
Tailoring the composition of ultrathin, ternary alloy PtRuFe nanowires for the methanol oxidation reaction and formic acid oxidation reaction†
Megan E.
Scofield
a,
Christopher
Koenigsmann
a,
Lei
Wang
a,
Haiqing
Liu
a and
Stanislaus S.
Wong
*ab
aDepartment of Chemistry, State University of New York at Stony Brook, Stony Brook, NY 11794-3400, USA. E-mail: stanislaus.wong@stonybrook.edu; sswong@bnl.gov
bCondensed Matter Physics and Materials Sciences Department, Brookhaven National Laboratory, Building 480, Upton, NY 11973, USA
First published on 25th November 2014
Abstract
In the search for alternatives to conventional Pt electrocatalysts, we have synthesized ultrathin, ternary PtRuFe nanowires (NW), possessing different chemical compositions in order to probe their CO tolerance as well as electrochemical activity as a function of composition for both (i) the methanol oxidation reaction (MOR) and (ii) the formic acid oxidation reaction (FAOR). As-prepared ‘multifunctional’ ternary NW catalysts exhibited both higher MOR and FAOR activity as compared with mono-metallic Pt NWs, binary Pt7Ru3 and Pt7Fe3 NWs, and commercial catalyst control samples. In terms of synthetic novelty, we utilized a sustainably mild, ambient wet-synthesis method never previously applied to the fabrication of crystalline, pure ternary systems in order to fabricate ultrathin, homogeneous alloy PtRuFe NWs with a range of controlled compositions. These NWs were subsequently characterized using a suite of techniques including XRD, TEM, SAED, and EDAX in order to verify not only the incorporation of Ru and Fe into the Pt lattice but also their chemical homogeneity, morphology, as well as physical structure and integrity. Lastly, these NWs were electrochemically tested in order to deduce the appropriateness of conventional explanations such as (i) the bi-functional mechanism as well as (ii) the ligand effect to account for our MOR and FAOR reaction data. Specifically, methanol oxidation appears to be predominantly influenced by the Ru content, whereas formic acid oxidation is primarily impacted by the corresponding Fe content within the ternary metal alloy catalyst itself.
Broader contextWe address both environmental and energy concerns in our current work. First, we demonstrate the successful synthesis of pure, crystalline ultrathin nanowires possessing a controlled ternary metal alloy chemical composition (i.e. PtRuFe) using a relatively benign and uncomplicated protocol. Second, our manuscript showcases the use of a relatively stable and durable ‘multifunctional’ nanowire catalyst, possessing promising methanol oxidation reaction (MOR) and formic acid oxidation reaction (FAOR) activity. Third, we have gained mechanistic insights into both the MOR and FAOR reaction, wherein we have found that methanol oxidation appears to be predominantly influenced by the Ru content, whereas formic acid oxidation is primarily impacted by the corresponding Fe content within the ternary metal alloy catalyst itself. Such advances are critical for fuel cell optimization and highlight the role of chemistry in improving electrochemical activity. Fourth and most importantly, we bridge energy and environmental sciences by creating chemically tailored, active nanoscale catalysts incorporating plentiful, cheap, and nontoxic Fe, which is an alternative replacement metal for comparatively more expensive and less accessible traditional Pt catalysts for fuel cell applications. Our novel ternary metal alloy nanowires are moreover synthesized using techniques that are milder and hence, arguably more ‘sustainable’ as compared with conventional methods. |
1. Introduction
With the growing demand for alternative energy sources, much research effort has focused on the development of direct methanol fuel cells (DMFCs) as a viable energy conversion device. Specifically, DMFCs consist of both an anode and a cathode at which the oxidation of methanol and the reduction of oxygen can respectively occur.1,2 Specifically, eqn (1) and (2) highlight the overall and half-cell reactions, respectively, for the DMFC process.CH3OH + 3/2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) O2 → CO2 + 2H2O O2 → CO2 + 2H2O | (1) |
| CH3OH + H2O → CO2 + 6H+ + 6e− | (2) |
In this work, we are primarily preoccupied with understanding the methanol oxidation reaction (MOR), occurring at the anode half-cell. Typically, Pt-based materials are used as MOR catalysts, due to their relatively high catalytic activities. However, this has proven to be problematic, since platinum is expensive because of its relative scarcity. Moreover, when Pt is incorporated into a catalyst, deleterious effects, such as surface poisoning, particle ripening, and dissolution, often arise,3,4 all of which lead to a decrease in the number of active sites available for MOR and thereby contribute to slow kinetics and poor durability.5
One strategy to enhance the catalytic activity of Pt has been to incorporate other electrochemically active, more plentiful metals, such as ruthenium for example, thereby forming homogeneous Pt1−xRux alloys. Specifically, outstanding MOR performance has already been observed in Pt1−xRux systems using a range of Ru concentrations (‘x’ = 0.07–0.33). The presence of 30% Ru dopant, as in the Pt7Ru3 system, yielded exceptionally high activities.6–8 As such, we used this particular binary alloy composition as our explicit ‘starting point’ control sample from which to seek out possible permutations of this overall 30% metal dopant for creating ever more electrochemically active catalysts.
In PtRu systems, the MOR reaction proceeds through a plausible ‘bifunctional mechanism’, wherein adsorbed hydroxide (OH−) species at the Ru site facilitate the oxidation and removal of the CO intermediate adsorbed onto the Pt active sites.9–12 Consequently, more Pt active sites become accessible for methanol oxidation, thereby enhancing catalytic performance at lower overpotentials.11–14 Although binary alloys clearly represent a positive step forward, there is a functional need for improvement, due to issues such as long-term catalytic durability, which ultimately hinder the current commercial viability of DMFCs.
As such, in recent years, efforts have shifted beyond the use of bimetallic catalysts to the study of more complex ternary ‘platforms’, incorporating three different transition metals. Specifically, ternary systems including but not limited to PtRuNi,3,10,15,16 PtRuFe,17–20 PtRuSn,3 PtRuMo,3 PtRuRh,21 and PtRuAu22 have been investigated as potential electrocatalysts for MOR. The addition of this extra metal not only decreases the overall cost of the catalyst by reducing the amount of Pt utilized but also improves catalytic activity through a favorable synergistic electronic interaction between the Pt active sites and the transition metal dopants.4,10,11
Specifically, the inherent structural coupling between the dopant metal lattice and the Pt lattice forces a compression strain upon the Pt lattice, due to the shorter interatomic distance of the dopant sites as compared with Pt. This so-called ‘ligand effect’ therefore increases the d-orbital overlap, contributing to a down-shift in energy for the weighted center of the d-band. In essence, electron density is withdrawn from the Pt d-band towards that of the dopant metals. In terms of practical consequences for catalysis, both the lower weighted center of the d-band and the reduced electron density collectively contribute to a lowered CO affinity as a result of a concomitant weakening of the overlap between the Pt d-orbitals and the CO π*-orbitals. Hence, CO coverage is effectively reduced, which correspondingly increases the number of exposed Pt active sites available for MOR.
In this light, utilizing a combinatorial computational approach, Nørskov and co-workers19 analyzed a wide array of ternary and quaternary alloy electrocatalysts, and determined that PtRuCo, PtRuNi, as well as PtRuFe catalysts exhibited significantly higher CO tolerance values as compared with binary PtRu catalysts. Experimentally, PtRuNi nanoparticles synthesized by Park et al.3 demonstrated that the incorporation of Ni into a PtRu alloy resulted in an increase in specific activity, which was as much as ∼2.2-fold higher as compared with that of the PtRu catalyst, an observation attributable to electron transfer from Ni to Pt. Similarly, Yu et al.18 synthesized and compared the activity of multiple ternary nanoparticle electrocatalysts, i.e. PtRuM (‘M’ = Co, Ni, Fe); in particular, both PtRuCo and PtRuNi electrocatalysts evinced a favorable 50 mV negative shift in the onset potential for MOR. Moreover, Sun and co-workers23 synthesized FePtPd nanowires, possessing different chemical compositions in order to analyze their MOR activity as compared with that of PtFe, PtPd, and Pt catalysts. They found that their Fe28Pt38Pd34 electrocatalysts also exhibited a favorable negative shift in onset potential for methanol oxidation as well as a corresponding increase in the peak current density as compared with both binary alloy and Pt analogues. All of these examples demonstrate and corroborate the notion that by incorporating a third transition metal, such as Fe, CO tolerance can be increased, and as a result, the activity can be enhanced due to the availability of active sites. In a previous Perspective article24 in Energy and Environmental Sciences, we had also discussed the idea that ternary metal Pt-based alloys can be considered as promising anodes for DMFCs.
