
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSyntheses, structural characterisation and electronic structures of some simple acyclic amino carbene complexes†‡
Samantha G.
Eaves
ab,
Dmitry S.
Yufit
a,
Brian W.
Skelton
c,
Judith A. K.
Howard
a and
Paul J.
Low
*ab
aDepartment of Chemistry, Durham University, South Rd, Durham, DH13LE, UK. E-mail: paul.low@uwa.edu.au
bSchool of Chemistry and Biochemistry, University of Western Australia, 35 Stirling Highway, Crawley, WA 6009, Australia
cCentre for Microscopy, Characterisation and Analysis, University of Western Australia, Crawley 6009, WA, Australia
First published on 30th June 2015
Abstract
Reactions of the vinylidene complexes [M(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CCH2)(PP)Cp′]PF6 with NH3 readily afford the acyclic amino carbene complexes [M{C(Me)NH2}(PP)Cp′]PF6 [M(PP)Cp′ = Fe(dppe)Cp, Ru(dppe)Cp, Ru(PPh3)2Cp, Ru(dppe)Cp*]. Crystallographic, spectroscopic and computational data support a description of these complexes in terms of a M-{C(Me)N+H2} mesomeric form, and simple σ-coordination of the singlet acyclic amino carbene.
CCH2)(PP)Cp′]PF6 with NH3 readily afford the acyclic amino carbene complexes [M{C(Me)NH2}(PP)Cp′]PF6 [M(PP)Cp′ = Fe(dppe)Cp, Ru(dppe)Cp, Ru(PPh3)2Cp, Ru(dppe)Cp*]. Crystallographic, spectroscopic and computational data support a description of these complexes in terms of a M-{C(Me)N+H2} mesomeric form, and simple σ-coordination of the singlet acyclic amino carbene.
Introduction
Heteroatom-stabilised carbene ligands have played an important role in the development of organometallic chemistry. Since the seminal work of Fischer identifying the earliest examples of this class of compound, extensive studies of the synthesis and properties of complexes bearing carbene ligands have seen carbenes rise to such prominence as supporting ligands that they now rival phosphines as the ubiquitous 2-electron σ-donor ligand. Indeed, many of the spectacular advances in the area of metal complexes of carbenes are correlated with the pioneering efforts that have made stable carbenes available as reagents.1Stable (i.e. isolable) carbenes have been known since 1988, when the Bertrand group reported the phosphino(silyl)carbene (iPr2N)2PCSiMe3.2 Since then, phosphino(phosphonio)-,3 phosphino(amino)-,4 acyclic diamino carbenes,5 dicarbene-,6 mono-phosphino-,7 mono-amino-8 and a wide range of cyclic heteroatom-stabilised carbenes, including of course N-heterocyclic carbenes (NHCs),9 have been explored. However, whilst cyclic heteroatom-stabilised carbenes have proven to be ligands of wide applicability,10 isolated acyclic carbenes have found less use in the preparation of metal complexes,11 although it should be noted that these are accessible through modular template synthetic routes, and have given rise to a number of useful catalysts.12 In the case of phosphoranyl carbenes, the relatively poor coordinating ability has been attributed to the wide carbene bond angle associated with the greater stability of the triplet form and the consequent significant reorganisation energy cost of complexation in addition to the higher carbene reactivity.13
Relatively wide carbene angles are also observed in acyclic mono- and di-amino carbenes such as tBuCN(iPr)2 (120.50(12)°)8a and (iPr)2NCN(iPr)2 (121.0(5)°),14 which can be attributed to the steric bulk of the groups necessary to allow isolation of the carbene. These angles contrast the much narrower angles calculated for the smaller parent singlet amino carbene HCNH2 (105.3°) but are closer to the structures of the corresponding triplet (124.1°).13b Whilst metal complexes of these bulky acyclic amino carbenes are known,8,13 the wide carbene angle decreases the σ-donor properties of the carbene, and steric factors can also destabilise the resulting complexes.10 The constrained geometries in NHCs such as the prototypical Arduengo system, 1,3-diadamantyl-imidazol-2-ylidene (102.2(2)°),15 1,3-dimesitylimidazol-2-ylidene (101.4(2)°)16 and the saturated analogue 1,3-dimesitylimidazolin-2-ylidene (104.7(3)°)16 are much closer to the singlet structure of the parent acyclic amino carbene. The propensity of these ligands to form metal complexes is perfectly well established.10
In contrast to the coordination reactions of isolated heteroatom-stabilised carbenes with metal complexes, the original Fischer synthesis of alkyloxy-stabilised carbenes involved nucleophilic attack of an alkyl group on a metal-bound carbonyl ligand to given an anionic acyl fragment, and subsequent alkylation.17 As a consequence of the electrophilic nature of the carbene carbon, these alkyloxy carbenes are readily converted to amino-, thio-, alkyl- or aryl-carbenes providing facile ‘on complex’ synthetic routes to a wide range of complexes featuring heteroatom-stabilised carbene ligands.18 The addition of nucleophiles such as alcohols, thiols or amines to the α-carbon of transition metal vinylidene or allenylidene complexes also affords Fischer-type (heteroatom-stabilised) carbene compounds.19,20
Here we describe the facile addition of ammonia to the α-carbon of the parent vinylidene ligand in complexes [M(CCH2)(PP)Cp′][PF6] (M = Fe, PP = dppe, Cp′ = Cp; M = Ru, PP = dppe, Cp′ = Cp; M = Ru, PP = (PPh3)2, Cp′ = Cp; M = Ru, PP = dppe, Cp′ = Cp*) to give rare examples of complexes bearing the simple, sterically unencumbered methyl(amino) carbene ligand, [M{C(Me)NH2}(PP)Cp′]PF6. These complexes have been spectroscopically and crystallographically characterised, and their geometric and electronic structures, together with those of the free ligand, explored with additional insight from density functional theory based calculations.
