
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A CH2Cl2 complex of a [Rh(pincer)]+ cation†
Gemma M.
Adams
,
F. Mark
Chadwick
,
Sebastian D.
Pike
and
Andrew S.
Weller
*
Department of Chemistry, Chemistry Research Laboratories, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: andrew.weller@chem.ox.ac.uk
First published on 5th March 2015
Abstract
The CH2Cl2 complex [Rh(tBuPONOP)(κ1-ClCH2Cl)][BArF4] is reported, that also acts as a useful synthon for other complexes such as N2, CO and H2 adducts; while the analogous PNP complex undergoes C–Cl activation.
Coordinatively and electronically unsaturated transition-metal pincer complexes, [M(pincer)], are key intermediates in alkane dehydrogenation processes,1 as well as other catalytic transformations.2 They have also played a major role in the elucidation of fundamental bond transformations, such as C–H, C–C and C–X breaking and making.3 Recently, Brookhart and co-workers reported the synthesis of transition-metal methane and ethane sigma complexes, by a low temperature (ca. −110 °C to −150 °C) protonation of the corresponding Rh(tBuPONOP)R precursors using [H(OEt2)2][BArF4] in CDF2Cl–CH2Cl2 solvent to give [Rh(tBuPONOP)(H-R)][BArF4] [tBuPONOP = 2,6-(tBu2PO)2C5H3N; R = Me, Et; ArF = 3,5-(CF3)2C6H3], Scheme 1.4 Such complexes are key, but transient, intermediates in C–H bond activation processes. On warming above −87 °C (R = Me) or −130 °C (R = Et) they lose alkane and generate complexes tentatively characterised in situ on the basis of 31P NMR spectroscopy as [Rh(tBuPONOP)(solv)][BArF4] (solv = CDF2Cl or CD2Cl2). These solvent adducts remain to be definitively characterised. They are particularly interesting given their role in alkane coordination chemistry, and more generally as latent-low coordinate intermediates in catalytic processes.
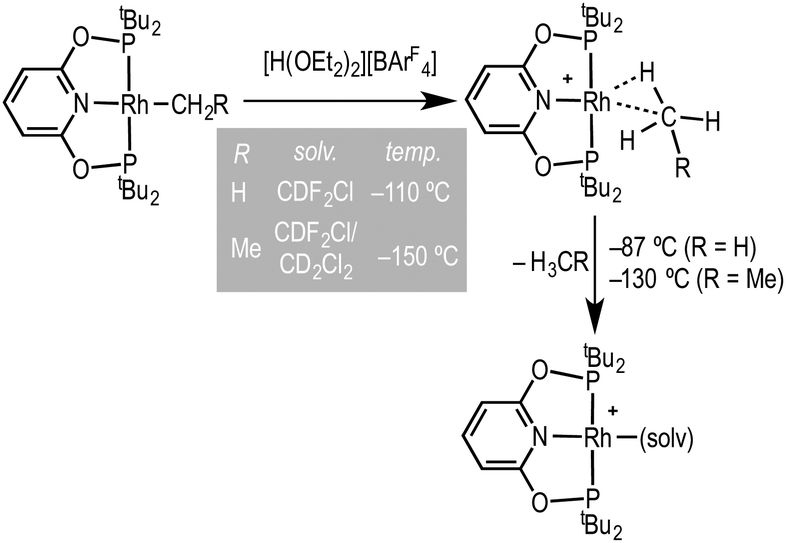 | ||
| Scheme 1 Formation of a sigma alkane complex and decomposition to give tentatively characterised solvent complexes (Brookhart and co-workers). [BArF4]− anions are not shown.4 | ||
We now report the full characterisation of the CH2Cl2 adduct accessed via a different, halide abstraction, route including a single crystal X-ray diffraction study and its onward reactivity. We also demonstrate that changing the pincer ligand to the more electron donating tBuPNP [2,6-(tBu2PCH2)2C5H3N] results in C–Cl bond activation of the solvent molecule.