Moreover, PtRuM electrocatalysts (wherein ‘M’ = Co, Ni, and Fe, i.e. incorporating first row transition metals) have become particularly appealing, because Fe, for instance, represents a more plentiful, lower cost, and reasonably less toxic alternative, especially as compared with bulk Pt. Hence, due to the attractiveness of Fe as a viable catalytic component and potential replacement for Ru, the purpose of this work has been to systematically explore Fe-based PtRuM electrocatalysts with the aim of correlating chemical composition with electrochemical activity in order to understand and fine tune the preparation of electrocatalysts for enhanced performance in both MOR and the formic acid oxidation reaction (FAOR). The other key point is that by incorporating Fe within this ternary alloy architecture, we have been able to demonstrate an increase in electrochemical stability by nearly 4-fold as compared with commercial PtRu standards. In so doing, we have potentially addressed and mitigated for a serious limitation preventing the widespread commercialization of anode materials, an issue which had been previously commented upon by our group and others.24,25
As an additional parameter to tailoring chemical composition, we have also been interested in exploring the effect of electrocatalyst morphology upon corresponding activity. Specifically, it is known that the use of anisotropic one-dimensional (1D) structures such as nanowires (NWs) and nanotubes (NTs) has led to significant improvements in electrocatalytic performance as compared with traditional, conventional zero-dimensional (0D) morphologies such as nanoparticles (NPs). The rationale is that crystalline 1D nanostructures possess (a) high aspect ratios, (b) fewer lattice boundaries, (c) long segments of smooth crystal planes, and (d) a low number of surface defect sites, all of which are desirable attributes for fuel cell catalysts.26 Recent results from our group confirm this hypothesis.20,27,28 Furthermore, when the diameter of the 1D wire is decreased toward the ultrathin size regime (<5 nm), we are able to minimize the presence of not only intrinsic defect sites, which tend to alter the surface energy of the Pt, but also deleterious lattice boundary imperfections. Both are more susceptible to removal through decomposition, because the lower coordination Pt surface atoms become exposed and are therefore more prone to irreversible oxidation. All of these factors suggest that ultrathin PtRuFe NWs represent a particularly favorable structural paradigm for the synthesis of high-performance electrocatalysts.
In this manuscript therefore, we have synthesized ultrathin, ternary PtRuFe NW electrocatalysts using an ambient, wet synthesis method that has not, to the best of our knowledge, ever before been applied to the successful generation of multi-functional ternary metal-based catalysts. Solution-based techniques are advantageous for the production of electrocatalysts, since they represent potentially straightforward, reasonably mild, high-yield, scalable, and cost-efficient processes. Specifically, the methodology29,30 used herein involves the reduction of metal precursors with sodium borohydride in the presence of a so-called ‘soft template’, created by cetyltrimethylammonium bromide (CTAB) surfactant within a two-phase water–chloroform system. One of the advantages of this technique is that because the nucleation and growth of the wires are fundamentally controlled by the size and shape of the ‘soft template’ pores, one can rather easily create porous, high surface area networks of interconnected, ‘wormlike’ metallic nanowires, possessing average diameters of as small as 1.9 nm. Utilizing this protocol,29,30 we were therefore able to tune the relative concentrations of Ru and Fe precursors, and fabricate a range of homogeneous alloyed ultrathin NWs, as confirmed by our structural characterization data.
We have investigated not only the CO tolerance but also the electrochemical activity of our NWs as a function of Ru and Fe content. Specifically, as discussed earlier, using Pt7Ru3 as our ‘starting’ catalyst composition, upon the addition of 10% Fe to replace Ru content, the resulting Pt7Ru2Fe NW catalyst gave rise to a negative shift of ∼230 mV in onset potentials for CO stripping as well as to ∼11 times higher MOR activity as compared with our mono-metallic Pt NW controls, rendering this as our best catalyst tested for MOR. Moreover, these results suggest significant improvements by comparison with currently available, commercial PtRu nanoparticle standards. Specifically, our ternary NWs yielded an order of magnitude higher activity of 1.52 mA cm−2versus 0.15 mA cm−2 for commercial NPs @ 0.7 V vs. RHE. Moreover, as we increased the amount of Fe present in the samples from 10% to 25% and correspondingly decreased the Ru content from 20% to 5%, we observed a shift in the onset for CO absorption towards higher potentials, thereby indicating the presence of delayed kinetics. As a result, from an analysis of our data based on the volcano-type trend observed in Fig. 6, the Pt7Ru2Fe NW catalyst represented a particularly attractive option for MOR.
To further explore the effects associated with varying the concentrations of the dopant metals, these catalysts were additionally analyzed for their performance in formic acid oxidation. It should be noted that ternary catalysts have rarely been tested for activity with respect to both complementary reactions, namely the methanol oxidation and formic acid oxidation reactions, which thereby renders this study as both significant and novel. In particular, when these catalysts were purposely used to oxidize formic acid, we noted that Pt7Ru1.5Fe1.5 NWs maintained the lowest onset potential as compared with the other ternary samples, even Pt7Ru2Fe NWs.
As a result, from our data for both MOR and FAOR, we have been able to demonstrate that the actual chemical composition of the catalyst counts, because this parameter can be specifically used to finely tune the activity of our NWs for enhanced performance. These data therefore highlight a promising strategy for using chemistry to rationally and controllably optimize activities for both MOR and FAOR.
2. Materials and methods
2.1. Synthesis of our binary PtRu and ternary PtRuFe NWs
The synthesis of ternary metal-based nanowire networks (i.e. Pt7Ru2Fe, Pt7Ru1.5Fe1.5, Pt7RuFe2, and Pt7Ru.5Fe2.5 NWs, respectively) has been accomplished by a solution technique based on confining metal growth within a soft template consisting of a network of inverse wormlike micelles.30 In typical experiments used to synthesize our series of PtRuFe nanowires as an example, for the relevant metal precursors, we prepared an aqueous mixture of hexachloroplatinic acid hydrate (H2PtCl6·6H2O, Alfa Aesar, 99.9%, 1.5 mM), ruthenium(III) chloride hydrate (RuCl3·xH2O, Acros Organics, 35–40% Ru, 1.5 mM) and iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O, Aldrich, 98+%, 1.5 mM) in appropriate, stoichiometrically determined quantities. These solutions were then combined with a solution of hexadecyltrimethylammonium bromide (CTAB, Fluka, 40 mM) in chloroform (Acros, 99.8%) under stirring. Subsequently, 40 mL of H2O was also added under stirring for an additional 30 min. An aqueous solution of sodium borohydride (NaBH4, Alfa Aesar, 98% powder) was later used to reduce the metal ions encapsulated within the inverse micellar network, as denoted by a color change from a dark green to a dark gray/black hue. After an additional 20 minutes of stirring, as-synthesized nanowires were centrifuged. The supernatant was discarded and subsequently washed three times with 2 mL of ethanol followed by centrifugation for 2 min at 6000 rpm. Binary PtRu and PtFe NW controls were prepared analogously, using precursor concentrations of 2 mM in the absence of the third precursor.The surfactant ‘contaminant’ was removed using an effective cleaning protocol.31 Upon isolation of the NWs, these nanomaterials were subsequently dispersed and ‘washed’ in n-butylamine (Acros Organics, 99+%, 0.5 mg catalyst per mL) by sonication for approximately 30 seconds. The solution was then left to stir for 3 days at 400 rpm. The NWs were subsequently collected upon centrifugation at 5000 rpm for 5 minutes. The product was dispersed in 10 mL methanol and sonicated for 15 minutes. The solution was centrifuged again with the entire process repeated for an additional 2 times. The catalyst was later re-dispersed into ethanol prior to further use. Finally, the n-butylamine residue was removed by a combination of selective CO adsorption and subsequent CO stripping techniques, as described in Section 2.3.