Results and discussion
Synthesis and characterisation
The preparation of vinylidene complexes from rearrangement reactions of terminal alkynes within the coordination sphere of metal complexes is very well established.19,21 For example, in the case of half-sandwich complexes MCl(L2)Cp′ (L = phosphine or L2 = chelating bis(phosphine), Cp′ = Cp, Cp* etc.), such reactions with HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR are typically performed in polar solvents such as methanol at reflux and in the presence of a salt, often NH4PF6, to enhance the ionisation of the M–Cl bond and provide a large anion to assist isolation of the product cation [M{CC(H)R}(L2)Cp′]+ (Scheme 1).19,22,23 Whilst the ancillary ligands and the R group provide a degree of steric protection for the electrophilic α-carbon, prolonged reaction leads to the conversion of the vinylidene to the corresponding methoxy carbene [M{C(OMe)CH2R}(L2)Cp′]+.24 In the case of the formation of terminal vinylidene complexes [M(CCH2)(L2)Cp*]+ from MCl(L2)Cp*, HCCSiMe3 and NH4PF6, methanol remains a suitable solvent;25 however, the less sterically restricted Cp derivatives require the use of either a bulkier alkyl alcohol (such as tBuOH)26 or prolonged reaction time in a non-nucleophilic solvent, such as CH2Cl2.27
CR are typically performed in polar solvents such as methanol at reflux and in the presence of a salt, often NH4PF6, to enhance the ionisation of the M–Cl bond and provide a large anion to assist isolation of the product cation [M{CC(H)R}(L2)Cp′]+ (Scheme 1).19,22,23 Whilst the ancillary ligands and the R group provide a degree of steric protection for the electrophilic α-carbon, prolonged reaction leads to the conversion of the vinylidene to the corresponding methoxy carbene [M{C(OMe)CH2R}(L2)Cp′]+.24 In the case of the formation of terminal vinylidene complexes [M(CCH2)(L2)Cp*]+ from MCl(L2)Cp*, HCCSiMe3 and NH4PF6, methanol remains a suitable solvent;25 however, the less sterically restricted Cp derivatives require the use of either a bulkier alkyl alcohol (such as tBuOH)26 or prolonged reaction time in a non-nucleophilic solvent, such as CH2Cl2.27
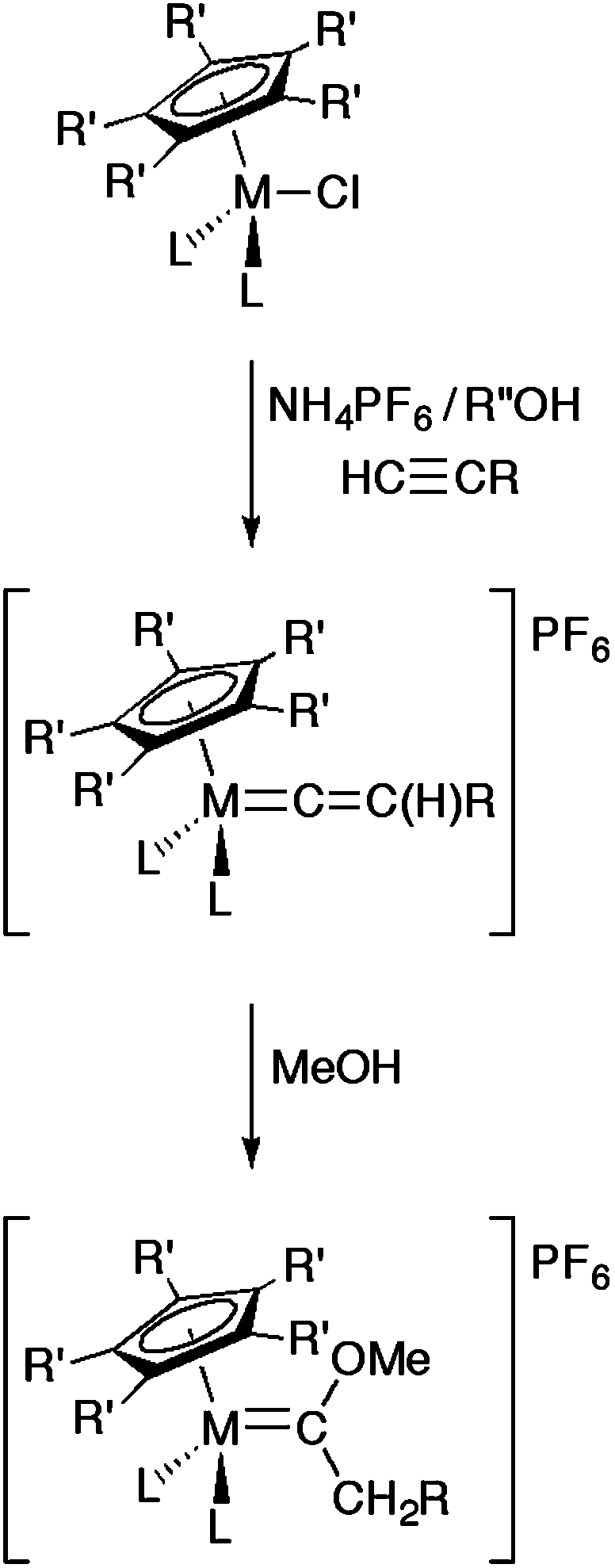 | ||
| Scheme 1 Representative syntheses of vinylidenes and methoxy carbenes from half-sandwich MCl(L2)Cp′ precursors. | ||
Interestingly, the prolonged (15 h) reaction of the η5-indenyl complex RuCl(dppm)(η5-C9H7) with HCCSiMe3 and NH4PF6 in CH2Cl2 at elevated temperatures (90 °C) in a sealed tube gave a methyl(amino) carbene complex, formulated as [Ru{C(Me)NH2}(dppm)(η5-C9H7)]PF6 (Scheme 2).28 To the best of our knowledge, this is the only example of a complex bearing the parent alkyl(amino) carbene fragment, and whilst spectroscopically well characterised,28 no crystallographic data are available through which to examine the effect of coordination on the key N–C–CMe bond angle.
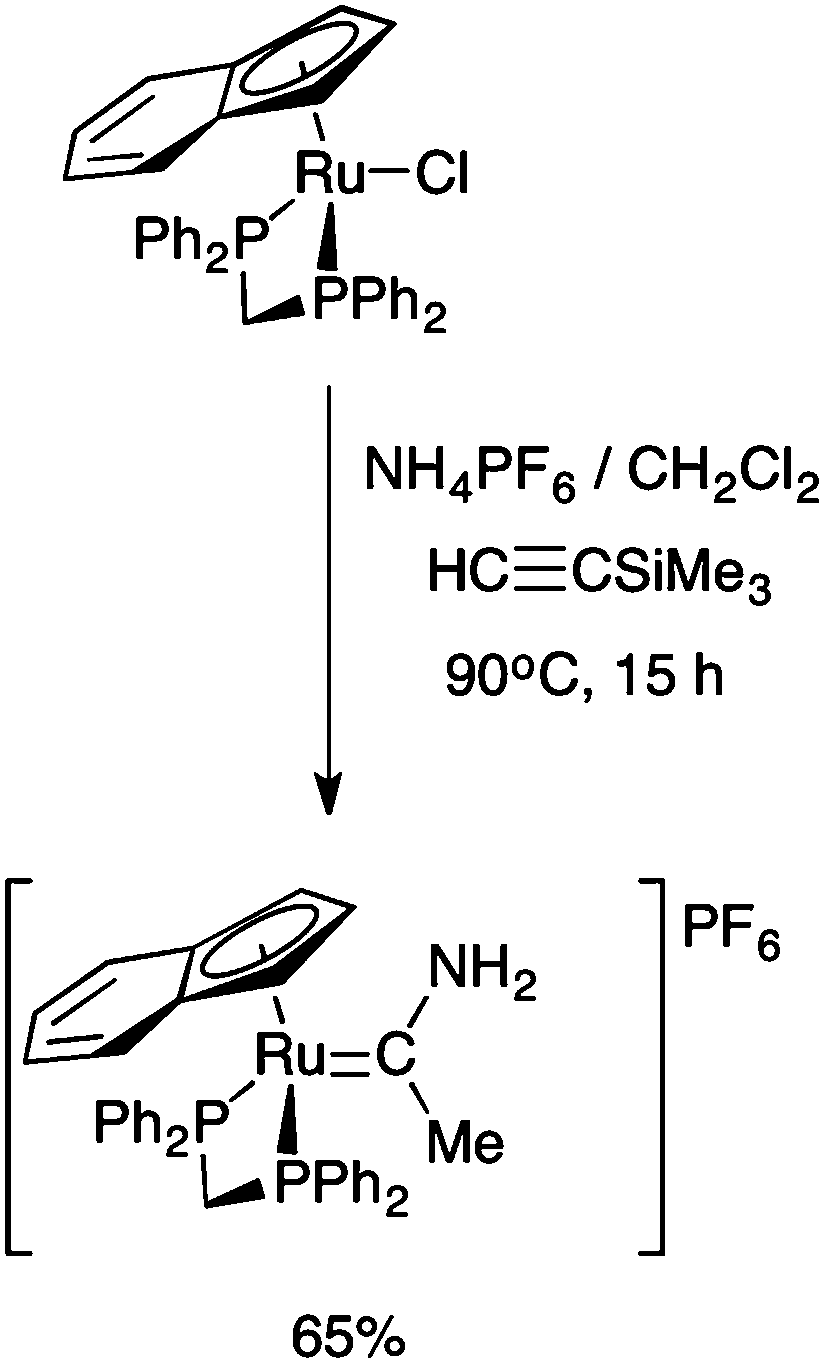 | ||
| Scheme 2 The prolonged reaction of RuCl(dppe)(η5-C9H7) with HCCSiMe3 and NH4PF6.28 | ||
Having observed similar products formed as minor by-products in preparations of vinylidene complexes [M(CCH2)(L2)Cp′]PF6 from MCl(L2)Cp′, HCCSiMe3 and NH4PF6, we pursued a more direct synthesis to complexes containing this simple acyclic amino carbene. The reactions of the half-sandwich complexes [Fe(CCH2)(dppe)Cp]PF6 ([1a]PF6), [Ru(CCH2)(dppe)Cp]PF6 ([1b]PF6), [Ru(CCH2)(PPh3)2Cp]PF6 ([1c]PF6) and [Ru(CCH2)(dppe)Cp*]PF6 ([1d]PF6) with NH3 proceeded smoothly at room temperature in CH2Cl2 to give the amino carbene complexes [Fe{C(Me)NH2}(dppe)Cp]PF6 ([2a]PF6), [Ru{C(Me)NH2}(dppe)Cp]PF6 ([2b]PF6), [Ru{C(Me)NH2}(PPh3)2Cp]PF6 ([2c]PF6) and [Ru{C(Me)NH2}(dppe)Cp*]PF6 ([2d]PF6) in 77–94% isolated yields (Scheme 3). Whilst reactions were complete in 1–4 h for the Cp complexes, in the case of [2d]PF6, overnight reaction was required to complete the conversion, no doubt a consequence of the additional steric protection afforded to the vinylidene α-carbon by the Cp* ligand.
