
A general framework for the assessment of solar fuel technologies†
Jeffrey A.
Herron
,
Jiyong
Kim‡
,
Aniruddha A.
Upadhye
,
George W.
Huber
and
Christos T.
Maravelias
*
Department of Chemical and Biological Engineering, University of Wisconsin – Madison, Madison, WI 53706, USA. E-mail: maravelias@wisc.edu; Fax: +1 608 262 5494; Tel: +1 608 265 9026
First published on 3rd October 2014
Abstract
The conversion of carbon dioxide and water into fuels in a solar refinery presents a potential solution for reducing greenhouse gas emissions, while providing a sustainable source of fuels and chemicals. Towards realizing such a solar refinery, there are many technological advances that must be met in terms of capturing and sourcing the feedstocks (namely CO2, H2O, and solar energy) and in catalytically converting CO2 and H2O. In the first part of this paper, we review the state-of-the-art in solar energy collection and conversion to solar utilities (heat, electricity, and as a photon source for photo-chemical reactions), CO2 capture and separation technology, and non-biological methods for converting CO2 and H2O to fuels. The two principal methods for CO2 conversion include (1) catalytic conversion using solar-derived hydrogen and (2) direct reduction of CO2 using H2O and solar energy. Both hydrogen production and direct CO2 reduction can be performed electro-catalytically, photo-electrochemically, photo-catalytically, and thermochemically. All four of these methods are discussed. In the second part of this paper, we utilize process modeling to assess the energy efficiency and economic feasibility of a generic solar refinery. The analysis demonstrates that the realization of a solar refinery is contingent upon significant technological improvements in all areas described above (solar energy capture and conversion, CO2 capture, and catalytic conversion processes).
Broader contextThe conversion of carbon dioxide and water into fuels (and chemicals) using solar energy presents a promising means of achieving energy security, promoting sustainability, and mitigating the effects of climate change. However, much of the technology involved in such a “solar refinery” is still at the basic research level and it is unclear which, and to what extent, these technologies must be improved to develop a competitive process. Towards this goal, we have developed a simple, flexible framework that allows for the systematic comparison between various technology alternatives and for the identification of the key technology gaps that must be overcome in order to realize a feasible process. Although the specific framework has been implemented for the assessment of solar refineries, the larger vision and methodology is applicable to a wider range of processes. The results of our analyses demonstrate that an economically and energetically feasible solar refinery is contingent upon fundamental research advances in a number of disparate areas, including CO2 sourcing (combustion technology), CO2 capture, CO2 (catalytic) conversion, separations, and solar energy capture/conversion to process utilities. |
1. Introduction
Reducing our greenhouse gas emissions, while improving the global standard of living, is one of the key fundamental challenges of the 21st century. One of the options that has been proposed to reduce greenhouse gas emissions is the conversion of carbon dioxide and water into fuels and chemicals.1–11 This conversion is challenging because the feedstocks have no energy content, carbon dioxide is usually present as a very dilute molecule (∼400 ppm in the air) or present as a mixture of gases, and this process requires substantial amounts of energy that must come from another source. Ideally, the energy for this reaction would come from the sun. Several approaches have been proposed for solar fuel production including solar driven electrochemical, photo-electrochemical and photo-catalytic CO2 conversion to fuels, CO2 catalytic conversion using solar-derived hydrogen (e.g. methanol synthesis for CO2 and H2 (ref. 12)), and solar-driven thermochemical processes. A number of ongoing projects are developing this technology including the Joint Center for Artificial Photosynthesis (JCAP, a DOE Energy Innovation Hub), several DOE-sponsored Energy Frontier Research Centers (EFRCs) (e.g. the Argonne-Northwestern Solar Energy Research Center, the Center for Solar Fuel), the Solar Fuels Institute,13 the Research Triangle Solar Fuels Institute,14 Sunshine to Petrol (S2P) at Sandia National Laboratories, the SUNCAT (sustainable energy through catalysis) Center for Interface Science and Catalysis, Liquid Light,15 ETOGAS,16 and SOLAR-JET.17While the field of solar fuels is in its infancy, we believe that evaluating the technologies at a conceptual process level is crucial to help evaluate the realistic viability of the proposed solar refinery and identifying the critical research that must be done to make a future refinery successful. In this review, we will describe and assess, from a systems-level perspective, various technologies involved in the renewable production of chemical fuel using solar energy and CO2 as the primary carbon source (see Fig. 1). In the first half of this review, we describe the important feedstocks for a solar refinery (solar energy, H2O and CO2), the state of technologies for capturing CO2 from different sources, and the methods of converting CO2 to fuels using solar energy. CO2 conversion follows two principal routes (1) catalytic conversion using solar-derived hydrogen, and (2) direct CO2 reduction with H2O. In the first route, hydrogen can be produced using solar energy through various technologies, including electrolysis, photo-electrochemical water splitting, photo-catalytic water splitting, and thermochemical methods. In the second route, CO2 is reduced with H2O using electrocatalytic, photo-electrochemical, photo-catalytic, or thermochemical methods. In the second half of this review, we demonstrate how these various technologies are integrated into a solar refinery and perform a techno-economic analysis, using methanol as the final product as a case study, to assess these technologies. Importantly, through our modeling and analysis, we have identified the critical challenges and key targets for improving the overall economic feasibility of the solar to chemical energy process. The objective of this study is to provide an overview of the current technological approaches being used for solar fuel production and to provide a vision as to where future research efforts should be focused. Importantly we will illustrate the fundamental laboratory discoveries that are needed to turn these technologies into commercial projects.
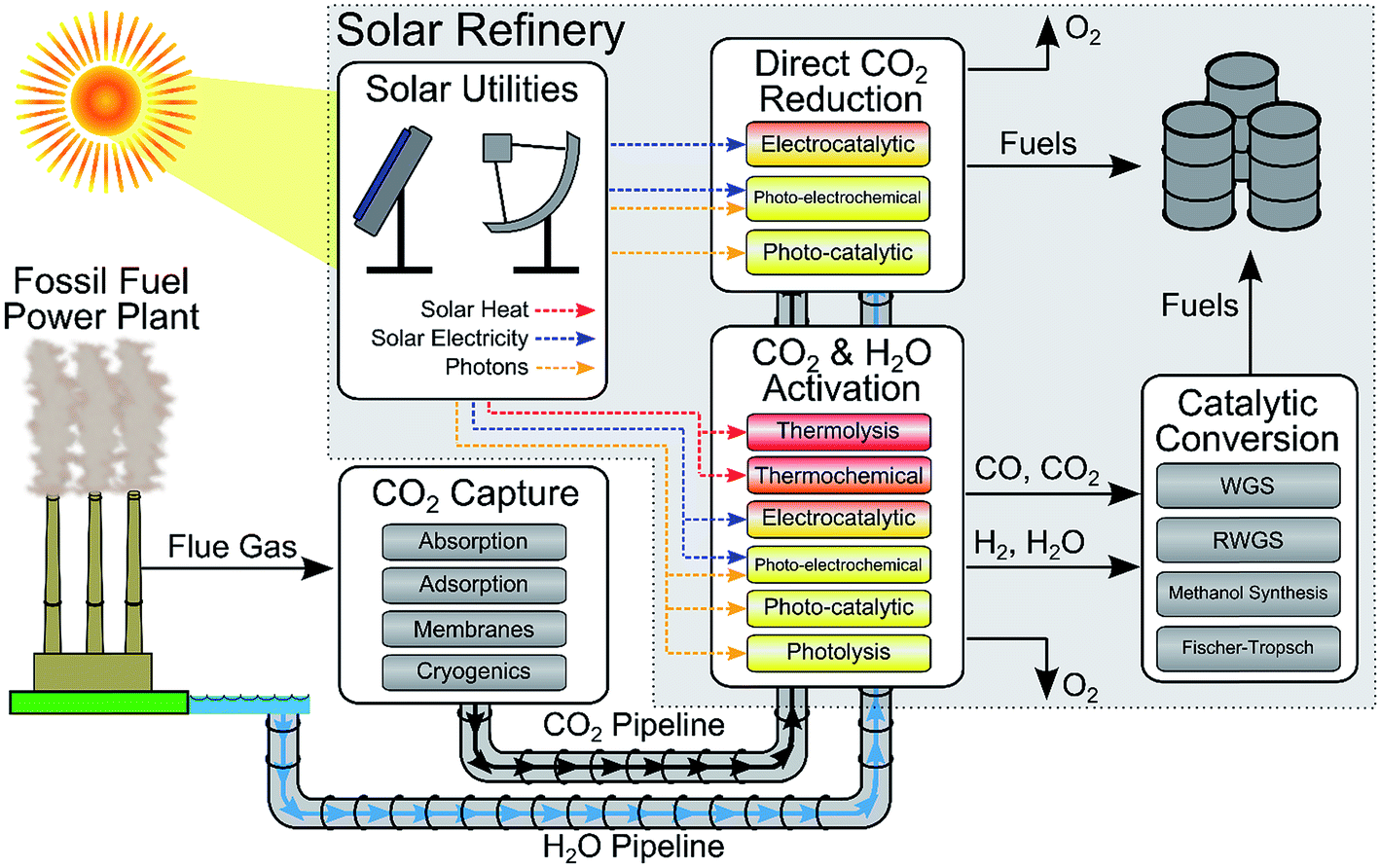 | ||
| Fig. 1 Schematic for solar fuels production. Solar fuel feedstocks (CO2, H2O, and solar energy) are captured on-site and/or transported to the solar refinery. Solar energy provides solar utilities in the form of heating, electricity, and photons which are used in the solar refinery to convert CO2 and H2O into fuels. CO2 and H2O are converted to fuels through two principal routes: (1) direct solar-driven CO2 reduction by H2O to fuels or (2) solar activation of CO2/H2O to CO/H2, respectively, and subsequent catalytic conversion to fuels via traditional processing (i.e. methanol synthesis or Fischer-Tropsch). The approximate temperature requirements for the solar-driven conversion processes are color-coded (red = high temperature, yellow = ambient temperature). | ||
2. Solar fuel feedstocks
In this section, we will describe the key resources that are required for solar fuels production: solar energy, water, and CO2. We will describe their availability and the costs to acquire those resources, from both energy and capital perspectives. We will first discuss the solar energy resources, then briefly comment on water resources, and finally describe CO2 resources (including CO2 capture and separation technologies).2.1. Solar energy resources
The solar radiation spectrum spans a wide range of wavelengths, and resembles black body radiation at 5777 K as seen in Fig. 2.18,19 The energy in solar radiation is proportional to the frequency (ν) of the light as given by Planck's relation (E = hν). More than 25% of the extraterrestrial radiation is absorbed by greenhouse gases such as O3, CO2 and H2O.20 Of the solar radiation reaching Earth's surface, about 52% of the energy is in the infrared region (>700 nm), 43% is visible-light radiation (400–700 nm) and about 5% is in the ultraviolet range (<400 nm). The amount of radiation incident on the earth's surface depends upon many factors such as (a) location (latitude), (b) time of the day, (c) inclination of the surface, (d) declination, (e) weather, etc.19 The resulting intermittent nature of solar radiation is one of the major challenges in designing solar fuels technology. Thus, storing solar energy is critical for continuous processing during these fluctuations. The monthly averaged daily radiation numbers can be calculated using correlations available in the literature.19,21 Furthermore, the National Renewable Energy Laboratory (NREL) has databanks and models available which can be used to estimate the solar radiation.22 | ||
| Fig. 2 Spectral distribution of solar radiation (i) extraterrestrial radiation outside earth's atmosphere, (ii) global total radiation incident on the 37° inclined surface and (iii) black body radiation at 5777 K. Absorption bands for O2, H2O, and CO2 are indicated. (Data adapted from ref. 18). | ||
Fig. 3 shows the localization of solar resources throughout the world.23 The amount of solar energy intercepted by the earth in 1 hour is more than the annual world energy consumption,3,24 though it is not uniformly distributed. As seen from the Fig. 3, solar energy resources are abundant in most parts of Africa, the Middle East, Australia, South America, the Indian subcontinent, and Central America. These areas receive more than 6 kWh m−2 of solar radiation per day. Northern parts of America, Europe and Asia receive relatively lower solar radiation in comparison. Furthermore, the fluctuations in solar energy availability over time are minimized near equatorial regions, hence these represent promising locations for solar fuels refineries. Though, one must also consider the availability of raw materials such as water and CO2.
 | ||
| Fig. 3 World map of global horizontal solar radiation (figure is reprinted with permission, SolarGIS© 2014 GeoModel Solar23). | ||
With flat surface collectors, the incident light is directly utilized without concentrating. Therefore, the intensity of the solar radiation is limited to a maximum of 1000 W m−2 (i.e. the solar energy intensity reaching the earth's surface). Flat surface collectors are often used for domestic solar heating and photovoltaic (PV) applications because of their low cost and ease of installation.25,26
Solar concentrators increase the solar intensity that reaches the receiving surface. Concentration ratios (ratio of radiation intensity at receiver to intensity at the collector) from 1 to 105 can be obtained using these devices.19 Concentrating the solar intensity helps to reduce the amount of expensive PV or catalyst material required in a given process and also allows for the generation of higher temperatures at the receiver. Unfortunately, for photo-catalytic systems as the solar intensity increases, the rate dependence on the solar intensity diminishes from 1 to 0.27,28 Solar concentrators can only utilize direct normal solar radiation, unlike flat plate collectors which can utilize diffuse radiation along with direct normal radiation. Reflectors and luminescent29 solar collectors are two of the most common concentrated solar collectors. The commercial applicability of these concentrators is limited because of issues with stability and lower collection efficiency.
Different configurations of reflective solar concentrators are possible,19 with parabolic trough and dish collectors being the most common.25 Parabolic trough collectors use reflectors that are curved around one axis in a parabolic shape, which concentrates solar radiation on one line. Parabolic dish reflector concentrators use 2-dimensional parabolic dishes to concentrate radiation over a small area. The current cost of concentrating solar collectors ranges between 150–250 USD m−2.30 Sphinx glass, Saint-Gobain, Guardian industries, and 3M are leading technology providers for concentrating solar collectors.
Solar heat is the most mature application for solar energy. Apart from industrial solar thermal energy,31 solar energy is widely used for domestic water heating and air heating.32 It is a low cost application with high energy conversion efficiency. Concentrated solar collectors can be used in industrial settings to reach temperatures as high as 2000 °C.33 The solar to heat efficiencies obtained from these systems highly depend upon the operating conditions such as the surface temperature and ambient temperature.19 Though, annual collection efficiencies of ∼45% have been reported.34 Unfortunately, a major drawback for solar heat systems (including solar heat reactors) is that they are highly sensitive to the intermittency (e.g. weather effects) of solar radiation.35
There are two main approaches to produce electricity from solar energy: (i) photovoltaics and (ii) solar thermal power.36 Photovoltaic devices use semiconductor materials to generate direct current electricity from incident light. In contrast, solar thermal power systems use solar energy as a heat source to drive heat engines, which are connected to electrical power generators.
In photovoltaics, when a photon having energy greater than the material's band gap (the separation between the bottom of the conduction band and the top of the valence band) is incident on the material, an electron–hole pair is generated. The recombination of electron–hole pairs is minimized by building a potential barrier through a thin-layer of junction, which is created by doping the semiconductor material with n-type (electron-rich) dopants on one side and p-type (hole-rich) dopants on the other side. Electrical current is produced when the sides are connected through an external circuit. The efficiency of photovoltaic cells depends on several factors such as operating temperature,37,38 operating load, intensity of the radiation, and materials properties.19 Recent reviews on photovoltaic cell technology are available in the literature.39–41
Fig. S1† shows the time-evolution of research-cell efficiencies.42 Based on the material characteristics, the PV cells can be classified into (i) crystalline Si cells,43 (ii) single-junction GaAs cells,44 (iii) multijunction cells,44 (iv) thin-film technologies45 and (v) emerging PV technologies. Crystalline silicon cells utilize pure silicon material which has a band gap of 1.12 eV. The best reported efficiency for such materials is 27.6% (the theoretical efficiency (Shockley–Queisser) limit is 29% (ref. 46)), and it was obtained with single crystal materials using concentrated solar radiation.47 GaAs based materials are characterized by a band gap of 1.424 eV. The highest efficiency achieved with these materials is about 29.1% using concentrated solar radiation, while the theoretical Shockley–Queisser limit is 33.5%.48 Multijunction cells utilize multiple layers of different semiconductors with varying band gaps to boost the overall efficiency of the cells. The materials are stacked in layers such that the incident radiation contacts the layers in order of decreasing band gap. The highest energy photons are absorbed in the first layer, while the lower energy photons pass through to successive layers where they are absorbed. This maximizes the energy efficiency of the system, and the highest reported efficiency of such a system is 44.4%.49 We note that the theoretical efficiency limit for a tandem solar cell with an infinite number of cells is 86.8%.50 Because of the high cost of manufacturing crystalline materials, alternative photovoltaic cells have received considerable attention. These include thin-film cells, dye-sensitized solar cells,51,52 organic photovoltaic cells,53 inorganic cells54 and quantum cells.55 Although the highest cell efficiencies obtained with such systems are lower compared with crystalline materials, these materials are very promising in terms of commercializing PV electricity. The current estimated levelized cost of electricity using PV is expected to be around 144.3 USD MWh−1.56
Solar thermal power, often referred to as concentrated solar power (CSP), is an alternative to PV technology.57,58 In these systems, concentrated solar energy is used to drive a heat engine at high temperatures to produce electricity.59,60 These systems can achieve 20–25% solar to electricity conversion efficiencies.61 Because of the need to operate at high temperature, these systems are typically large-scale, which results in higher capital costs. Thus, the estimated levelized cost of electricity from such systems is much higher than from PV, 261.3 USD MWh−1.56 Although the cost of electricity is higher, one of the advantages of these thermal systems is that the cost to store thermal energy is significantly lower than the cost of electrical energy storage in batteries. Energy storage is critical to overcome issues with solar energy intermittency.62–64
2.2. Water resources
Water presents a second key resource in the solar refinery. Water can be directly converted to hydrogen (and oxygen) through a number of different solar technologies as described in Section 3. Alternatively, water can be directly consumed (without forming hydrogen as an intermediate) during CO2 reduction to various fuels, as described in Section 4. In general, locations which have high solar resource availability may not have high water availability.In 2005, the estimated daily water withdrawal in the United States was 410 billion gallons per day (∼1.5 trillion kg per day).65 Of those withdrawals, approximately 49% was used for cooling in thermo-electric power plants, 33% for irrigation/agriculture, 11% for public supply, 4% in industry, 1% in mining, and 1% for domestic use. We note that the term “public supply” refers to water withdrawals that serve at least 25 people, and 86% of the U.S. population receives their water in this way. The majority of this water is returned to the source after use (e.g. once-through cooling of power-plant), though estimating the exact consumption is difficult. Even so, in the United States, an estimated 5.9 billion gallons (22.3 billion kg) of water are consumed daily in thermo-electric power plants.66
Due to these large withdrawals, there are areas of the United States which already exceed precipitation rates. These areas include the southwest United States, California, Florida and the high plains.67 Therefore, water may need to be transported to the solar refinery. Based on simulations, we have calculated a cost for water transportation of 0.007 USD MT−1 km−1, assuming 15 MT per year−1 water capacity. We note that based on the process modeling in Sections 5–8 that this translates into a very small fraction of the overall process cost, therefore we will not discuss this resource further.
2.3. CO2 sources, capture and separation
CO2 is available abundantly in the air, with 34 billion tons emitted globally in 2011.68 However, it is in low concentration, approximately 400 ppm (0.04%), which makes it unsuitable for most conversion processes without employing expensive capture and purification methods. Therefore, it is beneficial to acquire CO2 from more concentrated sources. In the United States, approximately 38% of CO2 emissions are from electric power plants, primarily coal-fired plants, and another 18% are from industry,69 presenting promising sources for CO2. Excluding CO2 capture from the atmosphere, there are three main routes for CO2 capture from these stationary sources: post-combustion, pre-combustion, and from oxy-combustion. Each of these routes have preferred CO2 capture methods because of the varying physical properties and chemical composition of the capture source (see Table 1).| Atmosphere | Post-combustion | Pre-combustion | Oxy-combustion | |
|---|---|---|---|---|
| Pressure (bar) | 1 | 1 | 15–40 | 1 |
| CO2 concentration | 400 ppm | 3–20% | 15–40% | 75–80% |
| Major impurities | N2, O2 | N2, O2, H2O | H2, CO | H2O |
| Lesser impurities | NOx, SOx, particulates | NOx, SOx, H2S | NOx, SOx | |
Post-combustion capture of CO2 refers to CO2 separation from flue gases that were created by combustion in air. The flue gas stream consists of 3–20% CO2 in nitrogen, oxygen and water vapor with some minor impurities including SOx, NOx, and particulate matter.70 Post-combustion capture is relatively easily retrofitted on existing fossil fuel power plants, and presents the most near-term solution to CO2 capture. Due to the relatively low concentration of CO2 in the stream, it is best captured through chemical absorption/adsorption and is industrially captured using absorption in monoethanolamine (MEA).
Pre-combustion capture of CO2 is performed from synthesis gas (syngas) streams prior to combustion/power production, as in integrated gasification combined cycle (IGCC) power plants.71–73 In these power plants, the fossil fuel is first gasified to produce syngas, which is converted via water–gas shift (WGS) to remove the CO and increase the quantity of H2. Carbon dioxide is removed from this stream prior to combustion of the hydrogen gas, the stream consists of 15–40% CO2 at elevated pressure (15–40 bar).70 Due to the high partial pressure of CO2 in these streams, physical absorbents/adsorbents and membranes present promising methods for capture.74
In an oxy-combustion process, the fuel is combusted in an atmosphere of O2/CO2, rather than air. The resulting effluent consists of 75–80% CO2 in water vapor, with trace amounts of NOx and SOx.70 Some of the CO2 must be captured and used as a diluent in the oxygen stream to control the temperature of the furnace. It is typical to separate the CO2 by lowering the temperature to condense the water vapor.75 Chemical Looping Combustion (CLC)76 is an alternative oxy-combustion process where the fuel is combusted upon contacting a solid oxygen carrier (e.g. a metal oxide). The depleted oxygen carrier must be re-oxidized, closing the overall loop. This strategy is advantageous because it avoids the high energy costs required to obtain purified oxygen (as in the oxy-combustion process, which is usually by cryogenic oxygen separation from air).