2.2. Structural characterization
Powder diffraction samples were prepared by dispersing the relevant NW samples into ethanol and drop casting the resulting slurry onto a glass microscope slide. Powder diffractograms of as-prepared NWs were obtained on a Scintag diffractometer, operating in the Bragg–Brentano configuration with Cu Kα radiation (λ = 1.54 Å). Diffraction patterns were acquired from 35° to 85° at a scanning rate of 0.25° in 2θ per minute.The structural morphology and crystallinity of as-prepared PtRu, PtFe, and PtRuFe NWs were characterized by transmission electron microscopy (TEM) obtained with a Technai12 BioTwinG2 TEM instrument, equipped with an AMT XR-60 CCD camera system. Energy dispersive X-ray spectroscopy (EDAX) was performed on a Leo 1550 field-emission scanning electron microscope (FE-SEM), operating at an accelerating voltage of 20 kV. High resolution transmission electron microscopy (HRTEM) and selected area electron diffraction (SAED) patterns were acquired on a JEOL 2100F instrument operating at accelerating voltages of 200 kV with a beam size of 2 Å. Additional HRTEM images and SAED patterns were collected using a JEOL 3000F microscope, equipped with a field-emission gun operating at an accelerating voltage of 300 kV.
2.3. Electrochemical characterization
Electrochemical characterization of both as-prepared binary and ternary NWs was performed with the NWs supported onto a glassy carbon electrode (GCE; 5 mm, Pine Instruments). Initially, the electrode was polished to a mirror finish using an aluminum oxide powder slurry (0.050 μm particle size). Prior to deposition of the catalyst, the GCE surface was pre-modified with a thin layer of Vulcan XC-72R carbon in order to serve as a carbonaceous support structure for the as-prepared NWs. The NWs dispersed in ethanol (at a concentration of 2 mg mL−1) were then loaded onto a modified GCE by adding two 5 μL drops of the catalyst dispersion onto the surface, which was subsequently allowed to dry in air. The GCE was later sealed with one 5 μL drop of an ethanolic 0.025% Nafion solution, prepared from a 5% stock solution.Prior to electrochemical analysis, the catalyst-loaded GCE was immersed into fresh aliquots of water, so as to remove any impurities. In order to prepare the commercial standard, alloy-type Pt1−xRux (‘x’ = 0.5) NPs with a 20% precious metal content (ETek) were rendered into catalyst ink dispersions (1 mg mL−1) in 25% isopropyl alcohol in water and deposited directly onto the surface of polished GCE for characterization. Electrochemical measurements were obtained in 0.1 M perchloric acid (Fisher Scientific, optima grade) solutions, created using high-purity water possessing a resistivity value of 18.2 MΩ cm. Pt foil and an Ag/AgCl combination (3 M Cl−) served as the counter and reference electrodes, respectively. All potentials have been reported with respect to the reversible hydrogen electrode (RHE).
The corresponding electrochemical properties of the binary and ternary catalysts were examined by cyclic voltammetry (CV) as well as using CO stripping voltammetry. CVs were obtained in the desired argon-saturated electrolyte at a scan rate of 20 mV s−1. The adsorption of a monolayer of CO was accomplished by immersing the electrodes into a CO-saturated perchloric acid (Fisher Scientific, Optima grade) electrolyte for a period of 30 min. Subsequently, the electrode was transferred to a deoxygenated electrolyte solution, so as to obtain the corresponding CO stripping CV. The CO stripping process was implemented in order to remove n-butylamine from the NW surface in order to expose available active sites. The presence of CO effectively displaces residual n-butylamine, since CO possesses a higher affinity for adsorption and can be subsequently removed upon cycling. The electrochemically accessible surface area (ECSA) was calculated from the amount of integrated hydrogen adsorption (Hads) determined in the cyclic voltammetry analysis, utilizing 210 μC cm−2 as the conversion factor. As-obtained ECSA values represent a reasonable estimate of the active Pt and Ru sites in the system.
2.4. Measurement of the MOR and FAOR kinetics
The MOR kinetics were measured by first obtaining CVs at a scan rate of 20 mV s−1 in a deoxygenated 0.5 M methanol (Fisher Scientific, Optima grade) solution, supported in a 0.1 M HClO4 electrolyte. Typically, a linear-sweep voltammogram (LSV) was obtained in the anodic sweep direction, so as to collect the MOR kinetics curves. The observed current was subsequently normalized to the Pt surface area, which can be determined from the Hads charge. After the initial LSV, collection of the MOR CVs was repeated to ensure that the surface of the catalyst was sufficiently stable in order to generate more reproducible CVs.Chronoamperometry was also put to use in order to test the stability of our as-prepared catalysts. Chronoamperograms were obtained in a de-oxygenated 0.5 M methanol solution, supported in a 0.1 M HClO4 electrolyte. The electrode was submerged and tested, while the potential was maintained at a value of 0.65 V for a period of one hour. This specific potential was used, due to the fact that it designates a potential that resides within the onset region of all of the catalysts tested herein, thereby allowing for an appropriate comparison of relative activity. The analogous formic acid oxidation kinetics data were acquired in a 0.1 M HClO4 electrolyte in the presence of a 0.5 M formic acid solution (EMD, 98% ACS reagent grade). The same electrochemical parameters described above were applied. All electrodes in this manuscript have been tested and run under identical conditions, in order to establish a self-consistent comparison amongst all of the electrodes. In particular, the activity of our novel PtRuFe NWs as well as of binary PtRu and PtFe NWs has been compared with respect to that of PtRu NP/C (Etek), serving as a commercial standard and control.
3. Results and discussion
3.1. Ambient synthesis and characterization of PtRuFe NWs
In this manuscript, for the first time, we have adapted an ambient wet synthesis technique to prepare ternary alloy nanowires under relatively mild reaction conditions. By appropriately manipulating the stoichiometric ratios of the metal precursors, we are able to correspondingly vary the constituent concentrations of each metal within the NWs, thereby allowing for the fabrication of homogeneous alloys with tunable compositions. The structure, purity, and crystallinity of our ultrathin ternary NWs were analyzed using a suite of structural characterization techniques including XRD, TEM, and HRTEM.Specifically, Fig. 1 highlights a number of powder XRD patterns obtained on our ternary PtRuFe systems (i.e. Pt7Ru2Fe NWs, Pt7Ru1.5Fe1.5 NWs, Pt7RuFe2 NWs, and Pt7Ru.5Fe2.5 NWs, respectively) as well as on controls consisting of binary Pt7Ru3, Pt7Fe3, and mono-metallic Pt NWs. All seven NW samples possessed peaks located at 39°, 46°, and 67°, which can be ascribed to the corresponding (111), (200), and (220) planes of an underlying Pt fcc framework structure (JCPDS database #04-0802). It is evident that the patterns are devoid of peaks that can be attributed to either the elemental ruthenium or iron phases (JCPDS database #06-0663 and #85-1410, respectively), thereby suggesting that reduction of the precursors can result in the formation of uniform alloys. Moreover, no apparent peaks ascribable to any possible impurities within the nanowires could be observed, indicative of the reasonably high purity of our samples. Nevertheless, for the series of PtRuFe NWs, slight shifts to higher 2θ are noted for the Pt (111) peak, likely due to the contraction of the Pt lattice by the incorporation of both Ru and Fe. The broadness of the peaks can potentially be attributed to both the inherently small sizes of the NWs tested (i.e. diameters under 5 nm) as well as to their intrinsic segmented texture.
