits much lower affinity (Ka = 1017.3 and pFe(III) = 16).45 Mathematical simulations support the hypothesis that P. aeruginosa strains with the capacity to switch between producing and utilizing these two siderophores in response to changing iron conditions have an enhanced degree of fitness over those that cannot.46
Although access to multiple siderophores can confer advantages related to iron acquisition, another logical proposal is that organisms may produce seemingly redundant siderophores because some of these molecules are actually serving functions other than iron delivery. Indeed, investigations into alternative, non-classical functions of siderophores have revealed that this scenario occurs. Studies approaching this problem from both chemical and biological perspectives have revealed novel bioinorganic chemistry. In this Perspective, we intend to provide the reader with an overview of non-classical siderophore functions and the connections that exist between them (Table 2). We also intend for this exposition to spark new initiatives that will address as-yet unconceived siderophore functions. We will limit our discussion to biological siderophore functions, and we refer the reader to a number of reviews on the technological potential of siderophores, which, although arguably distinct from the classical siderophore function, fall outside the scope of this Perspective.47–50
| Iron transport | The classical function of siderophores in which they are secreted to bind iron and transport it within bacteria to satisfy a nutritional iron requirement |
| Non-iron metal transport | Siderophores can bind other essential metals like Zn, Mo, V, and Mn to mediate their cellular uptake |
| Non-metal transport | Elements such as boron and silicon can bind siderophores and may be used by bacteria to take up these elements |
| Toxic metal sequestration | Heavy metals can be bound by siderophores to prevent their cellular entry |
| Protection from oxidative stress | Catecholate-bearing siderophores can scavenge harmful radicals and cupric yersiniabactin can function as a superoxide dismutase |
| Molecular signalling | Siderophores can regulate gene expression and potentially function in quorum sensing |
| Antibiotic activity | Siderophores can be conjugated to antibiotic warheads to increase efficacy |
In the following sections, we will begin with a non-classical function that is perhaps the simplest extension from iron transport: the transport of other essential metals such as zinc, manganese, molybdenum, and vanadium. The interaction of siderophores with copper and heavy metals will also be discussed. A digression from metal–siderophore interactions will summarize the role that marine siderophores may play in boron uptake. The focus will then shift from siderophores binding ions and influencing mass transport to the putative role of metal–siderophore complexes and apo siderophores as signalling molecules. The ability of siderophores to protect bacteria from oxidative stress will also be addressed. We will conclude with a return to ferric siderophore complexes and present sideromycins, antibiotic–siderophore conjugates that exploit bacterial iron uptake machinery to enhance efficacy. Investigations into this non-classical siderophore function may prove to be particularly valuable in the development of therapies targeting the antibiotic-resistant pathogenic bacteria that are infesting hospitals worldwide.
Siderophores as zincophores
Following iron, zinc is the most abundant transition metal in E. coli.15 It has been estimated that approximately 5% of the E. coli proteome comprises zinc-binding proteins.51 Given the important structural and catalytic roles that Zn(II) plays in bacterial proteins, it is not surprising that these organisms have machinery, such as the ZnuABC transporter, dedicated to zinc uptake.52 The importance of Zn(II) acquisition for bacterial pathogens is underscored by the fact that humans secrete zinc-sequestering proteins (e.g. calprotectin and psoriasin) to inhibit the progression of microbial infections.53,54 It is possible that zinc-withholding by the host applies an evolutionary pressure on pathogenic bacteria to develop high-affinity zinc uptake mechanisms akin to those in which siderophores play a role.55 Little is known, however, about the extent to which bacteria secrete high-affinity zinc chelators, termed zincophores or tsinkosphores.54After the genome of the filamentous soil-dwelling bacterium Streptomyces coelicolor was sequenced,57 two non-ribosomal peptide synthases (NRPSs) were identified and deduced to be responsible for the biosynthesis of two small molecules, namely coelichelin and coelibactin. The structures of these molecules were predicted on the basis of bioinformatics analyses, which included identification of the specificity-determining residues of the NRPS adenylation domains that select and activate monomeric precursors.58 Subsequently, the proposed structure of coelichelin was amended following NMR studies of its gallium complex.59 Comparison with known siderophore structures suggested that coelichelin and coelibactin also function in iron transport. During later studies on the effect of zinc on the production of antibiotics by S. coelicolor, it was observed that increased zinc levels decreased the expression of coelibactin, as determined by qRT-PCR of the coelibactin NRPS mRNA transcripts, and a putative zincophore activity was ascribed to this molecule.60 Next, a study of the effect of coelibactin on S. coelicolor sporulation confirmed that Zur, the zinc uptake repressor, regulates coelibactin biosynthesis.61 Although the biosynthesis of coelibactin has been investigated,56,62 this metallophore has not yet been isolated from S. coelicolor and structurally elucidated; thus, its coordination chemistry remains to be explored.
A variety of pseudomonads produce the small molecule metal chelator pyridine-2,6-dithiocarboxylic acid, or pdtc, which was first identified as a result of its ability to bind iron and was therefore described as a siderophore.63 Production of this molecule has since been observed in both environmental samples and in laboratory cultures of Pseudomonas stutzeri KC grown either aerobically or anaerobically.64–66 The soft character of the sulphur donor atoms of pdtc favours complex formation with soft metals, providing higher affinity for Fe(II) than Fe(III). Pdtc also complexes zinc,67 and the ability of pdtc to effect biological transport of zinc was demonstrated in Pseudomonas putida.68 Subsequent studies revealed that the zinc–pdtc complex is recognized and transported by the outer membrane receptor and inner membrane permease of the pdtc utilization machinery; however, transport of zinc–pdtc is much less efficient than transport of the iron–pdtc complex.69
Very recently, it has been demonstrated that yersiniabactin, a heterocyclic siderophore produced by Yersinia pestis and some pathogenic E. coli, can act as a zincophore.70 The ability of Y. pestis znu mutants lacking functional ZnuABC transport machinery to grow in response to supplementation of Chelex-treated medium with zinc suggested that an unidentified high-affinity zinc transporter was operational.71 In support of this proposition, none of the other divalent metal ion transport systems of this organism mediated zinc uptake.71,72 Mutations in the irp2 gene encoding the yersiniabactin synthetase HMWP2 produced severe growth defects when combined with a ΔznuBC mutation. Bacterial growth was restored by either complementation with the irp2 gene in trans or Zn(II) supplementation.70 Moreover, addition of purified exogenous apo-yersiniabactin stimulated growth in mutant Y. pestis strains lacking a functional ZnuABC system. Remarkably, uptake of the yersiniabactin-complexed zinc requires neither the outer membrane receptor nor the inner membrane ABC transporter that are required for yersiniabactin-mediated iron transport, indicating that a separate uptake pathway is operative.72 The interaction of yersiniabactin with Zn(II) may have important implications for human disease based on experiments in a mouse model of septicaemic plague. The ZnuABC transport system and HMWP2, likely via its role in yersiniabactin synthesis, both contribute to the development of lethal Y. pestis infections.70
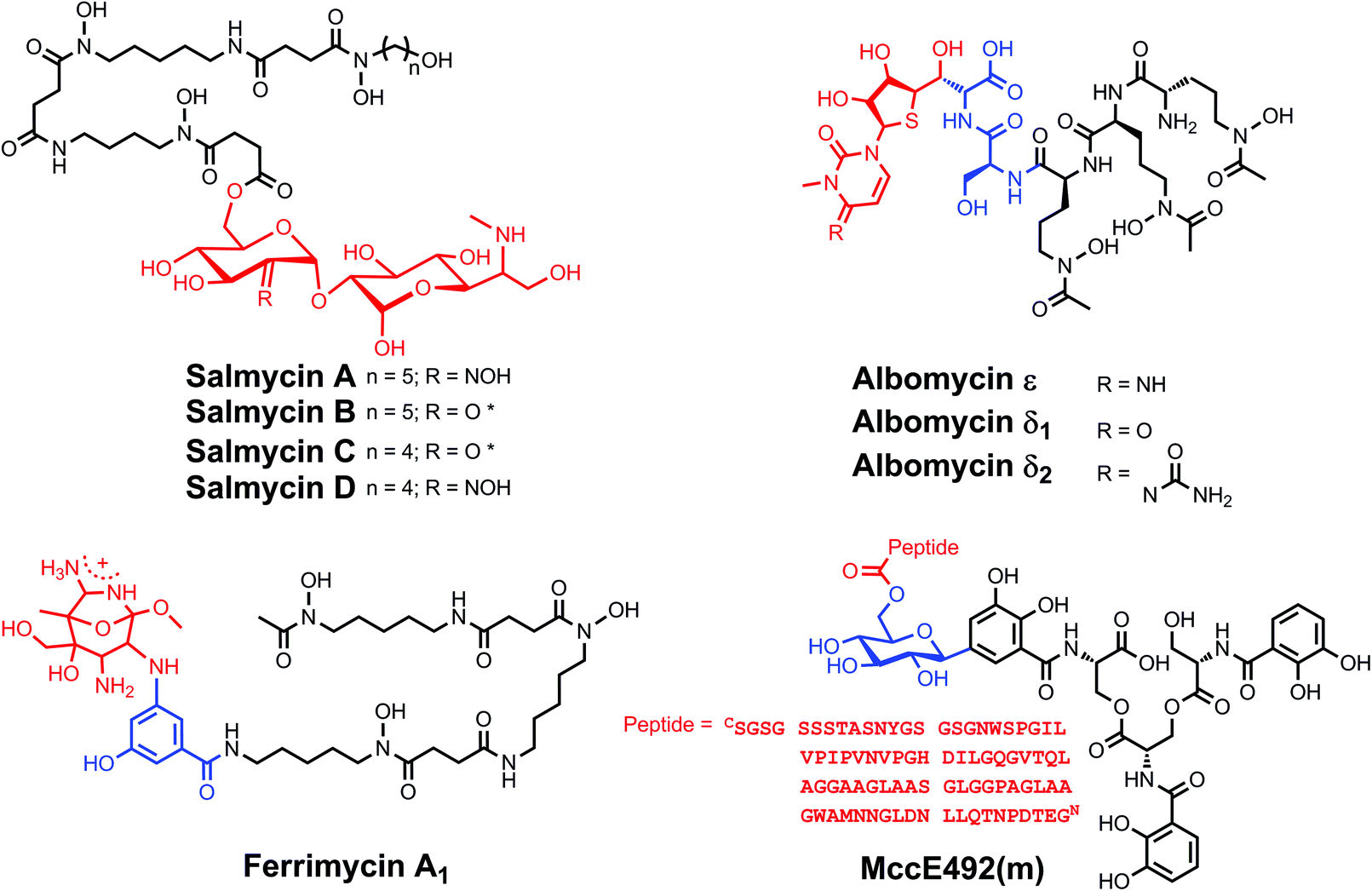
Explicit studies of the zinc coordination chemistry of yersiniabactin appear to be lacking in the literature. In this context, we note that the antibiotic micacocidin, produced by Pseudomonas sp. No. 57–250, complexes zinc as well as iron and copper.73,74 The structure of micacocidin is identical to that of yersiniabactin with the exception of methylation of the nitrogen atom in the saturated thiazolidine ring and substitution of the phenolic ring at the 3-position with an n-pentyl group. Micacocidin forms a complex with Fe(III), known as micacocidin C, but was not investigated as a siderophore until recently.75 In addition to the Fe(III) complex, a Cu(II) complex, micacocidin B, and a zinc complex, micacocidin A, were isolated from bacterial cultures.73 Micacocidin A was the dominant product obtained from bacterial culture and was characterized crystallographically.74,76 The crystal structure reveals a coordination geometry resembling that of ferric–yersiniabactin (Fig. 1).31 One key difference is that the deprotonated secondary alcohol in the structure of the Fe(III) complex remains protonated in the Zn(II) structure, resulting in charge neutrality for both compounds. The Zn(II)-bound structure of micacocidin corroborates a zinc-related non-classical function for yersiniabactin, and highlights the need for detailed investigations of the zinc coordination chemistry of the latter siderophore.