Once CO2 is captured, it must be transported to the solar refinery for conversion. The current state of CO2 pipeline infrastructure was recently reviewed,77 finding approximately 6500 km of CO2 pipeline worldwide, with the majority in the United States. (We note that these pipelines are mainly employed for enhanced oil recovery.) The cost for CO2 transportation has been studied by NETL, suggesting a cost of 3.65 USD MT−1 for 100 km transport range.77,78
The state of CO2 capture technology has recently been reviewed by Aaron,79 Choi,80 D'Alessandro,81 Espinal,68 Jones,82 Mondal,83 MacDowell,84 Spigarelli,70 and Folger.85 Here, we summarize the four main CO2 capture/separation technologies: absorption, adsorption, membrane separation, and cryogenic distillation with an emphasis on CO2 capture from post-combustion flue gases.
| CO2 + 2R1R2NH ↔ [R1R2NCO2− + R1R2NH2+] |
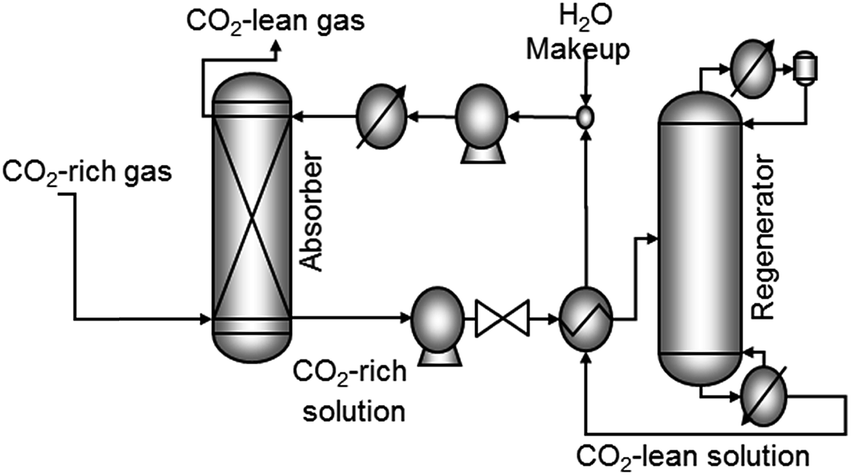 | ||
| Fig. 4 Generic absorption and regeneration process for CO2 capture. MEA is a typical absorbant used to capture CO2 from flue gas. | ||
The energy cost for commercial CO2 capture via the MEA wet-scrubbing has been reported to be as low as 0.37–0.51 MWh tonCO2−1,87 though the energy costs will vary depending on the capture conditions. The CO2 loading capacity in MEA is 0.40 kgCO2 kgMEA−1.70 Some of the main disadvantages of this process are the high energy cost for regenerating the solvent, the cost to compress the CO2 for transport and storage, and specifically for the MEA-process, the low degradation temperature of MEA.
Research in the field has primarily focused on finding improved solvents for the process: optimizing CO2 capacity, heat of CO2 absorption and rate of CO2 absorption. One class of promising chemical solvents are sterically hindered amines (e.g. 2-amino-2-methyl-1-propanol, AMP).84,88–91 Ionic liquids are a class of promising physical solvents that are advantageous due to their very low vapor pressure, thermal stability, non-flammability, low heats of adsorption (physisorption), and relative ease of tuning.92–95
Zeolites are microporous crystalline aluminosilicates composed of SiO4 and AlO4 groups. Substitution of an AlO4 groups with a SiO4 groups creates a negative charge, which is compensated by a cation (e.g. alkali) in the pores.97 Metal–organic frameworks are microporous crystalline networks composed of metal centers which are connected by organic ligands.98–100 MOFs can be produced of a wide variety of structures. As such, they can separate through several different mechanisms, including chemisorption, size-exclusion, and molecular sieving. Supported amines function similarly as solvent amines.82 There are many methods of supporting the amines: polymeric amines can be physically loaded into the support,101,102 they can be covalently linked the support,103–105 or by in situ polymerization of aminopolymers.101,106 Metal oxides (e.g. CaO, MgO) capture and release CO2 through reversible, carbonate looping. One of the most common materials for carbonate looping is CaO, which forms CaCO3 between 600–650 °C in the presence of CO2 and calcines to regenerate CaO between 800–850 °C:81,107,108 Finally, hydrotalcites are a class of anionic clays with the stoichiometry M1−x2+Mx3+(OH)2Ax/mm−·yH2O, where M2+ is typically Mg2+, Zn2+, or Ni2+; M3+ is typically Al3+, Ga3+, Fe3+, or Mn3+; and Am− includes CO32−, Cl−, SO42−, NO3−.109 Adsorption of CO2 occurs on the basic surface sites. See the ESI† for additional details on these materials.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 MT of CO2 per day for a 500 MW power plant),117 which must be scaled to process the high volume of CO2 that would be required to feed a solar refinery.70 Alternative absorbents and other technologies (adsorption, membranes, and cryogenics) provide a number of advantages in terms of energy of separation, capital cost, mechanical robustness, chemical stability, and CO2 capacity. Although, these technologies also present a number of issues that must be resolved. For adsorbents, the main challenge is to improve their stability and recyclability. Membranes have relatively low capital costs, but they require high partial pressure of CO2 in order to have achieve high selectivity and high rates of separation. Therefore, there may be significant operating costs to compress the CO2 stream. Similarly, cryogenic separation has high operating costs, and may be more viable from concentrated CO2 streams. Specific cost estimates for some of these CO2 capture methods are available (absorption,118–121 membranes,118,122 cryogenics118), though they vary significantly depending on the assumptions employed, capacity, CO2 concentration in the source, year of estimate, etc. Recently, Folger et al. reviewed commercial and pilot-scale demonstrations of these capture technologies, which utilize, almost exclusively, amine based absorption or physical absorption systems.85
000 MT of CO2 per day for a 500 MW power plant),117 which must be scaled to process the high volume of CO2 that would be required to feed a solar refinery.70 Alternative absorbents and other technologies (adsorption, membranes, and cryogenics) provide a number of advantages in terms of energy of separation, capital cost, mechanical robustness, chemical stability, and CO2 capacity. Although, these technologies also present a number of issues that must be resolved. For adsorbents, the main challenge is to improve their stability and recyclability. Membranes have relatively low capital costs, but they require high partial pressure of CO2 in order to have achieve high selectivity and high rates of separation. Therefore, there may be significant operating costs to compress the CO2 stream. Similarly, cryogenic separation has high operating costs, and may be more viable from concentrated CO2 streams. Specific cost estimates for some of these CO2 capture methods are available (absorption,118–121 membranes,118,122 cryogenics118), though they vary significantly depending on the assumptions employed, capacity, CO2 concentration in the source, year of estimate, etc. Recently, Folger et al. reviewed commercial and pilot-scale demonstrations of these capture technologies, which utilize, almost exclusively, amine based absorption or physical absorption systems.85
| Technology CO2 recovery | Material capacity (gCO2 gmaterial−1) | Conditions P (bar)/T (°C) | Energy required (MJ kgCO2−1) | Advantages | Disadvantages |
|---|---|---|---|---|---|
| Absorption 90–98% | MEA 0.4 | 1/40–60 | 4.0–6.0 | • Mature process | • High cost for regenerating |
| • Viable for low partial pressure of CO2 | • Degradation | ||||
| • Challenge to scale up | |||||
| Adsorption 80–95% | Zeolite 0.21 | 0.1–1/0–100 | 2.0–3.0 | • High capacity | • Impurities affect selectivity |
| • Stable | |||||
| • Fast adsorption kinetics | |||||
| MOFs 0.20 | 1/<100 | • High surface area | • Recyclability not demonstrated | ||
| • High void volume | |||||
| • Tunable | |||||
| • Stable | |||||
| Supported amines 0.24 | 0.05–1/50–75 | • Water tolerant | • Thermal degradation | ||
| • Fast adsorption kinetics | |||||
| • Viable for low partial pressure of CO2 | |||||
| Metal oxide 0.51 | 1/600–700 | • High capacity | • Low stability | ||
| • Inexpensive and abundant materials | |||||
| Hydrotalcite 0.29 | 1/500–600 | • Stable | • Lower capacity than metal oxides | ||
| Membranes 80–90% | Polymer | 15–20/100 | 0.5–6.0 | • Simple | • Requires high pressure |
| • No reagents | • Low purity product | ||||
| • Thermal degradation | |||||
| Cryogenic >95% | 6.0–10.0 | • No reagents | • High capital cost | ||
| • High energy cost | |||||
Advances in fuel combustion technology72,73,107 through oxy-combustion or IGCC, for example, may produce capture conditions which are suitable for alternative capture technologies. In particular, the high partial pressure of CO2 in pre-combustion streams is more suitable for membrane separation or physical absorption/adsorption.
3. Hydrogen production
The catalytic conversion of CO2 to liquid fuels and chemicals requires the addition of hydrogen. Hydrogen is produced industrially using hydrocarbon feeds through three main routes: steam reforming of hydrocarbons (e.g. methane), partial oxidation, and autothermal reforming.123 These reactions produce carbon monoxide, hydrogen and carbon dioxide. The amount and purity of hydrogen can be increased by water–gas shift and preferential oxidation of CO. NREL has calculated a levelized cost of hydrogen production from natural gas of 1.32 USD kg−1 (in 2005 USD), which achieves 71% process energy efficiency (using 2 MJ of process electricity per kilogram of hydrogen produced).124In this section, we review methods of producing hydrogen from water using renewable, solar energy. The methods that will be discussed are electrolysis, photo-electrochemical water splitting, photo-catalytic water splitting, and thermochemical water splitting. For reference, the stoichiometry and thermochemistry of these processes are outlined in Table 3.
| Reaction | Thermochemistry | ||
|---|---|---|---|
| ΔH° (kJ mol−1) | ΔG° (kJ mol−1) | E 0 (V) | |
| 2H2O → 2H2 + O2 | 483.6 | 457.2 | 1.19 |
| 2H2O(l) → 2H2 + O2 | 571.6 | 474.0 | 1.23 |
3.1. Electrolysis of water
Electrolysis126 uses electrical current to split water into hydrogen and oxygen, achieving hydrogen purity >99.999%. Oxygen is evolved at the system anode, while hydrogen is evolved at the cathode. The overall process is endothermic, requiring a cell voltage of at least 1.23 V, though, in practice, commercial electrolyzers require cell voltages around 1.9 V in order to overcome system inefficiencies and overpotential. The energy required for the electrolysis could be provided by solar-PV cells by connecting cells in series.127There are three main technologies for water electrolysis: alkaline electrolyzers, polymer electrolyte membrane (PEM) electrolyzers and solid-oxide electrolyzer cells (SOEC), details on the reaction chemistry for electrolyzers are provided in the ESI.† A survey of commercial electrolyzers shows that PEM electrolyzers are able to achieve system efficiencies of 56%, while the system efficiency of alkaline electrolyzers varies from 63–73%.128 Commercial solar PV systems have demonstrated solar to electricity efficiencies in the range of 11.5–17.5%, and optimized PV-electrolyzer systems have achieved overall solar-to-hydrogen efficiencies of 12%.129 A listing of commercial electrolyzer companies and their employed technologies are provided in Table 4. Capital costs for these systems have ranged from approximately $400 to $1600 per kg per day of hydrogen production capacity.130 The levelized cost of hydrogen from electrolysis was calculated as 4.50 USD kg−1 (in 2005 USD) by NREL.124
| Company | Technology |
|---|---|
| Avalence | Alkaline |
| NEL hydrogen | Alkaline |
| Proton OnSite | PEM |
| Teledyne energy systems | Alkaline |
| Hydrogenics | PEM |
| Industrie Haute technologie | Alkaline |
Alkaline electrolyzers are commercially available as shown in Table 4. Unfortunately, there are some drawbacks to the technology, including crossover of hydrogen and oxygen through the semi-permeable diaphragm138 and low current density due to high ohmic losses, thereby requiring large systems to produce high volumes of hydrogen.
PEM electrolyzers can be manufactured in a more compact design and have higher current densities than alkaline electrolyzers. Unfortunately, the membrane and precious metal electrocatalysts are expensive. Some possible alternative electrocatalysts for hydrogen evolution include metal carbides,144,145 metal nitrides,144 while oxygen evolution electrocatalysts include metal (Ir, Sn, Ru) oxides.146–150 Alternative electrolytes include solid acids, ceramic oxides, sulfonated aromatics, and heteropolyacids.142,151,152
The high temperature operation is advantageous because the ΔG of reaction decreases by 25% from 25 °C to 1000 °C, providing a significant electricity reduction for operation (though there is a cost to heat the water to the high temperature).126 The electrical efficiency of SOEC systems has been reported to be as high as 85–90%.159 In contrast, the mechanical robustness of the system is a challenge at high temperature operation.
3.2. Photo-electrochemical water splitting
Photo-electrochemical (PEC) water splitting7,160–165 follows similar principles as electrolysis, though the energy is supplied directly from light. In order to capture light, the anode, cathode, or both are made of a semiconductor material (referred to as a photo-electrode). A photo-anode is an n-type semiconductor; when a photon with energy greater than its band gap is adsorbed, an electron–hole pair is generated. The holes oxidize water to oxygen and the electrons travel through an external circuit to the cathode where hydrogen is evolved. A photo-cathode is a p-type semiconductor where water is reduced to hydrogen. In this case, oxygen evolution occurs on the counter electrode. Importantly, the photo-cathode or photo-anode must have a band gap which is at least as large as the thermodynamic potential, 1.23 eV. Though, in practice, due to energetic losses, a band gap of 1.6–2.0 eV is required.123 Furthermore, the bottom of the conduction band must be more negative than the redox potential of H+/H2 (0 V vs. NHE) and the top of the valence band must be more positive than the redox potential of O2/H2O (1.23 V vs. NHE), see Fig. 5. The most common material for the photo-anode is TiO2. Even though it has a high band gap of 3 eV, its high corrosion resistance makes it an attractive material.160 TiO2 is typically paired with a Pt cathode.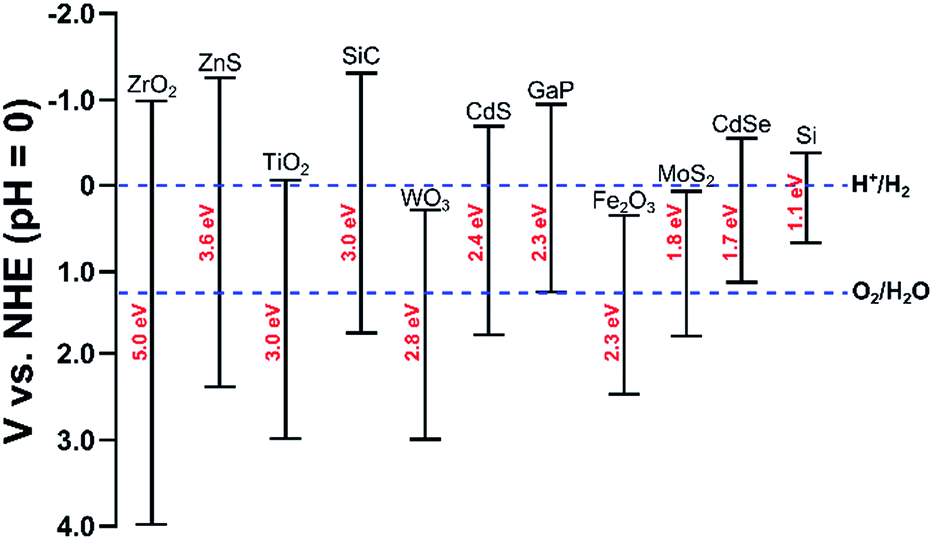 | ||
| Fig. 5 Band edges and band gaps for common semi-conductors and redox potentials for hydrogen and oxygen evolution reactions. Adapted from ref. 188. | ||
Since TiO2 has a wide band gap, it is unable to capture a majority of the solar spectrum (i.e. visible light). Therefore, it has a low solar to hydrogen efficiency of ∼0.4%.160 By assessing the energy lost per photon due to thermodynamic entropy of mixing and kinetic overpotential, Murphy and co-workers suggested that an optimal material would have a band gap of 2.03 eV, which would correspond to a solar to hydrogen efficiency of 16.8% for AM1.5 solar irradiation.166 Therefore, research has focused on identifying efficient, durable, and cheap photo-anodes and photo-cathodes for this reaction that maximize absorption of the solar spectrum. Promising photo-anode materials include metal oxides (doped-TiO2,167,168 WO3,169,170 BiVO4,171–173 Fe2O3 (ref. 174−176)), oxynitrides (TaON,177,178 LaTiO2N179), and GaAs-based materials.180,181 The conduction band levels of many of these visible light-active photo-anodes (WO3, BiVO4, Fe2O3) is too positive for hydrogen evolution because the O 2p valence band is very positive. This challenge can be overcome by applying an electrical bias to the system, though this lowers the overall energy efficiency of the system. Progress in the design of photo-cathodes has been less extensive, with promising materials including Cu2O182 and phosphides (InP, GaInP2)180
Another method to increase the solar to hydrogen efficiency is to utilize a multijunction stack.183 Multijunction stacks can be constructed using a combination of PEC and PV cells. The highest efficiency PV-PEC stack documented was developed at NREL.180 It is composed of a p-type GaInP2 PEC cell connected to GaAs PV cell and has shown a solar to hydrogen efficiency of 12%.180
Recently, Nocera and coworkers developed an “artificial leaf,” a triple-junction cell comprising earth abundant materials.184 It consists of an amorphous silicon photo-voltaic interfaced to a Co-oxygen evolution complex (Co-OEC)185,186 and a ternary NiMoZn alloy hydrogen evolution catalyst. The technology was once being commercially developed by Sun Catalytix, though even with the use of cheap, earth abundant materials, the engineering cost was deemed too high for commercialization.187 In particular, they estimated a hydrogen production cost of 6.50 USD kg−1.
3.3. Photo-catalytic water splitting
Photo-catalytic water splitting165,188–191 follows the same principles as photo-electrochemical water splitting, though both oxygen evolution and hydrogen evolution occur on the same photo-catalyst, not separately on an anode and cathode, respectively. This simplifies the process, allowing powder photo-catalyst materials to be dispersed in water to evolve oxygen and hydrogen. Unfortunately, as a consequence of this, the separation of hydrogen and oxygen becomes more difficult. We briefly describe design considerations for photo-catalytic reactors in the ESI.†Photo-catalyst materials have the same band structure requirements as photo-electrodes. The photo-catalyst must have few defects, be highly crystalline, and have a short pathway for electron and hole migration to the surface. There have been a variety of different materials studied for photo-catalytic water splitting including metal oxides, oxysulfides, and oxynitrides.188–191 Photo-catalyst particles are often accompanied by co-catalysts (e.g. metal nanoparticles) which are deposited onto the photo-catalysts. These provide active sites for the chemical evolution reactions with lower chemical barriers than on the semi-conductors.
However, even with all these materials tested, it is often difficult to find a material which absorbs visible light and has a conduction band level suitable for hydrogen evolution.165 The state-of-the-art visible light active photo-catalyst, a solid solution of gallium and zinc nitrogen oxide (Ga1−xZnxN1−xOx) with a mixed Rh–Cr oxide co-catalyst, only achieves a solar to hydrogen efficiency of 0.2%.192,193 One method to overcome this challenge, called the Z-scheme, involves splitting hydrogen evolution and oxygen evolution onto two different photo-catalysts which are connected via a shuttle redox couple (e.g. Fe3+/Fe2+, IO3−/I−) in the solution.194–196 This has the added advantage of separating the location of hydrogen and oxygen production, enabling easier separation. On the other hand, this system requires twice the number of photons as in the conventional system. Another recent method to utilize visible light is to incorporate plasmonic metal nanostructures (i.e. Au, Ag) into the photo-catalysts.197
3.4. Thermochemical water splitting
Solar energy can be collected and used for heat to thermochemically split water into hydrogen and oxygen. The direct thermolysis of water can be achieved at very high temperatures (>2500 K). H-ion Solar Company developed a prototype nozzle/skimmer reactor system operating at 2600–2900 K which split water with 1–2% solar to hydrogen efficiency.198 Though this is attractive due to its simplicity, reactor materials stability, difficulties in separating high temperature oxygen and hydrogen, and radiation losses made the system infeasible.34,199Alternatively, solar thermal energy can be used to drive a thermochemical cycle that have the net reaction of releasing hydrogen and oxygen.6,34,199,200 These “thermochemical water splitting cycles” usually consist of a high temperature endothermic reaction, followed by one or more exothermic steps.34,201 Over three hundred and fifty thermochemical water splitting cycles were investigated by the U.S. Department of Energy's Solar Thermochemical Hydrogen (STCH) program finding only nine that were deemed technically and practically feasible.202 Accounting for the efficiency of the solar thermal collection system and the efficiency of the splitting reaction, the overall solar-to-hydrogen efficiency can approach 20%.34
High temperature (>1000 °C) thermochemical water splitting cycles usually consist of metal-oxide (e.g. Zn/ZnO,203 FeO/Fe3O4,204,205 and others206–209) reduction/oxidation, generically:
| MOx → MOx−δ + δ/2O2 |
| MOx−δ + δH2O → δH2 + MOx |
These oxidation/reduction cycles have several advantages: only two reaction steps reduces overall inefficiency and allows easy interfacing with the daily solar cycle (light on versus light off), hydrogen and oxygen are produced in separate steps, and separating the solid oxide from vapor product is simple.34 An economic evaluation of a 100000 kgH2 per day Zn/ZnO solar-thermochemical process estimated a hydrogen selling price of 5.58 USD kg−1 assuming that 70% ZnO conversion is possible.210 Sandia National Laboratory has developed a CR5 reactor (counter-rotating ring receiver reactory recuperator) in which a cobalt ferrite/zirconia mixture is rotated through (1) a solar-irradiated high temperature receiver where it is reduced at 1400 °C and then (2) a low-temperature hydrolysis reactor where hydrogen is evolved.211–213
Lower temperature thermochemical cycles are advantageous because there is less energy lost by radiation. One of the most promising is the sulfur–iodine (SI)214,215 cycle because of the low temperature (900–1000 °C) required. Unfortunately, there are a number of drawbacks in this cycle. For one, H2SO4 must be separated from HIx to prevent sulfur formation during HI decomposition. Furthermore, the separation of H2 from HI and I2 is challenging. Another promising method cycles between Mn(II)/Mn(III) oxides using a Na+ shuttle.216 The main advantages of this approach are that the maximum temperature can be limited to 850 °C and it does not involve any corrosive components. On the other hand, the NaMnO2 must be cooled to 80 °C in order to extract the Na+, which may lead to overall high energy costs. Furthermore, the hydrogen must be separated from CO2 (for pure hydrogen), requiring additional costs.
3.5. Summary of solar hydrogen production methods