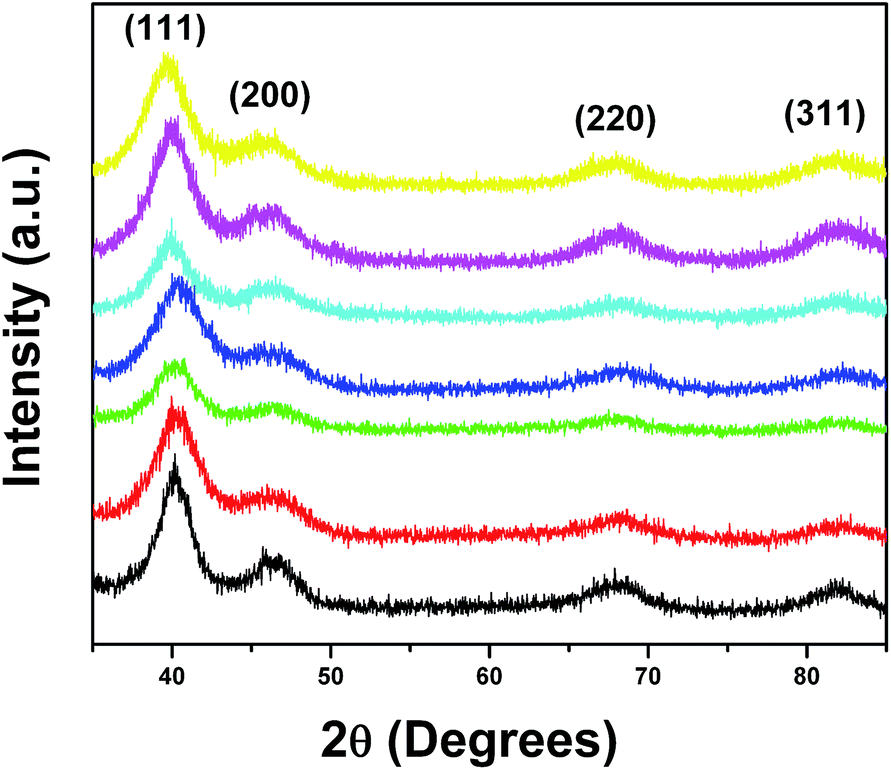 | ||
| Fig. 1 XRD patterns for Pt NWs (black), Pt7Ru3 NWs (red), Pt7Ru2Fe NWs (green), Pt7Ru1.5Fe1.5 NWs (blue), Pt7RuFe2 NWs (baby blue), Pt7Ru.5Fe2.5 NWs (magenta), and Pt7Fe3 NWs (yellow), respectively. All peaks are labeled and correspond to a Pt fcc structure (JCPDS database #04-0802). | ||
Fig. 2, 3, and S1 (ESI†) collectively illustrate the morphological analysis of all of our as-synthesized electrocatalysts using TEM, HRTEM, and SAED. Fig. 2 and 3 suggest that we were able to successfully synthesize the desired NW structural motif for all compositions prepared. All samples possess an average cross-sectional diameter in the range of 1.9 to 2.2 nm, as shown in Table S1.† The usefulness and relevant applicability of our wet synthesis method are evident, as the nanowires produced are effectively homogeneous and monodisperse, and moreover, their diameters are reasonably uniform, even while possessing a host of chemical compositions.
 | ||
| Fig. 2 Representative high resolution TEM images (A, D and G), higher magnification HRTEM images with measured d-spacings (B, E and H), and associated single area electron diffraction patterns (C, F and I) for Pt NWs (A–C), Pt7Ru3 NWs (D–F), and Pt7Fe3 NWs (G–I), respectively. | ||
 | ||
| Fig. 3 Representative high resolution TEM images (A, D, G and J), higher magnification HRTEM images with measured d-spacings (B, E, H and K), and associated single area electron diffraction patterns (C, F, I and L) for Pt7Ru2Fe NWs (A–C), Pt7Ru1.5Fe1.5 NWs (D–F), Pt7RuFe2 NWs (G–I), and Pt7Ru.5Fe2.5 NWs (J–L), respectively. | ||
Higher-resolution TEM images (Fig. 2 and 3) confirm that the interconnected nanowires are segmented and consist of constituent single-crystalline segments with overall lengths exceeding 100 nm.32 These data are consistent with the selected area electron diffraction patterns (Fig. 2 and 3), which collectively indicate that all of the samples maintain a polycrystalline texture. The corresponding lattice parameters were determined from the SAED data as well as deduced from the HRTEM images, and are summarized in Table S2† along with their calculated theoretical parameters. Specifically, the alloy-type NWs possessed measured d-spacings that correspond to the (111) plane of these various alloys, and these values are actually all within experimental error. The slight contraction in the lattice parameters with respect to that of elemental Pt is consistent with that of prior reports in the literature, due to the incorporation of Ru and Fe into the Pt lattice, thereby causing a slight contraction of the fcc lattice. As a result, the SAED patterns and the XRD data further confirm the notion that we can design and subsequently generate highly uniform and homogeneous alloyed nanowires, possessing a variety of desired compositions.
To complement these data, the actual chemical composition of our nanowire samples was determined using energy dispersive X-ray spectroscopy (EDAX). The elemental results are presented in Fig. S2 and Table S1 (ESI†), and are consistent with the expected chemical compositions based on the precursor concentrations used and the NWs we intended to synthesize. As a result, both Fe and Ru are present in these samples with their chemical compositions predicted within experimental error. We should note that an eighth sample (Pt7Ru2.5Fe.5 NW) was synthesized, as can be seen in the ESI section (Fig. S3†). Although the structural and atomic compositions were within error, this particular sample did not evince reliable, interpretable electrochemical results, which might have been due to the relatively small amount of Fe present in the sample. Specifically in this NW sample, as a result of preferential surface dissolution, the Fe may have chemically leached out during electrochemical data acquisition, thereby altering its inherent composition so as to become more similar to that of Pt8Ru2 NWs. Nevertheless, overall, the XRD data coupled with the TEM and SEM-EDAX measurements together confirm that there is a high degree of correlation between the chemical composition of the precursor solutions and the corresponding composition of the resulting NWs.
3.2. Correlating composition of homogeneous alloy catalysts with MOR
As shown in Fig. 4A and B, CVs were obtained for Pt NWs, Pt7Ru3 NWs, Pt7Fe3 NWs, as well as the series of four ternary PtRuFe NWs in order to investigate and correlate the impact of altering Ru and Fe ratios on electrochemical performance. Regarding the substructure of the CVs, the shape and location of the observed hydrogen and oxygen adsorption features associated with the Pt7Ru3 NW catalyst are consistent with prior reports of homogeneous Pt1−xRux alloy type nanowires.33,34 Interestingly, a significant upshift of the oxide reduction peak of ∼200 mV is noted, as the Fe content is increased from 10% to 25% in the Pt7Ru2Fe and Pt7Ru.5Fe2.5 catalysts, respectively. Fig. 5 depicts the linear trend and correlation between onset potential and corresponding alterations in atomic composition. This apparent shift in the surface oxide reduction peak suggests a weakening of the interaction with the oxygen adsorbate and has been observed by previous groups.7,34,35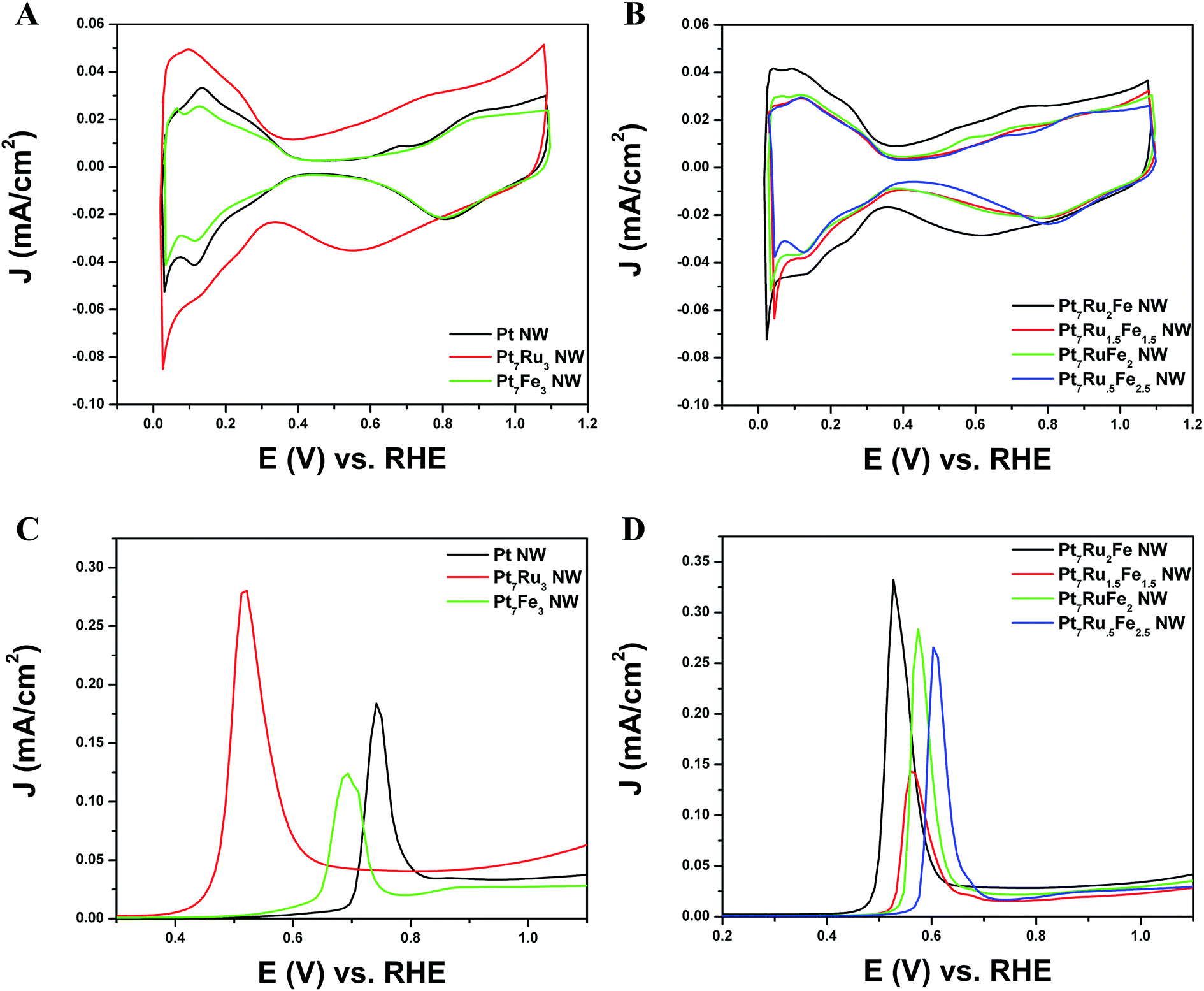 | ||