 | ||
| Fig. 1 Ball-and-stick representations of the metal complexes from the crystal structures of (A) yersiniabactin–Fe(III) (CCDC ID: 619878)31 and (B) micacocidin–Zn(II) (micacocidin A, CCDC ID: 130920).76 Non-polar hydrogen atoms have been omitted for clarity. Colour code: C gray, O red, N blue, H white, Fe orange, and Zn green. | ||
The interaction of siderophores with manganese
Manganese is another essential nutrient and, in prokaryotes, Mn-containing metalloproteins carry out functions ranging from photosynthesis to oxidative stress defence.77 The metal-withholding host-defence paradigm described above in relation to iron and zinc extends to manganese as well.53 Bacterial protein-based uptake systems for manganese have been described in Gram-negative and -positive strains, and further studies continue to reveal how these proteins contribute to manganese homeostasis.78,79 The bacterial manganese transport protein MntC and its homologues are extra-cytoplasmic solute-binding proteins that bind Mn(II) and interact with other members of the ABC transporter to bring the divalent metal ion into the cell.77,80 In addition, small molecules may also be exported by cells to bind Mn(II) with high affinity and facilitate cellular uptake.81 A molecule fulfilling such a nutritional role has yet to be definitively identified, however.Bacterial Mn metabolism contributes to the global redox cycling of this element.82 Mn(II) is readily soluble in water but more highly oxidized species typically deposit in the environment as insoluble Mn(III,IV) oxide minerals. Numerous microorganisms catalyse the oxidation of Mn(II), although whether this chemistry serves a biological function has yet to be definitively ascertained.83 The enzyme-catalysed oxidation is proposed to proceed through sequential one-electron transfer steps, generating Mn(III) intermediates.84–86 Indeed, such one-electron transfers are proposed to contribute to the persistent levels of soluble Mn(III) observed in suboxic zones.81 The most widely studied manganese-oxidizing bacteria are fluorescent pseudomonads.87 The luminescence of these microbes results from production of pyoverdine siderophores. These siderophores impact manganese metabolism, forming Mn-pyoverdine complexes. For instance, the Mn(II)-oxidizing strain P. putida MnB1 produces the pyoverdine PVDMnB1.88 This molecule has all of the properties expected of a siderophore, but has a reported binding affinity for Mn(III) that is almost 1000-fold greater than that for Fe(III).88 Mutant strains that overexpress pyoverdine demonstrate a reduced capacity to carry out Mn(II) to Mn(IV) oxidation.89 Similar results were obtained using P. putida GB-1, which produces the pyoverdine PVDGB-1.87
The investigation of siderophore–Mn(III) coordination chemistry was extended to other pyoverdines and rhizoferrin, and these molecules also coordinated Mn(III) with greater affinity than Fe(III).90 It was proposed that the large siderophore–Mn(III) stability constants derive from the ability of these siderophores to accommodate the Jahn–Teller distortion of the high-spin d4 Mn(III) centre.90
We also briefly note here, in relation to biogeochemical cycling, that various hydroxamate and catecholate siderophores facilitate the dissolution of oxyhydroxide minerals containing Mn as well as Co, Cr, and Fe.91–93
Fuelling nitrogenase: molybdenum and vanadium
Nitrogenases are enzymes produced by diazotrophs that carry out the chemically challenging six-proton, six-electron reduction of dinitrogen to two equivalents of ammonia in water under ambient conditions.94 The key to this transformation is an iron–sulphur-containing inorganic cofactor present within the enzyme. Whereas some bacterial strains produce nitrogenases with iron as the only metal in the cofactor, other strains incorporate molybdenum or vanadium into the cofactor as well.94–96 Early studies identified aminochelin as a catecholamine siderophore produced by the nitrogen-fixing diazatroph Azotobacter vinelandii that coordinates both iron and molybdenum.97 Using aminochelin, A. vinelandii was able to strip molybdenum from silicate samples.98 Further studies on this model organism revealed that the siderophores it produces under Mo- and V-limiting conditions form stable complexes of these metals.99 The biscatechol and triscatechol siderophores, protochelin and azotochelin, are secreted under these conditions and form 1:1 complexes with vanadate and molybdate as evidenced by tandem high performance liquid chromatography-inductively coupled plasma mass spectrometry.99,100 Isotopically-enriched metal–siderophore complexes were used to confirm cellular uptake of the Mo- and V-bound forms of the metallophores. A. vinelandii also produces a pyoverdine-like siderophore called azotobactin that contains each of the three metal-binding motifs typical of siderophores: hydroxamate, catecholate, and α-hydroxycarboxylate. Like protochelin and azotochelin, azotobactin coordinates vanadate and molybdate to form complexes that are transported into the bacterium.101 Experiments using mutant strains deficient in the production of either azotobactin or the catecholate siderophores suggest that the latter are preferentially used to take up iron and the former, molybdenum.102
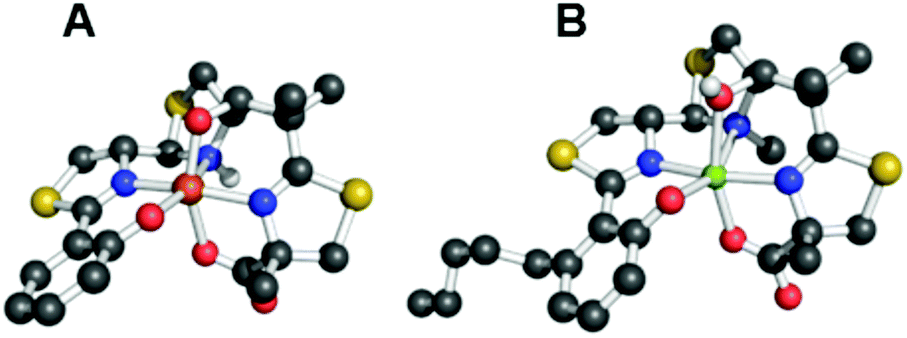
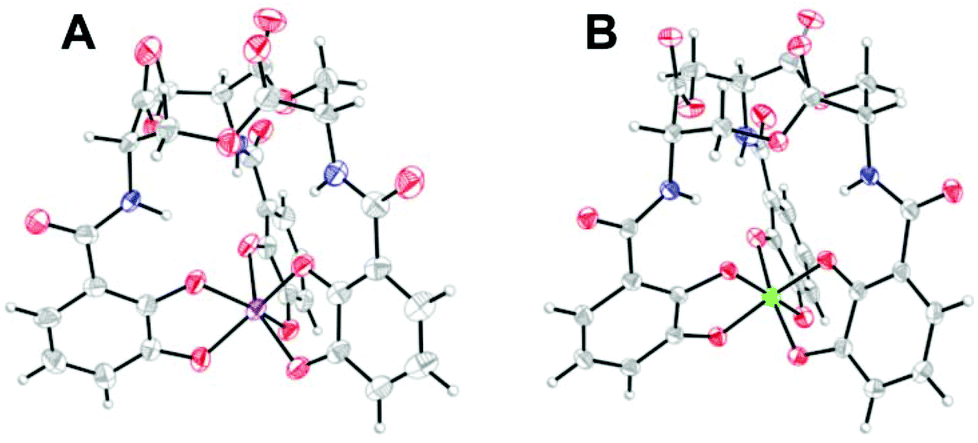
Although not related to an explicit biological function, we mention briefly here that, for many years, the vanadium complex of enterobactin was the closest analogue of the ferric complex of this archetypal siderophore to be characterized by atomic-resolution X-ray crystallography (Fig. 2).103,104 Recently, structures of the silicon (Fig. 2), germanium, and titanium complexes of enterobactin were reported.105 This work was motivated by the observation that enterobactin and salmochelin S4 bind silicon.106 Further insight into the structure of enterobactin can be obtained from crystal structures of ferric enterobactin complexed with different proteins. Early macromolecular structures of lipocalin-2 bound to enterobactin suggested that the trilactone was at least partially hydrolysed (Fig. 3).107,108 A crystal structure of lipocalin-2 bound to what appears to be a non-hydrolysed ferric enterobactin complex was deposited in the protein databank in 2008 (PDB ID: 3CMP, Fig. 3), but no follow-up publication has appeared in the literature. More recently, a crystal structure was obtained of ferric enterobactin complexed with FeuA, a component of the triscatecholate siderophore transport machinery of Bacillus subtilis (Fig. 3).109
 | ||
| Fig. 2 Thermal ellipsoid diagrams from the crystal structures of the (A) vanadium and (B) silicon complexes of enterobactin (CCDC ID: 624678 and 920703, respectively).103,105 Thermal ellipsoids are drawn at the 50% probability level and hydrogen atoms are shown as spheres of arbitrary radius. Colour code: C gray, O red, N blue, V purple, and Si green. | ||
 | ||
| Fig. 3 Lipocalin-2 bound to either (A) hydrolyzed or (B) non-hydrolyzed ferric enterobactin (PDB ID: 3BY0 and 3CMP, respectively).108 (C) Non-hydrolyzed ferric enterobactin complexed with FeuA (PDB ID: 2XUZ).109 In all panels, the protein is shown as a wheat-coloured surface. The ligands of the protein-bound small molecule are shown as sticks and the iron atom as a sphere. Colour code: C grey, O red, N blue, and Fe orange. | ||
The interaction of siderophores with copper
The concentration and localization of copper within cells is tightly regulated because of its propensity to catalyse deleterious Fenton-like chemistry.110 Moreover, cellular over-accumulation of copper leads to toxicity.111 Thus, bacterial copper transport proteins and chaperones are employed to control intracellular copper concentrations.112–116 Although numerous studies have uncovered many aspects of the mechanisms by which bacteria use proteins to chaperone, sequester, and efflux copper, less is known about the means by which bacteria import copper.116 Methanotrophic bacteria require particularly large supplies of copper to produce functional particulate methane monooxygenase.117,118 This enzyme allows the bacteria to derive energy and the carbon they need for anabolic processes from methane.119 Analysis of the growth medium in which methanotrophs were cultured under copper-limited conditions revealed the presence of small molecules that bind copper with high affinity.120,121 The compound identified in these studies that has received the most significant attention is called methanobactin.122,123 This peptidic compound has been alternately referred to as a copper-binding ligand (CBL), a copper-binding compound (CBC), or a chalkophore. These names all serve to evoke an identical impression: this molecule binds copper ions and transports them into the cell.123 Other molecules have been proposed to act as chalkophores, including coproporphyrin III, which is secreted by Paracoccus denitrificans under copper-limiting conditions to ensure an adequate supply of copper to the enzymes involved in denitrification.124 The ubiquitous small molecule glutathione avidly binds copper but is thought to play a role in detoxification as opposed to transport.125In addition to the small molecules described above, do siderophores bind copper? Similar to Zn(II), Cu(II) ions are softer than Fe(III) ions and so many of the siderophores that are well-tuned to complex ferric ions within a field of hard ligands will not interact as favourably with cupric ions. These two metal ions also prefer that their ligand fields assume different geometries, suggesting a lack of interaction between copper ions and siderophores pre-organized to bind Fe(III). Nonetheless, siderophores with softer donor atoms interact with copper. In the instances described below, this interaction is proposed have a specific biological purpose.
The pdtc metal-binding studies described above also identified copper as a metal that is coordinated by this siderophore.67 A number of early studies on pdtc focused on the ability of its metal complexes to dehalogenate carbon tetrachloride,64,65 and the cupric pdtc complex was identified as the most active species.126 Although pdtc can act as a siderophore, it does not appear to be used by bacteria to increase intracellular copper concentrations.127 The copper pdtc complex is capable of redox cycling, which may contribute to an as-yet undetermined biological function of this complex.67
Insight into the role that pseudomonal siderophores play in copper regulation was obtained by monitoring changes in the transcriptional profiles of P. aeruginosa following exposure to elevated copper concentrations.128 These studies indicated that production of high-affinity pyoverdine siderophores increased under high-copper conditions, whereas levels of the low-affinity siderophore pyochelin decreased. These results were interpreted to indicate that pyochelin may be involved in the uptake of copper.128 This proposal is in agreement with previous experiments, which revealed a decrease in pyochelin synthesis upon incubation of P. aeruginosa with copper as well as molybdenum, nickel, or cobalt.129 Studies of the coordination chemistry of pyochelin confirmed that it forms stable complexes with Cu(II) and Zn(II).45
Some small molecules, such as glutathione, that interact with copper in bacterial cells do so to mitigate the toxicity of this element.130 Copper, particularly Cu(I), is sufficiently toxic that mammals may use it as a bactericidal agent to combat microbial infection.131 For example, cultured RAW264.7 mouse macrophage cells and peritoneal macrophages from freshly sacrificed mice appear to pump copper into phagosomes containing E. coli in order to kill the bacteria.132,133 Above, an immune response was described in which a host fends off an invading pathogen by starving it of essential nutrients. The pathogen can respond by secreting molecules, such as siderophores, to bind the nutrient metal ion with high affinity and increase its uptake. The situation is reversed with copper given that the microbe is bombarded with an excess of a nutrient, but surprisingly an increase in siderophore production again appears to be a viable survival strategy, as described below.