The operating conditions, system efficiencies and advantages/disadvantages of these solar-based hydrogen production technologies are summarized in Table 5. Water electrolysis is a commercially demonstrated technology, and has reasonably high system efficiencies. Coupling with solar-PV technology is a near-term solution to producing clean hydrogen with reasonable solar to hydrogen efficiency. Photo-electrochemical and thermochemical methods are capable of achieving similar solar-to-hydrogen efficiencies as the electrolysis systems. Thermochemical cycles are more complex to engineer, have high radiative heat losses, and are capital intensive. Photo-electrochemical cells are only able to achieve these high solar efficiencies with complicated multi-junction systems which have been shown to degrade over time.7 Photo-catalytic systems are attractive due to their simplicity, though current solar to hydrogen conversion efficiencies are very low due to low quantum efficiency and inability to harness visible light.165 Additionally, separation of hydrogen and oxygen in these photo-catalytic systems remains an issue.| System | Operating Conditions | System Efficiency | Solar-to-H2 Efficiency | Advantages | Disadvantages | |
|---|---|---|---|---|---|---|
| P (bar) | T (°C) | |||||
| a Assuming 15% solar PV efficiency. b Does not account for thermal energy. c Assuming 45% solar to thermal efficiency. | ||||||
| Alkaline electrolysis | 25–30 | 80–90 | 63–73% | 10a% | • Commercial technology | • Low current density |
| • Low capital cost | • H2–O2 mixing | |||||
| PEM electrolysis | <85 | <100 | 56% | 8.5a% | • High current density | • High capital cost for membrane |
| • Compact design | • Precious metal catalyst | |||||
| • H2–O2 produced separately | ||||||
| Solid oxide electrolysis | 30 | 500–850 | 85–90b% | 12a,b% | • High electrical efficiency | • Brittle ceramics |
| • Non-noble catalyst | ||||||
| • H2–O2 produced separately | ||||||
| Photo-electrochemical | 1 | 25 | — | 12% | • High solar efficiency | • Degradation |
| • H2–O2 produced separately | ||||||
| Photo-catalytic | 1 | 25 | — | 0.20% | • Simple process | • H2–O2 are mixed |
| • Low solar efficiency | ||||||
| Thermolysis | 1 | 2200 | — | 1–2% | • Simple process | • Low materials stability |
| • H2–O2 are mixed | ||||||
| • High radiative losses | ||||||
| Thermochemical | — | >700 | 40% | 18c% | • High energy efficiency | • High capital cost |
| • H2–O2 produced separately | • Complex process design | |||||
4. CO2 conversion
CO2 can be converted to fuels using mature industrial processes with renewable, solar-derived hydrogen (as described in the previous section) and solar heat, as reviewed by Centi and coworkers.1,4 The reaction thermochemistry and stoichiometry for CO2 hydrogenation reactions are shown in Table 6. In particular, the reverse-water–gas-shift reaction (RWGS) can be used to convert CO2 and hydrogen to CO and water. The CO mixed with hydrogen produces syngas, which can be used to produce a variety of products, including methanol, dimethyl ether, or hydrocarbons through Fischer–Tropsch synthesis. There are also direct routes for hydrogenating CO2 to products including methanol, methane, and formic acid.| Product | Reaction | Thermochemistry | |
|---|---|---|---|
| ΔH° (kJ mol−1) | ΔG° (kJ mol−1) | ||
| Carbon monoxide | CO2 + H2 → CO + H2O | 41.2 | 28.6 |
| Methanol | CO2 + 3H2 → CH3OH + H2O | −49.3 | 3.5 |
| Methane | CO2 + 4H2 → CH4 + 2H2O | −164.7 | −113.3 |
| Formic acid | CO2 + H2 → HCOOH(l) | −31.5 | 33.0 |
Besides these hydrogenation routes, CO2 can also be converted to fuels using direct solar energy through electro-catalytic reduction, photo-electrochemical reduction, photo-catalytic reduction, and thermochemical reduction, as reviewed hereafter.
4.1. Electro-catalytic
The electro-catalytic reduction of CO2 (ref. 5,217 and 218) to fuels follows the same principles as electrolysis of water. Oxygen evolves at the cell anode, while CO2 reduction occurs at the system cathode. The product of the reduction depends on the electro-catalyst used, and each of these products has a different thermodynamic cell potential, examples are indicated in Table 7.| Product | Reaction | Thermochemistry | ||
|---|---|---|---|---|
| ΔH° (kJ mol−1) | ΔG° (kJ mol−1) | E 0 (V) | ||
| Carbon monoxide | CO2 → CO + 1/2O2 | 283.0 | 257.2 | 1.34 |
| Formic acid | CO2 + H2O(l) → HCOOH + 1/2O2 | 254.3 | 270.0 | 1.41 |
| Formaldehyde | CO2 + H2O(l) → HCHO + O2 | 570.7 | 528.9 | 1.38 |
| Methanol | CO2 + 2H2O(l) → CH3OH + 3/2O2 | 764.1 | 706.1 | 1.23 |
| Methane | CO2 + 2H2O(l) → CH4 + 2O2 | 890.5 | 817.9 | 1.06 |
| Ethylene | 2CO2 + 2H2O(l) → C2H4 + 3O2 | 1411.0 | 1331.2 | 1.16 |
Electrocatalytic reduction of CO2 can be performed under ambient conditions using similar technology as water electrolysis.219 The major obstacles to implementing this technology are catalytic in nature. That is, current electrocatalysts operate with high overpotentials, low faradaic efficiency (i.e. poor product selectivity), low current densities and are deactivated over time.219,220 Low faradaic efficiency occurs because the thermodynamic potential for CO2 reduction is similar to that of water splitting (1.23 V), which allows for hydrogen evolution to compete with CO2 reduction.
CO2 electro-reduction has been studied over a variety of different catalysts,221 including metal complexes,222 transition metals,218 and metal oxides.223 Of the metals studied, copper is the only one that is able to reduce CO2 to hydrocarbons (i.e. methane, ethylene) with significant current densities at moderate overpotentials and reasonable faradaic efficiency.224,225 The major product is CO on Au, Ag, Zn, and Pd, it is HCOO− on Pb, Hg, In, Sn, Cd, and Tl, while it is hydrogen on Ni, Fe, Pt, Ti, and Ga. For CO2 conversion to C2H4 on copper, the overall energy efficiency of the conversion is only 41% due to the high cathodic overpotential.217 Furthermore, copper dissolutes in acidic environment and deactivation due to surface poisoning limit its efficacy.219 Unlike on metal catalysts, CO2 reduction on metal oxide catalysts (RuO2 (ref. 226) and copper oxides, in particular) has shown methanol to be a major product.223,227,228 Other studies have shown that copper oxides have higher selectivity towards ethylene than pure copper.224
High-temperature CO2 reduction has been demonstrated in solid-oxide electrolyzer cells.153,229 As with water electrolysis, the high temperature operation decreases the electrical energy required to drive the reaction, and the kinetics are also improved. Interest in this technology began in the 1960s by NASA in order to produce O2 for life support and propulsion systems in spacecraft.230,231 Recent work has focused on developing electrode materials for efficient CO2 reduction to CO. Conventional Ni-YSZ electrodes are efficient for hydrogen evolution, but the activity is rather low for CO2 reduction229 and the stability is in question due to the formation of volatile Ni carbonyls.232 One promising electrode material is ceramic La0.8Sr0.2Cr0.5Mn0.5O3 (LSCM),233 which has been combined with a Pd–ceria/YSZ co-catalyst to achieve efficient reduction of CO2 at 800 °C.229 An extension of this technology is to perform high-temperature co-electrolysis of H2O and CO2 in a solid oxide electrolyzer cell to produce syngas at the cathode, and O2 at the anode.234–237 Graves et al. proposed a CO2 to fuels process involving co-electrolysis, calculating that the process could operate at 70% electricity to liquid fuels efficiency.238
The majority of the literature on CO2 reduction has been performed in batch systems with only a few reports for continuous operation for comparison.239–243 Li and Oloman described a continuous, laboratory-scale CO2 electro-reduction reactor which was able to achieve 20–80% conversion of CO2 over a Sn–Cu catalyst with 6–17% organic product yield.240 (This technology is being commercialized by Mantra Energy Alternatives, Ltd.) Xie and co-workers have developed a solid oxide electrolyzer operating at 614 °C which has demonstrated CO2 reduction to CO and CH4 with 60% conversion.242 In the US, Liquid light Inc.15,244,245 is commercializing an electrochemical process for conversion of CO2 to chemicals such as ethylene glycol using proprietary catalysts.
4.2. Photo-electrochemical
The direct photolysis of CO2 to CO and O2 has been demonstrated using deep UV light (wavelength below 200 nm). Deep UV irradiation can be produced renewably using solar PV-powered low pressure mercury (185 nm) or deuterium (165 nm) lamps, at an energy requirement of 0.3 GJ mol−1.246,247 Interestingly, CO2 photolysis in the presence of hydrogen has demonstrated high selectivity to methane.248 The hydrogen can be substituted with H2O at the cost of lower CO2 conversion due to competition with H2O for photons.The photo-electrochemical reduction of CO2 follows the same principles as photo-electrochemical water splitting, as outlined in Section 3.2. As with water splitting, the main challenge is to find suitable photo-cathodes that allow for reduction using visible light irradiation. An additional complication, here, is that hydrogen evolution competes with CO2 reduction. Therefore, there has been interest in using non-aqueous solvents for CO2 photo-electrochemical reduction. Kumar et al. recently reviewed the state-of-the-art in photo-cathodes for CO2 photo-electrochemical reduction in aqueous and non-aqueous solvents.249 Some of the most common photocathode materials are p-Si,250n-Si,251p-InP,252,253p-GaP,254,255p-GaAs,256 n-GaAs,256,257 and p-CdTe253 with typical products: HCOOH, CO, CH2O, and H2. The product selectivity and activity can be altered by incorporating metal co-catalysts such as Au, Ag, Cu, Pd, Ni, Ru, Zn, In and Pb250,255,258−260 or by covalently linking molecular catalysts to the surface.261,262
4.3. Photo-catalytic
In 1979, Inoue and co-workers first reported photo-catalytic CO2 reduction to HCOOH, CH2O, CH3OH, and CH4 on powdered TiO2, ZnO, CdS, SiC, and WO3 under Xe lamp irradiation.263 Since then, there have been investigations into a wide variety of materials, as recently reviewed.8,264 TiO2 has been one of the most widely studied materials due to its durability, stability and lack of toxicity.247,265–272 However, due to its wide band gap, it is inactive under visible light irradiation, leading to low solar energy conversion. Doping TiO2 (with e.g. Ag,267 Co,273 In,274 Cu–Fe,275 I,276 N277) is one possible method to overcome this deficiency, while incorporating plasmonic nanoparticles is another attractive option.197,278 Aside from these thermodynamic limitations of TiO2, other major challenges are catalytic in nature, namely low product selectivity, and low activity. To meet these challenges, metal co-catalysts have been incorporated into the photo-catalyst. Cu, Pd, Rh, Pt, and Au, added to TiO2 has been shown to increase CO2 conversion efficiency and selectivity.266,268,279 In another report, Cu decreased selectivity towards CH4, but lead to the formation of CH3OH.270 It has also been demonstrated that Hg improves the yield of CH2O.2804.4. Non-aqueous reduction
A major challenge in these three CO2 reduction technologies is that CO2 solubility is low in the aqueous phase, which limits reduction rates. Additionally, hydrogen evolution from water competes with CO2 reduction, lowering the solar-energy conversion efficiency. As a result of these two challenges, there has been interest in finding alternatives to aqueous phase reduction.There have been a number of approaches to overcoming these main issues. Ogura demonstrated an electro-catalytic system where CO2 is reduced selectively to ethylene at the three-phase (solid/liquid/vapor) interface of a metal-mesh electrode that is partially immersed in the solution.225 In this way, CO2 is directly supplied to the electrode from the vapor phase, overcoming much of the CO2 transport issue. A similar, common, approach is to utilize gas-diffusion electrodes.281–283 These porous electrodes are composed of hydrophobic materials which facilitates the formation of three-phase interfaces in the pores. Finally, non-aqueous solvents can be used in place of water. Some common examples include acetonitrile,284 methanol,285 dimethyl formamide,252 propylene carbonate,286 and ionic liquids.287–289
4.5. Thermochemical
One-step direct thermolysis of CO2 to CO and O2 is possible at extremely high temperature. At 3075 °C, the reaction is thermoneutral,290 allowing for 100% conversion, but even a modest 30% conversion is possible at 2400 °C.291 Traynor and Jensen developed a prototype solar reactor, operating at 2400 °C, that achieved a peak solar to chemical energy efficiency of 5% with 6% conversion of CO2.292 Assessment of the technology indicates that a mature system may achieve up to 20% solar to chemical energy efficiency. Although these results are promising, the high temperature environment presents stability issues, radiative energy loss is high, and the high temperature CO/O2 separation is a challenge.These problems can be circumvented by employing redox thermochemical cycles, as similarly described for water splitting in Section 3.4. Two-step thermochemical cycles, in particular, are reasonably simple to implement, generate CO and O2 in separate stages, and require lower operating temperature than direct thermolysis.199,293 Usually, these thermochemical cycles involve thermochemical reduction of a metal oxide followed by re-oxidation using CO2 as an oxidant. Generically, the reaction proceeds as follows:
| MOx → MOx−δ + δ/2O2 |
| MOx−δ + δCO2 → δCO + MOx |
The Zn/ZnO thermochemical cycle is one promising technology, which has a theoretical maximum solar to chemical energy conversion efficiency of 39%.291 The reduction of ZnO proceeds at 1600 °C and re-oxidation by CO2 occurs at 360 °C.294 One of the main drawbacks to this process is that the Zn is volatile and must be separated from O2. Ferrites (MxFe3−xO4) present another promising class of materials for thermochemical CO2 splitting, though sintering, formation of liquid phases, and metal vaporization leads to loss of activity over cycling.209,295–297 A recent innovation is the so-called “hercynite cycle” in which a cobalt ferrite spinel (CoFe2O4) decomposes simultaneously with alumina (Al2O3) at 1460 °C to form aluminates.293,298 Then, the alumina and cobalt ferrite spinel regenerate upon oxidation by CO2 at 1000 °C. This material has demonstrated stability over six thermochemical cycles without loss of surface area or activity.
Chueh et al. developed a prototype solar reactor utilizing a CeO2-based thermochemical cycle to simultaneously split CO2 and H2O to produce CO, H2 and O2.299,300 CeO2 reduction occurred between 1420–1640 °C and oxidation at ∼900 °C, and the fuel production was stable over 500 cycles. The solar to fuel efficiency reached 0.8% with the majority of loss from thermal radiation and conduction, while theoretical assessment shows that 16–19% efficiency is possible.
Sandia National Laboratory have adapted their prototype CR5 reactor (as described in Section 3.4) for CO2 splitting.212,213
4.6. Summary of CO2 conversion methods
Overall, there are a number of different routes and strategies for the conversion of carbon dioxide to fuels using solar energy as summarized in Fig. 1. In one broad category, CO2 is converted with hydrogen through mature industrial processes. The CO2 can be converted directly using renewable-solar hydrogen, by RWGS with renewable hydrogen to produce syngas, or by solar-conversion of CO2 to CO followed by reaction with renewable hydrogen (or WGS to generate the hydrogen). In a second broad category, CO2 is directly reduced to fuel through PV-electrocatalytic, photo-catalytic, or photo-electrochemical methods. The former category is advantageous due to the maturity of the conversion methods, yet the simplicity of the conversion process in the latter category is attractive. However, compared with water splitting, the technology and catalysts for CO2 reduction have been much less studied and remain much less efficient. In particular, conversion rates and selectivity are low.5. Process modeling and analysis
In Sections 2–4, we summarized the state of key technologies involved in solar fuels production. In this section, we illustrate how these technologies are integrated into the solar refinery and critically assess what technological innovations are needed in order to develop a feasible process. Towards this goal, we employ process synthesis, modeling, and sensitivity analysis of possible solar refinery configurations. We consider two main cases based on the broad categories, (1) CO2 conversion with hydrogen (Section 5.1) and (2) CO2 reduction using water (Section 5.2). For the first case, we have developed a process model based on mature technology and evaluated the technical feasibility of the process based on an energy balance. For the second case, we first derive the mass balance for a generic solar CO2 reduction process. Based on the mass balance, we have developed a generic energy cost model (that only examines the energy balance of this process) which can be used to evaluate the energy feasibility of a solar CO2 reduction process. These process models include not only reactant conversion processes, but also reactant acquisition (i.e. CO2 capture), and separation processes. The separation steps are important for the overall process because of incomplete conversion of reactants, imperfect reaction selectivity issues, and the high energy requirements for some of these separations. The utility of the general model is that it provides insight into how the various technologies in the solar refinery interact, and the extent to which changes to those individual technologies will affect the overall process mass and energy balances, as well as the process economics.Later, we consider three simple case studies (Section 6) to demonstrate the use of the general process model towards the production of methanol. Methanol was selected as the target product as it is one of the simplest liquid fuels and it can be converted to other chemicals or fuels using mature technologies. We note that this same framework can be easily modified to analyze other systems, or for the production of other fuels and chemicals. We perform a sensitivity analysis to develop targets for these technologies in order to achieve positive primary energy production (Section 7). Finally, we perform an economic assessment (Section 8), which allows us to determine the feasibility of the solar refinery and where future research should be focused.
5.1. Modified CAMERE process for methanol synthesis from solar-derived hydrogen
In this section, we consider the production of methanol using mature technology with solar-derived hydrogen via a modified “carbon dioxide hydrogenation to form methanol via reverse-water–gas shift reaction” (CAMERE) process.12,212,301 A simplified block flow diagram of the process is shown in Fig. 6. In the process, CO2 is captured from a flue gas source (possible technologies are described in Section 2.3) and pumped to the facility. Hydrogen gas, produced using solar energy through one of the many different methods described in Section 3, is combined with CO2.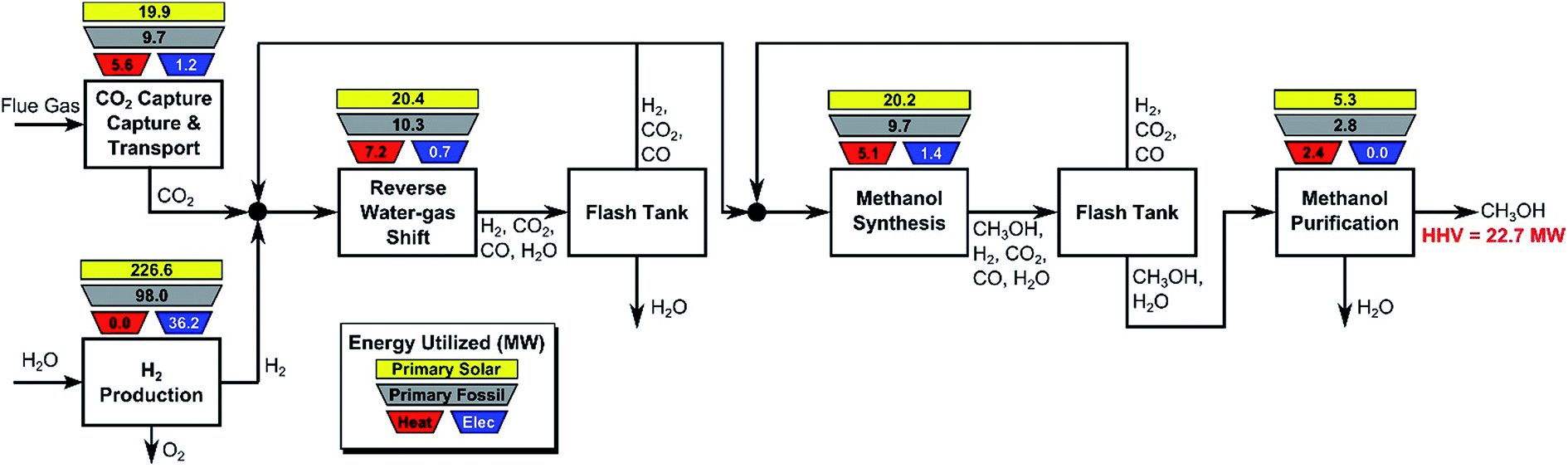 | ||
| Fig. 6 Block flow diagram for modified CAMERE process. The process heating (red trapezoids) and process electricity (blue trapezoids) are indicated for the major process blocks in units of MW. The higher heating value (HHV) of the methanol product is 22.7 MW. The process energy can be provided by fossil fuels or solar utilities (or a combination of both). If all fossil fuels are used, the required primary fossil fuel energy is indicated in the grey trapezoid above each block (assuming 86% fossil fuel to heating efficiency and 37% fossil fuel to electricity efficiency). Alternatively, if all the process energy is supplied from solar utilities, the required solar energy is indicated in the yellow square above each block (assuming 45% solar energy to heat conversion and 16% solar energy to electricity conversion). We do not provide numbers for the case where a mixture of fossil and solar utilities are used, in a given block. | ||
The stream is compressed to 17 bar and heated to 320 °C before entering the reverse water–gas shift reactor, in which CO2 and H2 are partially converted to CO and H2O. The stream is cooled to 35 °C to condense water which is removed in the flash tank, while a fraction of the vapor is recycled to increase overall CO2 conversion and adjust the H2:(CO + CO2) ratio. The remaining CO, CO2, and H2 is compressed to 47 bar and heated to 206 °C before entering the methanol synthesis reactor. The reactor effluent is cooled to 35 °C and flashed to separate volatile CO, CO2, and H2, which are partially recycled to improve methanol yield, with the remainder discarded. The condensed methanol/water stream is expanded to remove residual incondensable material and then purified by distillation. For additional details, see ESI.†
In Fig. 6 the process energy (and equivalent primary energy) required for the main unit operations is reported, with a 1 kg s−1 (28771 Mg per year assuming 333 days per year operation) methanol production basis. In this case, the hydrogen production energy is calculated by assuming a 70% efficient alkaline electrolysis system (1.76 V cell voltage). We assume CO2 is captured from flue gas. The energy required for this is assumed to be 3.1 MJ kgCO2−1 in process electricity and 0.7 MJ kgCO2−1 in process heat. (These numbers are based on Aspen simulations for MEA absorption.)
For a given process, energy is expended to move components, separate components, and convert species. In general, this process energy is derived from fossil fuels (i.e. primary energy). Conventional resources to electricity efficiency (ηE) ranges from 32–38% depending on the fuel mixture and we assume a value of 37% based on the following energy mixture (US 2011): coal (41%), natural gas (25%), nuclear (21%), petroleum (1%), and renewable (12%).302 Natural gas to heat efficiency (ηH) varies from 85–90%, and we assume a value of 86%. If the process energy were provided by solar utilities, we calculate the solar energy required by assuming a 45% solar energy to process heat conversion efficiency34 and a 16% solar energy to electricity conversion efficiency (typical for crystalline silicon).303 The goal of the process is to produce liquid fuel, which can be combusted to produce work or heating. We use the higher heating value (HHV) as a measure of its worth. For methanol, the HHV is 22.7 MJ kg−1. For the process to be feasible, the primary fossil fuel energy consumed should be less than the heating value of the product. Therefore, we define the energy incorporation efficiency (EIE) as:

In general, this efficiency varies from negative infinity to positive 100%. If the value is positive, the energy content in the fuel product is greater than the total fossil fuel energy expended. This is the feasibility point for our process and we refer to this as positive primary energy production. For clarity, at −100% energy incorporation efficiency, the primary energy consumed in the process is twice that of the heating value of the product.
We calculate the energy incorporation efficiency for three scenarios, as described hereafter and summarized in Table 8 and Fig. 7. In Scenario 1, all process energy is provided by conventional fossil fuel utilities. The energy incorporation efficiency for the process is calculated as −477%. Examining the individual blocks in the flow diagram (Fig. 6) it is clear that the energy cost for hydrogen production completely dominates the others. To illustrate this point, we consider a second scenario (Scenario 2) where all process utilities except for the hydrogen production system are provided by solar utilities. This scenario could also be thought of as the case where all other systems have been improved such that there is zero energy cost. Even in this extreme case, the energy incorporation efficiency would only increase to −332% because of the high energy cost for hydrogen production. Therefore, in order to achieve positive energy incorporation efficiency, hydrogen production must utilize solar-based technology (e.g. PV-alkaline electrolysis, photo-electrochemistry, photo-catalysis, thermal splitting). In Scenario 3, the energy of the hydrogen production system is provided by solar energy, while the energy for the other systems is provided by conventional fossil fuel utilities. However, even if all hydrogen production was via solar-energy, the energy incorporation efficiency would still be −43%.
| System block | Primary energy source | ||
|---|---|---|---|
| Scenario 1 | Scenario 2 | Scenario 3 | |
| H2 production | Fossil | Fossil | Solar |
| CO2 capture & transport | Fossil | Solar | Fossil |
| Reverse WGS | Fossil | Solar | Fossil |
| Methanol synthesis | Fossil | Solar | Fossil |
| Methanol purification | Fossil | Solar | Fossil |
| Energy incorporation efficiency | −477% | −332% | −43% |
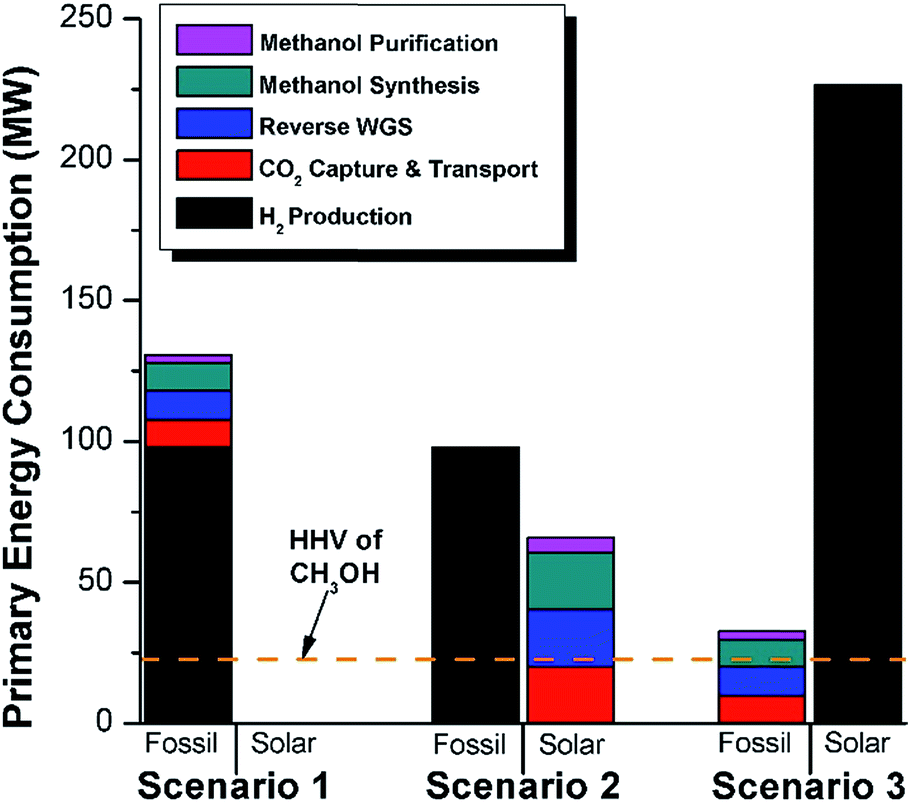 | ||
| Fig. 7 Primary energy consumption for modified CAMERE process scenarios (1, 2, and 3) for 1 kg s−1 methanol production basis. The consumption of primary fossil and primary solar energy for each process system is indicated for each scenario. For comparison, the heating value of the methanol product is 22.7 MW, as indicated in the horizontal dashed orange line. | ||
Overall, this analysis demonstrates that the modified CAMERE process described would only be energetically feasible if the hydrogen were produced using solar energy. Furthermore, even in this case, there must be improvements to reduce CO2 capture costs or incorporate additional solar utilities into the process.
5.2. Solar-driven CO2 reduction process
In the previous section, we demonstrated that a methanol synthesis process from CO2 could almost achieve positive energy incorporation efficiency if the hydrogen were produced using solar energy. An alternative to this process is to produce fuel directly from CO2 and H2O, without forming hydrogen as an intermediate product. Here, we present a generic process for CO2 reduction by water to fuels, as shown in Fig. 8. Possible technologies employed for this conversion were summarized in Section 4. In this reaction, the oxygen is removed as oxygen gas, which must be separated from the fuel product. We divide the generic process into five distinct sub-systems. In the CO2 capture & transport sub-system (A), CO2 is captured from some source (stream 1). The captured CO2 is mixed with purified, recycled CO2 (stream 9) and sent to CO2 reduction sub-system (B). Fresh water (stream 3) is mixed with recycled, purified water (stream 12) and fed to the CO2 reduction sub-system. (In the generic process, we assume that no other solvents are used). In the CO2 Reduction sub-system, CO2 is reduced in a solar reactor to various fuel products with a one-pass CO2 conversion of ξ. The one-pass water/CO2 conversion is denoted by φ/ξ (φ = ξ when there is a stoichiometric feed). The reaction stoichiometry is generically described by:with stoichiometric coefficients νi and products Zi. The sets IV and IL are subsets of I (the set of all species) which are vapor and liquid phase products, respectively. We will, in general, distinguish between vapor and liquid products because these two classes of products will be initially separated before further processing. (The vapor and liquid products are distinguished by their phases at standard conditions.) The carbon selectivity (Si) for species i is defined as:
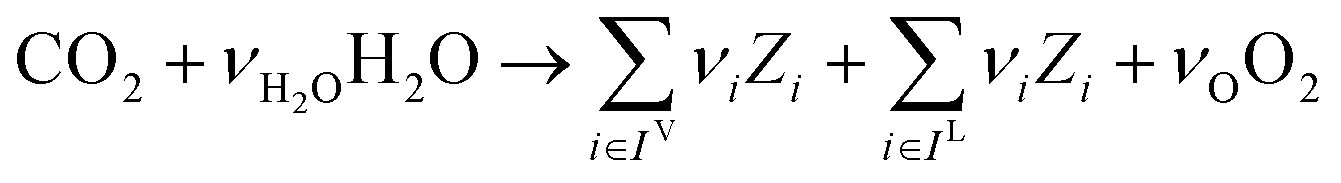
where NC,i is the number of moles of carbon in one mole of species i. Since the stoichiometry of the reaction was written for 1 mole of CO2, the denominator always equals 1. The products, unreacted H2O and unreacted CO2 exit the reactor (stream 5) and are separated into vapor (stream 6) and liquid (stream 7) streams in the gas/liquid separation sub-system (C). The vapor is sent to the Gas Product Purification sub-system (D) where the desired products (stream 8) are purified from by-products (stream 10) and the CO2 (stream 9) is separated and recycled. The ratio of the recovered CO2 to the amount of CO2 entering the system in stream 6 is denoted by ΨCO2. The liquid from the gas/liquid separation sub-system (stream 7) is sent to the liquid product purification sub-system (E) where the desired products (stream 11) are purified from by-products (stream 13) and the unreacted H2O is recycled (stream 12). The ratio of the recycled water to the unreacted water is denoted by ΨH2O.
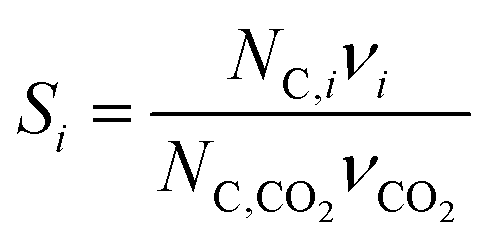
 | ||
| Fig. 8 Block flow diagram for generic solar-driven CO2 reduction process. Process variables indicated in the diagram. | ||
In designing a CO2 reduction process, there are a number of considerations including but not limited to: the source and method of CO2 capture, the technology and catalyst employed for CO2 conversion, and down-stream processing steps including methods of separating products from undesired by-products. A summary of the most important process considerations and options for each of the sub-systems are outlined in Table 9.
| Sub-system | Process considerations and options |
|---|---|
| A: CO2 capture and transport | • What is the source of CO2: atmosphere, post-combustion, pre-combustion, or oxy-combustion? |
| • Which CO2 capture technology should be employed (energy cost, capital cost) | |
| B: CO2 reduction | • What solar reduction technology to use: PV-electrochemical, photo-electrochemical, photo-catalytic, or thermochemical? |
| • What is the optimal catalyst: activity, product distribution (selectivity), and cost? | |
| • What are the reaction conditions: pressure, temperature, concentration of components (cell voltage)? | |
| • How should the oxygen by-product be handled? Can it be separated during reaction (i.e. separation of electrodes, Z-Scheme, etc.)? | |
| • How much catalyst should be used, what is the trade-off between capital cost of reactor and catalyst versus down-stream separation costs? | |
| • Should there be excess water or a solvent? How will product be separated from product? Is solubility of CO2 in the solvent an issue? | |
| C: gas/liquid separation | • What are the components that need to be separated? |
| • What is the best method for separating those components? | |
| D: gas product purification | • Are there desirable products and what are their purity specifications? |
| • Should CO2 be recycled and if so, what CO2 separation technology should be employed? | |
| E: liquid product purification | • Are there desirable products and what are their purity specifications? |
| • Are there any solvents or electrolytes that must be recovered? |
We note that both the gas and liquid product purification steps include only one product stream each (streams 8 and 11, respectively) and a single purge stream each (streams 10 and 13, respectively). In an actual process, there may be multiple liquid and multiple vapor phase products (some desirable and others undesirable) that are separated into purified components (or lumped components) in these sub-systems. Stream 8 (and 11) represents the total mass of multiple product streams, each one purified to a desired specification. Similarly, stream 10 (and 13) represents the sum total of discarded materials. This includes purged materials to prevent build-up of components, separated undesirable by-products and mass losses due to imperfect separations (in particular CO2 mass losses are included in the variable ΨCO2). In general, the separation blocks could be replaced with a purge (i.e. discarding a percentage of the materials without separating components). The advantage of such a scenario is that there is no direct capital or energy cost. However, indirectly, valuable product or reactants are lost, and the process must be scaled in order to reach a desired production basis. Also, since obtaining reactants requires energy (i.e. CO2 capture), the trade-off may be sub-optimal, as we will demonstrate later. There may be no choice but to separate materials in order to meet a product specification. In a related note, in the generic process, we include recycle streams for the CO2 (and also the water). Their existence is motivated by reported low conversions and the high cost for CO2 capture (which must be significantly increased if un-reacted CO2 is discarded). We will demonstrate the importance of the CO2 recycle later.
where Fj,i is the molar flow rate of component i in stream j and the set IP includes all product species which are deemed “valuable,” i.e. undesired by-products are not included in this set. For a different basis, HHV could be replaced with another quantity (e.g. molecular weight for mass of product basis, molar volume for volume of fuel basis, etc.). We define the variable θ as:

which describes how many moles/time of CO2 must be consumed in the reactor to meet a given production basis, and it functions as a scaling factor for the overall process (with units of moles/time). For a given value of X, the value of θ varies depending on the reaction stoichiometry and how each product is weighted. We then solved for the molar flow rates of the various species in each stream. The most important results are displayed in Table 10.

| Stream | Molar flow rate |
|---|---|
| 1 |

|
| 2 |
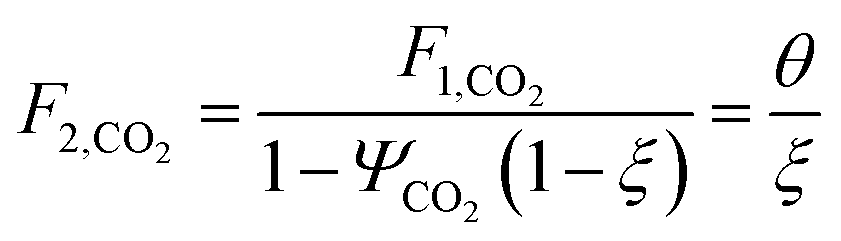
|
| 3 |

|
| 4 |

|
| 5 |
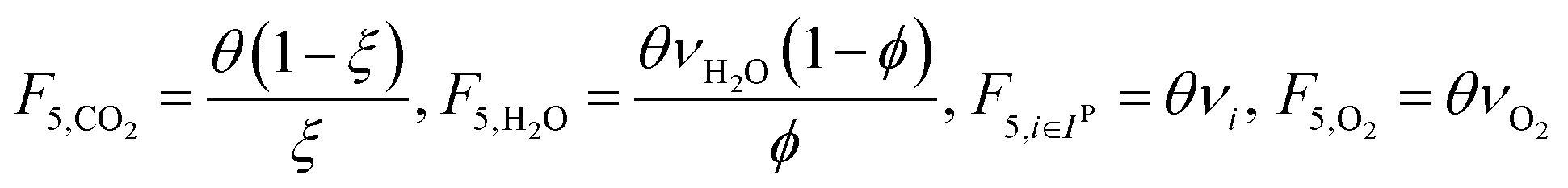
|
| 6 |

|
| 7 |

|
The mass of CO2 that must be captured and transported to the solar refinery is a function of the amount of CO2 consumed during the process. This includes the converted CO2 as well as the amount lost during purification/processing. Since we assumed that the amount of recovered unreacted CO2 is a fraction of the total amount unreacted, both the CO2 conversion and recovery ratio affect the amount of fresh CO2 that must be brought into the refinery. If all of the unreacted CO2 were recovered (ΨCO2 = 1) or all of the CO2 were converted (ξ = 1), then F1 = θ, and is a constant (for a given process size). As a result, there will always be a cost associated with CO2 capture and transport.
The fresh and recycled CO2 and water streams enter the solar reactor and are converted to products. The feed streams (2 and 4) may need to be heated or compressed before entering the reactor. The amount of material processed is inversely proportional to their respective conversions. At 100% conversion, the amount of material is a constant value.
After conversion, the vapor and liquid products are separated. In the simplest case, this could be accomplished through a series of flash tanks. However, in general this may require more advanced separation technology to achieve reasonable separation, requiring heating and/or compression. The relative masses of the products (vapor, liquid, and oxygen) are constant for a given process basis, while the amount of water and CO2 in the stream are functions of their respective conversions. Specifically, the amount of water and CO2 is inversely proportional to their respective conversions, though at 100% conversion the amount reaches zero.
The gas product must be separated from CO2 and O2. The amount of product and O2 are proportional to θ and their stoichiometric coefficients, while the amount of CO2 varies inversely with the conversion. Therefore, the composition of the gas could vary significantly with conversion. At low conversion, CO2 will be in high concentration, while at 100% conversion, there will be zero CO2 in the stream. The optimal product/CO2 separation strategy will likely change with respect to this composition. Even in the case of 100% conversion, the product would still need to be separated from a constant amount of O2. This could be avoided by choosing a CO2 reduction technology where the CO2 is reduced separately from oxygen evolution. This is possible by separating the anode and cathodes in electro-catalytic or photo-electrocatalytic systems. In photo-catalytic systems, separation of the two reactions has been demonstrated for water splitting (there, hydrogen evolution and oxygen evolution) using the Z-scheme, at the cost of solar efficiency.
The liquid product must be separated from water. The amount of product in the stream is proportional to θ and the stoichiometric coefficient. The amount of water is inversely proportional to the conversion of water, reaching zero at 100% conversion. Unlike the vapor product separation, there is not always an additional by-product (i.e. O2) that must be separated. Though, for certain CO2 conversion technologies, there may be additional components in the stream including electrolytes, other solvents, or other undesirable by-products that must be removed.