| Fig. 4 Representative CV curves in an argon-saturated 0.1 M HClO4 solution, obtained at a scan rate of 20 mV s−1 with the current normalized to ECSA for (A) Pt NWs, Pt7Ru3 NWs, and Pt7Fe3 NWs, respectively, as well as for (B) Pt7Ru2Fe NWs, Pt7Ru1.5Fe1.5 NWs, Pt7RuFe2 NWs, and Pt7Ru.5Fe2.5 NWs, respectively. (C) Representative CO stripping LSV curves for Pt NWs, Pt7Ru3 NWs, and Pt7Fe3 NWs, respectively, as well as for (D) Pt7Ru2Fe NWs, Pt7Ru1.5Fe1.5 NWs, Pt7RuFe2 NWs, and Pt7Ru.5Fe2.5 NWs, respectively. | ||
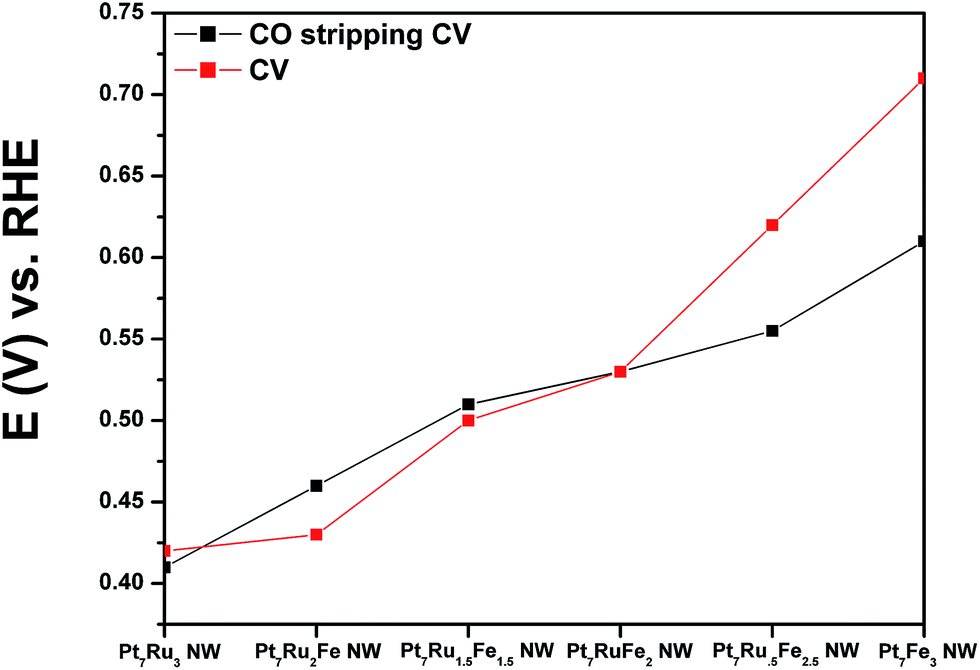 | ||
| Fig. 5 A plot investigating the trend in onset potential for CO stripping (black) and the corresponding onset of surface oxide reduction (red) as a function of systematically varying chemical composition from Pt7Ru3 NWs to Pt7Fe3 NWs. | ||
We attribute this observation to a restructuring effect associated with our as-processed nanowires. Specifically, dissolution is known to occur for a variety of transition metals, including Fe, Ni, and Co, especially when localized at the surface and exposed to anodic potentials.36 Therefore, as the amount of Ru decreases and is effectively substituted with increasing Fe content, any Fe present within these alloys would tend to be preferentially consolidated and incorporated as part of the nanowire core due to the likely dissolution and subsequent removal of Fe at the exposed surface. As a result, the dissolution of iron and concomitant formation of a Pt-rich surface typically results in improved catalytic performance, owing to the unique structural and electronic effects imparted by the interactions between the Pt-rich surface and the alloy-type core.37–39 Corroborating evidence for the enrichment of Pt at the catalytic interface is also apparent in the hydrogen adsorption region, which shows increasing Pt-like character as the Fe content is increased from 10 to 25%; such an observation has also been noted for Pt7Fe3 NWs.
It is evident from the Hads results that Pt is increasingly enriched at the catalytic interface as a function of the decreasing amount of Ru present within the catalyst due to preferential Fe surface dissolution. Nevertheless, it is interesting to note that while the onset of oxide formation for Pt7Ru3 NWs and for Fe-doped Pt7Ru2Fe NWs is similar, the corresponding observed onset for the Pt7Ru1.5Fe1.5 NWs as well as for the remaining ternary catalysts analyzed shifted slightly to higher potentials, denoting behavior analogous to that for the Pt7Fe3 NW catalyst.
To explain all of these data, it is known that in the case of Pt1−xRux alloys, the presence of Pt–Ru pair sites increases MOR catalytic performance by facilitating the oxidation of adsorbed CO species at potentials lower than those observed on elemental Pt as a result of a process referred to as ‘carbonyl-spillover’.40,41 That is, in the presence of Ru, adsorbed CO species generated by the rapid dehydrogenation of methanol at Pt active sites can functionally ‘spill over’ and react with RuOH species, thereby forming CO2. Thus, the CV results suggest that the Fe-doped PtRu NWs may actually benefit from the complementary beneficial effects of (i) the bifunctional mechanism, wherein –OH species dissociated from water and adsorbed onto the Ru surface catalyze the removal of CO adsorbed onto Pt active sites at lower potentials, thereby increasing the overall activity of Pt1−xRux-based alloys, as well as (ii) the favorable electronic effects associated with PtFe alloys, in which the presence of Fe functionally lowers the d-band center of the Pt, thereby resulting in more d-band vacancies and therefore, overall less susceptibility to poisoning of active sites by CO species formed as intermediates in the indirect oxidation of methanol.3,6,42,43
In order to evaluate the methanol oxidation performance, all seven nanowire catalysts were tested electrochemically by comparison with commercial PtRu/C serving as a standard and the resulting linear sweep voltammograms (LSVs) are shown in Fig. 6. For the sake of clarity, the complete MOR CVs, including the forward and reserve sweep scans, are shown in Fig. S4.† We note that Pt7Ru2Fe NWs evinced the highest MOR activity, generating an activity of 2.27 mA cm−2 at 0.8 V vs. RHE; analogous Pt7Ru1.5Fe1.5, Pt7RuFe2, Pt7Ru.5Fe2.5, Pt7Ru3, and Pt7Fe3 NWs yielded notably lower activities with values of 1.86 mA cm−2, 1.46 mA cm−2, 1.35 mA cm−2, 1.46 mA cm−2, and 0.67 mA cm−2, respectively.