An investigation of the response of pathogenic bacteria to increased copper concentrations revealed that the microbes increase expression of efflux proteins and oxidases, and secrete molecules to sequester reactive copper species.130 Glutathione and MymT, a bacterial metallothionein produced by Mycobacterium tuberculosis in response to high levels of copper, attenuate Cu(I) toxicity.113,114,125,134 Moreover, molecules traditionally viewed as siderophores are employed by bacteria to grow under toxic concentrations of copper. In particular, yersiniabactin binds Cu(II) ions and prevents reduction to the more toxic Cu(I) oxidation state.135 Conversely, catecholate-containing siderophores, such as enterobactin, reduce Cu(II), producing toxic cuprous ions.136 As described below, the redox-active catechol moieties of enterobactin can alternatively play a cytoprotective antioxidant role.
Uropathogenic E. coli UTI89 delicately balance the desired iron-binding and undesired copper-reducing chemistry of catecholate siderophores. In the current model,135E. coli UTI89 produce yersiniabactin and enterobactin to sequester iron from host proteins. To combat infection, the host increases Cu(II) concentrations in the vicinity of the bacteria and these cupric ions react with the catecholate moieties of enterobactin to produce toxic cuprous ions. Increased production of yersiniabactin allows Cu(II) to be sequestered before it is reduced.135 The cupric yersiniabactin complex is stable and has been detected in the urine of patients infected with uropathogenic E. coli.135 It should be noted that expression of CueO, typically designated as a cuprous oxidase, can also be increased to mitigate the toxic effects of copper by oxidizing the enterobactin precursor 2,3-dihydroxybenzoic acid to 2-carboxymuconate, thereby preventing catechol-mediated reduction of Cu(II).136
The copper–yersiniabactin complex is competent to effect superoxide dismutation.133 As noted above, expression of yersiniabactin confers a survival advantage upon pathogenic E. coli that are engulfed by macrophages. In addition to Cu(II) sequestration, the superoxide dismutase activity of copper–yersiniabactin may help to reduce the level of NADPH oxidase-derived superoxide that is produced by the macrophage to kill internalized bacteria.
Finally, we note that the ability of yersiniabactin and pyochelin to function in copper sequestration relies on the inability of the copper–siderophore complexes to interact with the receptors and transport proteins that mediate uptake of ferric siderophores. Nonetheless, experiments with the siderophore schizokinen indicate that this distinction is not universal. Production of this siderophore increases the sensitivity of Bacillus megaterium to copper by increasing intracellular Cu accumulation, but has the opposite effect in Anabaena spp.137,138 In addition to copper transport by schizokinen, it is possible that alteration of iron uptake dynamics may factor into the ultimate toxicity observed in these experiments.
Other metals: transport and sequestration
The coordination chemistry of a variety of siderophores has been studied using metals other than iron or those described above.67,139 In addition to fundamental investigations of the abiological inorganic chemistry of some siderophore complexes,140 a number of reports have focused on the broad-spectrum metal-binding capabilities of siderophores to probe the abilities of these ligands to mitigate or exacerbate the toxicity of heavy metals. The work described above on the protochelin-mediated transport of Fe, Mo, and V in A. vinelandii, which is important in evaluating the ability of this organism to satisfy the metal requirements of nitrogenases, was extended to include Cr and Co.141 Protochelin is unlikely to play a role in the intracellular accumulation of Cr and Co, but interactions of these metals with protochelin have implications for Cr/Co biogeochemical cycling.141The ability of the siderophores produced by Pseudomonas spp. to complex metals other than iron has received significant attention because of the prevalence of these bacteria in soil and marine environments as well as their classification as opportunistic human pathogens.145 The pseudomonal siderophores pyochelin and pyoverdine form complexes with over 15 different transition metal and main group metal ions.146,147 Moreover, these siderophore complexes were able to interact to varying degrees with their cognate receptors – the pyochelin complexes with FptA (Kd = 10 nM–4.8 μM) and the pyoverdine complexes with FpvA (Kd = 2.9 nM–13 μM).148,149 Despite the interaction that the non-iron pyochelin and pyoverdine complexes have with the appropriate outer membrane receptor, cellular accumulation was either absent or, in a few cases, present to a degree substantially less than that of the corresponding ferric complex.148 Subsequent investigation revealed that the outer membrane transporters both recognize the non-iron metal complexes and import the complexes into the cell.149 The lack of observed intracellular accumulation arises from the ATP-dependent efflux pump PvdRT-OpmQ, which exports pyoverdine complexes of unwanted metals. These properties have led to the investigation of Pseudomonas spp. as potential bioaugmentation agents in the phytoremediation of soils contaminated with environmental pollutants such as chromium, lead, and mercury.150,151
Pdtc, the small thiocarboxylate-containing tridentate siderophore described above, was also interrogated for its ability to complex a wide range of metals and metalloids.67 This ligand typically assumes a 1:2 metal-to-ligand ratio, affording an octahedral coordination sphere (Fig. 4). The ligand complexed 14 different metals and exerted a protective effect on Pseudomonas spp., Staphylococcus spp., an Arthobacter sp., Bacillus spp., E. coli, and Candida albicans that were incubated with mercury. Some of these organisms were also protected from cadmium and tellurium exposures.152
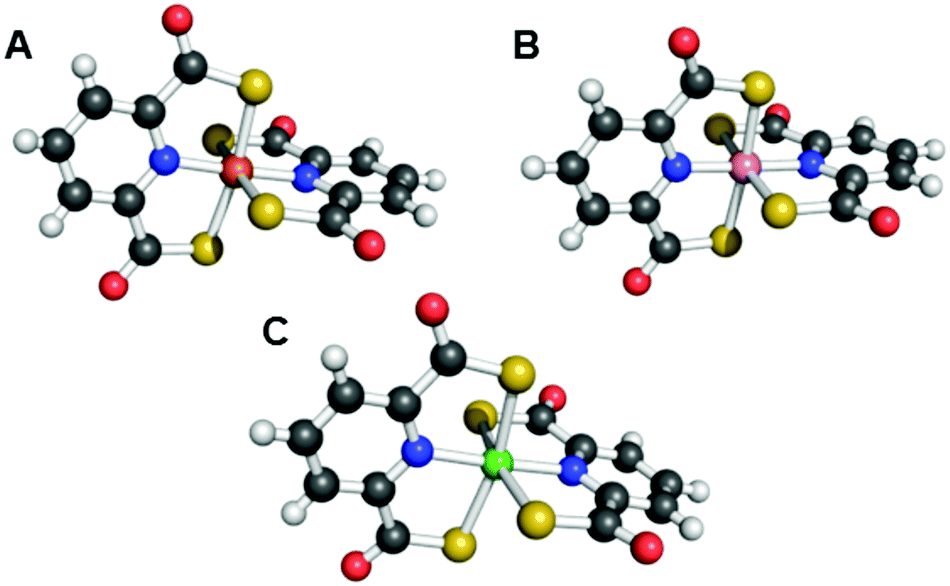 | ||
| Fig. 4 Ball-and-stick representations of the metal complexes from the crystal structures of (A) [Fe(pdtc)2]− (NBS ID: 557651),153 (B) [Co(pdtc)2]− (NBS ID: 596508),154 and (C) [Ni(pdtc)2]2− (NBS ID: 596506).154 Colour code: C gray, O red, N blue, H white, Fe orange, Co pink, and Ni green. | ||
Although the work described above illustrates the ability of some siderophores to interact with a range of soft, heavy metals, it is not surprising that many siderophores interact better with smaller, harder metal ions. For instance, desferrioxamine B (DFOB) binds Fe(III), Al(III), Ga(III), and In(III) with logβ110 values of 31.0, 24.5, 28.7, and 21.4, respectively.155 This tight binding interaction is exploited to treat iron overload disorders, such as thalassemia, and aluminium overload arising from ingestion of high levels of aluminium.156,157 The siderophore schizokinen and its N-deoxy derivative, produced by B. megaterium, also bind aluminium.158 The aluminium–siderophore complex was delivered into the cell through the siderophore transport receptor, leading to increased intracellular accumulation of aluminium.
In at least one instance, the strength of the interaction between a siderophore and aluminium resulted in the unintended complexation of the two.159,160 Initial studies of the interaction of pyoverdine with its cognate outer membrane transporter FpvA appeared to indicate that the transporter bound the apo-siderophore with high affinity.161 In depth investigation revealed that the strong transporter-small molecule interaction was in fact with the aluminium complex of pyoverdine, formed from trace amounts of aluminium in the buffers used for the experiments. The aluminium complex exhibits enhanced fluorescence as compared to the apo-siderophore,162 which confounded the initial interpretation of the fluorescence resonance energy transfer data that were used to investigate pyoverdine-FpvA binding. We also note in passing that the first siderophore to be isolated, mycobactin (later renamed mycobactin P),163 was initially isolated as an aluminium complex, the facile crystallization of which permitted detailed studies to be carried out on pure material.164–166 The aluminium most likely originated from the alumina column that was used in the chromatographic preparation of the bacterial cell extract under investigation and is not involved in the biological function of mycobactin P.
Pu(IV) exhibits coordination chemistry similar to that of Fe(III). The bioinorganic chemistry of plutonium is often studied with the aim of understanding its toxicity. Plutonium can bind transferrin and enter mammalian cells via the ferric transferrin uptake machinery.167 The interplay between plutonium and microorganisms has also been investigated. Desferrioxamine (DFO) siderophores display high affinity for Pu(IV);168 for Pu(IV)-DFOB, logβ110 = 30.8 and Pu(IV)-DFOE is sufficiently stable that its crystal structure was determined using X-ray diffraction methods (Fig. 5).169,170 Studies with the DFOB-producing bacterium Microbacterium flavescens JG-9 demonstrated that the Pu(IV)-DFOB complex was taken up by living, metabolically-active bacteria.171 As reviewed recently,172 a number of subsequent studies have shown that, through the use of siderophores, microorganisms can dissolve plutonium and significantly affect its subsurface and environmental distribution.