The energy function of these sub-systems depends on the specific technology being employed, the condition of the incoming streams, the specific components in those streams, and the mass of the components in those streams. For some systems, the cost may be proportional to the mass of a specific component. For instance, the cost of CO2 capture from MEA absorption has been calculated to be proportional to the mass of the CO2 captured.
We have developed energy cost functions for the five sub-systems, which are explained hereafter. In general, the energy cost for each sub-system, k, (Ek) was found to be proportional to the mass (volume) being processed by that sub-system. As a first approximation, the cost functions were written as the unit cost to process species i, multiplied by the flow rate of species i entering the sub-system in stream j (Fj,i):
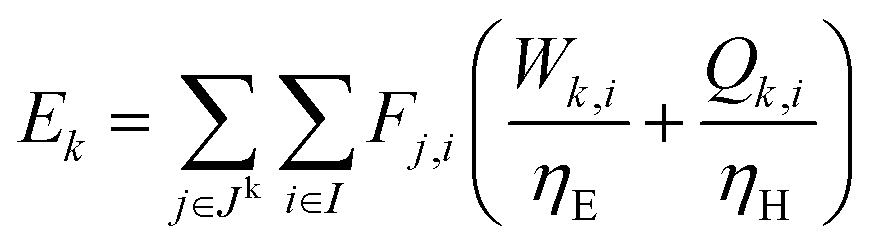
6. Case study: CO2 reduction to methanol using solar energy
The general framework that we developed in the previous section could be applied to analyze a variety of different systems. To illustrate this, we apply our process and energy models to a specific case: CO2 photo-catalytic reduction to methanol with a methane by-product. Methanol (and methane) is produced directly, in a single-step, by photo-catalytic reduction of CO2 based on results reported by Ikeue and co-workers using a Ti-containing porous silica photocatalyst.3046.1. Process model
We assume a CO2 one-pass conversion of 5% with a methanol selectivity of 40% (methane selectivity 60%). Based on the selectivity, the reaction stoichiometry is as follows:| CO2 + 2H2O → 0.6CH4 + 0.4CH3OH + 1.8O2 |
We assume the reactor is fed a stoichiometric CO2:H2O feed (implying that the water conversion is equal to the CO2 conversion). We consider three case studies (we refer to these as Case I, II, and III to distinguish from previous Scenarios 1, 2, and 3), which are summarized in Table 11. Block flow diagrams of these process models are provided in the ESI.†
| Sub-system | Case I | Case II | Case III | |||
|---|---|---|---|---|---|---|
| Technology | Process parameters | Technology | Process parameters | Technology | Process parameters | |
| a Designates a dimensionless quantity. b The energy cost function for the distillation system is a more complicated power-law expression, see Section 6.2. | ||||||
| A: CO2 capture and transport | MEA absorption from post-combustion flue gas | W A,CO2 = 0.7 | MEA absorption from post-combustion flue gas | W A,CO2 = 0.7 | Physical absorption from oxy-combustion process | W A,CO2 = 0.42 |
| Q A,CO2 = 3.1 | Q A,CO2 = 3.1 | Q A,CO2 = 0 | ||||
| B: CO2 reduction | Ti–SiO2 Photo-catalyst. | Q B,CO2 = 0.021 | Ti–SiO2 Photo-catalyst | Q B,CO2 = 0.021 | Ti–SiO2 Photo-catalyst | Q B,CO2 = 0.021 |
| Q B,H2O = 0.104 | Q B,H2O = 0.104 | Q B,H2O = 0.104 | ||||
| C: gas/liquid separation | Series of flash tanks | W C,CO2 = 0.19 | Series of flash tanks | W C,CO2 = 0.19 | Series of flash tanks | W C,CO2 = 0.19 |
| W C,O2 = 0.28 | W C,O2 = 0.28 | W C,O2 = 0.28 | ||||
| W C,CH4 = 0.55 | W C,CH4 = 0.55 | W C,CH4 = 0.55 | ||||
| D: gas product purification | None | Ψ CO2 = 0a | Physical absorption of CO2 | Ψ CO2 = 0.97a | Physical absorption of CO2 | Ψ CO2 = 0.97a |
| W D,CO2 = 0.42 | W D,CO2 = 0.42 | |||||
| E: liquid product purification | Distillation | m = 2.15b | Distillation | m = 2.15b | Distillation | m = 2.15b |
| b = −0.71a,b | b = −0.71a,b | b = −0.71a,b | ||||
In Case I, CO2 is captured from the dilute flue gas stream via MEA absorption and transported to the solar refinery. The CO2 stream is mixed with water and heated to 50 °C before entering the photo-reactor. In the solar-reactor, CO2 and H2O are converted using solar energy to methanol, methane and O2. A series of flash tanks operating from 20 to 3 atm separate the vapor (CO2, O2, CH4) from the condensables (CH3OH, H2O). The CO2/O2/CH4 stream is vented, while the methanol is purified to 99.8% purity using a series of distillation columns with the water recycled (please see ESI† for more details). In Case II, the process is generally the same as Case I, except that unreacted CO2 is recovered and recycled back to the solar reactor. The concentrated CO2 in the vapor stream is separated by physical absorption using Selexol™305,306 and recycled to the solar-reactor. The CH4/O2 stream is vented. Finally, Case III is the same as Case II except that the initial CO2 capture occurs from an oxy-combustion process through physical absorption using Selexol™.
6.2. Energy model
The specific functional forms and unit cost parameters (Wk,i and Qk,i) for the generic energy cost functions were evaluated for the systems in the case studies using Aspen simulations (please see ESI† for specific details) and data from the literature, and are shown in Table 11. We caution that, in general, the energy costs that we derive from these simplified models are likely under-estimating the actual energy required for a given process.where F1,CO2 is the molar flow rate of CO2 captured, MCO2 is the molecular weight of CO2, WA,CO2 is the unit work required per mass of CO2, and QA,CO2 is unit heating required per mass of CO2. Based on Aspen simulations of the MEA absorption process, we have calculated values WA,CO2 = 0.7 MJ kg−1 and QA,CO2 = 3.1 MJ kg−1. For physical absorption using Selexol™, we use literature results for a 97% CO2 recovery system, WA,CO2 = 420 kJ kg−1 and QA,CO2 = 0.307 We will refer to the term

 as the “specific CO2 capture cost”.
as the “specific CO2 capture cost”.
where F2,i is the molar flow rate of component i in stream 2, Mi is the molar mass of component i, WB,i is the work required per mass of i and QB,i is the heating required per mass of i. Assuming that the feed contains only CO2 and water, we have evaluated the heat and work required to perform CO2 reduction at 50 °C and 1 atm. From Aspen simulation, we calculate QB,CO2 = 21.5 kJ kg−1 and QB,H2O = 104.1 kJ kg−1, with all other parameters equal to zero (assuming no pumping or compression is required). In reality, due to imperfect separations there may be other components in the stream which may need to be included in the energy cost function. We note that because solar irradiation may be directly used in the solar reactor, it is possible that the heating costs could be derived from this irradiation. Also, this does not account for the solar energy that is consumed in the actual chemical transformation.

where F5,i is the molar flow rate of component i in stream 5, Mi is the molar mass of component i, WC,i is the work required per mass of i and QC,i is the heating required per mass of i. For the separation of CO2, CH4, and O2 from CH3OH and H2O, the stream is compressed to 22 atm to condense residual water. Using a multi-stage compressor, the work required for each of the components was calculated as WC,CO2 = 0.19 MJ kg−1, WC,O2 = 0.28 MJ kg−1 and WC,CH4 = 0.55 MJ kg−1, respectively. The compression work could also be estimated by assuming ideal gas behavior. The reversible adiabatic work of compression for species X is given by:

where γ is the “heat capacity ratio”, the ratio of the constant pressure heat capacity to constant volume heat capacity, T1 is the initial temperature, P1 is the initial pressure and P2 is the final pressure. For the species considered here, the percent error in these ideal gas values are <10% of the simulated results.
where WD,CO2 and QD,CO2 are the work and heat required per mass of CO2. Assuming that the separation is via physical absorption in Selexol™, we again assume WD,CO2 = 420 kJ kg−1 and QD,CO2 = 0.307 We note that the cost could be reduced by lowering the CO2 recovery ratio, but this would require additional CO2 capture to balance the mass that is lost.

where m and b were the results of the fitting, 2.15 MJ kg−1 and −0.71, respectively.

6.3. Model results
Using these cost functions and the mass balances, we evaluated the primary energy expenditures in each of the five sub-systems for the three case studies as shown in Fig. 9. The input parameters for the mass balances and cost model are given in Table 11. First, we note that all three cases have negative energy incorporation efficiencies, with Case I being the worst performer. Clearly, recycling unreacted CO2 is of tremendous benefit. The reason for this is that the energy cost to produce 1 unit of CO2 from dilute streams (i.e. via MEA absorption) is much higher than that from concentrated CO2 streams (i.e. in the gas product purification sub-system). Therefore, there will always be an energetic benefit of recycling CO2 as long as the cost per unit is less than the initial cost of capturing CO2. This simple calculation shows the importance of separations in terms of energy efficiency of the integrated process for solar fuels production. Comparing Case II with Case III, we see that changing the source of CO2 (i.e. lowering the energy cost per unit of CO2) has a significant effect on the overall energy efficiency, but the cost is relatively small compared with the energy costs for the other sub-systems. The reason for this is that the mass flow rates through the other sub-systems (B–E) is much more strongly affected by CO2 one-pass conversion. Since all of these cases are operating at relatively low conversion (5%), the energy costs for the other systems dominates. Interestingly, if we increase the CO2 one-pass conversion, we find that neither Case I nor Case II is able to achieve positive energy incorporation efficiency even at 100% conversion. This is due to the high specific cost of CO2 capture (see Table 11). In comparison, Case III achieves positive energy incorporation efficiency at 45% CO2 one-pass conversion. We will revisit the sensitivity of the process to conversion in the Section 7. | ||
| Fig. 9 Primary energy expenditure for each sub-system for the three case studies. The heating value of the methanol product is 22.7 MW. The energy incorporation efficiency (EIE) for each case study is written above the respective bars. | ||
7. Sensitivity analysis of case II
In this section, we build upon results from the previous case studies by performing sensitivity analysis on our Case II model (even though Case III had the most positive energy incorporation efficiency of the three cases studied), and will refer to it as the base case. We choose Case II as the basis because CO2 capture from flue gas is easily retrofitted on existing power plants, and represents the most near-term solution for CO2 capture. Therefore, unless otherwise indicated, the process parameters for the mass balance and energy balances are taken from that case.7.1. Sensitivity to CO2 one-pass conversion
First, we evaluated the sensitivity of the process to changes in CO2 one-pass conversion (ξ). The results of this analysis are shown in Fig. 10. We caution the reader that since the original process was developed for low conversion, the optimal separation methods and technologies may change with conversion (or selectivity, later), which is not captured by our model. | ||
| Fig. 10 Primary energy costs for photo-catalytic reduction of CO2 to methanol sub-systems as a function of CO2 one-pass conversion with a 1 kg s−1 methanol production basis (22.7 MW HHV). Corresponding energy incorporation efficiency is plotted in green dashed line on the secondary y-axis. | ||
First, one of the most important results of this analysis is the shape of these curves. The energy use scales inversely with conversion. Therefore, the energy incorporation efficiency improves as CO2 one-pass conversion increases, at a decreasing rate (i.e. diminishing returns). Secondly, the results show that at low CO2 conversion (<14%), the energy consumption in the gas product purification sub-system is the most significant cost. In the base case for this sub-system, CO2 is separated by physical absorption in Selexol™. The overall energy cost of this sub-system is high because (1) of the high specific cost of CO2 separation, and (2) the mass being separated is inversely proportional to the conversion. Both of these effects contribute to very high total energy costs at low conversion. The next most significant costs are for methanol purification and gas/liquid separation, which also scale inversely with conversion. Above 14% one-pass conversion, the most significant energy cost is the CO2 Capture & Transport sub-system. In the base case, CO2 is captured from flue gas using MEA absorption. The energy cost decreases slightly with increasing CO2 conversion due to fewer losses during separation, ultimately plateauing at a cost of 18.9 MW at 100% CO2 conversion (corresponding to 83% of the total heating value of the product). Therefore, even if CO2 conversion could be improved by reaction engineering and catalyst design, it would be difficult to achieve positive primary energy efficiency without reducing the energy cost for CO2 capture.
7.2. Sensitivity to CO2 capture costs
As the CO2 Capture & Transport sub-system represents the most significant energy cost at moderate to high conversion (ξ > 14%), we have calculated the total primary energy cost (Fig. 11) and energy incorporation efficiency (Fig. S6†) while also varying the specific CO2 capture cost, . (For reference, the specific CO2 capture cost for our simulated MEA absorption process is 5.5 MJ kgCO2−1).
. (For reference, the specific CO2 capture cost for our simulated MEA absorption process is 5.5 MJ kgCO2−1).
 | ||
| Fig. 11 Total primary energy cost for photo-catalytic reduction of CO2 to methanol process for 1 kg s−1 methanol basis (22.7 MW HHV) as a function of CO2 one-pass conversion and specific CO2 capture cost. A contour line for positive energy incorporation efficiency (22.7 MW primary energy cost) is indicated within and for positive energy incorporation efficiency while including the heating value of 25%, 50%, and 75% of the produced methane. | ||
As we noted from the previous analysis, the CO2 capture system encompasses a greater fraction of the overall energy cost with increasing CO2 one-pass conversion. Furthermore, the energy cost for the CO2 capture system asymptotes to a non-zero value quite rapidly with increasing CO2 conversion. In particular, the energy cost is only 10% higher than its asymptote value at 33% CO2 one-pass conversion. Therefore, at mid to high conversion, the overall energy cost for the system can be considered to be proportional to the specific CO2 capture cost. These results manifest in Fig. 11 by changes in the shape of the contour lines with conversion. At low conversion, improving CO2 capture has an insignificant change in the primary energy efficiency, and the contours are nearly parallel with the CO2 capture cost axis. At moderate and high conversion, the contour lines are sloped with respect to the CO2 capture cost axis, and they can significantly affect the efficiency (even though the magnitude of the change in the cost is smaller at higher conversion).
If 100% conversion can be achieved, the specific CO2 capture cost must be less than 4.4 MJ kgCO2−1 in order to achieve positive energy incorporation efficiency, which is within the range reported in the literature (see Section 2.3). However, even if the cost of CO2 capture were reduced to zero, a CO2 conversion of 39% would be required. CO2 capture costs as low as 0.5 MJ kg−1 have been reported for membrane separations, which would require a CO2 conversion of ∼42% to have positive primary energy production. Adsorption processes have been quoted in the 2–3 MJ kg−1 range, which require 54% and 69% conversion, respectively.
7.3. Sensitivity to product selectivity
As the CO2 capture system represents one of the key energy costs in the overall process, reaction selectivity can be considered one of the main drivers for the overall cost. The reason for this is that by converting CO2 to unwanted products, we not only waste the raw materials but we also waste energy to capture that material. However, if the by-product could be separated or combusted to provide process heating, the overall energy efficiency would improve. To quantify this effect, we first consider the case where the CH4/O2 stream is combusted to produce heat for the system. In Fig. 11, we plot contours corresponding to positive primary energy production, with the inclusion of a fraction of the methane heating value (25%, 50%, and 75%). The plot shows that recovering even a small portion of the methane heating value can significantly reduce the required CO2 conversion or capture costs. Additional details and discussion are provided in the ESI.†An alternative to methane combustion is to develop a photo-catalyst (or alternative CO2 reduction process) which is more selective for the desired product. In Fig. 12 we plot the primary energy costs for the five main sub-systems as functions of methanol selectivity at 10%, 40%, and 70% CO2 one-pass conversion. For our case, where only C1 species are produced, the carbon selectivity is equal to the stoichiometric coefficient, i.e. SCH3OH = νCH3OH. For a 1 kg s−1 methanol production basis, θ = 1 kg s−1/(SCH3OHMCH3OH) and therefore one might conclude that the molar flow rates of the streams should scale inversely with the methanol selectivity. For CO2 and H2O, this is true, and the costs follow the same dependence, as shown in Fig. 12. Reactor heating costs are also proportional to water and CO2 masses; inversely proportional to selectivity and the energy cost follows the same result. However, the dependence of methane and oxygen (and for alternative products, water as well) flow rates on selectivity is more nuanced. By mass balance, νO2 = (4 − SCH3OH)/2 and νCH4 = 1 − SCH3OHs. The corresponding flow rates are F6,O2 = θνO2 and F6,CH4 = θνCH4, respectively. Therefore, at low selectivity, they behave as if they scale inversely with selectivity, while at high selectivity they approach a non-zero constant value (since selectivity cannot exceed 1, none of the costs reach zero). In Fig. 12, it can be seen that for a given CO2 one-pass conversion, the relative ordering of energy costs for the sub-systems does not change with respect to selectivity, e.g. in Fig. 12a CO2 separation is the most costly regardless of selectivity. The main exception to this rule is the methanol purification system, which is a complicated function of selectivity. The cost of the purification varies with conversion, scaling as S−0.71 at low water conversion and as S0 (i.e. independent of selectivity) at high conversion (see ESI†).
 | ||
| Fig. 12 Primary energy costs for photo-catalytic reduction of CO2 to methanol sub-systems as a function of CH3OH selectivity with a 1 kg s−1 methanol production basis (22.7 MW HHV) for (a) 10% CO2 one-pass conversion, (b) 40% CO2 one-pass conversion, (c) 70% CO2 one-pass conversion. | ||
Overall, due to these scaling relationships, the energy costs for all of the process sub-systems can be significantly reduced by improving reaction selectivity. If the selectivity were doubled from 40% to 80%, the cost of the CO2 capture system, the reactor heating, and CO2 separation would all be halved. Similarly, the energy cost for gas/liquid separation would be reduced by slightly more than 50%, while the energy cost for methanol purification would be reduced by slightly less than 50% (depending on the conversion).
Finally, we consider the combined case where specific CO2 capture costs are reduced and reaction selectivity improves. For a given specific CO2 capture cost and reaction selectivity, we calculated the minimum CO2 one-pass conversion which would be required to achieve positive EIE. The results are shown in Fig. 13. In order to achieve positive energy incorporation efficiency, a minimum of 15% CO2 one-pass conversion is required. Beneath this value, even if the catalyst were 100% selective to the product, CO2 would need to be supplied at a negative energy cost. If catalyst selectivity and CO2 capture costs were improved by 25% with respect to the base case (i.e. 50% selectivity and 4.13 MJ kgCO2−1 capture cost), the CO2 conversion would need to be increased to 60% in order to achieve positive energy incorporation efficiency. With 50% improvement in both (60% selectivity and 2.75 MJ kgCO2−1 capture cost) conversion would need to exceed 37%.
 | ||
| Fig. 13 Minimum CO2 one-pass conversion required to achieve positive energy incorporation efficiency as a function of methanol selectivity and specific CO2 capture costs. Each contour line corresponds to a change in the minimum conversion of 5%. Since the maximum possible conversion is 100%, at high specific CO2 capture costs and low methanol selectivity (upper left corner of contour plot) it is impossible to reach positive energy incorporation efficiency. | ||
8. Economic analysis
In our previous analysis, we demonstrated the application of a simplified energy cost model towards the development of improved solar to fuels processes. In that analysis, we did not account for the economic (operating and capital) costs. Importantly, the cost for the solar energy is completely discounted (i.e. free). Therefore, to understand the role of process economics and identify the major cost drivers, we developed a simple economic model for the estimation of a methanol minimum selling price (MSP). Based on discounted cash flow analysis (see Table 12 for major assumptions and ESI† for additional details), we calculated the MSP; i.e., the price of methanol that covers exactly all operating costs, income tax and return on investment. The utilities costs were scaled to the process (as a function of conversion, selectivity, etc.) using the mass balance and energy cost models as presented in the previous sections. For the base-case scenario, the cost to transport CO2 and H2O to the solar refinery would be 0.01 USD kgmethanol−1 and 0.002 USD kgmethanol−1, respectively, assuming 100 km transportation distances. Since these costs are quite low, we do not consider them in the model. We used a low-pressure steam price of 10.50 USD MT−1 and an electricity price of 0.06 USD kWh−1.308 The direct capital costs for all of the sub-systems were also scaled using the mass balances and the following equation:where Co is the direct capital cost calculated for a given mass flow through the system (Vo), and V is the mass flow through the scaled system. The direct capital cost for the CO2 capture system and Selexol™ system were scaled using only the CO2 mass flow rates, while the other units were scaled with respect to the total mass flowing through the respective sub-system. Specific details of the capital costs for the five sub-systems, operating costs, and revenues are presented in the ESI.† All capital costs are scaled to a reference year of 2011 based on the Chemical Engineering Plant Cost Index (CEPCI).
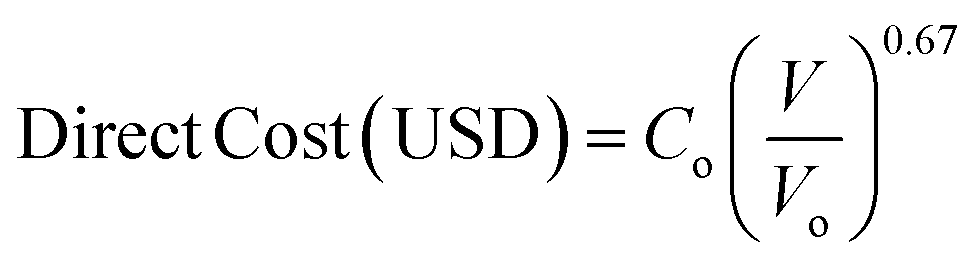
| Project economic life (year) | 30 |
| Methanol production rate (Mg per year) | 28771 |
| Rate of return (% per year) | 8% |
| Tax rate (% per year) | 35% |
| Depreciation method | Straight line |
First, we performed an economic evaluation of the base-case (Case II, see Table 11) while assuming that all costs (aside from the low-cost of pre-heating) associated with the CO2 reduction sub-system were zero. The results of the economic evaluation including operating costs, total project investment and minimum selling price are in Table 13. For comparison, the industrial price of methanol fluctuates considerably, with a price of 0.632 USD kgmethanol−1 in March 2014 (Methanex, non-discounted reference price in March 2014). The calculated minimum selling price is nearly 3 times higher than the industrial price, without including costs associated with the CO2 reduction sub-system. As was described in the previous sections, the low conversion (5%) leads to large recycles and therefore high costs for utilities and capital.