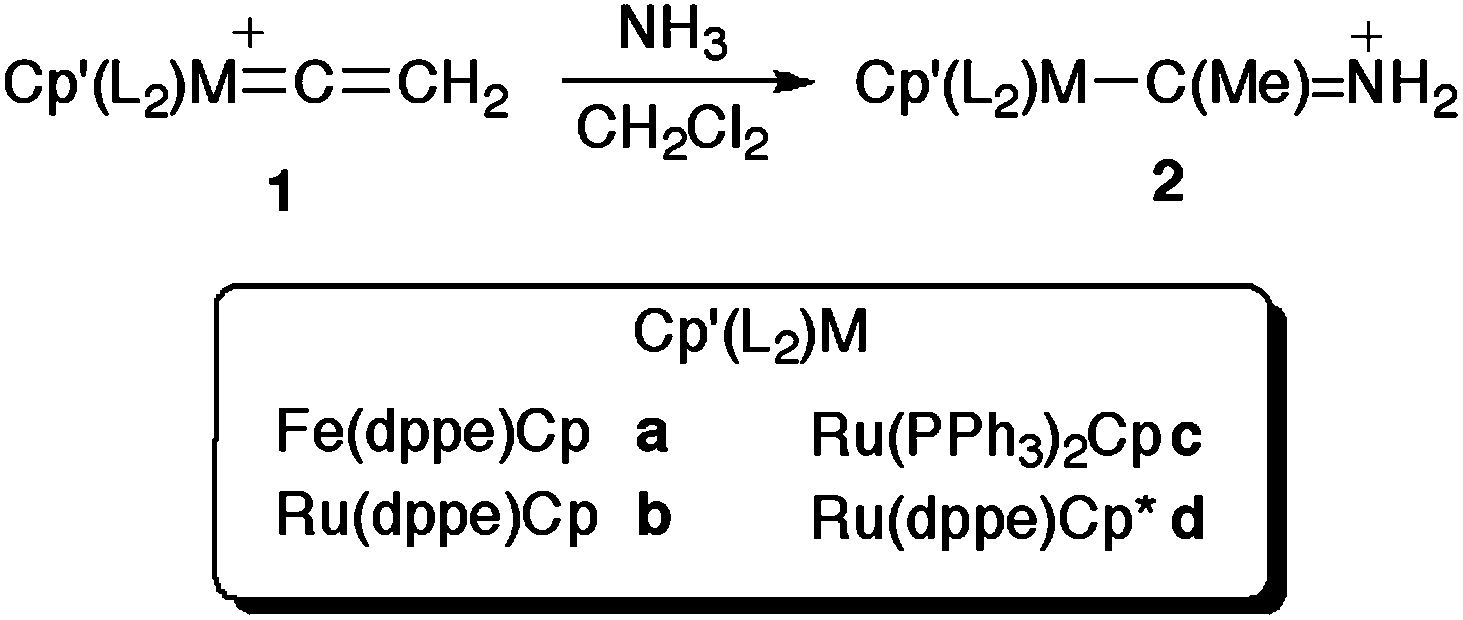 | ||
| Scheme 3 The reaction of [1a–d]+ with NH3 to afford [2a–d]+. | ||
The complexes were readily characterised by the usual spectroscopic methods, and single crystal X-ray diffraction studies. In NMR spectra (Table 1), triplets (unresolved in the case of [2c]+) in the 13C{1H} NMR spectra between 253–275 ppm confirmed the carbene-nature of the α-carbon, whilst two unresolved low-field doublets in the 1H NMR spectra, each integrating to a single proton, indicated the restricted rotation about the C(α)–N bond, supporting a CN(+)H2 valence description. The iminium description was further enhanced by the observation of the Cp resonances for [2a–c]PF6 between 4.37–4.80 ppm (cf. FeCl(dppe)Cp 4.76; RuCl(dppe)Cp 4.55;29 RuCl(PPh3)2Cp 5.99;23 [1a]PF6 5.25;26 [1b]PF6 5.37;26 [1c]PF6 5.0927 ppm). A singlet between 1.60–2.90 ppm (integrating to 3H) was assigned to the methyl group of the carbene ligand. In the positive ion electrospray (ES(+)) mass spectra, ions corresponding to the cation were clearly observed. In the IR spectra, bands in the range 1600–1650 cm−1 were assigned to δ(NH2) with the assistance of DFT based molecular models (vide infra).
Molecular structures
Single crystal X-ray diffraction studies were carried out on [2a–d]PF6 to explore the amino carbene ligand geometry and metal–carbene bond lengths. A representative plot of the cation of [2a]+ is shown in Fig. 1, and selected bond lengths and angles are summarised in Table 2. The Ru–C(1) bond lengths fall at the longer end of the range spanned by the crystallographically characterised examples of methyl(methoxy) carbene complexes: [Ru{C(OMe)Me}Ln]+ (RuLn = RuCl{P(CHCH2)Ph2}(η6-C6H2Me4) 1.963(7) Å;30 Ru{P(CHCH2)2Ph}2(η5-C5H4Me) 1.921(10) Å;31 RuCl{P(CHCH2)Ph2}(η6-C6Me6) 1.964(10) Å; RuCl(PPh3)(η6-C6Me6) 1.962(10) Å and RuCl(PMe3)(η6-C6Me6) 2.015(8) Å.32 The M–C(1), C(1)–N(1) and C(1)–C(2) distances are consistent with a predominant description in terms of a strongly N-stabilised carbene with a significant degree of CNH2 double bond character and a M–C single bond. There is little change in the carbene angles across the series [2a–d]+, suggesting π-interaction between the metal and carbene is not significant. The relatively precisely determined M–P bond lengths are sensitive to M–P back-bonding effects, and can be used as a proxy measure of the relative electron density at the metal centre.33 Here, the Fe–P and Ru–P bond distances in [2a]+, [2b]+, [2c]+ and [2d]+ compare with those of the vinylidene cations [Fe(CCH2)(dppe)Cp*]+ (2.2219(15), 2.2185(16) Å),34 [Ru(CCH2)(PPh3)2Cp*]+ (2.362(2), 2.355(2) Å),35 [Ru(CCH2)(dppe)Cp*]+ ([2d]+ 2.320(3), 2.317(3); 2.318(3), 2.308(3) Å)25 and the methyl(methoxy) carbene complex [Ru{C(OMe)CH2CO2Me}(dppe)Cp]+ (2.2890(9), 2.3043(10) Å).36 The shorter M–P bond lengths in the amino carbene complexes indicates more electron-density at the metal than in the vinylidenes, which is consistent with the poorer π-accepting character of the Fischer-type carbene.