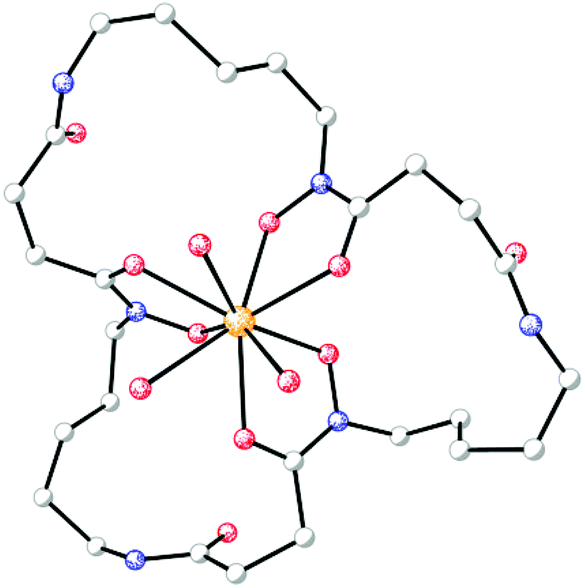 | ||
| Fig. 5 Molecular diagram of the siderophore complex from the crystal structure of desferrioxamine E-triaquaplutonium(IV) (CCDC ID: 136339).170 Non-hydrogen atoms are shown as shaded spheres of arbitrary radius and hydrogen atoms have been removed for clarity. Colour code: C gray, O red, N blue, and Pu orange. | ||
The last example we will present in this section involves the delftibactins. These siderophore-like molecules are produced by the bacterium Delftia acidovorans and are products of NRPS machinery. The genes encoding the NRPS are clustered with genes of other siderophore synthases as well as siderophore transporters and regulators.142 Like Cupriavidus metallidurans, D. acidovorans colonizes the surface of gold nuggets.173 The delftibactins produced by D. acidovorans protect the bacterium from the toxic effects of gold and enable biomineralization of the metal as gold nanoparticles.142 Enterobactin, yersiniabactin, and aerobactin were also tested for their ability to form gold nanoparticles.143 Yersiniabactin and enterobactin were able to form small and large amounts of colloidal gold particles, respectively. In neither instance, however, were the particles formed as well or in a similar amount as with delftibactin A or B.143
Marine siderophores and boron
The estuarine enteropathogen Vibrio parahaemolyticus is associated with episodes of gastroenteritis following consumption of contaminated seafood.174 This bacterium produces vibrioferrin,175 a citrate-based siderophore that is photoreactive and displays a relatively low affinity for Fe(III) (logβ110 = 24.0) as compared to other marine siderophores.144,176 Vibrioferrin was also identified in a search for siderophores in Marinobacter spp., and was unexpectedly observed as the borate complex (Fig. 6).177 Given that boron was not added to the solutions from which the siderophore was isolated, the authors proposed that vibrioferrin was stripping boron from the borosilicate glassware used during the isolation procedure. Further investigation revealed that a number of different citrate and catecholate siderophores form stable borate complexes.177 Although borate is not typically present at very high concentrations in terrestrial environments, it is common in marine systems.178 It is not yet clear whether the borate–siderophore complex has biological relevance; however, functions as either a boronophore or a signalling molecule have been proposed.177,179–181
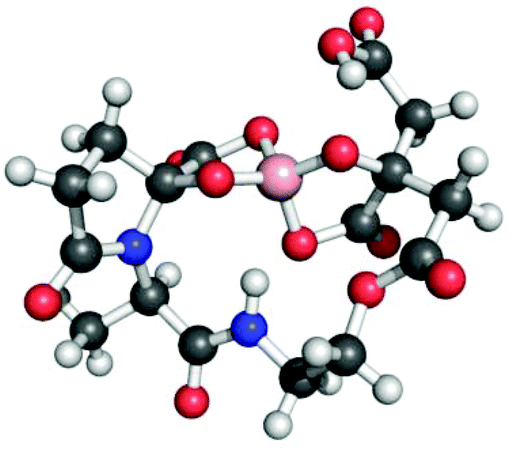 | ||
| Fig. 6 Ball-and-stick model of the proposed structure of the vibrioferrin–borate complex. The structure was minimized with GaussView based on that depicted in ref. 177. Color code: C gray, O red, N blue, H white, and B pink. | ||
Recent studies with Marinobacter algicola DG893 revealed that boron significantly influences the expression of at least 23 genes in this organism.182 A number of the influenced genes are related to iron homeostasis. With the exception of FbpA, a periplasmic iron-binding protein, the expression of all of the iron uptake-related proteins examined increased with increasing boron concentration. The authors proposed that boron influences the iron uptake regulator, Fur.182 Investigation into the interaction of borate with FbpA revealed that the protein synergistically binds Fe(III) and borate, effectively facilitating sequestration of the metal.183 The mode of synergistic borate binding was proposed to be analogous to that of carbonate.184
We note that the Marinobacter genus was chosen as the subject of the aforementioned studies because it is frequently associated with toxic dinoflagellates such as Gymnodinium catenatum and Scrippsiella trochoidea. These algae are proposed to have a mutualistic relationship with Marinobacter whereby photolysis of ferric vibrioferrin releases iron to the benefit of the phytoplankton and the bacteria make use of the carbon fixed by the eukaryote.185 This situation provides an example of a siderophore being used to feed an organism other than the producer to the benefit of the producer, in contrast to the well-established ability of bacteria to compete with one another by consuming xenosiderophores.186,187
Siderophores as signalling molecules
The iron that is transported into cells by siderophores can act as a signalling agent, notably in the regulation of genes related to iron homeostasis.188 It is also plausible that the siderophore itself, or a metal complex thereof, acts directly as a signalling molecule.189 For example, the boron complex of vibrioferrin may act as either a traditional signalling molecule or a mediator of quorum sensing as described above.181 The use of siderophores as signalling agents by Pseudomonas spp. is likely the most widely investigated manifestation of this chemical communication strategy.190–192 The pyoverdines produced by P. aeruginosa strains serve not only to supply the bacteria with essential iron, and possibly carry out functions related to its binding to other metals as described above, but also regulate the expression of three virulence factors: exotoxin A, an endoprotease, and pyoverdines themselves.43,193 Because the siderophore is secreted, it can signal within one bacterium as well as mediate communication between bacteria.194 As a consequence of the regulatory effects that pyoverdines have on these virulence factors, mutants of the plant pathogen Pseudomonas syringae pv. tabaci 6605 deficient in their capacity to produce this siderophore exhibited reduced virulence in host tobacco infection.195 Enhanced swarming ability and increased biosurfactant production were also phenotypic effects of the pyoverdine deficiency.An early example of siderophores being used as signalling molecules came from the observation that some marine bacteria use exogenous siderophores to stimulate the production of endogenous siderophores.196 For example, growth of the α-proteobacterium strain V0210 was stimulated by the addition of N,N′-bis(2,3-dihydroxybenzoyl)-O-serylserine, a siderophore produced by Vibrio spp.196
Members of the Bordetella genus, including the pathogen Bordetella pertussis that causes whooping cough in humans, employ a range of alternative iron acquisition mechanisms to adapt to fluctuations in iron concentration during the course of infection.197 In Bordetella spp., the siderophores alcaligin and enterobactin function as inducers that activate expression of their cognate transport systems.198 Because of this signalling capacity, these complexes have been referred to as ferrimones.198
In Y. pestis, yersiniabactin can activate the transcription of the yersiniabactin outer membrane receptor (psn), the yersiniabactin synthetase HMWP2 (irp2), and a transcriptional regulator (ybtP). It also decreases expression of ybtA, another transcriptional regulator.199,200 Importantly, yersiniabactin influenced transcription at concentrations 500-fold lower than those at which it exerted a nutritional effect, indicating that its signalling role functions independently of its nutritional role.199 These genes, as well as yersiniabactin-mediated modulation of gene expression, were essential for Y. pestis to cause pneumonic and bubonic plagues in murine models.201 We note that yersiniabactin-like molecules of unconfirmed structure produced by mutants with non-functional salicylate synthase YbtS or mutants that lack all ybt genes except for ybtD, a putative phosphopantetheinyl transferase, also modulated gene transcription in a similar manner.202 Although these yersiniabactin analogues are not biologically relevant per se, such structure–activity relationship studies may provide insight into the nature of yersiniabactin signalling.
Protection from oxidative stress
During an investigation of the antibiotic properties of pyochelin, enterobactin was observed to play a role in mitigating the deleterious effects of oxidative stress.203 Addition of pyochelin to E. coli increased levels of reactive oxygen species (ROS). The deleterious effects of these ROS were abrogated as a result of enterobactin production. Moreover, exogenous enterobactin supplementation provided protection to an entE mutant that could not biosynthesize enterobactin. Notably, this function is associated with the redox activity of the enterobactin catecholates rather than iron transport.204 Moreover, E. coli strains that cannot produce enterobactin are unable to form colonies on minimal medium. This effect could not be reversed with iron supplementation; however, addition of enterobactin or a reducing agent such as ascorbic acid to the agar allowed the colonies to form, as did anaerobic incubation. On the basis of these results, the catecholate moieties of the siderophore are proposed to scavenge radicals.204A similar siderophore-mediated protective effect was observed in Salmonella enterica serovar Typhimurium.207 This enteric pathogen produces enterobactin and a series of glucosylated enterobactin derivatives known as salmochelins. The primary means by which these siderophores confer virulence is acquisition of iron from the host.42 Recent studies indicate that these catecholate-based siderophores may also protect Salmonella from the oxidative stress encountered upon entering the macrophage. The enhanced survival of Salmonella producing salmochelins, as compared to mutant strains deficient in salmochelin biosynthesis and uptake, occurred primarily during the early stage of infection when the macrophage typically unleashes a cytotoxic oxidative burst. In vitro studies confirmed that catecholate siderophores, but not yersiniabactin or aerobactin, could protect Salmonella against oxidative stress and that the siderophore needed to be taken up into the cell to have this effect.207
The redox-mediated cytoprotective function of catecholate siderophores is reminiscent of the ability of cupric yersiniabactin to act as a superoxide dismutase and protect macrophage-internalized pathogens from oxidative stress.133 As described above, however, results from the yersiniabactin studies put forward a role for the catecholate moieties of enterobactin in the generation of toxic cuprous ions that are used by the host organism to stem bacterial infections.135 The combined results of these experiments indicate that catecholate siderophores can switch between cytoprotective and cytotoxic roles depending on the nature of the molecules in the environment surrounding the bacterium.
Staphylococcus aureus produces two citrate-based siderophores, staphyloferrin A and staphyloferrin B.208 The SirABC protein system mediates uptake of the siderophore staphyloferrin B and contributes to virulence.209 Under oxidative or nitrosative stress, S. aureus increases expression of sirA, as detected by qRT-PCR,210 which conferred enhanced resistance to oxidative stress. It remains unclear whether this effect simply results from enhanced iron uptake or some other process.210 Staphyloferrin B does not contain catecholate moieties,205,211,212 so a direct analogy to the protective role that enterobactin plays in this regard is unwarranted.
Sideromycins: siderophores as antibiotics
Sideromycins are a class of antibiotics in which the bactericidal warhead is attached to a siderophore.213 The siderophore moiety performs targeting and delivery functions and thereby affords enhanced cellular uptake of the antibiotic for strains expressing the appropriate transport machinery. This “Trojan horse” drug delivery strategy has been employed in the design of a number of synthetic constructs aimed at enhancing therapeutic efficiency or overcoming antibiotic resistance.214–218 Examples include catecholate,219,220 hydroxamate,221 and carboxylate222 siderophore conjugates. The significant, and growing, body of work on synthetic siderophore-antibiotic conjugates is a fertile ground for further investigation, and we refer the reader to the aforecited recent reviews on the topic. Here, we will restrict our discussion to those siderophore-antibiotic conjugates that occur naturally.Naturally occurring sideromycins were discovered before siderophores,223 with the identification of a substance called grisein produced by Streptomyces griseus.224 This compound is a member of a class of sideromycins called albomycins (Chart 9). These sideromycins feature a hydroxamate siderophore unit consisting of three N5-acetyl-N5-hydroxyornithine moieties, reminiscent of those found in fungal ferrichromes.20 The C-terminus of the siderophore tripeptide motif harbours a serine that is linked to a nonproteinogenic α-amino acid with a 4-thioxylofuranose side-chain. The modified furanose ring is bound to a cytosine, and modifications to the thioribosyl pyrimidine antibiotic group distinguish the different members of this family.225 In all three albomycins, the pyrimidine is methylated at the N3 position. In albomycin δ1, the cytosine N4 is carbamoylated and in albomycin ε it is not. In albomycin δ2, the N3-methylcytosine is replaced with N3-methyluracil.25 These conjugates display a broad-spectrum of antibiotic activity against both Gram-negative and Gram-positive bacteria because of the widespread nature of transport proteins capable of binding ferrichrome-type siderophores.213,226 The antibiotic activity stems from the ability of the pyrimidine unit to inhibit seryl-tRNA synthetase.227 One intriguing feature of the albomycins is that the protein synthesis inhibitor is cleaved from the siderophore unit by intracellular peptidases to unleash antibiotic activity.228,229 We briefly note that this feature of natural sideromycins can be used to inform the design of synthetic siderophore-antibiotic conjugates, in which a cleavable linker may be required.215,217,219
Salmycins (Chart 9), produced by a particular strain of Streptomyces violaceus, are another class of well-studied sideromycins.213 They comprise a ferrioxamine siderophore unit conjugated to an aminodisaccharide.230 Four salmycins, salmycins A–D, have been identified. In each case, the siderophore unit is danoxamine. Salmycins A and D are oximes of salmycins B and C, respectively. The aminodisaccharide unit is presumed to inhibit protein synthesis similarly to other aminoglycosides.174 Danomycins A and B, the former of which is a carbamate of the latter, also feature a danoxamine iron-binding unit and an aminodisaccharide that inhibits protein synthesis,231 but the detailed structures of these compounds remain unknown.232
Although three ferrimycins, A1, A2, and B, were isolated from Streptomyces griseoflavus ETH 9578, a comprehensive structural analysis appears to only be present for ferrimycin A1 (Chart 9).213,223,232–234 This compound features a DFOB siderophore unit linked to an iminoester-substituted lactam, via a 4-amino-5-hydroxybenzoate linker.26 The heterobicyclic antibiotic warhead inhibits protein synthesis as demonstrated by studies monitoring the cellular incorporation of radiolabeled phenylalanine by S. aureus.235 In contrast to the albomycins, which exhibit broad-spectrum antimicrobial activity, the salmycins, danomycins, and ferrimycins are typically active only against Gram-positive bacteria.213
The final class of sideromycins that will be discussed is the siderophore-modified microcins. Microcins are small, ribosomally synthesized antimicrobial peptides that are produced by Gram-negative bacteria and used as narrow-spectrum antibiotics.236 A number of microcins have been described, and different classification schemes have been put forth to categorize these molecules. A recently devised schema distinguishes between (i) class I microcins, which exhibit extensive backbone post-translational modification and are <5 kDa; (ii) class IIa microcins, which have no post-translational modifications except disulfide bonds and molecular weights in the 5–10 kDa range; and (iii) class IIb microcins that are linear polypeptides and may carry a C-terminal post-translational modification.237 In this classification scheme, the class IIb microcins correspond to the catecholate microcins. These peptides are post-translationally modified with glucosylated catecholate siderophores, presumably to enhance cellular uptake by strains expressing catecholate siderophore receptors.237 The class IIb microcins include MccE492, MccH47, MccI47, MccG492, and MccM. MccE492 was the first discovered to exhibit the catecholate siderophore modification, producing a form named MccE492m.238 The post-translational modification is a linear trimer of 2,3-dihydroxybenzoylserine attached to the serine-84 carboxylate at the C-terminus of the peptide via a β-D-glucose linker (Chart 9). Effectively, the microcin is modified with a linearized and monoglucosylated enterobactin moiety.32 Subsequently, biochemical studies confirmed that MccM and MccH47, which were putatively assigned as siderophore–microcin conjugates based on bioinformatics analyses,239 each contain C-terminal salmochelin-like post-translational modifications.240 Chemical verification of the structures of MccG492 and MccI47 remains to be obtained.