Addition of Na[BArF4] to a CH2Cl2 solution of Rh(tBuPONOP)Cl, 1,4a results in the formation of orange [Rh(tBuPONOP)(κ1-ClCH2Cl)][BArF4], 2 (Scheme 2). Filtration and removal of the solvent affords 2 in good isolated yield as a powder. Complex 2 can be recrystallised from CH2Cl2–pentane under an Ar atmosphere to give crystals suitable for an X-ray diffraction study. Under these conditions, orange 2 crystallises alongside the dinitrogen adduct, [Rh(tBuPONOP)(κ1-N2)][BArF4], 3, in an approximate 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (as measured by 31P NMR spectroscopy, vide infra). Single crystals of 2 suitable for an X-ray diffraction study were obtained by mechanical separation from orange/brown 3.‡ Presumably the exogenous N2 comes from trace (1–2 ppm) levels of N2 present in the argon, as has been noted previously,5 and is driven by relative solubilities of 2 and 3; as in neat CD2Cl2 under the same Ar atmosphere 2 does not go onto to form 3 to the detection limit of 31P{1H} NMR spectroscopy. The solid-state structure (Fig. 1A) shows a pseudo square planar cationic [Rh(tBuPONOP)]+ centre coordinated in the fourth position by a CH2Cl2 molecule. The Rh–Cl1 distance [2.350(2) Å] is significantly shorter than reported for related [RhCp*(PMe3)(Ph)(κ1-ClCH2Cl)][BArF4],6 2.512(2) Å, and [RhCp*(PMe3)(Me)(κ1-ClCH2Cl)][BArF4], 2.488(1) Å Cp* = η5−C5Me5).7 Complex 2 adds to the relatively small number of CH2Cl2 complexes that have been crystallographically characterised, and in particular CH2Cl2 adducts of pincer, or closely related, complexes.8
:1 ratio (as measured by 31P NMR spectroscopy, vide infra). Single crystals of 2 suitable for an X-ray diffraction study were obtained by mechanical separation from orange/brown 3.‡ Presumably the exogenous N2 comes from trace (1–2 ppm) levels of N2 present in the argon, as has been noted previously,5 and is driven by relative solubilities of 2 and 3; as in neat CD2Cl2 under the same Ar atmosphere 2 does not go onto to form 3 to the detection limit of 31P{1H} NMR spectroscopy. The solid-state