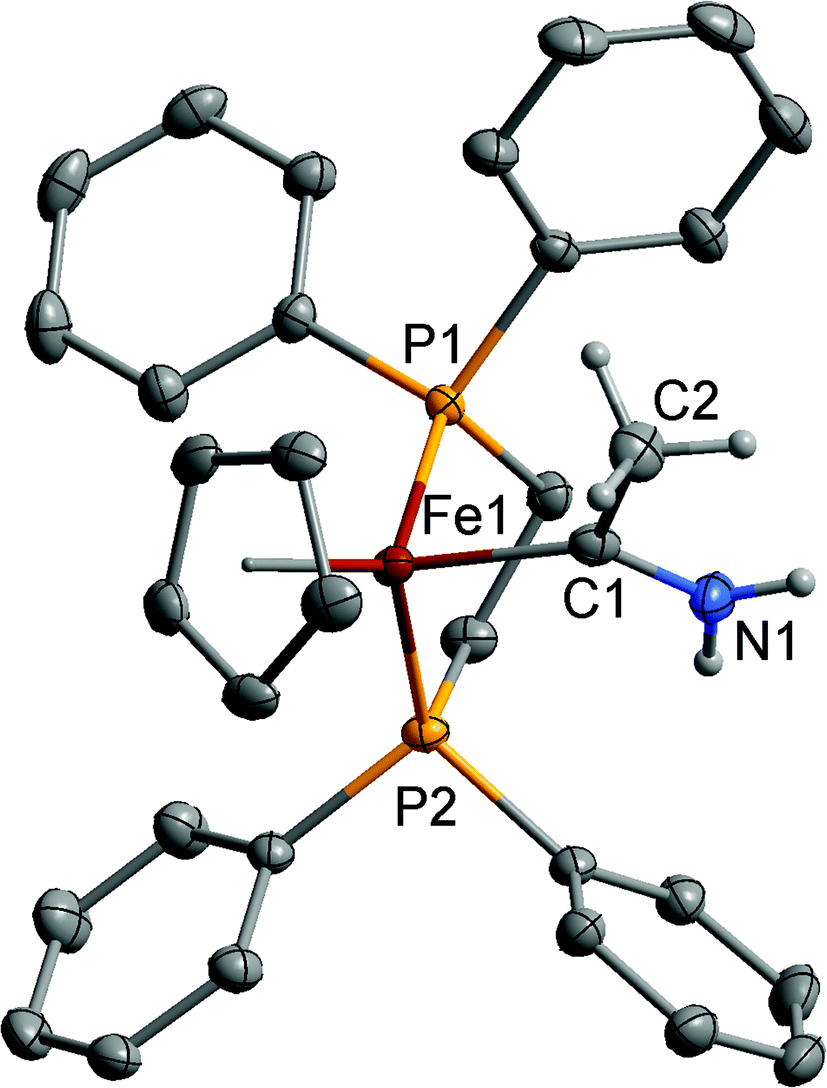 | ||
| Fig. 1 Structure of one of the cations of [2a]+ with selected hydrogen atoms omitted for clarity. Ellipsoids have been drawn at the 50% probability level. | ||
N(iPr)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8a and the DFT (B3LYP/LANL2DZ (Ru, Fe), 6-31G**/COSMO (CH2Cl2)) optimised geometries of [2a]+ and [2b]+ and gas phase (B3LYP/6-31G**) geometries of the free singlet carbenes MeCNH2 and tBuCN(iPr)2 (italics)
8a and the DFT (B3LYP/LANL2DZ (Ru, Fe), 6-31G**/COSMO (CH2Cl2)) optimised geometries of [2a]+ and [2b]+ and gas phase (B3LYP/6-31G**) geometries of the free singlet carbenes MeCNH2 and tBuCN(iPr)2 (italics)
| MeCNH2 |
t
BuCN(iPr)2 |
[2a]PF6 | [2b]PF6 | [2c]PF6 | [2d]PF6 | |
|---|---|---|---|---|---|---|
| a Methyl group C(2) disordered over two sites. | ||||||
| M–P1 | 2.1756(4), 2.1829(4) 2.1860 | 2.2657(7), 2.2733(7) 2.3740 | 2.326(1) | 2.2910(7) | ||
| M–P2 | 2.2032(4), 2.1860(4) 2.1829 | 2.2566(7), 2.2704(7) 2.3356 | 2.328(1) | 2.2819(7) | ||
| M–C1 | 1.9272(15), 1.9251(15) 1.9250 | 2.020(3), 2.021(3) 2.0265 | 2.014(4) | 2.016(3) | ||
| C1–N1 | 1.3266 | 1.2976(17) 1.3214 | 1.312(2), 1.310(2) 1.3099 | 1.305(4), 1.307(4) 1.3209 | 1.309(5) | 1.368(4) |
| C1–C2 | 1.5071 | 1.5392(18) 1.5452 | 1.518(2), 1.519(2) 1.5186 | 1.518(4), 1.515(4) 1.5157 | 1.506(6) | 1.454(6), 1.477(5)a |
| N1–C1–C2 | 112.30 | 120.50(12) 122.62 | 109.4(1), 109.5(1) 109.51 | 110.0(3), 110.4(2) 111.84 | 111.9(4) | 109.5(4), 108.7(3)a |
Density functional modelling
To explore the electronic structure of these complexes we turned to DFT calculations (B3LYP/LANL2DZ Ru, Fe; 6-31G** all other atoms/COSMO(CH2Cl2)) with gas-phase calculations also performed on the free ligand (B3LYP/6-31G**) for comparison. The free carbene MeCNH2 has a singlet ground state (ΔES–T = 33.8 kcal mol−1), with an appreciably bent (112.30°) structure, which compares with the corresponding triplet (127.59°). Interestingly, the bulkier analogue tBuCN(iPr)2, for which a singlet ground state is also calculated (ΔES–T = 35.8 kcal mol−1), demonstrates a significant additional steric influence on the ground state structures in both the singlet (122.62°) and triplet (137.40°) configurations.
The compounds [2a]+ and [2b]+ were chosen as representative examples of the metal complexes of methyl(amino) carbene, allowing exploration of any influence that the 3d vs. 4d metal might play on the electronic structure of the complexes. Key bond lengths and angles from the optimised structures are summarised in Table 2, and the composition of selected frontier orbitals are summarised in Table 3, with supporting plots given in Fig. 2. There is little change between the calculated geometry of the gas-phase carbene MeCNH2 and the amino carbene ligand in the complexes. The electronic structures of [2a]+ and [2b]+ are remarkably similar, with the HOMO−2 to LUMO+1 being well described as the metal dz2, dx2−y2, dxy, dyz and dxz admixed with the NC π* system. There is no π-type interaction between the metal and the carbene carbon, the NC π bonding combination lying as low as the HOMO−21 ([2a]+) and HOMO−20 ([2b]+). The amino carbene moiety therefore serves as a simple σ-donor ligand to the metal centre, with the singlet carbene being stabilised exclusively by π-donation from the N-atom.