Although MccE492 is potent in its unmodified form, with minimal inhibitory concentrations of 0.3 μM in assays with various E. coli and Salmonella strains, the antimicrobial activity increases ca. 10-fold upon catecholate siderophore modification.238 The increased activity of the modified microcin relies on the presence of siderophore uptake machinery.238,241 Although the class I microcin MccJ25 does not become post-translationally modified with a siderophore, it interacts with the ferrichrome receptor FhuA.242 Recent X-ray crystallographic characterization of the complex of MccJ25 and FhuA revealed that the peptide mimics the binding of the siderophore to its receptor.243
Conclusions and outlook
The study of siderophores has provided an opportunity for fundamental studies in coordination chemistry to make important contributions to understanding biological systems. Elucidation of the multifaceted role of a bacterial siderophore in metal homeostasis and bacterial physiology is a multidisciplinary endeavour that requires input from chemists, biochemists, and microbiologists. Approaching siderophores from a chemical standpoint is important for studies of non-classical siderophore functions. Undoubtedly, one of the most stunning attributes of siderophores is their ability to bind iron with remarkable affinity. Nevertheless, siderophores have a multitude of other chemical properties, and we now appreciate that bacteria will employ a siderophore for a secondary function provided that the siderophore can perform the requisite chemistry.In the coming years, we expect that initiatives focused on the ability of siderophores to acts as metallophores for other elements will continue on several fronts. Of particular interest is how this coordination chemistry contributes to metal-sequestering host-defence strategies and bacterial pathogenesis. Additionally, the influence of bacteria on the biogeochemical movement and environmental distribution of heavy metals via siderophore production is a rich area for continued investigation. Concerns over the fate of nuclear waste, particularly in the wake of natural disasters, will likely drive investigation into interactions of f-block elements with the siderophores of environmental microbes. With regards to sideromycins, many of the early discoveries of natural sideromycins require follow-up investigations. Such efforts have largely been eclipsed by initiatives to produce synthetic siderophore-drug conjugates to combat bacterial pathogens, including antibiotic-resistant strains. Although it lay outside the scope of this Perspective, we anticipate increased investigation into the use of siderophores as medicines to treat various human diseases. For instance, although DFOB is well-established for the treatment of iron-overload in conditions such as thalassemia and myelodysplastic syndrome, its use to treat other diseases, such as malaria, remains contentious.244
In addition to more focused investigations on the interactions of siderophores and siderophore-producing bacteria with non-iron metals, we expect that increased attention will be paid to larger mechanistic questions related to the means by which organisms employ siderophores for non-classical purposes without interference from the Fe-transport functions of these molecules. Also, better criteria will need to be established to delineate the difference between situations in which a siderophore simply exhibits the ability to interact with other metals and those situations in which these interactions are biologically relevant.
In closing, the chemistry of siderophores is vast, even when restricted to iron transport. The wide expanse of chemical space representing the interactions of siderophores with non-iron elements is certain to contain a wealth of discoveries in chemistry and biology. We anticipate that a great increase in the number of non-classical functions attributed to these molecules is forthcoming as we come to better understand the subtleties of siderophore chemistry.
Acknowledgements
Research on siderophore-based therapeutics in the Nolan lab is currently supported by the NIH (R21 A1101784).References
- F. Archibald, FEMS Microbiol. Lett., 1983, 19, 29–32 CrossRef CAS PubMed.
- J. E. Posey and F. C. Gherardini, Science, 2000, 288, 1651–1653 CrossRef CAS.
- N. N. Greenwood and A. Earnshaw, Chemistry of the elements, Butterworth-Heinemann, Oxford, Boston, 2nd edn, 1997 Search PubMed.
- H. B. Gray and J. R. Winkler, Annu. Rev. Biochem., 1996, 65, 537–561 CrossRef CAS PubMed.
- C. E. Tinberg and S. J. Lippard, Acc. Chem. Res., 2011, 44, 280–288 CrossRef CAS PubMed.
- M. Y. Darensbourg, E. J. Lyon, X. Zhao and I. P. Georgakaki, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3683–3688 CrossRef CAS PubMed.
- M. Costas, M. P. Mehn, M. P. Jensen and L. Que Jr., Chem. Rev., 2004, 104, 939–986 CrossRef CAS PubMed.
- S. Shaik, H. Hirao and D. Kumar, Acc. Chem. Res., 2007, 40, 532–542 CrossRef CAS PubMed.
- K. N. Raymond and C. J. Carrano, Acc. Chem. Res., 1979, 12, 183–190 CrossRef CAS.
- S. C. Andrews, A. K. Robinson and F. Rodríguez-Quiñones, FEMS Microbiol. Rev., 2003, 27, 215–237 CrossRef CAS.
- K. N. Raymond, E. A. Dertz and S. S. Kim, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3584–3588 CrossRef CAS PubMed.
- W. M. Latimer, The oxidation states of the elements and their potentials in aqueous solutions, Prentice-Hall, Inc., New York, 1938, p. 211 Search PubMed.
- J. R. Chipperfield and C. Ratledge, BioMetals, 2000, 13, 165–168 CrossRef CAS.
- C. Ratledge and L. G. Dover, Annu. Rev. Microbiol., 2000, 54, 881–941 CrossRef CAS PubMed.
- M. A. Rouf, J. Bacteriol., 1964, 88, 1545–1549 CAS.
- R. Milo, P. Jorgensen, U. Moran, G. Weber and M. Springer, Nucleic Acids Res., 2010, 38, D750–D753 CrossRef CAS PubMed.
- S. Sundararaj, A. Guo, B. Habibi-Nazhad, M. Rouani, P. Stothard, M. Ellison and D. S. Wishart, Nucleic Acids Res., 2004, 32, D293–D295 CrossRef CAS PubMed.
- R. B. Martin, J. Savory, S. Brown, R. L. Bertholf and M. R. Wills, Clin. Chem., 1987, 33, 405–407 CAS.
- C. E. Lankford, Crit. Rev. Microbiol., 1973, 2, 273–331 CrossRef CAS.
- H. Haas, M. Eisendle and B. G. Turgeon, Annu. Rev. Phytopathol., 2008, 46, 149–187 CrossRef CAS PubMed.
- J. W. Kloepper, J. Leong, M. Teintze and M. N. Schroth, Nature, 1980, 286, 885–886 CrossRef CAS.
- L.-C. Königsberger, E. Königsberger, P. M. May and G. T. Hefter, J. Inorg. Biochem., 2000, 78, 175–184 CrossRef.
- H. Boukhalfa and A. L. Crumbliss, BioMetals, 2002, 15, 325–339 CrossRef CAS.
- M. Miethke and M. A. Marahiel, Microbiol. Mol. Biol. Rev., 2007, 71, 413–451 CrossRef CAS PubMed.
- S. Bertrand, Siderophore Base: The Web Data Base of Microbial Siderophores, Accessed: 2014 Search PubMed, 〈http://bertrandsamuel.free.fr/siderophore_base/siderophores.php〉.
- R. C. Hider and X. Kong, Nat. Prod. Rep., 2010, 27, 637–657 RSC.
- R. C. Scarrow, D. J. Ecker, C. Ng, S. Liu and K. N. Raymond, Inorg. Chem., 1991, 30, 900–906 CrossRef CAS.
- Z. Hou, K. N. Raymond, B. O'Sullivan, T. W. Esker and T. Nishio, Inorg. Chem., 1998, 37, 6630–6637 CrossRef CAS PubMed.
- J. H. Crosa and C. T. Walsh, Microbiol. Mol. Biol. Rev., 2002, 66, 223–249 CrossRef CAS.
- C. T. Walsh and E. M. Nolan, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5655–5656 CrossRef CAS PubMed.
- M. C. Miller, S. Parkin, J. D. Fetherston, R. D. Perry and E. DeMoll, J. Inorg. Biochem., 2006, 100, 1495–1500 CrossRef CAS PubMed.
- B. Bister, D. Bischoff, G. J. Nicholson, M. Valdebenito, K. Schneider, G. Winkelmann, K. Hantke and R. D. Süssmuth, BioMetals, 2004, 17, 471–481 CrossRef CAS.
- H. Lin, M. A. Fischbach, D. R. Liu and C. T. Walsh, J. Am. Chem. Soc., 2005, 127, 11075–11084 CrossRef CAS PubMed.
- K. Hantke, Mol. Gen. Genet., 1983, 191, 301–306 CrossRef CAS.
- A. J. Bäumler and K. Hantke, Mol. Microbiol., 1992, 6, 1309–1321 CrossRef PubMed.
- K. Poole, L. Young and S. Neshat, J. Bacteriol., 1990, 172, 6991–6996 CAS.
- B. Ghysels, U. Ochsner, U. Möllman, L. Heinisch, M. Vasil, P. Cornelis and S. Matthijs, FEMS Microbiol. Lett., 2005, 246, 167–174 CrossRef CAS PubMed.
- J. H. Brock, P. H. Williams, J. Licéaga and K. G. Wooldridge, Infect. Immun., 1991, 59, 3185–3190 CAS.
- K. G. Wooldridge and P. H. Williams, FEMS Microbiol. Rev., 1993, 12, 325–348 CrossRef CAS PubMed.
- M. Valdebenito, A. L. Crumbliss, G. Winkelmann and K. Hantke, Int. J. Med. Microbiol., 2006, 296, 513–520 CrossRef CAS PubMed.
- K. Hantke, G. Nicholson, W. Rabsch and G. Winkelmann, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3677–3682 CrossRef CAS PubMed.
- M. A. Fischbach, H. Lin, D. R. Liu and C. T. Walsh, Nat. Chem. Biol., 2006, 2, 132–138 CrossRef CAS PubMed.
- J. Ravel and P. Cornelis, Trends Microbiol., 2003, 11, 195–200 CrossRef CAS.
- A.-M. Albrecht-Gary, S. Blanc, N. Rochel, A. Z. Ocaktan and M. A. Abdallah, Inorg. Chem., 1994, 33, 6391–6402 CrossRef CAS.
- J. Brandel, N. Humbert, M. Elhabiri, I. J. Schalk, G. L. A. Mislin and A.-M. Albrecht-Gary, Dalton Trans., 2012, 41, 2820–2834 RSC.
- Z. Dumas, A. Ross-Gillespie and R. Kümmerli, Proc. R. Soc. London, B, 2013, 280, 20131055 CrossRef PubMed.
- N. Jain and D. K. Sharma, Geomicrobiol. J., 2004, 21, 135–144 CrossRef CAS.
- G. M. Gadd, Geoderma, 2004, 122, 109–119 CrossRef CAS PubMed.
- T. Zheng and E. M. Nolan, Metallomics, 2012, 4, 866–880 RSC.
- E. Ahmed and S. J. M. Holmström, Microb. Biotechnol., 2014, 7, 196–208 CrossRef CAS PubMed.