structure (Fig. 1A) shows a pseudo square planar cationic [Rh(tBuPONOP)]+ centre coordinated in the fourth position by a CH2Cl2 molecule. The Rh–Cl1 distance [2.350(2) Å] is significantly shorter than reported for related [RhCp*(PMe3)(Ph)(κ1-ClCH2Cl)][BArF4],6 2.512(2) Å, and [RhCp*(PMe3)(Me)(κ1-ClCH2Cl)][BArF4], 2.488(1) Å Cp* = η5−C5Me5).7 Complex 2 adds to the relatively small number of CH2Cl2 complexes that have been crystallographically characterised, and in particular CH2Cl2 adducts of pincer, or closely related, complexes.8
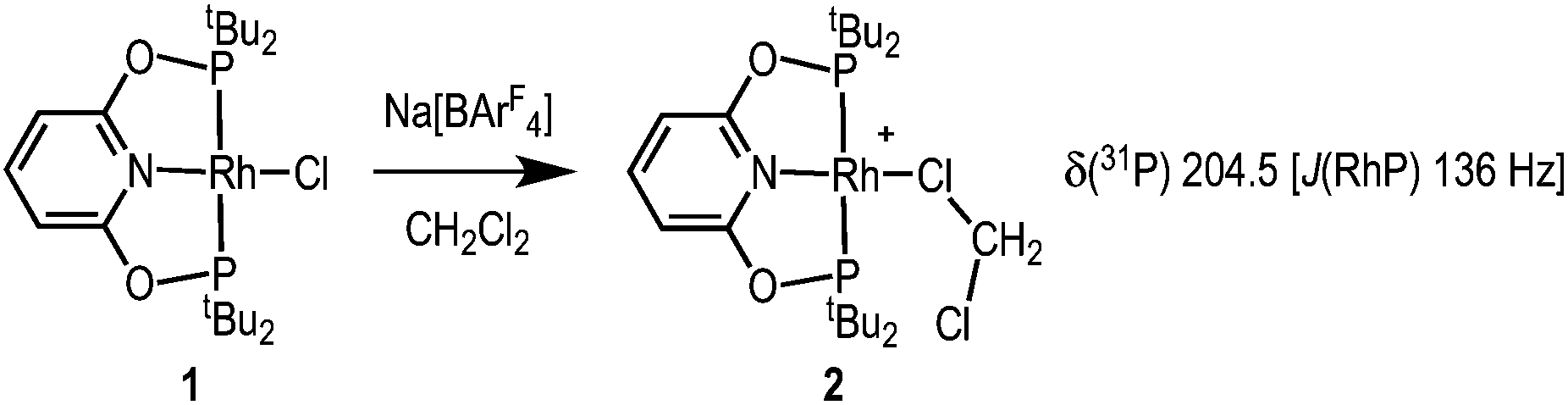 | ||
| Scheme 2 Synthesis of complex 2. [BArF4]− anion is not shown. | ||
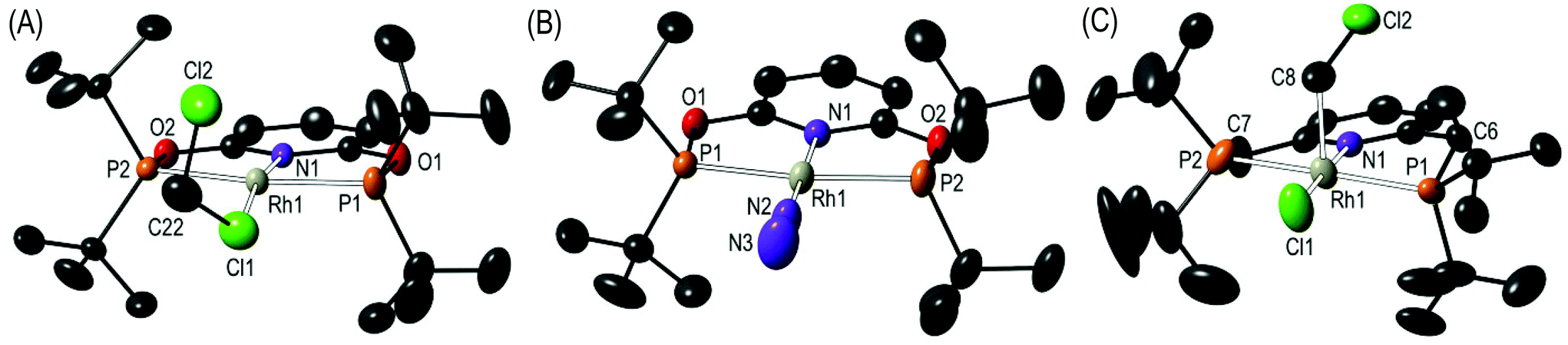 | ||
| Fig. 1 Solid-state structures of: (A) Complex 2; (B) Complex 3; (C) Complex 5. Displacement ellipsoids are shown at the 50% probability level, hydrogen atoms and the [BArF4]− anions are not shown. Selected bond lengths (Å) and angles (°): (2) Rh1–Cl1, 2.350(2); Rh1–N1, 2.011(4); Rh1–P1, 2.272(1); Rh1–P2, 2.285(1); Cl1–C22, 1.710(8); Cl2–C22, 1.758(7); Cl1–C22–Cl2, 114.3(4); N1–Rh1–Cl1, 169.65(11). (3) Rh1–N1, 2.018(3); Rh1–N2, 1.967(3); Rh1–P1, 2.2745(8); Rh1–P2, 2.2724(8); N2–N3, 1.063(5); Rh1–N2–N3, 179.3(4); N1–Rh1–N2, 179.37(13). (5) Rh1–Cl1, 2.311(2); Rh1–N1, 2.066(6); Rh1–P1, 2.335(2); Rh1–P2, 2.339(2); Rh1–C8, 2.196(15); C8–Cl2, 1.79(2); Rh1–C8–Cl2, 112.5(9). Complex 5 co-crystallises with [Rh(tBuPNP)(H)Cl][BArF4], 6, at the same lattice position in a 50:50 ratio.‡ | ||
Although the short Rh–Cl distance might suggest a stronger interaction in 2, in solution (vide infra) rapid exchange between solvent and bound CH2Cl2 occurs. The two C–Cl distances in the bound solvent molecule are similar, 1.710(8) [C22–Cl1] and 1.758(7) [C22–Cl2] Å, although the distal C–Cl bond is the slightly longer of the two. This is in contrast to other reported CH2Cl2 complexes in which the bound C–Cl bond is longer.8,9 We suggest that the slight lengthening of C22–Cl2 may be due to a number of weak C–H⋯Cl hydrogen bonds between proximal tBu groups and Cl2.10