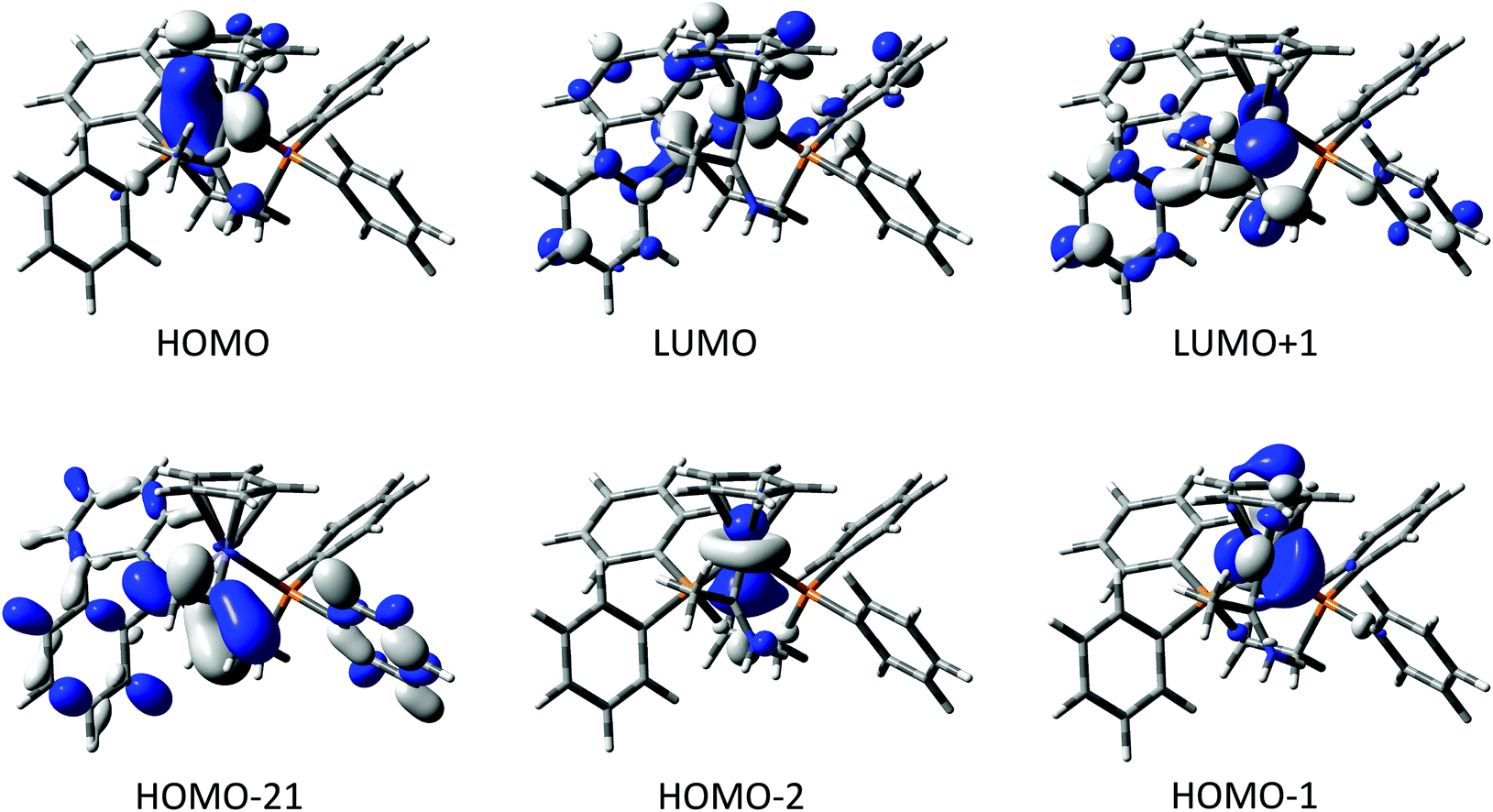 | ||
| Fig. 2 Selected frontier orbitals of [2a]+ (contour ± 0.04 (e bohr−3)1/2). | ||
| Energy | Fe | C(1) | NH2 | Me | dppe | Cp | |
|---|---|---|---|---|---|---|---|
| LUMO+1 | −1.29 | 8 | 27 | 10 | 3 | 51 | 1 |
| LUMO | −1.54 | 37 | 1 | 0 | 0 | 50 | 13 |
| HOMO | −6.10 | 68 | 2 | 3 | 1 | 9 | 16 |
| HOMO−1 | −6.21 | 72 | 2 | 0 | 0 | 9 | 16 |
| HOMO−-2 | −6.90 | 84 | 0 | 3 | 0 | 8 | 4 |
| Energy | Ru | C(1) | NH2 | Me | dppe | Cp | |
|---|---|---|---|---|---|---|---|
| LUMO+1 | −1.28 | 14 | 27 | 8 | 3 | 40 | 7 |
| LUMO | −1.55 | 28 | 0 | 0 | 0 | 56 | 16 |
| HOMO | −5.97 | 60 | 2 | 2 | 2 | 16 | 19 |
| HOMO−1 | −6.14 | 56 | 4 | 1 | 0 | 12 | 26 |
| HOMO−2 | −6.73 | 78 | 1 | 5 | 0 | 11 | 5 |
Conclusion
Reactions of the terminal vinylidene complexes [M(CCH2)(PP)Cp′]PF6 with NH3 provide a convenient entry point to complexes bearing the simple acyclic amino carbene ligand. Structural and electronic structure calculations indicate that the ligand acts as a simple singlet carbene, with good σ-donor character but little M(d)–C(p) π-interaction. Gas-phase density functional calculations on the free ligand give a singlet–triplet energy gap of ΔES–T = 33.8 kcal mol−1, with an appreciably bent (112.30°) structure. The related, more sterically encumbered acyclic amino carbene tBuCN(iPr)2 also offers a large singlet–triplet energy gap (ΔES–T = 35.8 kcal mol−1), and a more linear structure (N–C–C 122.62°) demonstrating the steric influence on the ground state structure of the singlet. The difficulties in formation and stability of metal complexes of mono(amino) carbenes therefore seems to be due in no small part to the significant steric bulk of the supporting groups necessary to stabilise the singlet carbene. The formation of these ligands through ‘on-complex’ synthetic methods therefore appears a more suitable entry point for further explorations.
Experimental
General conditions
All reactions were carried out under an atmosphere of dry nitrogen using standard Schlenk techniques. Dichloromethane was dried over CaH2, all other solvents were standard reagent grade and used as received. No special precautions were taken to exclude air or moisture during workup. The compounds [Fe(CCH2)(dppe))Cp]PF6 ([1a]PF6),26 [Ru(CCH2)(dppe)Cp]PF6 ([1b]PF6),26 [Ru(CCH2)(PPh3)2Cp]PF6 ([1c]PF6)27 and [Ru(CCH2)(dppe)Cp*]PF6 ([1d]PF6)25 were synthesised by literature methods. All other reagents were commercially available and used as received. NMR spectra were recorded in CDCl3 solutions at 23 °C on Bruker and Varian Mercury-400 (1H, 399.97 MHz; 31P, 161.10 MHz), Bruker Avance 600 (1H, 600.1 MHz; 13C, 150.9 MHz; 31P, 242.9 MHz) or Varian VNMRS-700 (1H, 699.73 MHz, 13C, 175.95 MHz; 31P, 279.89 MHz) spectrometers. Chemical shifts were determined relative to internal residual solvent signals (1H, 7.26 ppm; 13C, 77.2 ppm) or external H3PO4 (31P, 0.0 ppm). FT-IR spectra were measured on an Agilent Technologies Cary 660 spectrometer or a Nicolet Avatar 360 spectrometer from solutions in dichloromethane in a thin layer cell fitted with CaF2 windows. Positive and negative ion electrospray ionization (ES(+), ES(−)) mass spectra were recorded on a Waters LCT Premier XE mass spectrometer from solutions in methanol.