| Electricity (USD per year) [USD kgmethanol−1] | 22, 156, 218 [0.77] |
| Low pressure steam (USD per year) [USD kgmethanol−1] | 8, 511, 073 [0.29] |
| Fixed operating costs (USD per yr) [USD kgmethanol−1] | 10, 279, 745 [0.36] |
| Total project investment (USD) [USD kgmethanol−1] | 108, 588, 856 [0.13] |
| Minimum selling price (USD kgmethanol−1) | $1.87 |
8.1. Sensitivity analysis
Next, we performed sensitivity analysis on the process to understand how this impacts the economics. Here, we consider the effect of improvements to CO2 one-pass conversion and methanol selectivity (with all other parameters given by the base-case scenario). In Fig. 14a, we show the calculated minimum selling price of methanol as a function of CO2 one-pass conversion and methanol selectivity. The point in the bottom left corner of the plot (5% CO2 one-pass conversion and 40% methanol selectivity) corresponds to the base-case analysis, discussed previously. The results show that by increasing the one-pass conversion to ∼12.5%, the minimum selling price of methanol can be halved from the base-case scenario. In contrast, the change in minimum selling price with respect to improvements in methanol selectivity are more modest. The selectivity would need to be improved to ∼90% (at 5% CO2 one-pass conversion) in order to halve the minimum selling price. In order to meet the industrial selling price of methanol, 0.63 USD kg−1, the CO2 one-pass conversion must be improved to 22% (without improving methanol selectivity). In contrast, by only improving selectivity, this price cannot be met. As we showed earlier, the process scales inversely with both conversion and selectivity, rationalizing the large improvements in process economics with small changes in conversion. Furthermore, this scaling explains why only modest improvements in minimum selling price can be accomplished by improving selectivity. | ||
| Fig. 14 (a) The minimum selling price of methanol (USD kg−1) for base-case scenario with improvements in methanol selectivity and CO2 one-pass conversion. The upper right region, demarcated with a white line, shows the conditions (selectivity and conversion) that achieves positive energy incorporation efficiency. (b) The minimum selling price of methanol (USD kg−1) calculated as in (a), but the conventional fossil fuel utilities are replaced with just enough solar utilities such that the energy incorporation efficiency is non-negative at all points in the phase-space. The inset of each plot shows the low-conversion and low-selectivity region with higher resolution (40–60% CH3OH selectivity, 5–15 % conversion). All capital and energy costs associated with the CO2 reduction (B) sub-system are set to zero. | ||
8.2. Solar utilities
In the previous sensitivity analysis, we demonstrated how improvements in methanol selectivity and CO2 one-pass conversion affect the minimum selling price of methanol. An important thing we did not consider in the previous analysis was the process energetics. In Fig. 14a, we demarcate the process feasibility region, the region where the energy incorporation efficiency is non-negative. Interestingly, the minimum selling price of methanol is less than 0.30 USD kg−1 for all points (selectivity, conversion) in this region. As the goal is to produce methanol at a competitive price, with positive energy efficiency, and with minimum improvements in process technology, we can afford to expand the feasibility region outward (to lower conversion and selectivity) by trading monetary costs for energy costs by employing solar utilities.In our original economic analysis, we used fossil fuel utilities prices of 0.060 USD kWh−1 for electricity and 10.50 USD MT−1 (∼0.017 USD kWh−1) for process steam. In comparison, the levelized cost of solar-PV electricity has been studied quite extensively, and we will assume a value 0.144 USD kWh−1.56 In contrast, the cost of solar process heating has not been studied as extensively, we will use a literature value of 0.05 € kWh−1 (0.069 USD kWh−1 assuming a 1.38 USD to 1 € exchange rate).309 Based on these prices, we found that it is more economical to first replace fossil fuel electricity with solar-PV electricity; and then to replace fossil fuel heating with solar heating, see the ESI† for more details.
With this result, we revisited the selectivity study from the previous section and re-calculated the minimum selling price as a function of methanol selectivity and CO2 one-pass conversion. However, we allowed for solar utilities to be used in addition to fossil fuel utilities, and imposed the constraint that the energy incorporation efficiency is non-negative. The results are shown in Fig. 14b. Based on the assessment of cost of primary energy avoidance, at each point in the plot (selectivity, conversion) the minimum selling price is achieved by first replacing fossil fuel-derived electricity with solar electricity and then fossil fuel-derived heating with solar heating until the energy incorporation efficiency becomes non-negative. For regions in the phase-space where the conversion and selectivity are both low, energy costs present a high fraction of the overall costs (please see ESI† for cost distribution) and therefore replacing those with solar utilities greatly increases the minimum selling price (see inset of Fig. 14b). For example, for the base-case scenario (5% conversion, 40% selectivity), the MSP of methanol increases from 1.87 USD kg−1 to 3.39 USD kg−1. If the CO2 one-pass conversion increases (with 40% selectivity fixed), the industrial price of methanol (0.63 USD kg−1) is achievable at 38% conversion, whereas with just fossil fuels utilities, this is reached at 22% conversion (the MSP with solar utilities is 0.97 USD kg−1 at 22% conversion and 40% selectivity).
8.3. CO2 reduction sub-system
In the previous economic analyses, operational and capital costs associated with of the CO2 reduction sub-system were omitted. The reason for this omission is that there are a wide variety of technologies available for CO2 conversion and types of solar reactor architectures, as well as limited available literature on continuous processes. For a specific CO2 reduction process, the minimum selling price for methanol (or a different product) can be found by calculating the levelized cost of that specific system with respect to the amount of product produced (i.e. cost per unit produced) and adding that value to the calculated MSP from the previous section(s), which is a function of CO2 one-pass conversion, product selectivity, and solar energy utilization. To demonstrate this procedure and provide some benchmarks of costs for this sub-system we consider two simplified cases that are meant to illustrate the state of the technology. First, we consider the case of CO2 electro-reduction using PV-solar electricity. In the second case, we consider CO2 photo-catalytic reduction.The equilibrium cell potential for CO2 electro-reduction to methanol is 1.23 V, see Table 7. Assuming 100% selectivity (no energy is wasted to produce by-product) and 100% electrolyzer efficiency (i.e. no overpotential losses), 22 MW of electricity is required to produce 1 kg s−1 of methanol. Using a levelized cost of solar electricity of 144.3 USD MWh−1 from the literature,56 the cost of the PV-solar electricity is 0.88 USD kgmethanol−1 (corresponding to a capital cost of $759,554,400). Therefore, the MSP of methanol would be at least 0.88 USD kgmethanol−1 more than is indicated in Fig. 14, at every point (though this value would be under-estimating the electricity cost at lower selectivity). We note that, assuming 6 kWh m−2 average daily incidence with 16% solar to electricity conversion (typical for crystalline silicon),303 this would require 551, 562 m2 (136 acres) of incidence area. In comparison, the levelized cost of electricity from a natural gas-fired conventional combined cycle is 67.1 USD MWh−1.56 Therefore, if the electrolyzer were powered by natural-gas energy, the minimum selling price would be increased by 0.55 USD kgmethanol−1. More realistically, CO2 electro-reduction to methanol has been demonstrated with ∼60% faradaic efficiency at −0.8 VSCE on RuO2 electrodes.226 Including an additional assumed 0.5 V oxygen evolution overpotential, the overall electricity cost would be 66 MW, or 2.64 USD kgmethanol−1 for the solar-PV electricity. We note that this does not include costs associated with the actual electrolyzer unit or any electrocatalysts. As the cost of solar electricity completely dominates the overall process cost, improvements in PV technology would be paramount towards achieving a low minimum selling price. Additionally, improvements in electrocatalyst performance through high faradaic efficiency and low overpotentials would have a tremendous effect on the economics.
Photo-catalytic systems may have lower costs for the solar energy because it does not need to be converted into electricity. However, reported photo-catalytic reaction rates are very low, on the order of ∼0.1–10 μmolproduct gcat−1 h−1.8,304,310,311 In contrast, methanol synthesis rates on commercial Cu/Zn/Al2O3 catalysts (498 K, 50 bar, gas hourly space velocity (GHSV) = 10000) range from 0.3–1.5 kgmethanol Lcat−1 h−1 (using a bulk catalyst density of 1.2 g cm−3, the rates are 7800–39000 μmol gcat−1 h−1).312 As a result, though the cost of the solar energy may be lower, the cost of the photo-catalyst will be quite significant. Assuming a catalyst activity of 1 μmolproduct gcat−1 h−1 and that the required amount of photo-catalyst scales linearly to meet the 1 kg s−1 methanol production rate, 1.1 × 108 kg of TiO2 catalyst would be required. At a price of 1 USD kgTiO2−1, assuming a catalyst lifetime of one year, the additional operating cost would be 3.91 USD kgmethanol−1 ($112500000 per year), thereby increasing the minimum selling prices reported in Fig. 14 by 3.91 USD kgmethanol−1. At a more ambitious catalyst lifetime of 10 years, the additional operating cost would still be 0.39 USD kgmethanol−1. Though these parameters could be off by ∼1 order of magnitude, the calculated cost serves to illustrate the importance of improving the photo-catalytic activity towards developing an economical process.
To assess any other CO2 reduction process, if the levelized cost of the CO2 reduction sub-system were known (or calculated), the minimum selling price of methanol (or some other product) could be calculated by adding that levelized cost to the minimum selling price shown in Fig. 14a or Fig. 14b (depending if solar utilities will be used).
9. Summary and outlook
In this publication, we reviewed the state of the art in solar conversion of carbon dioxide to fuels and demonstrated two conceptual processes for the production of methanol using these technologies. In the first example, we showed how the energy efficiency of mature CO2 conversion processes could be significantly improved by utilizing solar-derived hydrogen for hydrogenation. The current state of water-splitting technology is significantly more advanced than analogous CO2 reduction technology, with solar to hydrogen efficiencies >10% achievable. Therefore, this presents a promising near-term method for leveraging solar resources. In the second example, we considered the case of a one-step, solar CO2 reduction with H2O process where methanol and methane are produced (with oxygen). We developed a general cost model and applied the model to a methanol production process by using results from simulation and the literature. We performed sensitivity analysis to determine the influence of different parameters (i.e. conversion, selectivity, and CO2 capture energy costs) on the energy efficiency of the process.One-pass CO2 conversion and carbon selectivity have dramatic effects on the overall process energy efficiency. As a first approximation, the amount of material flowing through the various sub-systems is inversely proportional to both of these parameters. Therefore, improving both of these through reaction engineering and catalyst design will be essential towards achieving high primary energy efficiency (and low capital costs). Thus, reactions that can only obtain low one-pass CO2 conversion are likely to never be economically beneficial and will always require more energy than they produce because of the high energy costs to recover/recycle CO2 and to separate the products from the oxygen and unreacted H2O. As the selectivity and one-pass conversion increase, the marginal improvement on energy incorporation efficiency diminishes. Additionally, CO2 capture presents an essential area for research. The energy required for CO2 capture via MEA absorption from dilute flue gas currently accounts for ∼80% of the total energy of the methanol product at 100% CO2 one-pass conversion and 40% methanol selectivity. Therefore, even with dramatic improvements in conversion and selectivity, it will be impossible to achieve positive primary energy efficiency without significant improvements to the technology. Efforts in developing solid adsorbents, membranes, or by capturing from more concentrated CO2 streams (pre-combustion or oxy-combustion capture) could decrease energy cost of CO2 capture. We calculate that if the energy cost of CO2 capture were reduced by 50% and the selectivity of the catalyst were improved by 50% (to 60% methanol selectivity), the minimum CO2 one-pass conversion to achieve positive energy incorporation efficiency is 37%.
Through a simplified economic analysis, we calculated the minimum selling price for methanol as a function of the methanol selectivity and CO2 one-pass conversion, assuming, first, that the capital and operating costs for the CO2 reduction sub-system were all zero. For the base-case scenario (40% selectivity and 5% conversion), we find a minimum selling price of 1.87 USD kgmethanol−1, which is approximately three times the industrial selling price. The minimum selling price can be reduced to the industrial price by improving CO2 one-pass conversion to 22% (with 40% selectivity). However, we find that even if the industrial selling price can be met, the energy incorporation efficiency would still be negative. In fact, the process must be improved (selectivity and conversion) such that the minimum selling price is less than 0.30 USD kgmethanol−1 for the energy incorporation efficiency to be non-negative. Since this price is below the industrial selling price, one can afford to trade increased costs, by purchasing solar utilities to replace fossil fuels utilities, for improvements in overall energy incorporation efficiency. This expands the feasible region to lower selectivity and conversion. To meet the industrial selling price with non-negative energy incorporation efficiency, the conversion must be improved to 38% (with 40% selectivity).
To assess the costs of the CO2 reduction sub-system, we considered two simplified cases: (1) CO2 electro-reduction and (2) photo-catalytic reduction of CO2. Based on current estimates, even at 100% faradaic efficiency and no overpotential, the cost of the solar electricity to power the electrolyzer is 0.88 USD kgmethanol−1, (increasing the minimum selling price by 0.88 USD kgmethanol−1 from the results of the previous analyses). This does not even include costs associated with the electrolyzer, itself, or any electrocatalysts. To compete with the industrial methanol selling price (0.632 USD kgmethanol−1 in March 2014), the cost of solar electricity would need to be decreased dramatically (over 50%). While photo-catalytic systems would benefit from reduced solar energy costs (non-electrical), photo-catalytic activities are very low, on the order of ∼1 μmol gcat−1 h−1, which presents an even higher catalyst operating cost of 3.91 USD kgmethanol−1 (assuming 1 year catalyst lifetime) This does not include the cost of the solar reactor, which would need to be massive (on the order of 105 m3) to contain all of the photo-catalyst. Therefore, the activity of the photo-catalyst would need to be improved by several orders of magnitude.
As we move towards more sustainable energy resources and try to reduce our dependence on fossil fuel reserves, our society is faced with one of the biggest technological challenges of solar energy conversion. With a concerted collaborative effort among researchers from the fields of chemical engineering, material science, physics, chemistry, we can realize our goal of a sustainable carbon based energy economy. Modeling and analysis of these solar fuels processes would play a pivotal part in the development of these technologies. By reviewing the state of the art technologies in the field and identifying key areas of future research through conceptual process design, we hope that our study helps accelerate the growth of solar fuels processes towards commercialization.
Nomenclature
Sets
| i ∈ I | Species |
| j ∈ J | Process streams |
| k ∈ K | Process sub-systems |
Subsets
| I L | Liquid phase product species |
| I P | Valued product species |
| I V | Vapor phase product species |
| J k | Process streams entering sub-system k |
Parameters
| b | Power law exponent for energy cost of distillation system |
| C o | Direct capital cost of reference equipment |
| E k | Total primary energy cost of sub-system k |
| EIE | Energy incorporation efficiency |
| F j | Total molar flow rate in process stream j |
| F j,i | Molar flow rate of species i in process stream j |
| HHVi | Heating value of species i |
| m | Power law coefficient for energy cost of distillation system |
| M i | Molecular weight of species i |
| n | Total moles |
| N C,i | Moles of carbon in species i per 1 mole of species i |
| P | Pressure |
| Q k,i | Unit heat required for sub-system k to process species i |
| R | Gas constant |
| Si | Carbon selectivity of species i |
| T | Absolute temperature |
| V | Volume of piece of equipment |
| Vo | Volume of reference piece of equipment |
| W k,i | Unit work required for sub-system k to process species i |
| X | Production basis for process |
| Y i | Yield of species i |
| Z i | Name of product species i |
| ν i | Stoichiometric coefficient of species i |
| η E | Conventional resource electricity conversion efficiency |
| η H | Natural gas to process heat conversion efficiency |
| θ | Process scaling factor |
| γ | Heat capacity ratio |
| φ | One-pass H2O conversion |
| ξ | One-pass CO2 conversion |
| ψ CO2 | Ratio of unreacted CO2 that is recycled |
| ψ H2O | Ratio of unreacted H2O that is recycled |
Acknowledgements
This research was funded by the Advanced Research Projects Agency-Energy (ARPA-E), U.S. Department of Energy, under Award Number DE-AR0000329.References
- G. Centi and S. Perathoner, Catal. Today, 2009, 148, 191–205 CrossRef CAS
.
-
N. S. Lewis, E. Warren, J. McKone, H. B. Gray, P. Agbo, B. D. Stubbert, K. Yokoyama, H. A. Atwater and A. J. Leenheer, Artificial Photosynthesis: Membrane Supported Assemblies That Use Sunlight to Split Water, 2011 Search PubMed
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed
- G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711–1731 CAS
- D. T. Whipple and P. J. A. Kenis, J. Phys. Chem. Lett., 2010, 1, 3451–3458 CrossRef CAS
- A. Steinfeld, Sol. Energy, 2005, 78, 603–615 CrossRef CAS
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. B. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum, S. Ardo, H. L. Wang, E. Miller and T. F. Jaramillo, Energy Environ. Sci., 2013, 6, 1983–2002 CAS
- S. C. Roy, O. K. Varghese, M. Paulose and C. A. Grimes, ACS Nano, 2010, 4, 1259–1278 CrossRef CAS PubMed
-
N. S. Lewis and G. Crabtree, Basic Research Needs for Solar Energy Utilization, B. E. Sciences, Department of Energy, 2005 Search PubMed
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed
- J. Newman, P. G. Hoertz, C. A. Bonino and J. A. Trainham, J. Electrochem. Soc., 2012, 159, A1722–A1729 CrossRef CAS
- O. S. Joo, K. D. Jung, I. Moon, A. Y. Rozovskii, G. I. Lin, S. H. Han and S. J. Uhm, Ind. Eng. Chem. Res., 1999, 38, 1808–1812 CrossRef CAS
- Solar Fuels Institute, http://www.solar-fuels.org/, accessed June 2014.
- Research Triangle Solar Fuels Institute, http://www.solarfuels.org/, accessed June 2014.
- Liquid Light, http://llchemical.com/technology, accessed June 2014.
- ETOGAS: Smart Energy Conversion, http://www.etogas.com/, accessed June 2014.
- SOLAR-JET: Zero-carbon Jet Fuel from Sunlight, http://www.solar-jet.aero/, accessed June 2014.
- Reference Solar Spectral Irradiance: Air Mass 1.5, http://rredc.nrel.gov/solar/spectra/am1.5/, accessed June 2014.
-
J. Duffie and W. Beckman, Solar Engineering of Thermal Processes, Wiley, Hoboken, NJ, 2006 Search PubMed
-
M. Iqbal, An Introduction to Solar Radiation, Academic Press, Toronto, 1983 Search PubMed
- D. G. Erbs, S. A. Klein and J. A. Duffie, Sol. Energy, 1982, 28, 293–302 CrossRef
- Solar Resource Information, http://www.nrel.gov/rredc/solar_resource.html, accessed June 2014.
- World Map of Global Horizontal Irradiation, http://solargis.info/doc/_pics/freemaps/1000px/ghi/SolarGIS-Solar-map-World-map-en.png, accessed June 2014.
- World Energy Assessment Report: Energy and the Challenge of Sustainability, U. N. D. Program, United Nations, New York, 2003 Search PubMed.
- Y. Tian and C. Y. Zhao, Appl. Energy, 2013, 104, 538–553 CrossRef CAS
- M. A. Alghoul, M. Y. Sulaiman, B. Z. Azmi and M. A. Wahab, Anti-Corros. Methods Mater., 2005, 52, 199–206 CrossRef CAS
- D. F. Ollis, E. Pelizzetti and N. Serpone, Environ. Sci. Technol., 1991, 25, 1522–1529 CrossRef CAS
- M. R. Hoffmann, S. T. Martin, W. Y. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS
- R. Reisfeld, Opt. Mater., 2010, 32, 850–856 CrossRef CAS
- Concentrating Solar Power, International Renewable Energy Agency, 2012 Search PubMed.
- S. Mekhilef, R. Saidur and A. Safari, Renewable Sustainable Energy Rev., 2011, 15, 1777–1790 CrossRef
- M. Thirugnanasambandam, S. Iniyan and R. Goic, Renewable Sustainable Energy Rev., 2010, 14, 312–322 CrossRef CAS
- S. Kalogirou, Appl. Energy, 2003, 76, 337–361 CrossRef CAS
- C. Perkins and A. W. Weimer, AIChE J., 2009, 55, 286–293 CrossRef CAS
- T. Kodama, Prog. Energy Combust. Sci., 2003, 29, 567–597 CrossRef CAS
- V. Fthenakis, J. E. Mason and K. Zweibel, Energy Policy, 2009, 37, 387–399 CrossRef
- E. Skoplaki and J. A. Palyvos, Sol. Energy, 2009, 83, 614–624 CrossRef CAS
- A. Royne, C. J. Dey and D. R. Mills, Sol. Energy Mater. Sol. Cells, 2005, 86, 451–483 CrossRef CAS
- B. Parida, S. Iniyan and R. Goic, Renewable Sustainable Energy Rev., 2011, 15, 1625–1636 CrossRef CAS
- L. El Chaar, L. A. lamont and N. El Zein, Renewable Sustainable Energy Rev., 2011, 15, 2165–2175 CrossRef CAS
- T. M. Razykov, C. S. Ferekides, D. Morel, E. Stefanakos, H. S. Ullal and H. M. Upadhyaya, Sol. Energy, 2011, 85, 1580–1608 CrossRef CAS
- Best Research-Cell Efficiencies, http://www.nrel.gov/ncpv/images/efficiency_chart.jpg, accessed June 2014.
- M. A. Green, J. Zhao, A. Wang and S. R. Wenham, Sol. Energy Mater. Sol. Cells, 2001, 65, 9–16 CrossRef CAS
- H. Cotal, C. Fetzer, J. Boisvert, G. Kinsey, R. King, P. Hebert, H. Yoon and N. Karam, Energy Environ. Sci., 2009, 2, 174–192 CAS
- S. Gall and B. Rech, Sol. Energy Mater. Sol. Cells, 2013, 119, 306–308 CrossRef CAS
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS
- Eight Generations of Advancements Lead to Multijunction Solar Cell & Photovoltaic Technology Integration, http://amonix.com/content/eight-generations, accessed May 2014.
-
X. F. Wang, M. R. Khan, M. A. Alam and M. Lundstrom, 38th IEEE Photovoltaic Specialists Conference (Pvsc), 2012, pp. 2117–2121 Search PubMed
- Sharp Develops Concentrator Solar Cell with World's Highest Conversion Efficiency of 44.4%, http://sharp-world.com/corporate/news/130614.html, accessed June 2014.
- A. Devos, J. Phys. D: Appl. Phys., 1980, 13, 839–846 CrossRef
- M. Grätzel, Inorg. Chem., 2005, 44, 6841–6851 CrossRef PubMed
- M. Grätzel, J. Photochem. Photobiol., C, 2003, 4, 145–153 CrossRef
- H. Hoppe and N. S. Sariciftci, J. Mater. Res., 2004, 19, 1924–1945 CrossRef CAS
- C. M. Fella, Y. E. Romanyuk and A. N. Tiwari, Sol. Energy Mater. Sol. Cells, 2013, 119, 276–277 CrossRef CAS
- A. J. Nozik, Nano Lett., 2010, 10, 2735–2741 CrossRef CAS PubMed
- Annual Energy Outlook, U.S. Energy Information Administration, 2013 Search PubMed.
- D. Barlev, R. Vidu and P. Stroeve, Sol. Energy Mater. Sol. Cells, 2011, 95, 2703–2725 CrossRef CAS
- H. L. Zhang, J. Baeyens, J. Degreve and G. Caceres, Renewable Sustainable Energy Rev., 2013, 22, 466–481 CrossRef
- D. Mills, Sol. Energy, 2004, 76, 19–31 CrossRef
- H. L. Zhang, J. Baeyens, J. Degrève and G. Cacères, Renewable Sustainable Energy Rev., 2013, 22, 466–481 CrossRef
- V. Siva Reddy, S. C. Kaushik, K. R. Ranjan and S. K. Tyagi, Renewable Sustainable Energy Rev., 2013, 27, 258–273 CrossRef
- A. Gil, M. Medrano, I. Martorell, A. Lazaro, P. Dolado, B. Zalba and L. F. Cabeza, Renewable Sustainable Energy Rev., 2010, 14, 31–55 CrossRef CAS
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed
- R. Sioshansi and P. Denholm, IEEE Trans. Sustainable Energy, 2010, 1, 173–183 CrossRef
-
J. F. Kenny, N. L. Barber, S. S. Hutson, K. S. Linsey, J. K. Lovelace and M. A. Maupin, Estimated use of water in the United States in 2005, U.S. Geological Survey, Reston, VA, 2009 Search PubMed
-
K. Averyt, J. Fisher, A. Huber-Lee, A. Lewis, J. Macknick, N. Madden, J. Rogers and S. Tellinghuisen, Freshwater use by U.S. power plants: Electricity's thirst for a precious resource, Union of Concerned Scientists, Cambridge, MA, 2011 Search PubMed
- Energy demands on water resources, U. S. Department of Energy, 2006 Search PubMed.
- L. Espinal, D. L. Poster, W. Wong-Ng, A. J. Allen and M. L. Green, Environ. Sci. Technol., 2013, 47, 11960–11975 CrossRef CAS PubMed