Complex 2 is stable in the solid-state under an Ar atmosphere, and in solution (CD2Cl2) for at least 1 week. In the 31P{1H} NMR spectrum (CD2Cl2) a single resonance is observed at δ 204.5 [J(RhP) 136 Hz]. These data are identical to those previously reported by Brookhart and co-workers for the complex tentatively characterised as [Rh(tBuPONOP)(CH2Cl2)][BArF4], i.e.2. The tBu groups are observed as a single environment in the 1H NMR spectrum. The bound CH2Cl2 ligand is not observed, even at −80 °C in the 13C{1H} NMR spectrum, presumably as it is undergoing fast exchange with the solvent.11 The electrospray ionisation mass spectrum of 2 using N2 as a desorption gas showed only 3 as the molecular ion.
Complex 2 is a useful synthon for the preparation of other pincer complexes (Scheme 3). Addition of H2 to a CD2Cl2 solution of 2 forms the previously reported dihydrogen complex [Rh(tBuPONOP)(η2-H2)][BArF4]12 [δ(1H) −8.27, lit. −8.26]. Addition of N2 forms the new complex [Rh(tBuPONOP)(κ1-N2)][BArF4], 3, for which a solid-state structure is shown in Fig. 1B. This demonstrates an end-on bound, monomeric, N2 adduct [N–N, 1.063(5); Rh–N2, 1.967(3) Å]. The 31P{1H} NMR spectrum displays a single environment at δ 211.0 [J(RhP) 132 Hz], while in the IR spectrum the N–N stretch is observed at 2201.9 cm−1. The N–N bond length is very similar (albeit a little shorter) than that in free N2 [1.09 Å], suggesting only a small degree of activation. Complex 3 can also be compared with previously reported [Rh(tBuPNP)(κ1-N2)][OTf] which shows a slightly longer N–N bond, a shorter Rh–N bond and a more red-shifted N–N stretch: 1.116(4), 1.898(3) Å, and 2153 cm−1 respectively; suggesting greater N2 activation for this more electron rich pincer ligand.13 This greater metal-based basicity in the tBuPNP complexes is reflected in the CO stretching frequencies of the corresponding CO-adducts: [Rh(tBuPONOP)(CO)][BArF4], 4 [2020 cm−1] and [Rh(tBuPNP)(CO)][BArF4] [1982 cm−1].14 Complex 4 was prepared by adding CO to a CH2Cl2 solution of 2, further demonstrating the utility of complex 2 in synthesis.