- C. Andreini, L. Banci, I. Bertini and A. Rosato, J. Proteome Res., 2006, 5, 3173–3178 CrossRef CAS PubMed.
- K. Hantke, Curr. Opin. Microbiol., 2005, 8, 196–202 CrossRef CAS PubMed.
- T. E. Kehl-Fie and E. P. Skaar, Curr. Opin. Chem. Biol., 2010, 14, 218–224 CrossRef CAS PubMed.
- M. I. Hood and E. P. Skaar, Nat. Rev. Microbiol., 2012, 10, 525–537 CrossRef CAS PubMed.
- A. M. Prentice, H. Ghattas and S. E. Cox, J. Nutr., 2007, 137, 1334–1337 CAS.
- B. Zhao, S. C. Moody, R. C. Hider, L. Lei, S. L. Kelly, M. R. Waterman and D. C. Lamb, Int. J. Mol. Sci., 2012, 13, 8500–8513 CrossRef CAS PubMed.
- S. D. Bentley, K. F. Chater, A.-M. Cerdeño-Tárraga, G. L. Challis, N. R. Thomson, K. D. James, D. E. Harris, M. A. Quail, H. Kieser, D. Harper, A. Bateman, S. Brown, G. Chandra, C. W. Chen, M. Collins, A. Cronin, A. Fraser, A. Goble, J. Hidalgo, T. Hornsby, S. Howarth, C.-H. Huang, T. Kieser, L. Larke, L. Murphy, K. Oliver, S. O'Neil, E. Rabbinowitsch, M.-A. Rajandream, K. Rutherford, S. Rutter, K. Seeger, D. Saunders, S. Sharp, R. Squares, S. Squares, K. Taylor, T. Warren, A. Wietzorrek, J. Woodward, B. G. Barrell, J. Parkhill and D. A. Hopwood, Nature, 2002, 417, 141–147 CrossRef PubMed.
- G. L. Challis and J. Ravel, FEMS Microbiol. Lett., 2000, 187, 111–114 CrossRef CAS PubMed.
- S. Lautru, R. J. Deeth, L. M. Bailey and G. L. Challis, Nat. Chem. Biol., 2005, 1, 265–269 CrossRef CAS PubMed.
- A. Hesketh, H. Kock, S. Mootien and M. Bibb, Mol. Microbiol., 2009, 74, 1427–1444 CrossRef CAS PubMed.
- D. Kallifidas, B. Pascoe, G. A. Owen, C. M. Strain-Damerell, H.-J. Hong and M. S. B. Paget, J. Bacteriol., 2010, 192, 608–611 CrossRef CAS PubMed.
- Y.-R. Lim, M.-K. Hong, J.-K. Kim, T. T. N. Doan, D.-H. Kim, C.-H. Yun, Y.-J. Chun, L.-W. Kang and D. Kim, Arch. Biochem. Biophys., 2012, 528, 111–117 CrossRef CAS PubMed.
- W. Ockels, A. Römer, H. Budzikiewicz, H. Korth and G. Pulverer, Tetrahedron Lett., 1978, 19, 3341–3342 CrossRef.
- M. J. Dybas, G. M. Tatara and C. S. Criddle, Appl. Environ. Microbiol., 1995, 61, 758–762 CAS.
- M. J. Dybas, M. Barcelona, S. Bezborodnikov, S. Davies, L. Forney, H. Heuer, O. Kawka, T. Mayotte, L. Sepúlveda-Torres, K. Smalla, M. Sneathen, J. Tiedje, T. Voice, D. C. Wiggert, M. E. Witt and C. S. Criddle, Environ. Sci. Technol., 1998, 32, 3598–3611 CrossRef CAS.
- C.-H. Lee, T. A. Lewis, A. Paszczynski and R. L. Crawford, Biochem. Biophys. Res. Commun., 1999, 261, 562–566 CrossRef CAS PubMed.
- M. S. Cortese, A. Paszczynski, T. A. Lewis, J. L. Sebat, V. Borek and R. L. Crawford, BioMetals, 2002, 15, 103–120 CrossRef CAS.
- T. A. Lewis, L. Leach, S. Morales, P. R. Austin, H. J. Hartwell, B. Kaplan, C. Forker and J.-M. Meyer, Environ. Microbiol., 2004, 6, 159–169 CrossRef CAS.
- L. H. Leach, J. C. Morris and T. A. Lewis, BioMetals, 2007, 20, 717–726 CrossRef CAS PubMed.
- A. G. Bobrov, O. Kirillina, J. D. Fetherston, M. C. Miller, J. A. Burlison and R. D. Perry, Mol. Microbiol., 2014, 93, 759–775 CrossRef CAS PubMed.
- D. C. Desrosiers, S. W. Bearden, I. Mier Jr., J. Abney, J. T. Paulley, J. D. Fetherston, J. C. Salazar, J. D. Radolf and R. D. Perry, Infect. Immun., 2010, 78, 5163–5177 CrossRef CAS PubMed.
- R. D. Perry, A. G. Bobrov, O. Kirillina, E. R. Rhodes, L. A. Actis and J. D. Fetherston, in Advances in Yersinia Research, ed. A. M. P. de Almeida and N. C. Leal, Springer, New York, 2012, vol. 954, pp. 267–279 Search PubMed.
- S. Kobayashi, S. Hidaka, Y. Kawamura, M. Ozaki and Y. Hayase, J. Antibiot., 1998, 51, 323–327 CrossRef CAS.
- S. Kobayashi, H. Nakai, Y. Ikenishi, W.-Y. Sun, M. Ozaki, Y. Hayase and R. Takeda, J. Antibiot., 1998, 51, 328–332 CrossRef CAS.
- M. F. Kreutzer, H. Kage, P. Gebhardt, B. Wackler, H. P. Saluz, D. Hoffmeister and M. Nett, Appl. Environ. Microbiol., 2011, 77, 6117–6124 CrossRef CAS PubMed.
- H. Nakai, S. Kobayashi, M. Ozaki, Y. Hayase and R. Takeda, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, 54–56 Search PubMed.
- N. S. Jakubovics and H. F. Jenkinson, Microbiology, 2001, 147, 1709–1718 CAS.
- D. G. Kehres and M. E. Maguire, FEMS Microbiol. Rev., 2003, 27, 263–290 CrossRef CAS.
- K. M. Papp-Wallace and M. E. Maguire, Annu. Rev. Microbiol., 2006, 60, 187–209 CrossRef CAS PubMed.
- V. V. Bartsevich and H. B. Pakrasi, EMBO J., 1995, 14, 1845–1853 CAS.
- R. E. Trouwborst, B. G. Clement, B. M. Tebo, B. T. Glazer and G. W. Luther III, Science, 2006, 313, 1955–1957 CrossRef CAS PubMed.
- B. M. Tebo, J. R. Bargar, B. G. Clement, G. J. Dick, K. J. Murray, D. Parker, R. Verity and S. M. Webb, Annu. Rev. Earth Planet. Sci., 2004, 32, 287–328 CrossRef CAS.
- T. G. Spiro, J. R. Bargar, G. Sposito and B. M. Tebo, Acc. Chem. Res., 2010, 43, 2–9 CrossRef CAS PubMed.
- R. Caspi, B. M. Tebo and M. G. Haygood, Appl. Environ. Microbiol., 1998, 64, 3549–3555 CAS.
- J. P. M. de Vrind, G. J. Brouwers, P. L. A. M. Corstjens, J. den Dulk and E. W. de Vrind-de Jong, Appl. Environ. Microbiol., 1998, 64, 3556–3562 CAS.
- G.-J. Brouwers, J. P. M. de Vrind, P. L. A. M. Corstjens, P. Cornelis, C. Baysse and E. W. de Vrind-de Jong, Appl. Environ. Microbiol., 1999, 65, 1762–1768 CAS.
- D. L. Parker, S.-W. Lee, K. Geszvain, R. E. Davis, C. Gruffaz, J.-M. Meyer, J. W. Torpey and B. M. Tebo, Front. Microbiol., 2014, 5, 202 Search PubMed.
- D. L. Parker, G. Sposito and B. M. Tebo, Geochim. Cosmochim. Acta, 2004, 68, 4809–4820 CrossRef CAS PubMed.
- D. L. Parker, T. Morita, M. L. Mozafarzadeh, R. Verity, J. K. McCarthy and B. M. Tebo, Geochim. Cosmochim. Acta, 2007, 71, 5672–5683 CrossRef CAS PubMed.
- J. M. Harrington, D. L. Parker, J. R. Bargar, A. A. Jarzecki, B. M. Tebo, G. Sposito and O. W. Duckworth, Geochim. Cosmochim. Acta, 2012, 88, 106–119 CrossRef CAS PubMed.
- Y. Bi, D. L. Hesterberg and O. W. Duckworth, Geochim. Cosmochim. Acta, 2010, 74, 2915–2925 CrossRef CAS PubMed.
- O. W. Duckworth, M. M. Akafia, M. Y. Andrews and J. R. Bargar, Environ. Sci.: Processes Impacts, 2014, 16, 1348–1359 CAS.
- M. M. Akafia, J. M. Harrington, J. R. Bargar and O. W. Duckworth, Geochim. Cosmochim. Acta, 2014, 141, 258–269 CrossRef CAS PubMed.
- L. C. Seefeldt, B. M. Hoffman and D. R. Dean, Annu. Rev. Biochem., 2009, 78, 701–722 CrossRef CAS PubMed.
- R. L. Robson, R. R. Eady, T. H. Richardson, R. W. Miller, M. Hawkins and J. R. Postgate, Nature, 1986, 322, 388–390 CrossRef CAS.
- A. Müller, K. Schneider, K. Knüttel and W. R. Hagen, FEBS Lett., 1992, 303, 36–40 CrossRef.
- W. J. Page and M. von Tigerstrom, J. Gen. Microbiol., 1988, 134, 453–460 CAS.
- L. J. Liermann, R. L. Guynn, A. Anbar and S. L. Brantley, Chem. Geol., 2005, 220, 285–302 CrossRef CAS PubMed.
- J. P. Bellenger, T. Wichard, A. B. Kustka and A. M. L. Kraepiel, Nat. Geosci., 2008, 1, 243–246 CrossRef CAS.
- M. Deicke, J.-P. Bellenger and T. Wichard, J. Chromatogr. A, 2013, 1298, 50–60 CrossRef CAS PubMed.
- T. Wichard, J.-P. Bellenger, F. M. M. Morel and A. M. L. Kraepiel, Environ. Sci. Technol., 2009, 43, 7218–7224 CrossRef CAS.
- F. Yoneyama, M. Yamamoto, W. Hashimoto and K. Murata, J. Appl. Microbiol., 2011, 111, 932–938 CrossRef CAS PubMed.
- T. B. Karpishin and K. N. Raymond, Angew. Chem., Int. Ed. Engl., 1992, 31, 466–468 CrossRef.
- T. B. Karpishin, T. M. Dewey and K. N. Raymond, J. Am. Chem. Soc., 1993, 115, 1842–1851 CrossRef CAS.
- T. Baramov, K. Keijzer, E. Irran, E. Mösker, M.-H. Baik and R. Süssmuth, Chem. – Eur. J., 2013, 19, 10536–10542 CrossRef CAS PubMed.
- T. Schmiederer, S. Rausch, M. Valdebenito, Y. Mantri, E. Mösker, T. Baramov, K. Stelmaszyk, P. Schmieder, D. Butz, S. I. Müller, K. Schneider, M.-H. Baik, K. Hantke and R. D. Süssmuth, Angew. Chem., Int. Ed., 2011, 50, 4230–4233 CrossRef CAS PubMed.
- D. H. Goetz, M. A. Holmes, N. Borregaard, M. E. Bluhm, K. N. Raymond and R. K. Strong, Mol. Cell, 2002, 10, 1033–1043 CrossRef CAS.
- R. J. Abergel, M. C. Clifton, J. C. Pizarro, J. A. Warner, D. K. Shuh, R. K. Strong and K. N. Raymond, J. Am. Chem. Soc., 2008, 130, 11524–11534 CrossRef CAS PubMed.
- F. Peuckert, A. L. Ramos-Vega, M. Miethke, C. J. Schwörer, A. G. Albrecht, M. Oberthür and M. A. Marahiel, Chem. Biol., 2011, 18, 907–919 CrossRef CAS PubMed.
- D. Osman and J. S. Cavet, in Advances in Applied Microbiology, ed. A. I. Laskin, S. Sariaslani and G. M. Gadd, 2008, vol. 65, pp. 217–247 Search PubMed.