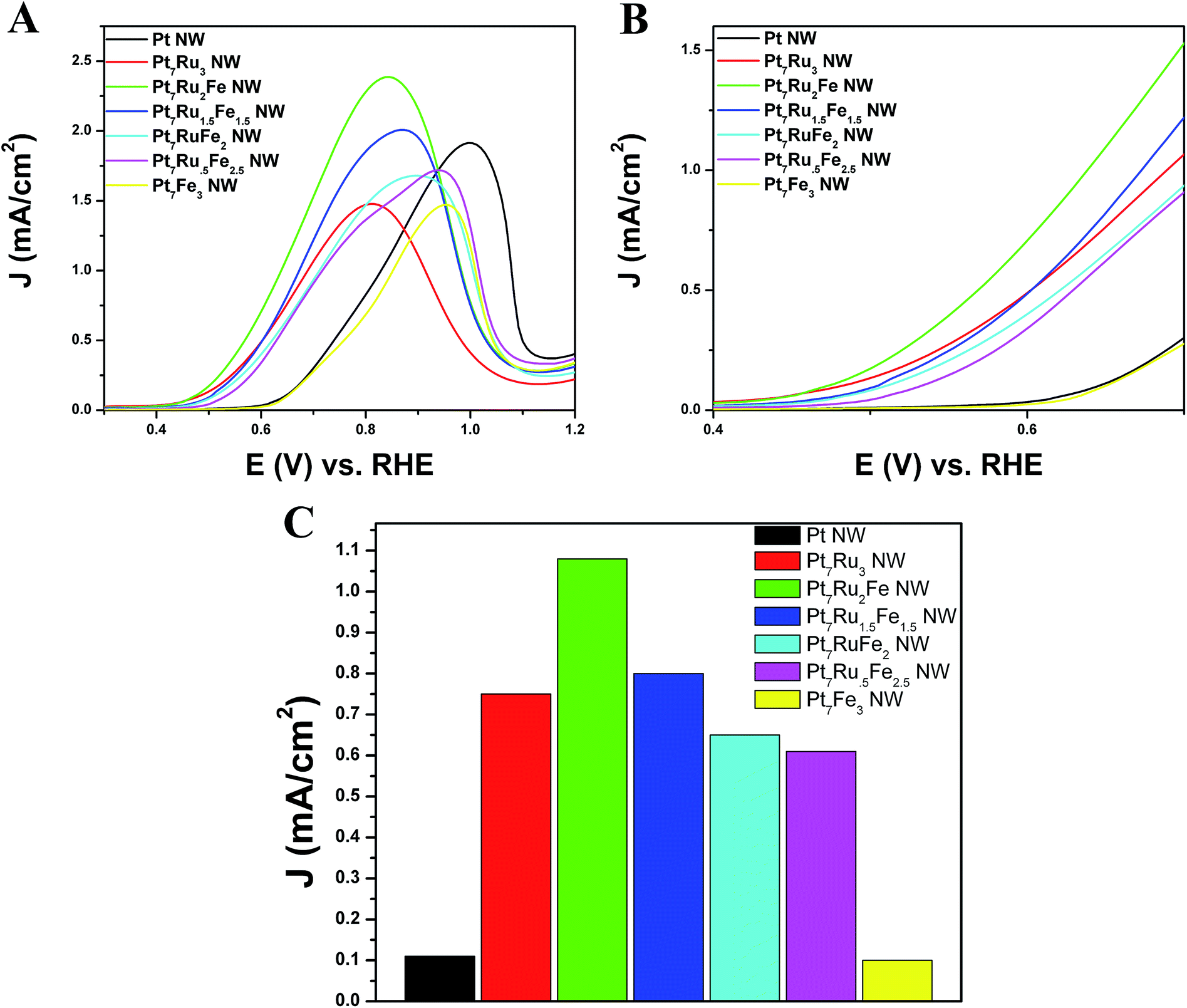 | ||
| Fig. 6 (A) Cyclic voltammograms for the methanol oxidation reaction in an argon-saturated 0.1 M HClO4 + 0.5 M CH3OH solution, obtained at a scan rate of 20 mV s−1 with the current normalized to ECSA. (B) Magnification of the MOR onset region highlighted between 0.4–0.7 V vs. RHE. (C) Bar graph highlighting MOR activity at E (V) vs. RHE = 0.65 V for Pt NWs, Pt7Ru3 NWs, Pt7Ru2Fe NWs, Pt7Ru1.5Fe1.5 NWs, Pt7RuFe2 NWs, Pt7Ru.5Fe2.5 NWs, and Pt7Fe3 NWs, respectively. | ||
Analyzing the onset region shown in Fig. 6B, it is apparent that Pt7Ru2Fe possessed the lowest onset potential of all the NWs tested with a potential of ∼0.44 V. Fig. 6C depicts a bar graph comparing the MOR activity at a representative potential of 0.65 V, which was in the onset region of the LSV. From these data, we measured a specific activity of 1.10 mA cm−2 for our best MOR catalyst tested, namely Pt7Ru2Fe NWs, which is 11-fold higher than that of Pt7Fe3 and Pt NWs (0.10 mA cm−2) and almost 2-times greater as compared with our ‘starting point’, i.e. our as-synthesized Pt7Ru3 NWs (0.75 mA cm−2). More importantly, we demonstrated a volcano-type trend in activity across our series of samples, further supporting our claim that chemical composition is crucial to electrocatalytic performance. Moreover, the numbers reproducibly obtained with our ternary NWs are higher in magnitude as compared with other prior reports of PtRuFe catalysts,6,20,27 and even significantly outperformed conventional commercial standards, such as PtRu NP/C, as can be observed in Fig. S5.†
3.3. Correlating composition of alloy catalysts with MOR and FAOR mechanisms
One of the aims of our study has been to gain insights into the observed enhancements, i.e. encouraging shifts in potential and activity, in our trimetallic Fe-containing PtRuFe NWs and to determine if these phenomena are dependent upon and can therefore be tuned by predictively tailoring chemical composition. Therefore, in experiments aimed at completing our activity analysis and understanding the overall potential of our catalysts, we investigated the performance of our nanowires toward the catalytic oxidation of CO and formic acid, which represent two potential critical intermediates in the oxidation of methanol.A number of factors can influence the principal pathway, such as potential, flow rate, and methanol concentration.47 Currently, experimental evidence for both mechanisms relies primarily on the measured MOR kinetics and an ex situ spectroscopic analysis of electrocatalysts. Therefore, it is necessary to study the oxidation behavior of both methanol and formic acid in order to pinpoint the effects that promote enhanced performance.
Initially, CO stripping LSVs were collected to analyze the CO tolerance of each catalyst, as observed in Fig. 4C and D. The currents measured for each sample were normalized to the electrochemically active surface area (ECSA), which was determined from the hydrogen adsorption region (Hads). As can be observed, the CO onset potential for the Pt NWs (black line) occurs at ∼0.69 V. As more Ru is added (30 atom%), the onset is shifted to a lower potential (i.e. ∼0.41 V), consistent with the bi-functional mechanism; the presence of additional Ru sites enables OH species to be adsorbed at lower potentials, thereby facilitating the removal of adsorbed CO species from Pt active sites. The CO stripping onset values for the various ternary catalysts are shown as a function of NW composition in Fig. 5 and highlight an approximately linear, monotonic correlation between NW composition and the CO stripping onset potential.
This apparent and proportional increase in the CO onset potential with a concomitant rise in Fe content in the composition of ternary NW catalysts is consistent with the relative enrichment of Pt at the interface, due to selective dissolution of Fe sites which correspondingly promotes the formation of a Pt surface layer with an alloy core. Such behavior can also be explained by the corresponding decrease in Ru content, which is responsible for adsorbing OH species that assist in subsequent CO oxidation on the Pt surface. All of these trends are consistent with prior reports, although the onset potentials measured in the Fe-doped, ultrathin NWs are measurably lower as compared with values observed for analogous systems.20,27
Although this trend in CO onset potential (i.e. systematic decrease with increasing Ru metal content) supports the presence of the bifunctional mechanism, it does not fully describe the complicated and synergistic interactions between Pt, Ru, and Fe, and their combined effect upon the resulting MOR mechanism. However, as previously mentioned during the analysis of the CV data, the catalyst possessing only 10% Fe dopant to replace Ru gave rise to a similar onset potential for oxide adsorption as that of control Pt7Ru3 NWs, an observation which further corroborates the viability of a ligand effect, in which the presence of Fe is less conducive to the formation of an adsorbed CO intermediate. In effect, the Pt7Ru2Fe nanowires were found to give rise to the lowest onset potential and highest MOR activity (Fig. 6), presumably due to a diminished affinity towards CO and the corresponding presence of more available Pt active sites for MOR.