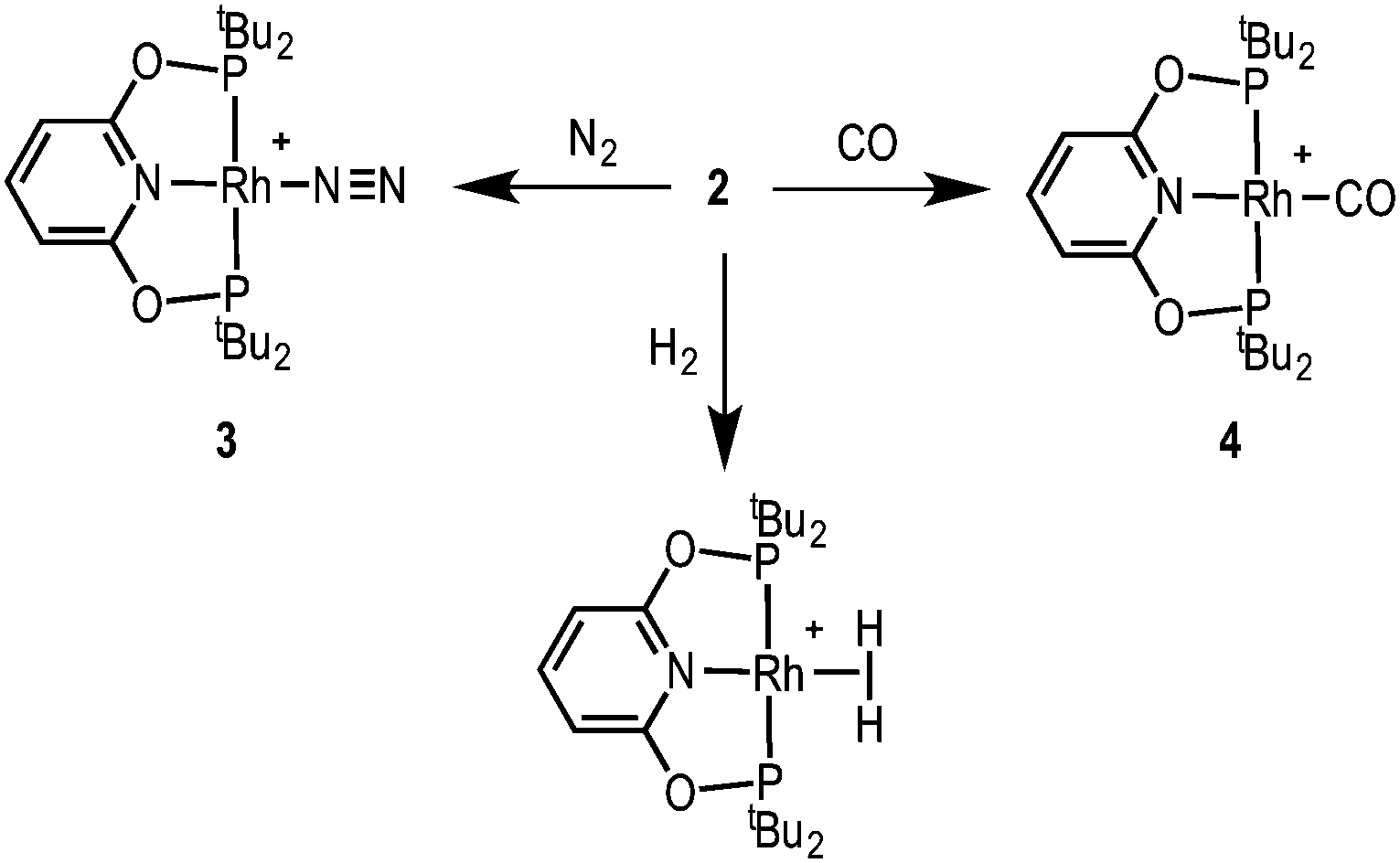 | ||
| Scheme 3 Reactivity of complex 2. CH2Cl2 solvent. [BArF4]− anions are not shown. | ||
The difference in electron-donating power of the tBuPONOP versustBuPNP ligands can also been shown by the attempted synthesis of the CH2Cl2 adduct of the {Rh(tBuPNP)}+ fragment, analogous to complex 2. Rather than simple coordination, this resulted in a number of products as measured by 31P{1H} NMR spectroscopy. Analysis of single crystals suitable for an X-ray diffraction study, obtained from recrystallisation of the reaction mixture, demonstrated co-crystallisation of two complexes [Rh(tBuPNP)(CH2Cl)Cl][BArF4], 5, and [Rh(tBuPNP)(H)Cl][BArF4], 6, in an approximate 50:50 ratio (Scheme 4); for which the solid-state structure of 5 is shown in Fig. 1C. Because of this co-crystallisation the metrical data associated with 5 should be treated with caution. The 1H NMR spectrum of these crystals showed a broad hydride signal at δ −15.48 (relative integral relative to [BArF4] of ∼0.5 H) which is assigned to 6. Given the number of products formed we are reluctant to speculate on mechanism of formation of 6, but protonation of 5 by trace acid arising from other decomposition pathways could form 6. Addition of H2 to this mixture of 5 and 6 in CD2Cl2 afforded mixture of products, from which [Rh(tBuPNP)(η2-H2)][BArF4] could be identified as the major species present.16