- Inventory of U.S. Greenhouse Gas Emissions and Sinks 1990–2011, U.S. Environmental Protection Agency, 2013 Search PubMed.
- B. P. Spigarelli and S. K. Kawatra, Journal of CO2 Utilization, 2013, 1, 69–87 CrossRef CAS
- K. Damen, M. van Troost, A. Faaij and W. Turkenburg, Prog. Energy Combust. Sci., 2006, 32, 215–246 CrossRef CAS
- M. Kanniche, R. Gros-Bonnivard, P. Jaud, J. Valle-Marcos, J. M. Amann and C. Bouallou, Appl. Therm. Eng., 2010, 30, 53–62 CrossRef CAS
- T. F. Wall, Proc. Combust. Inst., 2007, 31, 31–47 CrossRef
- H. W. Pennline, D. R. Luebke, K. L. Jones, C. R. Myers, B. I. Morsi, Y. J. Heintz and J. B. Ilconich, Fuel Process. Technol., 2008, 89, 897–907 CrossRef CAS
- D. Berstad, R. Anantharaman and P. Neksa, International Journal of Refrigeration-Revue Internationale Du Froid, 2013, 36, 1403–1416 CrossRef CAS
- J. Adanez, A. Abad, F. Garcia-Labiano, P. Gayan and L. F. de Diego, Prog. Energy Combust. Sci., 2012, 38, 215–282 CrossRef CAS
-
P. Noothout, F. Wiersma, O. Hurtado, P. Roelofsen and D. Macdonald, CO2 Pipeline Infrastructure, International Energy Agency, 2013 Search PubMed
-
T. Grant, D. Morgan and K. Gerdes, Carbon dioxide transport and storage costs in NETL studies, N. E. T. Laboratory, U.S. Department of Energy, 2013 Search PubMed
- D. Aaron and C. Tsouris, Sep. Sci. Technol., 2005, 40, 321–348 CrossRef CAS
- S. Choi, J. H. Drese and C. W. Jones, Chemsuschem, 2009, 2, 796–854 CrossRef CAS PubMed
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed
- C. W. Jones, Annu. Rev. Chem. Biomol. Eng., 2011, 2, 31–52 CrossRef CAS PubMed
- M. K. Mondal, H. K. Balsora and P. Varshney, Energy, 2012, 46, 431–441 CrossRef CAS
- N. MacDowell, N. Florin, A. Buchard, J. Hallett, A. Galindo, G. Jackson, C. S. Adjiman, C. K. Williams, N. Shah and P. Fennell, Energy Environ. Sci., 2010, 3, 1645–1669 CAS
-
P. Folger, E. S. Rubin, A. Marks, H. Mantripragada, P. Versteed and J. Kitchin, Carbon capture: a technology assessment, Carnegie Mellon University, 2013 Search PubMed
- O. Aschenbrenner and P. Styring, Energy Environ. Sci., 2010, 3, 1106–1113 CAS
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed
- A. Dibenedetto, M. Aresta, C. Fragale and M. Narracci, Green Chem., 2002, 4, 439–443 RSC
- J. T. Yeh and H. W. Pennline, Energy Fuels, 2001, 15, 274–278 CrossRef CAS
- P. D. Vaidya and E. Y. Kenig, Chem. Eng. Technol., 2007, 30, 1467–1474 CrossRef CAS
- W. Conway, X. G. Wang, D. Fernandes, R. Burns, G. Lawrance, G. Puxty and M. Maeder, Environ. Sci. Technol., 2013, 47, 1163–1169 CrossRef CAS PubMed
- J. E. Bara, T. K. Carlisle, C. J. Gabriel, D. Camper, A. Finotello, D. L. Gin and R. D. Noble, Ind. Eng. Chem. Res., 2009, 48, 2739–2751 CrossRef CAS
- J. E. Bara, D. E. Camper, D. L. Gin and R. D. Noble, Acc. Chem. Res., 2010, 43, 152–159 CrossRef CAS PubMed
- D. Wappel, G. Gronald, R. Kalb and J. Draxler, Int. J. Greenhouse Gas Control, 2010, 4, 486–494 CrossRef CAS
- M. Hasib-ur-Rahman, M. Siaj and F. Larachi, Chem. Eng. Process., 2010, 49, 313–322 CrossRef CAS
- Z. Yong, V. Mata and A. E. Rodrigues, Sep. Purif. Technol., 2002, 26, 195–205 CrossRef CAS
- R. V. Siriwardane, M. S. Shen, E. P. Fisher and J. A. Poston, Energy Fuels, 2001, 15, 279–284 CrossRef CAS
- S. Keskin, T. M. van Heest and D. S. Sholl, Chemsuschem, 2010, 3, 879–891 CrossRef CAS PubMed
- J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC
- A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43, 58–67 CrossRef CAS PubMed
- J. C. Hicks, J. H. Drese, D. J. Fauth, M. L. Gray, G. G. Qi and C. W. Jones, J. Am. Chem. Soc., 2008, 130, 2902–2903 CrossRef CAS PubMed
- X. C. Xu, C. S. Song, J. M. Andresen, B. G. Miller and A. W. Scaroni, Energy Fuels, 2002, 16, 1463–1469 CrossRef CAS
- Y. Belmabkhout and A. Sayari, Adsorption, 2009, 15, 318–328 CrossRef CAS
- P. J. E. Harlick and A. Sayari, Ind. Eng. Chem. Res., 2007, 46, 446–458 CrossRef CAS
- T. Tsuda and T. Fujiwara, J. Chem. Soc., Chem. Commun., 1992, 1659–1661 RSC
- J. H. Drese, S. Choi, R. P. Lively, W. J. Koros, D. J. Fauth, M. L. Gray and C. W. Jones, Adv. Funct. Mater., 2009, 19, 3821–3832 CrossRef CAS
- E. J. Anthony, Ind. Eng. Chem. Res., 2008, 47, 1747–1754 CrossRef CAS
- G. Grasa, B. GonzAlez, M. Alonso and J. C. Abanades, Energy Fuels, 2007, 21, 3560–3562 CrossRef CAS
- A. Hanif, S. Dasgupta, S. Divekar, A. Arya, M. O. Garg and A. Nanoti, Chem. Eng. J., 2014, 236, 91–99 CrossRef CAS
- A. D. Ebner and J. A. Ritter, Sep. Sci. Technol., 2009, 44, 1273–1421 CrossRef CAS
- C. E. Powell and G. G. Qiao, J. Membrane Sci., 2006, 279, 1–49 CrossRef CAS
- J. Zou and W. S. W. Ho, J. Membrane Sci., 2006, 286, 310–321 CrossRef CAS
- P. Scovazzo, J. Kieft, D. A. Finan, C. Koval, D. DuBois and R. Noble, J. Membrane Sci., 2004, 238, 57–63 CrossRef CAS
- T. S. Chung, L. Y. Jiang, Y. Li and S. Kulprathipanja, Prog. Polym. Sci., 2007, 32, 483–507 CrossRef CAS
- X. Q. Pan, D. Clodic and J. Toubassy, Greenhouse Gases: Sci. Technol., 2013, 3, 8–20 CrossRef CAS
-
D. Clodic and M. Younes, A new method for CO2 capture: Frosting CO2 at atmospheric pressure, Kyoto, Japan, 2002 Search PubMed
- eGRID, http://www.epa.gov/cleanenergy/energy-resources/egrid/, accessed May 2014.
- M. J. Tuinier, H. P. Hamers and M. V. Annaland, Int. J. Greenhouse Gas Control, 2011, 5, 1559–1565 CrossRef CAS
- J. van Straelen, F. Geuzebroek, N. Goodchild, G. Protopapas and L. Mahony, Int. J. Greenhouse Gas Control, 2010, 4, 316–320 CrossRef CAS
- M. R. M. Abu-Zahra, J. P. M. Niederer, P. H. M. Feron and G. F. Versteeg, Int. J. Greenhouse Gas Control, 2007, 1, 135–142 CrossRef CAS
- Carbon Capture & Storage: Assessing the Economics, M. C. C. Initiative, McKinsey & Company, 2008 Search PubMed.
- T. C. Merkel, H. Q. Lin, X. T. Wei and R. Baker, J. Membr. Sci., 2010, 359, 126–139 CrossRef CAS
- J. D. Holladay, J. Hu, D. L. King and Y. Wang, Catal. Today, 2009, 139, 244–260 CrossRef CAS
-
T. Ramsden, D. Steward and J. Zuboy, Analyzing the levelized cost of centralized and distributed hydrogen production using the H2A production model, version 2, N. R. E. Laboratory, U.S. Department of Energy, 2009 Search PubMed
- Q. A. Wang, J. Z. Luo, Z. Y. Zhong and A. Borgna, Energy Environ. Sci., 2011, 4, 42–55 CAS
- A. Ursua, L. M. Gandia and P. Sanchis, Proc. IEEE, 2012, 100, 410–426 CrossRef CAS
-
C. A. Grimes, O. K. Varghese and S. Ranjan, Light, Water, Hydrogen: The Solar generation of Hydrogen by Water Photoelectrolysis, Springer, US, 2008 Search PubMed
-
J. Ivy, Summary of Electrolytic Hydrogen Production, N. R. E. Laboratory, U.S. Department of Energy, 2004 Search PubMed
- T. L. Gibson and N. A. Kelly, Int. J. Hydrogen Energy, 2008, 33, 5931–5940 CrossRef CAS
-
J. E. Genovese, K. Harg, M. Paster and J. A. Turner, Current state-of-the-art hydrogen production cost estimate using water electrolysis, N. R. E. Laboratory, U.S. Department of Energy, 2009 Search PubMed
- K. Zeng and D. K. Zhang, Prog. Energy Combust. Sci., 2010, 36, 307–326 CrossRef CAS
- M. J. Giz, J. C. P. Silva, M. Ferreira, S. A. S. Machado, E. A. Ticianelli, L. A. Avaca and E. R. Gonzalez, Int. J. Hydrogen Energy, 1992, 17, 725–729 Search PubMed
- F. C. Crnkovic, S. A. S. Machado and L. A. Avaca, Int. J. Hydrogen Energy, 2004, 29, 249–254 CrossRef CAS
- I. A. Raj, J. Mater. Sci., 1993, 28, 4375–4382 CrossRef CAS
- M. S. El-Deab, M. I. Awad, A. M. Mohammad and T. Ohsaka, Electrochem. Commun., 2007, 9, 2082–2087 CrossRef CAS
- R. N. Singh, D. Mishra, Anindita, A. S. K. Sinha and A. Singh, Electrochem. Commun., 2007, 9, 1369–1373 CrossRef CAS
- H. Wendt, H. Hofmann and V. Plzak, Mater. Chem. Phys., 1989, 22, 27–49 CrossRef CAS
- M. Carmo, D. L. Fritz, J. Merge and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS
- S. A. Grigoriev, V. I. Porembsky and V. N. Fateev, Int. J. Hydrogen Energy, 2006, 31, 171–175 CrossRef CAS
- P. Millet, N. Mbemba, S. A. Grigoriev, V. N. Fateev, A. Aukauloo and C. Etievant, Int. J. Hydrogen Energy, 2011, 36, 4134–4142 CrossRef CAS
- F. Barbir, Sol. Energy, 2005, 78, 661–669 CrossRef CAS
- A. Goni-Urtiaga, D. Presvytes and K. Scott, Int. J. Hydrogen Energy, 2012, 37, 3358–3372 CrossRef CAS
- H. Ito, T. Maeda, A. Nakano and H. Takenaka, Int. J. Hydrogen Energy, 2011, 36, 10527–10540 CrossRef CAS
- W. F. Chen, J. T. Muckerman and E. Fujita, Chem. Commun., 2013, 49, 8896–8909 RSC
- D. V. Esposito, S. T. Hunt, A. L. Stottlemyer, K. D. Dobson, B. E. McCandless, R. W. Birkmire and J. G. G. Chen, Angew. Chem., Int. Ed., 2010, 49, 9859–9862 CrossRef CAS PubMed
- C. Felix, T. Maiyalagan, S. Pasupathi, B. Bladergroen and V. Linkov, Int. J. Electrochem. Sci., 2012, 7, 12064–12077 CAS
- E. Mayousse, F. Maillard, F. Fouda-Onana, O. Sicardy and N. Guillet, Int. J. Hydrogen Energy, 2011, 36, 10474–10481 CrossRef CAS
- C. B. Xu, L. R. Ma, J. L. Li, W. Zhao and Z. X. Gan, Int. J. Hydrogen Energy, 2012, 37, 2985–2992 CrossRef CAS
- A. Marshall, B. Borresen, G. Hagen, M. Tsypkin and R. Tunold, Electrochim. Acta, 2006, 51, 3161–3167 CrossRef CAS
- J. M. Spurgeon and N. S. Lewis, Energy Environ. Sci., 2011, 4, 2993–2998 CAS
- K. D. Kreuer, Solid State Ionics, 1997, 97, 1–15 CrossRef CAS
- C. A. Linkous, H. R. Anderson, R. W. Kopitzke and G. L. Nelson, Int. J. Hydrogen Energy, 1998, 23, 525–529 CrossRef CAS
- R. J. Gorte and J. M. Vohs, Annu. Rev. Chem. Biomol. Eng., 2011, 2, 9–30 CrossRef CAS PubMed
- M. A. Laguna-Bercero, J. Power Sources, 2012, 203, 4–16 CrossRef CAS
- M. Ni, M. K. H. Leung and D. Y. C. Leung, Int. J. Hydrogen Energy, 2008, 33, 2337–2354 CrossRef CAS
- H. S. Hong, U. S. Chae, S. T. Choo and K. S. Lee, J. Power Sources, 2005, 149, 84–89 CrossRef CAS
- M. D. Liang, B. Yu, M. F. Wen, J. Chen, J. M. Xu and Y. C. Zhai, J. Power Sources, 2009, 190, 341–345 CrossRef CAS
- C. H. Yang, A. Coffin and F. L. Chen, Int. J. Hydrogen Energy, 2010, 35, 3221–3226 CrossRef CAS
- The Hydrogen Economy: Opportunities, Costs, Barriers, and R&D Needs, Natrional Research Council and National Academy of Engineering, Washington D.C., 2004 Search PubMed.
- T. Bak, J. Nowotny, M. Rekas and C. C. Sorrell, Int. J. Hydrogen Energy, 2002, 27, 991–1022 CrossRef CAS
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed
- A. J. Nozik, Annu. Rev. Phys. Chem., 1978, 29, 189–222 CrossRef CAS
- D. A. Tryk, A. Fujishima and K. Honda, Electrochim. Acta, 2000, 45, 2363–2376 CrossRef CAS
- M. Gratzel, Nature, 2001, 414, 338–344 CrossRef CAS PubMed
- R. Abe, J. Photochem. Photobiol., C, 2010, 11, 179–209 CrossRef CAS
- A. B. Murphy, P. R. F. Barnes, L. K. Randeniya, I. C. Plumb, I. E. Grey, M. D. Horne and J. A. Glasscock, Int. J. Hydrogen Energy, 2006, 31, 1999–2017 CrossRef CAS
- A. K. Ghosh and H. P. Maruska, J. Electrochem. Soc., 1977, 124, 1516–1522 CrossRef CAS
- J. F. Houlihan, D. B. Armitage, T. Hoovler, D. Bonaquist, D. P. Madacsi and L. N. Mulay, Mater. Res. Bull., 1978, 13, 1205–1212 CrossRef CAS
- C. Santato, M. Ulmann and J. Augustynski, J. Phys. Chem. B, 2001, 105, 936–940 CrossRef CAS
- H. L. Wang, T. Lindgren, J. J. He, A. Hagfeldt and S. E. Lindquist, J. Phys. Chem. B, 2000, 104, 5686–5696 CrossRef CAS
- A. Iwase and A. Kudo, J. Mater. Chem., 2010, 20, 7536–7542 RSC
- K. Sayama, A. Nomura, Z. G. Zou, R. Abe, Y. Abe and H. Arakawa, Chem. Commun., 2003, 2908–2909 RSC
- J. Z. Su, L. J. Guo, S. Yoriya and C. A. Grimes, Cryst. Growth Des., 2010, 10, 856–861 CAS
- A. Duret and M. Gratzel, J. Phys. Chem. B, 2005, 109, 17184–17191 CrossRef CAS PubMed
- J. H. Kennedy and K. W. Frese, J. Electrochem. Soc., 1977, 124, C130 CrossRef
- C. Sanchez, M. Hendewerk, K. D. Sieber and G. A. Somorjai, J. Solid State Chem., 1986, 61, 47–55 CrossRef CAS
- R. Abe, T. Takata, H. Sugihara and K. Domen, Chem. Lett., 2005, 34, 1162–1163 CrossRef CAS
- R. Nakamura, T. Tanaka and Y. Nakato, J. Phys. Chem. B, 2005, 109, 8920–8927 CrossRef CAS PubMed
- N. Nishimura, B. Raphael, K. Maeda, L. Le Gendre, R. Abe, J. Kubota and K. Domen, Thin Solid Films, 2010, 518, 5855–5859 CrossRef CAS
- O. Khaselev and J. A. Turner, Science, 1998, 280, 425–427 CrossRef CAS PubMed
- S. Licht, B. Wang, S. Mukerji, T. Soga, M. Umeno and H. Tributsch, J. Phys. Chem. B, 2000, 104, 8920–8924 CrossRef CAS
- J. N. Nian, C. C. Hu and H. Teng, Int. J. Hydrogen Energy, 2008, 33, 2897–2903 CrossRef CAS
-
R. van de Krol and M. Gratzel, Photoelectrochemical Hydrogen Production, Springer, New York, 2012 Search PubMed
- S. Y. Reece, J. A. Hamel, K. Sung, T. D. Jarvi, A. J. Esswein, J. J. H. Pijpers and D. G. Nocera, Science, 2011, 334, 645–648 CrossRef CAS PubMed
- D. A. Lutterman, Y. Surendranath and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 3838–3839 CrossRef CAS PubMed
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed
- R. van Noorden, Nature News, 2012 Search PubMed
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC
- X. B. Chen, S. H. Shen, L. J. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed
- A. Kudo, Int. J. Hydrogen Energy, 2006, 31, 197–202 CrossRef CAS
- K. Maeda and K. Domen, J. Phys. Chem. C, 2007, 111, 7851–7861 CAS
- K. Maeda, K. Teramura, D. L. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed
- J. Kubota and K. Domen, Electrochem. Soc. Interface, 2013, 22, 57–62 CAS
- R. Abe, K. Sayama and H. Sugihara, J. Phys. Chem. B, 2005, 109, 16052–16061 CrossRef CAS PubMed
- K. Maeda, M. Higashi, D. L. Lu, R. Abe and K. Domen, J. Am. Chem. Soc., 2010, 132, 5858–5868 CrossRef CAS PubMed
- Y. Sasaki, A. Iwase, H. Kato and A. Kudo, J. Catal., 2008, 259, 133–137 CrossRef CAS
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed
-
W. R. Pyle, M. H. Hayes and A. L. Spivak, Direct solar-thermal hydrogen production from water using nozzle/skimmer and glow discharge, 1996 Search PubMed
- C. Perkins and A. W. Weimer, Int. J. Hydrogen Energy, 2004, 29, 1587–1599 CrossRef CAS
- C. C. Agrafiotis, C. Pagkoura, S. Lorentzou, M. Kostoglott and A. G. Konstandopoulos, Catal. Today, 2007, 127, 265–277 CrossRef CAS
- T. Kodama and N. Gokon, Chem. Rev., 2007, 107, 4048–4077 CrossRef CAS PubMed
-
R. Perret, Solar Thermochemical Hydrogen Production Research (STCH): Thermochemical Cycle Selection and Investment Priority, F. C. T. P. Office of Energy Efficiency & Renewable Energy, U.S. Department of Energy, Livermore, 2011 Search PubMed
- A. Steinfeld, Int. J. Hydrogen Energy, 2002, 27, 611–619 CrossRef CAS
- P. Charvin, S. Abanades, G. Flamant and F. Lemort, Energy, 2007, 32, 1124–1133 CrossRef CAS
- A. Steinfeld, S. Sanders and R. Palumbo, Sol. Energy, 1999, 65, 43–53 CrossRef CAS
- C. L. Muhich, B. W. Evanko, K. C. Weston, P. Lichty, X. H. Liang, J. Martinek, C. B. Musgrave and A. W. Weimer, Science, 2013, 341, 540–542 CrossRef CAS PubMed
- S. Abanades and G. Flamant, Sol. Energy, 2006, 80, 1611–1623 CrossRef CAS
- Y. Tamaura, A. Steinfeld, P. Kuhn and K. Ehrensberger, Energy, 1995, 20, 325–330 CrossRef CAS
- T. Kodama, Y. Kondoh, R. Yamamoto, H. Andou and N. Satou, Sol. Energy, 2005, 78, 623–631 CrossRef CAS
-
A. W. Weimer, C. Perkins, P. Lichty, H. Funke, J. Zartman, D. Hirsch, C. Bingham, A. Lewandowski, S. Haussener and A. Steinfeld, Development of a solar-thermal ZnO/Zn water-splitting thermochemical cycle, N. R. E. Laboratory, U.S. Department of Energy, 2009 Search PubMed
- R. B. Diver, J. E. Miller, M. D. Allendorf, N. P. Siegel and R. E. Hogan, J. Sol. Energ-T ASME, 2008, 130, 041001 CrossRef
- J. Kim, C. A. Henao, T. A. Johnson, D. E. Dedrick, J. E. Miller, E. B. Stechel and C. T. Maravelias, Energy Environ. Sci., 2011, 4, 3122–3132 CAS
- J. E. Miller, M. D. Allendorf, R. B. Diver, L. R. Evans, N. P. Siegel and J. N. Stuecker, J. Mater. Sci., 2008, 43, 4714–4728 CrossRef CAS
- S. Kasahara, S. Kubo, R. Hino, K. Onuki, M. Nomura and S. Nakao, Int. J. Hydrogen Energy, 2007, 32, 489–496 CrossRef CAS
- K. Onuki, S. Kubo, A. Terada, N. Sakaba and R. Hino, Energy Environ. Sci., 2009, 2, 491–497 CAS
- B. J. Xu, Y. Bhawe and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9260–9264 CrossRef CAS PubMed
-
Y. Hori, in Handbook of Fuel Cells – Fundamentals, technology, and applications, ed. W. Vielstich, H. A. Gasteiger, A. Lamm and H. Yokokawa, John Wiley & Sons, Ltd., 2005 Search PubMed
- Y. Hori, Mod. Aspects Electrochem., 2008, 42, 89–189 CAS
- N. S. Spinner, J. A. Vega and W. E. Mustain, Catal. Sci. Technol., 2012, 2, 19–28 CAS
- Basic Research Needs: Catalysis for Energy, B. E. Sciences, U.S. Department of Energy, 2007 Search PubMed.
- J. L. Qiao, Y. Y. Liu, F. Hong and J. J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC
- A. Bandi, J. Electrochem. Soc., 1990, 137, 2157–2160 CrossRef CAS
- J. Yano and S. Yamasaki, J. Appl. Electrochem., 2008, 38, 1721–1726 CrossRef CAS
- K. Ogura, Journal of CO2 Utilization, 2013, 1, 43–49 CrossRef CAS
- J. P. Qu, X. G. Zhang, Y. G. Wang and C. X. Xie, Electrochim. Acta, 2005, 50, 3576–3580 CrossRef CAS
- K. W. Frese, J. Electrochem. Soc., 1991, 138, 3338–3344 CrossRef CAS
- M. Le, M. Ren, Z. Zhang, P. T. Sprunger, R. L. Kurtz and J. C. Flake, J. Electrochem. Soc., 2011, 158, E45–E49 CrossRef CAS
- F. Bidrawn, G. Kim, G. Corre, J. T. S. Irvine, J. M. Vohs and R. J. Gorte, Electrochem. Solid-State Lett., 2008, 11, B167–B170 CrossRef CAS
-
J. Weissbart and W. H. Smart, Study of electrolytic dissociation of CO2–H2O using a solid oxide electrolyte, National Aeronautics and Space Administration, 1967 Search PubMed
- K. R. Sridhar and B. T. Vaniman, Solid State Ionics, 1997, 93, 321–328 CrossRef CAS
- K. Morikawa, T. Shirasaki and M. Okada, Adv. Catal., 1969, 20, 97–133 CAS
- S. S. Xu, S. S. Li, W. T. Yao, D. H. Dong and K. Xie, J. Power Sources, 2013, 230, 115–121 CrossRef CAS
- C. Graves, S. D. Ebbesen and M. Mogensen, Solid State Ionics, 2011, 192, 398–403 CrossRef CAS
- S. D. Ebbesen, R. Knibbe and M. Mogensen, J. Electrochem. Soc., 2012, 159, F482–F489 CrossRef CAS
- C. Stoots, J. O'Brien and J. Hartvigsen, Int. J. Hydrogen Energy, 2009, 34, 4208–4215 CrossRef CAS
- Z. Zhan, W. Kobsiriphat, J. R. Wilson, M. Pillai, I. Kim and S. A. Barnett, Energy Fuels, 2009, 23, 3089–3096 CrossRef CAS
- C. Graves, S. D. Ebbesen, M. Mogensen and K. S. Lackner, Renewable Sustainable Energy Rev., 2011, 15, 1–23 CrossRef CAS
- E. J. Dufek, T. E. Lister, S. G. Stone and M. E. McIlwain, J. Electrochem. Soc., 2012, 159, F514–F517 CrossRef CAS
- H. Li and C. Oloman, J. Appl. Electrochem., 2005, 35, 955–965 CrossRef CAS
- D. T. Whipple, E. C. Finke and P. J. A. Kenis, Electrochem. Solid-State Lett., 2010, 13, D109–D111 CrossRef
- K. Xie, Y. Q. Zhang, G. Y. Meng and J. T. S. Irvine, J. Mater. Chem., 2011, 21, 195–198 RSC
- B. X. Hu, V. Stancovski, M. Morton and S. L. Suib, Appl. Catal., A, 2010, 382, 277–283 CrossRef CAS
-
N. Sivansakar, J. J. Kaczur and E. B. Cole, US Pat., 8641885, 2014 Search PubMed
-
E. B. Cole, K. Teamey, A. B. Bocarsly and N. Sivansakar, US Pat., 8592633, 2013 Search PubMed
- B. H. Mahan, J. Chem. Phys., 1960, 33, 959 CrossRef CAS
- A. Corma and H. Garcia, J. Catal., 2013, 308, 168–175 CrossRef CAS
- F. Sastre, A. Corma and H. Garcia, J. Am. Chem. Soc., 2012, 134, 14137–14141 CrossRef CAS PubMed
- B. Kumar, M. Llorente, J. Froehlich, T. Dang, A. Sathrum and C. P. Kubiak, Annu. Rev. Phys. Chem., 2012, 63, 541–569 CrossRef CAS PubMed
- R. Hinogami, Y. Nakamura, S. Yae and Y. Nakato, J. Phys. Chem. B, 1998, 102, 974–980 CrossRef CAS
- K. Hara, A. Kudo and T. Sakata, J. Electroanal. Chem., 1995, 391, 141–147 CrossRef
- I. Taniguchi, B. Aurianblajeni and J. O. M. Bockris, Electrochim. Acta, 1984, 29, 923–932 CrossRef CAS
- H. Yoneyama, K. Sugimura and S. Kuwabata, J. Electroanal. Chem., 1988, 249, 143–153 CrossRef
- Y. Taniguchi, H. Yoneyama and H. Tamura, Bull.
Chem. Soc. Jpn., 1982, 55, 2034–2039 CrossRef CAS
- S. Ikeda, Y. Saito, M. Yoshida, H. Noda, M. Maeda and K. Ito, J. Electroanal. Chem., 1989, 260, 335–345 CrossRef CAS
- W. M. Sears and S. R. Morrison, J. Phys. Chem., 1985, 89, 3295–3298 CrossRef CAS
- K. W. Frese and D. Canfield, J. Electrochem. Soc., 1984, 131, 2518–2522 CrossRef CAS
-
Y. Nakamura, R. Hinogami, S. Yae and Y. Nakato, Advances in Chemical Conversions for Mitigating Carbon Dioxide, 1998, vol. 114, pp. 565–568 Search PubMed
- S. Kaneco, H. Katsumata, T. Suzuki and K. Ohta, Appl. Catal., B, 2006, 64, 139–145 CrossRef CAS
- H. Flaisher, R. Tenne and M. Halmann, J. Electroanal. Chem., 1996, 402, 97–105 CrossRef
- B. Aurianblajeni, I. Taniguchi and J. O. Bockris, J. Electroanal. Chem., 1983, 149, 291–293 CAS
- C. R. Cabrera and H. D. Abruna, J. Electroanal. Chem., 1986, 209, 101–107 CrossRef CAS
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS
- P. Usubharatana, D. McMartin, A. Veawab and P. Tontiwachwuthikul, Ind. Eng. Chem. Res., 2006, 45, 2558–2568 CrossRef CAS
- S. Y. Qin, F. Xin, Y. D. Liu, X. H. Yin and W. Ma, J. Colloid Interface Sci., 2011, 356, 257–261 CrossRef CAS PubMed
- Q. H. Zhang, W. D. Han, Y. J. Hong and J. G. Yu, Catal. Today, 2009, 148, 335–340 CrossRef CAS
- K. Koci, K. Mateju, L. Obalova, S. Krejcikova, Z. Lacny, D. Placha, L. Capek, A. Hospodkova and O. Solcova, Appl. Catal., B, 2010, 96, 239–244 CrossRef CAS
- Y. Li, W. N. Wang, Z. L. Zhan, M. H. Woo, C. Y. Wu and P. Biswas, Appl. Catal., B, 2010, 100, 386–392 CrossRef CAS
- K. Koci, L. Obalova, L. Matejova, D. Placha, Z. Lacny, J. Jirkovsky and O. Solcova, Appl. Catal., B, 2009, 89, 494–502 CrossRef CAS
- M. Anpo, H. Yamashita, Y. Ichihashi and S. Ehara, J. Electroanal. Chem., 1995, 396, 21–26 CrossRef
- K. Mori, H. Yamashita and M. Anpo, RSC Adv., 2012, 2, 3165–3172 RSC
- M. Kitano, M. Matsuoka, M. Ueshima and M. Anpo, Appl. Catal., A, 2007, 325, 1–14 CrossRef CAS
- M. Iwasaki, M. Hara, H. Kawada, H. Tada and S. Ito, J. Colloid Interface Sci., 2000, 224, 202–204 CrossRef CAS PubMed
- M. Tahir and N. S. Amin, Appl. Catal., A, 2013, 467, 483–496 CrossRef CAS
- T. V. Nguyen and J. C. S. Wu, Sol. Energy Mater. Sol. Cells, 2008, 92, 864–872 CrossRef CAS
- Q. Y. Zhang, Y. Li, E. A. Ackerman, M. Gajdardziska-Josifovska and H. L. Li, Appl. Catal., A, 2011, 400, 195–202 CrossRef CAS
- Z. H. Zhao, J. M. Fan, J. Y. Wang and R. F. Li, Catal. Commun., 2012, 21, 32–37 CrossRef CAS
- W. B. Hou, W. H. Hung, P. Pavaskar, A. Goeppert, M. Aykol and S. B. Cronin, ACS Catal., 2011, 1, 929–936 CrossRef CAS
- O. Ishitani, C. Inoue, Y. Suzuki and T. Ibusuki, J. Photochem. Photobiol., A, 1993, 72, 269–271 CrossRef CAS
- K. Tennakone, Sol. Energy Mater., 1984, 10, 235–238 CrossRef CAS
- M. L. Perry, J. Newman and E. J. Cairns, J. Electrochem. Soc., 1998, 145, 5–15 CrossRef CAS
- N. Furuya, T. Yamazaki and M. Shibata, J. Electroanal. Chem., 1997, 431, 39–41 CrossRef CAS
- K. Hara and T. Sakata, J. Electrochem. Soc., 1997, 144, 539–545 CrossRef CAS
- Y. Tomita, S. Teruya, O. Koga and Y. Hori, J. Electrochem. Soc., 2000, 147, 4164–4167 CrossRef CAS
- S. Kaneco, K. Iiba, N. Hiei, K. Ohta, T. Mizuno and T. Suzuki, Electrochim. Acta, 1999, 44, 4701–4706 CrossRef CAS
- K. Ogura and N. Endo, J. Electrochem. Soc., 1999, 146, 3736–3740 CrossRef CAS
- D. R. MacFarlane, N. Tachikawa, M. Forsyth, J. M. Pringle, P. C. Howlett, G. D. Elliott, J. H. Davis, M. Watanabe, P. Simon and C. A. Angell, Energy Environ. Sci., 2014, 7, 232–250 CAS
- M. V. Fedorov and A. A. Kornyshev, Chem. Rev., 2014, 114, 2978–3036 CrossRef CAS PubMed
- P. Hapiot and C. Lagrost, Chem. Rev., 2008, 108, 2238–2264 CrossRef CAS PubMed
- L. R. Martin, Sol. Energy, 1980, 24, 271–277 CrossRef CAS
- M. E. Galvez, P. G. Loutzenhiser, I. Hischier and A. Steinfeld, Energy Fuels, 2008, 22, 3544–3550 CrossRef CAS
- A. J. Traynor and R. J. Jensen, Ind. Eng. Chem. Res., 2002, 41, 1935–1939 CrossRef CAS
- D. Arifin, V. J. Aston, X. H. Liang, A. H. McDaniel and A. W. Weimer, Energy Environ. Sci., 2012, 5, 9438–9443 CAS
- S. Abanades and M. Chambon, Energy Fuels, 2010, 24, 6667–6674 CrossRef CAS
- N. Gokon, H. Murayama, A. Nagasaki and T. Kodama, Sol. Energy, 2009, 83, 527–537 CrossRef CAS
- T. Kodama, N. Gokon and R. Yamamoto, Sol. Energy, 2008, 82, 73–79 CrossRef CAS
- M. D. Allendorf, R. B. Diver, N. P. Siegel and J. E. Miller, Energy Fuels, 2008, 22, 4115–4124 CrossRef CAS
- J. R. Scheffe, J. H. Li and A. W. Weimer, Int. J. Hydrogen Energy, 2010, 35, 3333–3340 CrossRef CAS
- W. C. Chueh and S. M. Haile, Philos. Trans. R. Soc., A, 2010, 368, 3269–3294 CrossRef CAS PubMed
- W. C. Chueh, C. Falter, M. Abbott, D. Scipio, P. Furler, S. M. Haile and A. Steinfeld, Science, 2010, 330, 1797–1801 CrossRef CAS PubMed
- J. Kim, T. A. Johnson, J. E. Miller, E. B. Stechel and C. T. Maravelias, Energy Environ. Sci., 2012, 5, 8417–8429 CAS
- Annual Energy Review, U.S. Energy Information Administration, 2011.
- Solar Photovoltaic Cell/Module Shipments Report 2012, U.S. Energy Information Administration, 2013.
- K. Ikeue, S. Nozaki, M. Ogawa and M. Anpo, Catal. Lett., 2002, 80, 111–114 CrossRef CAS
- UOP Selexol Technology for Acid Gas Removal, UOP LLC, 2009.
- B. Burr and L. Lyddon, A Comparison of Physical Solvents for Acid Gas Removal, Bryan Research & Engineering, Inc, 2008 Search PubMed.
-
C. Chen, Ph.D. thesis, Carnegie Mellon, 2005
- Electric Sales, Revenue, and average price, U.S. Energy Information Administration, 2013.
- R. Silva, M. Berenguel, M. Perez and A. Fernandez-Garcia, Appl. Energy, 2014, 113, 603–614 CrossRef
- J. C. S. Wu, T. H. Wu, T. C. Chu, H. J. Huang and D. P. Tsai, Top. Catal., 2008, 47, 131–136 CrossRef CAS
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazabal and J. Perez-Ramirez, Energy Environ. Sci., 2013, 6, 3112–3135 CAS
-
C. H. Bartholomew and R. J. Farrauto, Fundamentals of Industrial Catalytic Processes, Wiley-AIChE, Hoboken, NJ, 2005 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ee01958j |
| ‡ Current address: Department of Energy and Chemical Engineering, Incheon National University, Incheon 406-772, Korea. |
| This journal is © The Royal Society of Chemistry 2015 |