Synthesis of [Fe{C(Me)NH2}(dppe)Cp]PF6 [2a]PF6
The compound [Fe(CCH2)(dppe)Cp]PF6 (0.10 g, 0.14 mmol) was added to dry degassed CH2Cl2 (15 ml) in a dry, degassed Schlenk flask connected to a gas bubbler. To a separate dried, degassed flask, fitted with a Dreschel head connected to a N2 line, was added 35% NH3(aq) (25 ml). The output of the Dreschel head was connected by a gas-tight transfer tube in the first flask, fitted below the CH2Cl2 solvent level. NH3(g) was then generated by bubbling N2 gas through the NH3(aq) solution, which was then subsequently bubbled through the vinylidene solution for 4 h. The orange solution colour lightened over time. After this period, the 35% NH3(l) flask is removed and reaction flask flushed with N2 for at least 10 minutes to remove any excess NH3(g). The orange solution was concentrated to dryness by rotary evaporation leaving an orange residue. The orange residue was extracted with CH2Cl2 and filtered into vigorously stirred hexane, resulting in the instantaneous precipitation of an orange solid. The solid was collected by filtration, washed with hexane (3 × 10 ml) and diethyl ether (3 × 10 ml) then dried under vacuum (0.079 g, 77%). Crystals suitable for single crystal X-ray diffraction were grown from CDCl3/pentane layer diffusion.
IR (CH2Cl2/cm−1): 1651 δ(N–H2). 1H NMR (CDCl3, 700 MHz) δ/ppm: 1.75 (s, 3H, CH3), 2.39–2.57 (m, 2H, CH2, dppe), 2.75–2.93 (m, 2H, CH2, dppe), 4.37 (s, 5H, Cp), 7.17 (t, J = 7 Hz, 4H, Hm, dppe), 7.30 (s, br., 1H, NH2), 7.38 (t, J = 7 Hz, 4H, Hm, dppe), 7.44 (t, J = 7 Hz, 2H, Hp, dppe), 7.46–7.52 (m, 6H, Hp and Ho, dppe), 7.53–7.60 (m, 4H, Ho, dppe), 8.17 (s, br., 1H, NH2). 31P{1H} NMR (CDCl3, 400 MHz) δ/ppm: 106.9 (s, Fe-dppe). 13C{1H} NMR (CDCl3, 700 MHz) δ/ppm: 26.8 (t, J = 23 Hz, CH2, dppe), 43.2 (s, CH3), 83.1 (s, Cp), 128.8 (t, J = 4 Hz, Cm, dppe), 129.5 (t, J = 4 Hz, Co, dppe), 130.4 (s, Cp, dppe), 131.3 (s, Cp, dppe), 131.8 (t, J = 4 Hz, Cm, dppe), 132.5 (t, J = 4 Hz, Co, dppe), 133.4–133.7 (m, Ci, dppe), 139.2–139.6 (m, Ci, dppe), 274.1 (t, J = 22 Hz, Cα). ES (+)-MS (m/z): 562 [M − PF6]+. Calculated for C33H34NF6P3Fe: C, 56.00; H, 4.85. Found: C, 56.12; H, 4.91.
Synthesis of [Ru{C(Me)NH2}(dppe)Cp]PF6 [2b]PF6
In a manner as described above, NH3(g) was bubbled through a solution of [Ru(CCH2)(dppe)Cp]PF6 (0.061 g, 0.082 mmol) in dry degassed CH2Cl2 (6 ml) for 4 h. The yellow solution colour lightened over time. Work up as described above gave the product as a yellow powder (0.049 g, 79%). Crystals suitable for single crystal X-ray diffraction were grown from CH2Cl2/hexane layer diffusion.
IR (CH2Cl2/cm−1): 1605 δ(N–H2). 1H NMR (CDCl3, 400 MHz) δ/ppm: 1.77 (s, 3H, CH3), 2.40–2.58 (m, 2H, dppe), 2.77–3.00 (m, 2H, dppe), 4.80 (s, 5H, Cp), 7.06 (s, br., 1H, NH2), 7.15 (t, J = 8 Hz, 4H, Hm, dppe), 7.37 (t, J = 8 Hz, 4H, Hm, dppe), 7.38 (t, J = 8 Hz, 2H, Hp, dppe), 7.46–7.51 (m, 6H, Ho and Hp, dppe), 7.59 (t, J = 8 Hz, 4H, Ho, dppe), 7.89 (s, br., 1H, NH2). 31P{1H} NMR (CDCl3, 400 MHz) δ/ppm: 87.9 (s, Ru-dppe). 13C{1H} NMR (CDCl3, 700 MHz) δ/ppm: 43.9 (s, CH3), 27.0 (t, J = 23 Hz, CH2, dppe), 87.2 (s, Cp), 128.7 (t, J = 5 Hz, Cm, dppe), 129.4 (t, J = 5 Hz, Co, dppe), 130.3 (s, Cp, dppe), 131.1 (t, J = 5 Hz, Cm, dppe), 131.3 (s, Cp, dppe), 132.3–132.5 (m, Ci, dppe), 132.7 (t, J = 5 Hz, Co, dppe), 139.7–140.1 (m, Ci), 256.6 (t, J = 13 Hz, Cα). ES (+)-MS (m/z): 608 [M − PF6]+. ES (−)-MS (m/z): 145 [PF6]−. Calculated for C33H34NF6P3Ru: C, 52.58; H, 4.55. Found: C, 52.54; H, 4.64.
Synthesis of [Ru{C(Me)NH2}(PPh3)2Cp]PF6 [2c]PF6
In a manner similar to that described above, NH3(g) was bubbled through a solution of [Ru(CCH2)(PPh3)2Cp]PF6 (0.10 g, 0.12 mmol) in dry degassed CH2Cl2 (6 ml) for 1 h. The orange solution colour lightened over time. Work-up as described above gave the product as an orange powder (0.095 g, 94%). Crystals suitable for single crystal X-ray diffraction were grown from CH2Cl2/ethyl acetate layer diffusion.
IR (CH2Cl2/cm−1): 1604 δ(N–H2). 1H NMR (CDCl3, 600 MHz) δ/ppm: 2.90 (s, 3H, CH3), 4.53 (s, 5H, Cp), 6.96 (s, br. 1H, NH2), 7.05 (t, J = 8 Hz, 12H, Ho, PPh3), 7.30 (t, J = 8 Hz, 12H, Hm, PPh3), 7.43 (t, J = 8 Hz, 6H, Hp, PPh3), 8.40 (s, br., 1H, NH2). 31P NMR (CDCl3, 400 MHz) δ/ppm: 48.2 (s, Ru-(PPh3)2). 13C NMR (CDCl3, 600 MHz) δ/ppm: 44.3 (s, CH3), 89.1 (s, Cp), 128.7 (t, J = 5 Hz, Cm, PPh3), 130.5 (s, Cp, PPh3), 133.6 (t, J = 5 Hz, Co, PPh3), 135.9–136.3 (m, Ci, PPh3), 253.4 (m, Cα). ES (+)-MS (m/z): 734 [M − PF6]+, 472 [M − PPh3 – PF6]+. ES (−)-MS (m/z): 145 [PF6]−. Calculated for C43H40NF6P3Ru: C, 58.65; H, 4.61. Found: C, 58.65; H, 4.61.
Synthesis of [Ru{![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) C(Me)NH2}(dppe)Cp*]PF6 [2d]PF6
C(Me)NH2}(dppe)Cp*]PF6 [2d]PF6
In a manner as described above, NH3(g) was bubbled through a solution of [Ru(CCH2)(dppe)Cp*]PF6 (0.11 g, 0.14 mmol) in dry degassed CH2Cl2 (15 ml) for 24 h. The solution colour changed from yellow to pale green gradually over time. Work up as described above gave the product as a pale green powder (0.090 g, 78%). Crystals suitable for single crystal X-ray diffraction were grown from CDCl3/pentane layer diffusion.