- L. Macomber and J. A. Imlay, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 8344–8349 CrossRef CAS PubMed.
- I. Bagai, C. Rensing, N. J. Blackburn and M. M. McEvoy, Biochemistry, 2008, 47, 11408–11414 CrossRef CAS PubMed.
- N. J. Robinson, Nat. Chem. Biol., 2008, 4, 582–583 CrossRef CAS PubMed.
- B. Gold, H. Deng, R. Bryk, D. Vargas, D. Eliezer, J. Roberts, X. Jiang and C. Nathan, Nat. Chem. Biol., 2008, 4, 609–616 CrossRef CAS PubMed.
- M. Zimmermann, S. R. Udagedara, C. M. Sze, T. M. Ryan, G. J. Howlett, Z. Xiao and A. G. Wedd, J. Inorg. Biochem., 2012, 115, 186–197 CrossRef CAS PubMed.
- Z. Ma, F. E. Jacobsen and D. P. Giedroc, Chem. Rev., 2009, 109, 4644–4681 CrossRef CAS PubMed.
- J. D. Semrau, A. A. DiSpirito and S. Yoon, FEMS Microbiol. Rev., 2010, 34, 496–531 CAS.
- R. Balasubramanian, S. M. Smith, S. Rawat, L. A. Yatsunyk, T. L. Stemmler and A. C. Rosenzweig, Nature, 2010, 465, 115–119 CrossRef CAS PubMed.
- A. S. Hakemian and A. C. Rosenzweig, Annu. Rev. Biochem., 2007, 76, 223–241 CrossRef CAS PubMed.
- A. A. DiSpirito, J. A. Zahn, D. W. Graham, H. J. Kim, C. K. Larive, T. S. Derrick, C. D. Cox and A. Taylor, J. Bacteriol., 1998, 180, 3606–3613 CAS.
- C. M. Téllez, K. P. Gaus, D. W. Graham, R. G. Arnold and R. Z. Guzman, Appl. Environ. Microbiol., 1998, 64, 1115–1122 Search PubMed.
- H. J. Kim, D. W. Graham, A. A. DiSpirito, M. A. Alterman, N. Galeva, C. K. Larive, D. Asunskis and P. M. A. Sherwood, Science, 2004, 305, 1612–1615 CrossRef CAS PubMed.
- G. E. Kenney and A. C. Rosenzweig, ACS Chem. Biol., 2012, 7, 260–268 CrossRef CAS PubMed.
- J. Anttila, P. Heinonen, T. Nenonen, A. Pino, H. Iwaï, E. Kauppi, R. Soliymani, M. Baumann, J. Saksi, N. Suni and T. Haltia, Biochim. Biophys. Acta, Bioenerg., 2011, 1807, 311–318 CrossRef CAS PubMed.
- K. Helbig, C. Bleuel, G. J. Krauss and D. H. Nies, J. Bacteriol., 2008, 190, 5431–5438 CrossRef CAS PubMed.
- T. A. Lewis, A. Paszczynski, S. W. Gordon-Wylie, S. Jeedigunta, C.-H. Lee and R. L. Crawford, Environ. Sci. Technol., 2001, 35, 552–559 CrossRef CAS.
- L. H. Leach and T. A. Lewis, Microbiology, 2006, 152, 3157–3166 CrossRef CAS PubMed.
- G. M. Teitzel, A. Geddie, S. K. De Long, M. J. Kirisits, M. Whiteley and M. R. Parsek, J. Bacteriol., 2006, 188, 7242–7256 CrossRef CAS PubMed.
- P. Visca, G. Colotti, L. Serino, D. Verzili, N. Orsi and E. Chiancone, Appl. Environ. Microbiol., 1992, 58, 2886–2893 CAS.
- K. S. Chaturvedi and J. P. Henderson, Front. Cell. Infect. Microbiol., 2014, 4, 3 Search PubMed.
- L. S. Tufft, C. F. Nockels and M. J. Fettman, Avian Dis., 1988, 32, 779–786 CrossRef CAS.
- C. White, J. Lee, T. Kambe, K. Fritsche and M. J. Petris, J. Biol. Chem., 2009, 284, 33949–33956 CrossRef CAS PubMed.
- K. S. Chaturvedi, C. S. Hung, D. E. Giblin, S. Urushidani, A. M. Austin, M. C. Dinauer and J. P. Henderson, ACS Chem. Biol., 2014, 9, 551–561 CrossRef CAS PubMed.
- R. A. Festa, M. B. Jones, S. Butler-Wu, D. Sinsimer, R. Gerads, W. R. Bishai, S. N. Peterson and K. H. Darwin, Mol. Microbiol., 2011, 79, 133–148 CrossRef CAS PubMed.
- K. S. Chaturvedi, C. S. Hung, J. R. Crowley, A. E. Stapleton and J. P. Henderson, Nat. Chem. Biol., 2012, 8, 731–736 CrossRef CAS PubMed.
- G. Grass, K. Thakali, P. E. Klebba, D. Thieme, A. Müller, G. F. Wildner and C. Rensing, J. Bacteriol., 2004, 186, 5826–5833 CrossRef CAS PubMed.
- J. E. L. Arceneaux, M. E. Boutwell and B. R. Byers, Antimicrob. Agents Chemother., 1984, 25, 650–652 CrossRef CAS.
- S. E. Clarke, J. Stuart and J. Sanders-Loehr, Appl. Environ. Microbiol., 1987, 53, 917–922 CAS.
- I. J. Schalk, M. Hannauer and A. Braud, Environ. Microbiol., 2011, 13, 2844–2854 CrossRef CAS PubMed.
- P. B. Chatterjee and D. C. Crans, Inorg. Chem., 2012, 51, 9144–9146 CrossRef CAS PubMed.
- J. M. Harrington, J. R. Bargar, A. A. Jarzecki, J. G. Roberts, L. A. Sombers and O. W. Duckworth, BioMetals, 2012, 25, 393–412 CrossRef CAS PubMed.
- C. W. Johnston, M. A. Wyatt, X. Li, A. Ibrahim, J. Shuster, G. Southam and N. A. Magarvey, Nat. Chem. Biol., 2013, 9, 241–243 CrossRef CAS PubMed.
- M. A. Wyatt, C. W. Johnston and N. A. Magarvey, J. Nanopart. Res., 2014, 16, 2212 CrossRef.
- S. A. Amin, D. H. Green, F. C. Küpper and C. J. Carrano, Inorg. Chem., 2009, 48, 11451–11458 CrossRef CAS PubMed.
- M. T. Madigan, J. M. Martinko, D. Stahl and D. P. Clark, Brock Biology of Microorganisms, Benjamin Cummings, San Francisco, 13th edn, 2012 Search PubMed.
- A. Braud, M. Hannauer, G. L. A. Mislin and I. J. Schalk, J. Bacteriol., 2009, 191, 3517–3525 CrossRef CAS PubMed.
- A. Braud, F. Hoegy, K. Jezequel, T. Lebeau and I. J. Schalk, Environ. Microbiol., 2009, 11, 1079–1091 CrossRef CAS PubMed.
- A. Braud, V. Geoffroy, F. Hoegy, G. L. A. Mislin and I. J. Schalk, Environ. Microbiol. Rep., 2010, 2, 419–425 CrossRef CAS PubMed.
- M. Hannauer, A. Braud, F. Hoegy, P. Ronot, A. Boos and I. J. Schalk, Environ. Microbiol., 2012, 14, 1696–1708 CrossRef CAS PubMed.
- A. Braud, K. Jézéquel, E. Vieille, A. Tritter and T. Lebeau, Water, Air, Soil Pollut., 2006, 6, 261–279 CrossRef CAS PubMed.
- A. Braud, K. Jézéquel and T. Lebeau, J. Hazard. Mater., 2007, 144, 229–239 CrossRef CAS PubMed.
- A. M. Zawadzka, A. J. Paszczynski and R. L. Crawford, in Advances in Applied Bioremediation, ed. A. Singh, R. C. Kuhad and O. P. Ward, Springer-Verlag, Berlin Heidelberg, 2009, vol. 17, pp. 221–238 Search PubMed.
- U. Hildebrand, J. Lex, K. Taraz, S. Winkler, W. Ockels and H. Budzikiewicz, Z. Naturforsch., 1984, 39b, 1607–1613 CAS.
- H.-J. Krüger and R. H. Holm, J. Am. Chem. Soc., 1990, 112, 2955–2963 CrossRef.
- A. Evers, R. D. Hancock, A. E. Martell and R. J. Motekaitis, Inorg. Chem., 1989, 28, 2189–2195 CrossRef CAS.
- H. H. Malluche, A. J. Smith, K. Abreo and M.-C. Faugere, N. Engl. J. Med., 1984, 311, 140–144 CrossRef CAS PubMed.
- G. Crisponi, V. M. Nurchi, V. Bertolasi, M. Remelli and G. Faa, Coord. Chem. Rev., 2012, 256, 89–104 CrossRef CAS PubMed.
- X. Hu and G. L. Boyer, Appl. Environ. Microbiol., 1996, 62, 4044–4048 CAS.
- J. Greenwald, G. Zeder-Lutz, A. Hagege, H. Celia and F. Pattus, J. Bacteriol., 2008, 190, 6548–6558 CrossRef CAS PubMed.
- P. Cornelis, J. Bacteriol., 2008, 190, 6541–6543 CrossRef CAS PubMed.
- I. J. Schalk, J. Inorg. Biochem., 2008, 102, 1159–1169 CrossRef CAS PubMed.
- A. del Olmo, C. Caramelo and C. SanJose, J. Inorg. Biochem., 2003, 97, 384–387 CrossRef CAS.
- G. A. Snow, Biochem. J., 1965, 94, 160–165 CAS.
- J. Francis, H. M. Macturk, J. Madinaveitia and G. A. Snow, Biochem. J., 1953, 55, 596–607 CAS.
- G. A. Snow, J. Chem. Soc., 1954, 2588–2596 RSC.
- G. A. Snow, J. Chem. Soc., 1954, 4080–4093 RSC.
- A. Jeanson, M. Ferrand, H. Funke, C. Hennig, P. Moisy, P. L. Solari, C. Vidaud and C. Den Auwer, Chem. – Eur. J., 2010, 16, 1378–1387 CrossRef CAS PubMed.
- M. P. Neu, Los Alamos Sci., 2000, 416–417 CAS.
- N. V. Jarvis and R. D. Hancock, Inorg. Chim. Acta, 1991, 182, 229–232 CrossRef CAS.
- M. P. Neu, J. H. Matonic, C. E. Ruggiero and B. L. Scott, Angew. Chem., Int. Ed., 2000, 39, 1442–1444 CrossRef CAS.
- S. G. John, C. E. Ruggiero, L. E. Hersman, C.-S. Tung and M. P. Neu, Environ. Sci. Technol., 2001, 35, 2942–2948 CrossRef CAS.
- D. T. Reed, R. P. Deo and B. E. Rittmann, in The Chemistry of the Actinide and Transactinide Elements, ed. L. R. Morss, N. M. Edelstein and J. Fuger, Springer, Netherlands, 2010, pp. 3595–3663 Search PubMed.
- F. Reith, L. Fairbrother, G. Nolze, O. Wilhelmi, P. L. Clode, A. Gregg, J. E. Parsons, S. A. Wakelin, A. Pring, R. Hough, G. Southam and J. Brugger, Geology, 2010, 38, 843–846 CrossRef CAS PubMed.
- K. J. Ryan, C. G. Ray and J. C. Sherris, Sherris Medical Microbiology: An Introduction to Infectious Diseases, McGraw-Hill, New York, 4th edn, 2004 Search PubMed.
- S. Yamamoto, N. Okujo, T. Yoshida, S. Matsuura and S. Shinoda, J. Biochem., 1994, 115, 868–874 CAS.
- A. Butler and R. M. Theisen, Coord. Chem. Rev., 2010, 254, 288–296 CrossRef CAS PubMed.
- S. A. Amin, F. C. Küpper, D. H. Green, W. R. Harris and C. J. Carrano, J. Am. Chem. Soc., 2007, 129, 478–479 CrossRef CAS PubMed.
- C. J. Carrano, S. Schellenberg, S. A. Amin, D. H. Green and F. C. Küpper, Mar. Biotechnol., 2009, 11, 431–440 CrossRef CAS PubMed.