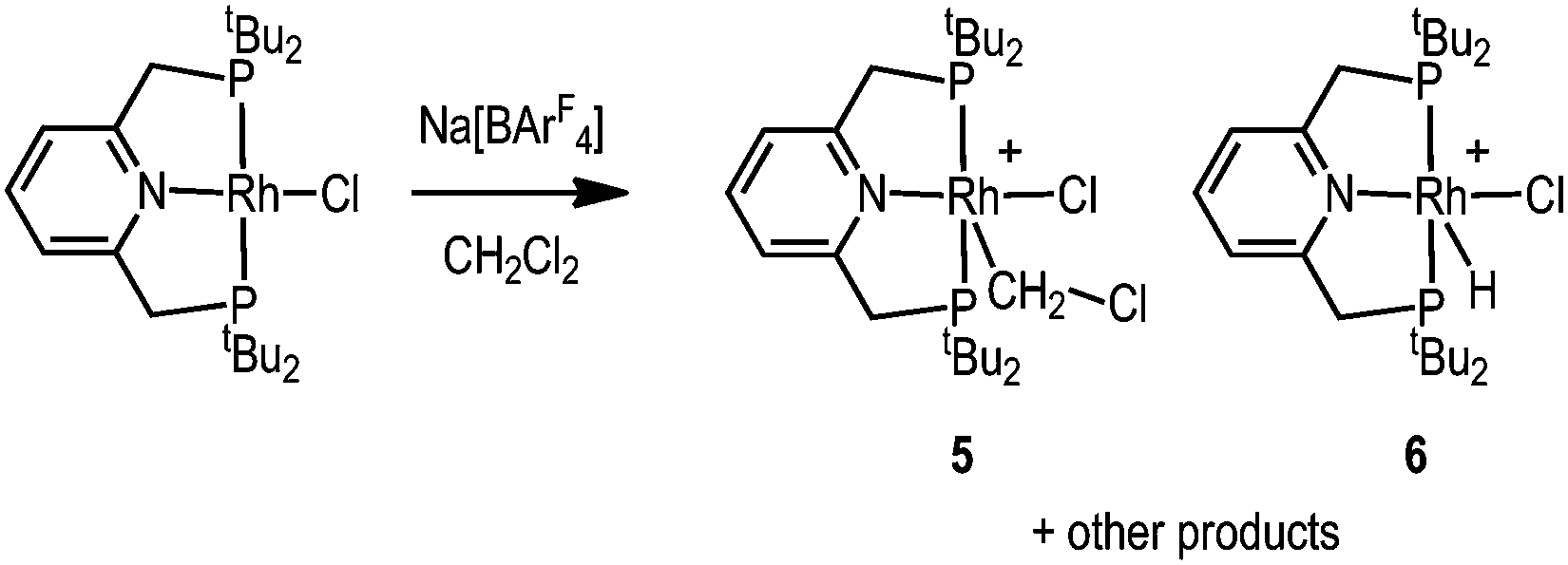 | ||
| Scheme 4 Reactivity of Rh(tBuPNP)Cl15 with Na[BArF4]. CH2Cl2 solvent. [BArF4]− anions are not shown. | ||
Conclusions
The CH2Cl2 complex [Rh(tBuPONOP)(κ1-ClCH2Cl)][BArF4] has been isolated, confirming its formation in the decomposition of the corresponding alkane adduct at low temperature, itself formed from protonation of an alkyl precursor.4 Synthesis has been achieved by an alternative halide-abstraction route in CH2Cl2 solvent, starting from a readily available chloride precursor. This complex, with its weakly bound CH2Cl2 ligand, also acts as a useful synthon for other complexes such as N2, CO and H2 adducts. The corresponding PNP ligand complex undergoes C–Cl activation to form a mixture of products, highlighting the difference in electron donating properties of these two ligands.Acknowledgements
The EPSRC for funding (EP/K035908/1) and Dr Adrian Chaplin for the initial synthesis of complex 5.Notes and references