In terms of the effect of the ultrathin NW morphology, recent work by our group48 and others49–51 has shown that the segmented texture results in the production of well-ordered smooth crystalline planes along the single crystalline segments as well as defect sites present at the interconnects between segments. Typically, CO binds rather strongly to well-ordered active sites, thereby requiring a higher potential for its removal. Defect sites, on the other hand, require a lower potential for the elimination of CO species.46,52 As a result, elemental ultrathin Pt NWs are particularly active toward alcohol oxidation due to their overall enhanced ability to oxidize CO. That is, since CO can be oxidized at relatively lower potentials as compared with their larger diameter nanowire analogues, ultrathin Pt nanowires provide for more active sites to be available for MOR. Hence, on the basis of our CO stripping results, we show that the CO oxidation performance of ultrathin nanowires can be purposely enhanced by tailoring chemical composition.
| HCOOH → CO2 + 2e− + 2H+ | (3) |
| HCOOH → CO + H2O → CO2 + 2e− + 2H+ | (4) |
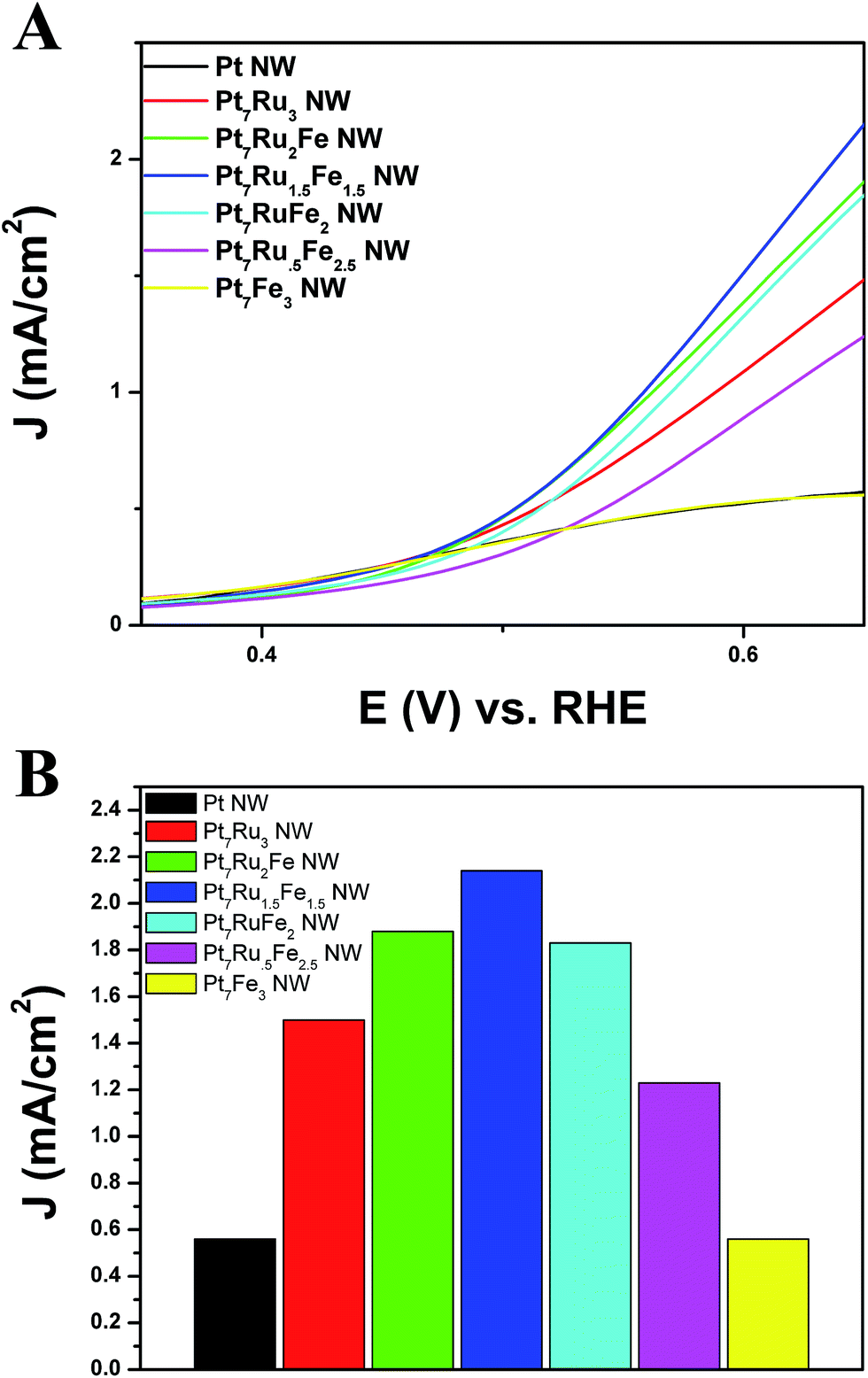 | ||
| Fig. 7 (A) Cyclic voltammograms for the formic acid oxidation reaction in an argon-saturated 0.1 M HClO4 + 0.5 M HCOOH solution, obtained at a scan rate of 20 mV s−1 with the current normalized to ECSA. (B) Bar graph demonstrating FAOR activity at E (V) vs. RHE = 0.65 V for Pt NWs, Pt7Ru3 NWs, Pt7Ru2Fe NWs, Pt7Ru1.5Fe1.5 NWs, Pt7RuFe2 NWs, Pt7Ru.5Fe2.5 NWs, and Pt7Fe3 NWs, respectively. | ||
Fig. 7A depicts the onset region for formic acid oxidation. As can be observed, for all nanowires analyzed, our Pt7Ru1.5Fe1.5 NW catalyst not only possesses the lowest onset potential (0.38 V) for the oxidation of formic acid but also exhibits the fastest kinetics for the reaction, as indicated by its steepest slope. Fig. 7B highlights a bar graph, describing the overall activity of each catalyst towards FAOR measured at a potential of 0.65 V.
Of the samples tested, the Pt7Ru1.5Fe1.5 NW catalyst yielded the highest activity observed (2.15 mA cm−2), which is nearly four times that of as-prepared Pt NWs (0.58 mA cm−2). We noted that the activities of the remaining catalysts also follow a volcano-type trend, similar to what had been previously found for the MOR data in Fig. 6. Specifically, these catalysts achieved FAOR activities of 1.90 mA cm−2 (Pt7Ru2Fe NWs), 1.83 mA cm−2 (Pt7RuFe2 NWs), 1.22 mA cm−2 (Pt7Ru.5Fe2.5 NWs), 1.50 mA cm−2 (Pt7Ru3 NWs), and 0.55 mA cm−2 (Pt7Fe3 NWs), respectively, all of which fall slightly below that of the ‘peak’ value observed for Pt7Ru1.5Fe1.5 NWs. Moreover, our ternary catalysts also outperformed that of commercial PtRu NP/C, as can be observed in Fig. S6.†
A similar onset potential value for both Pt7Ru2Fe NW and Pt7Ru1.5Fe1.5 NW catalysts indicated that the parameter of onset potential was not necessarily influenced by their corresponding CO stripping abilities. More specifically, we found that catalysts possessing the lowest onset for the methanol oxidation reaction and formic acid oxidation reaction did not necessarily give rise to the lowest onset potential for CO stripping.
In Fig. 4, in effect, we were able to observe two distinctive trends, one specifically related to Ru content and another associated with the Fe content. In the cathodic sweep, a shift to lower potentials occurred with the addition of more Ru dopant within a PtRu catalyst as compared with pure Pt, possibly as a result of the bi-functional mechanism.9 Upon the subsequent addition of 10% Fe to replace Ru, no apparent shift in the oxide reduction peak was visible, as compared with the Pt7Ru3 catalysts, suggesting that there may have been the same amount of –OH species absorbed onto the surface.
Such an observation would suggest the presence of a ligand effect.37 By contrast, by adding in 15–30% Fe and correspondingly decreasing the overall Ru content, the catalysts became more ‘noble’ as compared with Pt7Ru3 NW catalysts, due to the relative increase of Pt at the surface (i.e. through the ligand effect) as noted by the shift to higher potentials for the reduction of oxide species. The fact that the Pt7Fe3 NW catalyst evinced a similar onset as compared with Pt can be explained in terms of this phenomenon. Overall, based solely on the surface structure analysis of these catalysts by CV, the Pt7Ru2Fe NW catalyst appeared to exhibit the highest catalytic activity towards MOR.
As a result herein, we have demonstrated that both the oxidation of methanol and formic acid are governed by two contrasting trends. Specifically, methanol oxidation appears to be predominantly influenced by the Ru content, whereas formic acid oxidation is primarily impacted by the corresponding Fe content within the ternary metal alloy catalyst itself. Therefore, by carefully optimizing and tuning chemical composition, we have tried to create a synergistic balance between these two competing behaviors to generate the best compromise catalyst for the overall methanol oxidation reaction, in this case, the Pt7Ru2Fe NW catalyst.