- W. R. Harris, S. A. Amin, F. C. Küpper, D. H. Green and C. J. Carrano, J. Am. Chem. Soc., 2007, 129, 12263–12271 CrossRef CAS PubMed.
- M. Sandy and A. Butler, Chem. Rev., 2009, 109, 4580–4595 CrossRef CAS PubMed.
- V. M. Dembitsky, A. A. A. Al Quntar and M. Srebnik, Chem. Rev., 2011, 111, 209–237 CrossRef CAS PubMed.
- A. Romano, L. Trimble, A. R. Hobusch, K. J. Schroeder, S. A. Amin, A. D. Hartnett, R. A. Barker, A. L. Crumbliss and C. J. Carrano, Metallomics, 2013, 5, 1025–1030 RSC.
- A. J. Weerasinghe, S. A. Amin, R. A. Barker, T. Othman, A. N. Romano, C. J. Parker Siburt, J. Tisnado, L. A. Lambert, T. Huxford, C. J. Carrano and A. L. Crumbliss, J. Am. Chem. Soc., 2013, 135, 14504–14507 CrossRef CAS PubMed.
- S. R. Shouldice, R. J. Skene, D. R. Dougan, G. Snell, D. E. McRee, A. B. Schryvers and L. W. Tari, J. Bacteriol., 2004, 186, 3903–3910 CrossRef CAS PubMed.
- S. A. Amin, D. H. Green, M. C. Hart, F. C. Küpper, W. G. Sunda and C. J. Carrano, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17071–17076 CrossRef CAS PubMed.
- M. F. Traxler, M. R. Seyedsayamdost, J. Clardy and R. Kolter, Mol. Microbiol., 2012, 86, 628–644 CrossRef CAS PubMed.
- H. Lv, C. S. Hung and J. P. Henderson, J. Proteome Res., 2014, 13, 1397–1404 CrossRef CAS PubMed.
- M. U. Muckenthaler, B. Galy and M. W. Hentze, Annu. Rev. Nutr., 2008, 28, 197–213 CrossRef CAS PubMed.
- A. Roux, S. M. Payne and M. S. Gilmore, Cell Host Microbe, 2009, 6, 115–124 CAS.
- P. Visca, F. Imperi and I. L. Lamont, Trends Microbiol., 2007, 15, 22–30 CrossRef CAS PubMed.
- P. Williams and M. Cámara, Curr. Opin. Microbiol., 2009, 12, 182–191 CrossRef CAS PubMed.
- P. Nadal Jimenez, G. Koch, J. A. Thompson, K. B. Xavier, R. H. Cool and W. J. Quax, Microbiol. Mol. Biol. Rev., 2012, 76, 46–65 CrossRef PubMed.
- I. L. Lamont, P. A. Beare, U. Ochsner, A. I. Vasil and M. L. Vasil, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 7072–7077 CrossRef CAS PubMed.
- A. Buckling, F. Harrison, M. Vos, M. A. Brockhurst, A. Gardner, S. A. West and A. Griffin, FEMS Microbiol. Ecol., 2007, 62, 135–141 CrossRef CAS PubMed.
- F. Taguchi, T. Suzuki, Y. Inagaki, K. Toyoda, T. Shiraishi and Y. Ichinose, J. Bacteriol., 2010, 192, 117–126 CrossRef CAS PubMed.
- L. L. Guan, K. Kanoh and K. Kamino, Appl. Environ. Microbiol., 2001, 67, 1710–1717 CrossRef CAS PubMed.
- J. A. Melvin, E. V. Scheller, J. F. Miller and P. A. Cotter, Nat. Rev. Microbiol., 2014, 12, 274–288 CrossRef CAS PubMed.
- T. J. Brickman and S. K. Armstrong, BioMetals, 2009, 22, 33–41 CrossRef CAS PubMed.
- R. D. Perry, J. Abney, I. Mier Jr., Y. Lee, S. W. Bearden and J. D. Fetherston, in Genus Yersinia: Entering the Functional Genomic Era, ed. M. Skurnik, J. A. Bengoechea and K. Granfors, Springer, 2003, vol. 529, pp. 275–283 Search PubMed.
- R. Anisimov, D. Brem, J. Heesemann and A. Rakin, FEMS Microbiol. Lett., 2005, 250, 27–32 CrossRef CAS PubMed.
- J. D. Fetherston, O. Kirillina, A. G. Bobrov, J. T. Paulley and R. D. Perry, Infect. Immun., 2010, 78, 2045–2052 CrossRef CAS PubMed.
- M. C. Miller, J. D. Fetherston, C. L. Pickett, A. G. Bobrov, R. H. Weaver, E. DeMoll and R. D. Perry, Microbiology, 2010, 156, 2226–2238 CrossRef CAS PubMed.
- C. Adler, N. S. Corbalán, M. R. Seyedsayamdost, M. F. Pomares, R. E. de Cristóbal, J. Clardy, R. Kolter and P. A. Vincent, PLoS One, 2012, 7, e46754 CAS.
- C. Adler, N. S. Corbalan, D. R. Peralta, M. F. Pomares, R. E. de Cristóbal and P. A. Vincent, PLoS One, 2014, 9, e84734 Search PubMed.
- M. Münzinger, K. Taraz and H. Budzikiewicz, Z. Naturforsch., 1999, 54c, 867–875 Search PubMed.
- F. C. Beasley and D. E. Heinrichs, J. Inorg. Biochem., 2010, 104, 282–288 CrossRef CAS PubMed.
- M. E. S. Achard, K. W. Chen, M. J. Sweet, R. E. Watts, K. Schroder, M. A. Schembri and A. G. McEwan, Biochem. J., 2013, 454, 543–549 CrossRef CAS PubMed.
- N. D. Hammer and E. P. Skaar, Annu. Rev. Microbiol., 2011, 65, 129–147 CrossRef CAS PubMed.
- S. E. Dale, M. T. Sebulsky and D. E. Heinrichs, J. Bacteriol., 2004, 186, 8356–8362 CrossRef CAS PubMed.
- L. S. Nobre and L. M. Saraiva, Curr. Microbiol., 2014, 69, 164–168 CrossRef CAS PubMed.
- H. Drechsel, S. Freund, G. Nicholson, H. Haag, O. Jung, H. Zähner and G. Jung, BioMetals, 1993, 6, 185–192 CrossRef CAS.
- H. Haag, H.-P. Fiedler, J. Meiwes, H. Drechsel, G. Jung and H. Zähner, FEMS Microbiol. Lett., 1994, 115, 125–130 CrossRef CAS PubMed.
- V. Braun, A. Pramanik, T. Gwinner, M. Köberle and E. Bohn, BioMetals, 2009, 22, 3–13 CrossRef CAS PubMed.
- U. Möllmann, L. Heinisch, A. Bauernfeind, T. Köhler and D. Ankel-Fuchs, BioMetals, 2009, 22, 615–624 CrossRef PubMed.
- C. Ji, R. E. Juárez-Hernández and M. J. Miller, Future Med. Chem., 2012, 4, 297–313 CrossRef CAS PubMed.
- R. E. Juárez-Hernández, H. Zhu and M. J. Miller, in Iron Acquisition by the Genus Mycobacterium, ed. B. R. Byers, Springer International Publishing, 2013, pp. 65–88 Search PubMed.
- G. L. A. Mislin and I. J. Schalk, Metallomics, 2014, 6, 408–420 RSC.
- A. Górska, A. Sloderbach and M. P. Marszałł, Trends Pharmacol. Sci., 2014, 35, 442–449 CrossRef PubMed.
- T. Zheng, J. L. Bullock and E. M. Nolan, J. Am. Chem. Soc., 2012, 134, 18388–18400 CrossRef CAS PubMed.
- T. Zheng and E. M. Nolan, J. Am. Chem. Soc., 2014, 136, 9677–9691 CrossRef CAS PubMed.
- S. Yoganathan, C. S. Sit and J. C. Vederas, Org. Biomol. Chem., 2011, 9, 2133–2141 CAS.
- S. J. Milner, A. Seve, A. M. Snelling, G. H. Thomas, K. G. Kerr, A. Routledge and A.-K. Duhme-Klair, Org. Biomol. Chem., 2013, 11, 3461–3468 CAS.
- J. Nüesch and F. Knüsel, in Mechanism of Action, ed. D. Gottlieb and P. D. Shaw, Springer, Berlin, 1967, pp. 499–541 Search PubMed.
- D. M. Reynolds and S. A. Waksman, J. Bacteriol., 1948, 55, 739–752 CAS.
- Y. Zeng, A. Kulkarni, Z. Yang, P. B. Patil, W. Zhou, X. Chi, S. Van Lanen and S. Chen, ACS Chem. Biol., 2012, 7, 1565–1575 CrossRef CAS PubMed.
- K. D. Krewulak and H. J. Vogel, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 1781–1804 CrossRef CAS PubMed.
- A. L. Stefanska, M. Fulston, C. S. V. Houge-Frydrych, J. J. Jones and S. R. Warr, J. Antibiot., 2000, 53, 1346–1353 CrossRef CAS.
- A. Hartmann, H.-P. Fiedler and V. Braun, Eur. J. Biochem., 1979, 99, 517–524 CrossRef CAS PubMed.
- V. Braun, K. Günthner, K. Hantke and L. Zimmermann, J. Bacteriol., 1983, 156, 308–315 CAS.
- L. Vértesy, W. Aretz, H.-W. Fehlhaber and H. Kogler, Helv. Chim. Acta, 1995, 78, 46–60 CrossRef.
- P. Huber, H. Leuenberger and W. Keller-Schierlein, Helv. Chim. Acta, 1986, 69, 236–245 CrossRef CAS.
- G. Winkelmann and C. J. Carrano, Transition metals in microbial metabolism, Harwood Academic Publishers, Amsterdam, 1997 Search PubMed.
- V. Prelog, Pure Appl. Chem., 1963, 6, 327–338 CAS.
- H. Bickel, P. Mertens, V. Prelog, J. Seibl and A. Walser, Tetrahedron, 1966, 22, 171–179 CrossRef.
- A. Urban, S. Eckermann, B. Fast, S. Metzger, M. Gehling, K. Ziegelbauer, H. Rübsamen-Waigmann and C. Freiberg, Appl. Environ. Microbiol., 2007, 73, 6436–6443 CrossRef CAS PubMed.
- F. Baquero and F. Moreno, FEMS Microbiol. Lett., 1984, 23, 117–124 CrossRef CAS PubMed.
- S. Duquesne, D. Destoumieux-Garzón, J. Peduzzi and S. Rebuffat, Nat. Prod. Rep., 2007, 24, 708–734 RSC.
- X. Thomas, D. Destoumieux-Garzón, J. Peduzzi, C. Afonso, A. Blond, N. Birlirakis, C. Goulard, L. Dubost, R. Thai, J.-C. Tabet and S. Rebuffat, J. Biol. Chem., 2004, 279, 28233–28242 CrossRef CAS PubMed.
- M. E. Poey, M. F. Azpiroz and M. Laviña, Antimicrob. Agents Chemother., 2006, 50, 1411–1418 CrossRef CAS PubMed.
- G. Vassiliadis, D. Destoumieux-Garzón, C. Lombard, S. Rebuffat and J. Peduzzi, Antimicrob. Agents Chemother., 2010, 54, 288–297 CrossRef CAS PubMed.
- T. Zheng, Ph.D. Thesis, Massachusetts Institute of Technology, 2014.
- D. Destoumieux-Garzón, S. Duquesne, J. Peduzzi, C. Goulard, M. Desmadril, L. Letellier, S. Rebuffat and P. Boulanger, Biochem. J., 2005, 389, 869–876 CrossRef PubMed.
- I. Mathavan, S. Zirah, S. Mehmood, H. G. Choudhury, C. Goulard, Y. Li, C. V. Robinson, S. Rebuffat and K. Beis, Nat. Chem. Biol., 2014, 10, 340–342 CrossRef CAS PubMed.
- H. J. Smith and M. M. Meremikwu, Cochrane Database Syst. Rev., 2003, CD001474 CAS.
- J. S. Buyer, V. de Lorenzo and J. B. Neilands, Appl. Environ. Microbiol., 1991, 57, 2246–2250 CAS.
Footnote |
| † The pFe(III) is the negative of the decadic logarithm of the concentration of free Fe(III) in solution under the following fixed conditions: pH = 7.4, total ligand concentration = 10 μM, and total Fe(III) concentration = 1 μM. |
| This journal is © The Royal Society of Chemistry 2015 |