- (a) J. Choi, A. H. Roy MacArthur, M. Brookhart and A. S. Goldman, Chem. Rev., 2011, 111, 1761–1779 CrossRef CAS PubMed; (b) M. C. Haibach, S. Kundu, M. Brookhart and A. S. Goldman, Acc. Chem. Res., 2012, 45, 947–958 CrossRef CAS PubMed.
- (a) The Chemistry of Pincer Compounds, ed. D. Morales-Morales and C. M. Jensen, Elsevier, Amsterdam, 2006 Search PubMed; (b) Organometallic Pincer Chemistry, ed. G. van Koten and D. Milstein, Springer, Heidelberg, 2013 Search PubMed.
- (a) B. Rybtchinski and D. Milstein, Angew. Chem., Int. Ed., 1999, 38, 870–883 CrossRef; (b) N. Selander and K. J. Szabó, Chem. Rev., 2011, 111, 2048–2076 CrossRef CAS PubMed; (c) C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS PubMed.
- (a) W. H. Bernskoetter, C. K. Schauer, K. I. Goldberg and M. Brookhart, Science, 2009, 326, 553–556 CrossRef CAS PubMed; (b) M. D. Walter, P. S. White, C. K. Schauer and M. Brookhart, J. Am. Chem. Soc., 2013, 135, 15933–15947 CrossRef CAS PubMed.
- H. Aneetha, M. Jiménez-Tenorio, M. C. Puerta, P. Valerga and K. Mereiter, Organometallics, 2002, 21, 628–635 CrossRef CAS.
- B. K. Corkey, F. L. Taw, R. G. Bergman and M. Brookhart, Polyhedron, 2004, 23, 2943–2954 CrossRef CAS.
- F. L. Taw, H. Mellows, P. S. White, F. J. Hollander, R. G. Bergman, M. Brookhart and D. M. Heinekey, J. Am. Chem. Soc., 2002, 124, 5100–5108 CrossRef CAS PubMed.