IR (CH2Cl2/cm−1): 1639 δ(N–H2). 1H NMR (CDCl3, 700 MHz) δ/ppm: 1.49 (s, 15H, Cp*), 1.60 (s, 3H, CH3), 2.20–2.40 (m, 2H, CH2, dppe), 2.73–2.91 (m, 2H, CH2, dppe), 7.07 (t, J = 8 Hz, 4H, Hm, dppe), 7.18 (s, br., 1H, NH2), 7.36 (t, J = 8 Hz, 4H, Hm, dppe), 7.43 (t, J = 8 Hz, 4H, Hp, dppe), 7.45–7.57 (m, 8H, Ho, dppe), 8.44 (s, br, 1H, NH2). 31P{1H} NMR (CDCl3, 400 MHz) δ/ppm: 86.6 (s, Ru-dppe). 13C{1H} NMR (CDCl3) δ/ppm: 10.5 (s, CH3, Cp*), 28.1 (t, J = 23 Hz, CH2, dppe), 41.8 (s, CH3), 96.9 (s, Cp*), 128.2 (t, J = 5 Hz, Cm, dppe), 129.4 (t, J = 5 Hz, Co, dppe), 130.5 (s, Cp, dppe), 131.3 (s, Cp, dppe), 132.6 (t, J = 5 Hz, Co, dppe), 132.9 (t, J = 5 Hz, Cm, dppe), 259.6 (t, J = 13 Hz, Cα). ES (+)-MS (m/z): 678 [M − PF6]+, 677 [M − PF6 − H]+. ES (−)-MS (m/z): 145 [PF6]−. Calculated for C38H44NF6P3Ru.CHCl3: C, 49.72; H, 4.81. Found: C, 50.73; H, 5.14. Compound [2d]PF6 crystallises as a bis(chloroform) solvate. The analysis suggests one molecule is tenaciously retained.
Crystallography
Diffraction data were collected at 120(2) K (100(2) K for [2b]PF6) on a Bruker SMART CCD 6000 diffractometer ω-scan, 0.3°/frame) or on an Oxford Diffraction Xcalibur diffractometer ([2b]PF6) (sealed tubes, graphite monochromators, λMo-Kα, λ = 0.71073 Å). Following absorption corrections and solution by direct methods, the structures were refined against F2 with full matrix least-squares using the program SHELXL37 and OLEX2.38 Non-hydrogen atoms were refined with anisotropic displacement parameters with hydrogen atoms added at calculated positions and refined by use of a riding model with isotropic displacement parameters based on those of the parent atoms. Disordered atoms in the structures [2c]PF6 and [2b]PF6 were refined in isotropic approximation with fixed site occupation factors.Crystal data
779, unique = 18220, Rint = 0.0318. Data/restraints/parameters = 18220/0/811, GooF = 1.036. Final R indices: R1 = 0.0302 (14367 > 2σ(I)), wR2 = 0.0803 (all data). Δρmax,min = 0.578, −0.302 e Å−3. CCDC 1063656.
852, unique = 14611, Rint = 0.0420. Data/restraints/parameters = 14611/0/809. GooF = 1.043. Final R indices, R1 = 0.0389 (12339 > 2σ(I)), wR2 = 0.0971 (all data). Δρmax,min = 3.132, −0.869 e Å−3. CCDC 1063659.
442(3) Å3, Z = 12, μ = 0.600 mm−1, Dcalc = 1.530 Mg m−3, 2θmax = 58°. Reflections collected = 158766, unique = 30397, Rint = 0.0800. Data/restraints/parameters = 30397/66/1455. GooF = 1.030. Final R indices, R1 = 0.0528 (25573 > 2σ(I)), wR2 = 0.1265 (all data). Δρmax,min = 1.666, −1.266 e Å−3. CCDC 1063657.
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 10.9534(6), b = 13.4096(8), c = 15.8099(9) Å, α = 83.8710(10), β = 76.3000(10), γ = 81.0140(10)°, V = 2222.5(2) Å3, Z = 2, μ = 0.878 mm−1, Dcalc = 1.586 Mg m−3, 2θmax = 58°. Reflections collected = 43327, unique = 11826, Rint = 0.0228. Data/restraints/parameters = 11826/44/520. GooF = 1.077. Final R indices, R1 = 0.0526 (10787 > 2σ(I)) and wR2 = 0.1356 (all data). Δρmax,min = 2.371, −1.956 e Å−3. CCDC 1063658.
, a = 10.9534(6), b = 13.4096(8), c = 15.8099(9) Å, α = 83.8710(10), β = 76.3000(10), γ = 81.0140(10)°, V = 2222.5(2) Å3, Z = 2, μ = 0.878 mm−1, Dcalc = 1.586 Mg m−3, 2θmax = 58°. Reflections collected = 43327, unique = 11826, Rint = 0.0228. Data/restraints/parameters = 11826/44/520. GooF = 1.077. Final R indices, R1 = 0.0526 (10787 > 2σ(I)) and wR2 = 0.1356 (all data). Δρmax,min = 2.371, −1.956 e Å−3. CCDC 1063658.
Computational
All the calculations were performed with the Gaussian 09 program package,39 using the B3LYP functional,40 LANL2DZ basis set for Ru or Fe, and 6-31G** for all other atoms.41 A conductor-like polarization continuum model CPCM of CH2Cl2 solvent was applied to all calculations, and results analyzed further with GaussSum.42 Structures obtained were confirmed as true minima by the absence of imaginary frequencies.Acknowledgements
S.G.E. gratefully acknowledges funding from the University of Durham in the form of a Durham Doctoral Scholarship, and the University of Western Australia. P.J.L. holds an Australian Research Council Future Fellowship [FT120100073]. The authors acknowledge the facilities, scientific and technical assistance of the Australian Microscopy & Microanalysis Research Facility at the Centre for Microscopy, Characterisation & Analysis, The University of Western Australia, a facility funded by the University, State and Commonwealth Governments.References
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef CAS PubMed.
- A. Igau, H. Grützmacher, A. Baceiredo and G. Bertrand, J. Am. Chem. Soc., 1988, 110, 6463 CrossRef CAS.
- M. Soleilhavoup, A. Caceiredo, O. Treutler, R. Ahlrichs, M. Nieger and G. Bertrand, J. Am. Chem. Soc., 1992, 114, 10959 CrossRef CAS.
- (a) N. Merceron, K. Miqueu, A. Baceiredo and G. Bertrand, J. Am. Chem. Soc., 2002, 124, 6806 CrossRef CAS PubMed; (b) N. Merceron-Saffon, A. Baceiredo, H. Gornitzka and G. Bertrand, Science, 2003, 301, 1223 CrossRef CAS PubMed.
- L. M. Slaughter, Comments Inorg. Chem., 2008, 29, 46 CrossRef CAS PubMed.
- (a) C. A. Dyker, V. Lavallo, C. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2008, 47, 3206 CrossRef CAS PubMed; (b) S. Klein, R. Tonner and G. Frenking, Angew. Chem., Int. Ed., 2010, 16, 10160 CAS.
- (a) E. Despagnet, H. Gornitzka, A. B. Rozhenko, W. W. Schoeller, D. Bourissou and G. Bertrand, Angew. Chem., Int. Ed., 2002, 41, 2835 CrossRef CAS; (b) E. Despagnet-Ayoub, S. Sole, H. Gortnitzka, A. B. Rozhenko, W. W. Schoeller, D. Bourissou and G. Bertrand, J. Am. Chem. Soc., 2003, 125, 124 CrossRef CAS PubMed.
- (a) V. Lavallo, J. Mafhouz, Y. Canac, B. Donnadieu, W. W. Schoeller and G. Bertrand, J. Am. Chem. Soc., 2004, 126, 8670 CrossRef CAS PubMed; (b) X. Cattoën, H. Gornitzka, D. Bourissou and G. Bertrand, J. Am. Chem. Soc., 2004, 126, 1342 CrossRef PubMed.
- (a) C. M. Crudden and D. P. Allen, Coord. Chem. Rev., 2004, 248, 2247 CrossRef CAS PubMed; (b) O. Schuster, L. R. Yang, H. G. Raubenheimer and M. Albrecht, Chem. Rev., 2009, 109, 3445 CrossRef CAS PubMed; (c) R. H. Crabtree, J. Organomet. Chem., 2005, 690, 5451 CrossRef CAS PubMed; (d) P. L. Arnold and S. Pearson, Coord. Chem. Rev., 2007, 251, 596 CrossRef CAS PubMed.