- (a) J. Zhang, K. A. Barakat, T. R. Cundari, T. B. Gunnoe, P. D. Boyle, J. L. Petersen and C. S. Day, Inorg. Chem., 2005, 44, 8379–8390 CrossRef CAS PubMed; (b) A. R. Chianese, M. J. Drance, K. H. Jensen, S. P. McCollom, N. Yusufova, S. E. Shaner, D. Y. Shopov and J. A. Tendler, Organometallics, 2014, 33, 457–464 CrossRef CAS; (c) P. Ren, S. D. Pike, I. Pernik, A. S. Weller and M. C. Willis, Organometallics, 2015, 34, 711–723 CrossRef CAS.
- For example see: (a) J. Schaefer, A. Kraft, S. Reininger, G. Santiso-Quinones, D. Himmel, N. Trapp, U. Gellrich, B. Breit and I. Krossing, Chem. – Eur. J., 2013, 19, 12468–12485 CrossRef CAS PubMed; (b) J. Huhmann-Vincent, B. L. Scott and G. J. Kubas, J. Am. Chem. Soc., 1998, 120, 6808–6809 CrossRef CAS.
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley & Sons, Chichester, 2nd edn, 2009 Search PubMed.
- J. Huhmann-Vincent, B. L. Scott and G. J. Kubas, Inorg. Chem., 1999, 38, 115–124 CrossRef CAS.
- M. Findlater, K. M. Schultz, W. H. Bernskoetter, A. Cartwright-Sykes, D. M. Heinekey and M. Brookhart, Inorg. Chem., 2012, 51, 4672–4678 CrossRef CAS PubMed.
- S. Kloek Hanson, D. M. Heinekey and K. I. Goldberg, Organometallics, 2008, 27, 1454–1463 CrossRef.
- M. Feller, E. Ben-Ari, T. Gupta, L. J. W. Shimon, G. Leitus, Y. Diskin-Posner, L. Weiner and D. Milstein, Inorg. Chem., 2007, 46, 10479–10490 CrossRef CAS PubMed.
- D. Hermann, M. Gandelman, H. Rozenberg, L. J. W. Shimon and D. Milstein, Organometallics, 2002, 21, 812–818 CrossRef CAS.
- A. B. Chaplin and A. S. Weller, Organometallics, 2011, 30, 4466–4469 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental, characterisation and X-ray crystallography details. CCDC 1044741, 1044743, 1044744 and 1044745. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00481k |
| ‡ Crystal data: (2) RhP2O2NCl2C22H41·C32H12BF24, Monoclinic (C2/c), a = 16.9996(5) Å, b = 18.1716(4) Å, c = 39.8254(10) Å, α = γ = 90°, β = 96.458(2)°, volume = 12224.4(5) Å3, Z = 8, λ = 0.71073 Å, T = 150(2) K, μ = 0.53 mm−1, 16021 independent reflections [R(int) = 0.029], R1 = 0.0814, wR2 = 0.1692 [I > 2σ(I)]. CCDC: 1044744; (3): RhP2O2N3C21H39·C32H12BF24, Monoclinic (C2/c), a = 16.8578(4) Å, b = 18.1533(3) Å, c = 39.7792(7) Å, α = γ = 90°, β = 95.9972(17)°, volume = 12106.8(4) Å3, Z = 8, λ = 1.54180 Å, T = 150(2) K, μ = 3.83 mm−1, 12215 independent reflections [R(int) = 0.031], R1 = 0.0483, wR2 = 0.1183 [I > 2σ(I)]. CCDC: 1044745; (5/6) RhP2NCl2C24H45·C32H12BF24:RhP2NClC23H44·C32H12BF24, Monoclinic (P21/c), a = 13.8327(2) Å, b = 23.4907(3) Å, c = 20.1051(2) Å, α = γ = 90°, β = 97.5982(11)°, volume = 6475.59(4) Å3, Z = 2, λ = 1.54180 Å, T = 150(2) K, μ = 4.12 mm−1, 12878 independent reflections [R(int) = 0.029], R1 = 0.1064, wR2 = 0.2958 [I > 2σ(I)]. CCDC: 1044741. |
| This journal is © The Royal Society of Chemistry 2015 |