- (a) D. S. Weinberger, M. Melaimi, C. E. Moore, A. L. Rheingold, G. Frenking, P. Jerabek and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 8964 CrossRef CAS PubMed; (b) D. Martin, M. Melaimi, M. Soleilhavoup and G. Bertrand, Organometallics, 2011, 30, 5304 CrossRef CAS PubMed; (c) M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810 CrossRef CAS PubMed; (d) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122 CrossRef CAS PubMed.
- (a) J. Vignolle, X. Cattoen and D. Bourissou, Chem. Rev., 2009, 109, 3333 CrossRef CAS PubMed; (b) M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810 CrossRef CAS PubMed.
- (a) A. S. K. Hashmi, T. Hengst, C. Lothschütz and F. Rominger, Adv. Synth. Catal., 2010, 352, 1315 CrossRef CAS PubMed; (b) A. S. K. Hashmi, T. Häffner, M. Rudolph and F. Rominger, Eur. J. Org. Chem., 2011, 667 CrossRef CAS PubMed; (c) M. C. Blanco Jaimes, C. R. N. Bohling, J. M. Serrano-Becerra and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2013, 52, 7963 CrossRef CAS PubMed.
- (a) W. W. Schoeller, D. Eisner, S. Grigoleit, A. B. Rozhenko and A. Alijah, J. Am. Chem. Soc., 2000, 122, 10115 CrossRef CAS; (b) W. W. Schoeller, A. R. Rozhenko and A. Alijah, J. Organomet. Chem., 2001, 617–618, 435 CrossRef CAS; (c) W. A. Herrmann, K. Ofele, D. von Preysing and E. Herdtweck, J. Organomet. Chem., 2003, 684, 235 CrossRef CAS; (d) G. D. Frey, E. Herdtwerk and W. A. Herrmann, J. Organomet. Chem., 2006, 691, 2465 CrossRef CAS PubMed.
- R. W. Alder, P. R. Allen, M. Murray and A. G. Orpen, Angew. Chem., Int. Ed. Engl., 1996, 35, 1121 CrossRef CAS PubMed.
- A. J. Arduengo III, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361 CrossRef.
- A. J. Arduengo III, J. R. Goerlich and W. J. Marshall, J. Am. Chem. Soc., 1995, 117, 11027 CrossRef.
- E. O. Fischer and A. Maasböl, Angew. Chem., Int. Ed. Engl., 1964, 3, 580 CrossRef PubMed.
- (a) M. A. Schwindt, J. R. Miller and L. S. Hegedus, J. Organomet. Chem., 1991, 413, 143 CrossRef CAS; (b) J. M. Moretó, S. Ricart, K. H. Dötz and E. Molins, Organometallics, 2000, 20, 62 CrossRef; (c) R. Aumann and J. Schröder, Chem. Ber., 1990, 123, 2053 CrossRef CAS PubMed; (d) C. F. Bernasconi, K. W. Kittredge and F. X. Flores, J. Am. Chem. Soc., 1999, 121, 6630 CrossRef CAS; (e) G. Seidel, B. Gabor, R. Goddard, B. Heggen, W. Thiel and A. Fuerstner, Angew. Chem., Int. Ed., 2014, 53, 879 CrossRef CAS PubMed; (f) M. J. Winter and S. Woodward, J. Chem. Soc., Chem. Commun., 1989, 457 RSC; (g) C. F. Bernasconi and M. Ali, Organometallics, 2004, 23, 6134 CrossRef CAS.
- M. I. Bruce, Chem. Rev., 1991, 91, 197 CrossRef CAS.
- (a) H. Kopf, B. Holzberger, C. Pietraszuk, E. Hübner and N. Burzlaff, Organometallics, 2008, 27, 5894 CrossRef CAS; (b) C. Bianchini, D. Masi, A. Romerosa, F. Zanobini and M. Peruzzini, Organometallics, 1999, 18, 2376 CrossRef CAS; (c) H. Fischer, G. Roth, D. Reindl and C. Troll, J. Organomet. Chem., 1993, 454, 133 CrossRef CAS; (d) M. Serrano-Ruiz, C. Lidrissi, S. Manas, M. Peruzzini and A. Romerosa, J. Organomet. Chem., 2014, 751, 654 CrossRef CAS PubMed; (e) I. Hyder, M. Jinenez-Tenorio, M. C. Puerta and P. Valerga, Organometallics, 2011, 30, 726 CrossRef CAS; (f) G. Consiglio, F. Morandini, G. F. Ciani and A. Sironi, Organometallics, 1986, 5, 1976 CrossRef CAS.
- J. M. Lynam, Chem. – Eur. J., 2010, 16, 8238 CrossRef CAS PubMed.
- M. I. Bruce and R. C. Wallis, Aust. J. Chem., 1979, 32, 1471 CrossRef CAS.
- M. I. Bruce, C. Hameister, A. G. Swincer and R. C. Wallis, Inorg. Synth., 1982, 21, 78 CrossRef CAS PubMed.
- M. I. Bruce and A. G. Swincer, Aust. J. Chem., 1980, 33, 1471 CrossRef CAS.
- M. I. Bruce, B. G. Ellis, P. J. Low, B. W. Skelton and A. H. White, Organometallics, 2003, 22, 3184 CrossRef CAS.
- M. I. Bruce, K. Costuas, B. G. Ellis, J.-F. Halet, P. J. Low, B. Moubaraki, K. S. Murray, N. Ouddaï, G. J. Perkins, B. W. Skelton and A. H. White, Organometallics, 2007, 26, 3735 CrossRef CAS.
- M. I. Bruce and G. A. Koutsantonis, Aust. J. Chem., 1991, 44, 207 CrossRef CAS.
- M. P. Gamasa, J. Gimeno, B. M. Martìn-Vaca, J. Borge, S. García-Granda and E. Perez-Carreño, Organometallics, 1994, 13, 4045 CrossRef CAS.
- A. G. Alonso and L. B. Reventós, J. Organomet. Chem., 1988, 338, 249 CrossRef.
- K. Y. Ghebreyessus and J. H. Nelson, Inorg. Chim. Acta, 2003, 350, 12 CrossRef CAS.
- D. Duraczynska and J. H. Nelson, Dalton Trans., 2003, 449 RSC.
- H. D. Hansen and J. H. Nelson, Organometallics, 2000, 19, 4740 CrossRef CAS.
- R. L. Cordiner, D. Albesa-Jove, R. L. Roberts, J. D. Farmer, H. Puschmann, D. Corcoran, A. E. Goeta, J. A. K. Howard and P. J. Low, J. Organomet. Chem., 2005, 690, 4908 CrossRef CAS PubMed.
- G. Argouarch, P. Thominot, F. Paul, L. Toupet and C. Lapinte, C. R. Chim., 2003, 6, 209 CrossRef CAS.
- Y. Kawata and M. Sato, Organometallics, 1997, 16, 1093 CrossRef CAS.
- J. H. Bowie, M. I. Bruce, M. A. Buntine, A. S. Gentleman, D. C. Graham, P. J. Low, G. F. Metha, C. Mitchell, C. R. Parker, B. W. Skelton and A. H. White, Organometallics, 2012, 31, 5262 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed; (b) P. J. Stephens, C. F. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS.
- (a) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS PubMed; (b) G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081 CrossRef CAS PubMed.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839 CrossRef PubMed.
Footnotes |
| † Dedicated to the memory of Professor Kenneth Wade, FRS, an inspirational teacher, mentor, colleague and friend. |
| ‡ CCDC 1063656–1063659. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt01806d |
| This journal is © The Royal Society of Chemistry 2015 |