To take these results a step further, chronoamperometric experiments (Fig. 8) were run in order to probe the stability of our as-prepared nanowire catalysts during methanol oxidation. We were able to demonstrate that by using this unique ternary alloy system, we could readily increase catalyst stability as compared with a commercial PtRu NP standard. Overall, once these catalysts became stable, our ternary NW catalyst maintained higher steady state current densities of 0.605 mA cm−2 over the whole time range of 60 minutes as compared with a conventional PtRu NP/C catalyst (i.e. current density of 0.051 mA cm−2), thereby suggesting not only increased stability but also greater catalytic activity for our NW species.
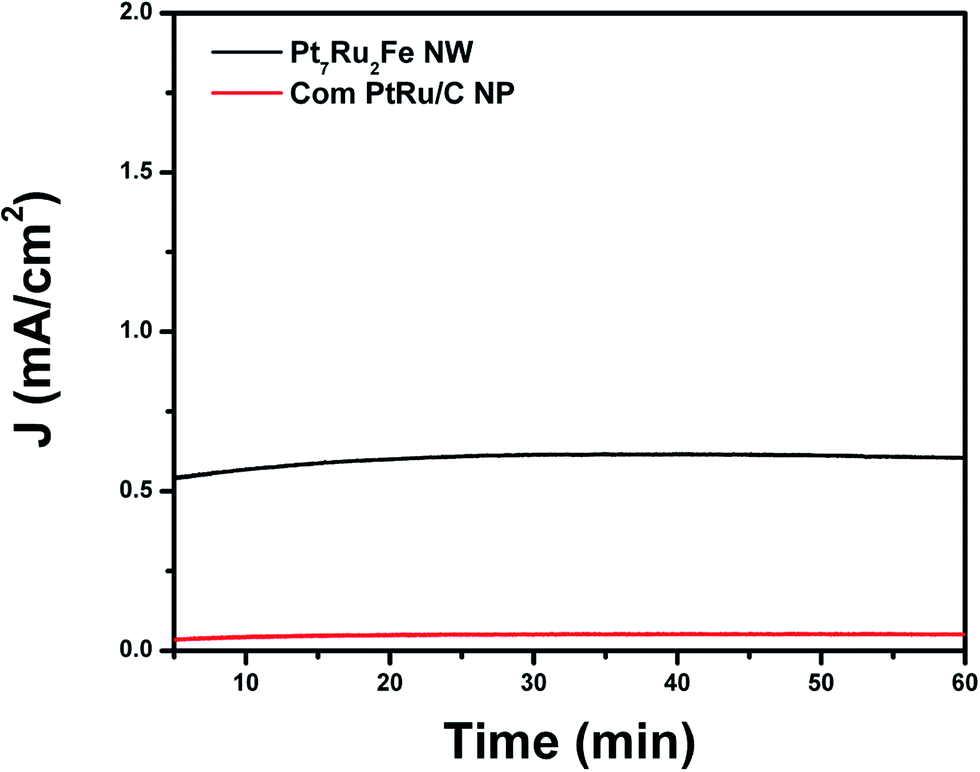 | ||
| Fig. 8 Chronoamperometry measurements of optimized Pt7Ru2Fe NW catalysts as compared with commercial standards (i.e. PtRu NP/C) in an argon-saturated 0.1 HClO4 + 0.5 M CH3OH solution, obtained at a potential of 0.65 V vs. RHE for a period of 60 min. | ||
The enhanced stability of NWs versus NPs has been previously ascribed to stronger NW interactions with the carbon support as well as to an inherently greater NW structural stability, especially under acidic MOR conditions.23 It should also be noted that commercial Pt NP/C achieve comparable steady state current densities as compared with that of the PtRu NP/C controls.25 These results simply reinforce the fact that our ternary Pt7Ru2Fe system represents a more active and stable catalyst for MOR as compared with conventional and even commercial choices.
4. Conclusions and future work
In a previous Perspective article in Energy and Environmental Sciences, we had provided evidence that ternary PtRuM (M = Ni, Co and W) alloys could be considered as promising anodes for DMFCs.24 Therefore, herein, as a variation of that theme, starting from a high-performance binary Pt7Ru3 sample as a ‘base control’, we have successfully synthesized a series of ternary PtRuM nanowire catalysts possessing various controlled atomic compositions of metal dopants (Ru and Fe), utilizing an inverse micellar protocol that had not as yet been previously applied to such chemically complex electrocatalytic systems. As comparative controls, we also reproducibly generated Pt7Ru3 NWs, Pt7Fe3 NWs, and Pt NWs to probe the effect of tailoring chemical composition upon the corresponding electrochemical activity. As-prepared ultrathin NW catalysts possessed diameters of approximately 2 nm and expected chemical compositions, as determined from EDAX data. HRTEM demonstrated that our as-synthesized nanowires were polycrystalline in nature and consisted of short segments of crystalline planes, as further corroborated by SAED patterns.The collected CVs gave rise to shifts in the oxide region, suggesting that interactions between Pt, Ru, and Fe can be explained in the context of the bifunctional mechanism (associated with the alloying of Ru) and the ligand effect (ascribed to the presence of Fe in the alloy core). Specifically, the Pt7Ru2Fe and Pt7Ru3 NW catalysts possessed the lowest onset of formation of Ru–OH species even with a mere 10% loss of Ru and a corresponding 10% increase in Fe content, suggestive of a ligand induced effect, likely as a result of the presence of Fe lowering the d-band center of Pt and thereby altering the electronic properties of the overall alloy. However, for ternary catalytic compositions possessing either a 15%, 20%, or even 25% addition of Fe with the concomitant loss of Ru, the onset potential perceptibly shifted to higher potentials, implying that the presence of 10% Fe content may actually represent the optimal PtRuFe composition for enhanced activity, since it evinced the lowest onset for MOR activity.
Such a finding would also suggest that the CO tolerance of the catalyst is not necessarily correlated with its corresponding MOR activity, since we determined that the Pt7Ru3 NW catalyst possessed a better CO tolerance as compared with the Pt7Ru2Fe NW catalyst but maintained a lower MOR activity. However, we noted that the Pt7Ru1.5Fe1.5 NW catalyst represents the most optimized catalyst amongst the ones tested for formic acid oxidation reaction, and the observed electrochemical enhancement may be due to the synergistic interactions between Fe and Ru, since no significant changes in the CVs, such as shifts to lower potentials in the oxide region, were observed. The stability and durability of our optimized Pt7Ru2Fe NW catalyst as compared with a commercial PtRu NP standard were subsequently probed by chronoamperometry. We noted that our NW system evinced a higher stability, further demonstrating its practicality and real potential as a stable, active, and viable MOR catalyst.
Overall, our multifunctional catalysts not only demonstrated a decreased affinity towards CO as compared with our as-synthesized Pt catalyst control and commercial PtRu standards but also exhibited both higher MOR and FAOR activity as compared with as-prepared binary Pt7Ru3 and Pt7Fe3 NWs, mono-metallic Pt NWs, and commercial catalyst samples. We will continue to investigate the effect of purposely tailoring the chemical compositions of ternary metal catalysts by utilizing additional, less expensive, and comparatively more plentiful first row transition metals to enhance electrocatalytic performance. Nevertheless, our work on this new class of reasonably sustainably produced ternary nanowire catalysts represents a promising avenue for designing new architectural motifs with relevance for fuel cell applications.
Acknowledgements
Research for all authors was supported by the U.S. Department of Energy, Basic Energy Sciences, Materials Sciences and Engineering Division. Experiments for this manuscript were performed in part at the Center for Functional Nanomaterials located at Brookhaven National Laboratory, which is supported by the U.S. Department of Energy under Contract no. DE-AC02-98CH10886. We thank James Quinn (SUNY Stony Brook) for assistance with EDAX measurements.References
- W. C. Choi, M. K. Jeon, Y. J. Kim, S. I. Woo and W. H. Hong, Catal. Today, 2004, 93–95, 517–522 CrossRef CAS PubMed
.
- S. Wasmus and A. Küver, J. Electroanal. Chem., 1999, 461, 14–31 CrossRef CAS
- D. K. Kang, C. S. Noh, N. H. Kim, S.-H. Cho, J. M. Sohn, T. J. Kim and Y.-K. Park, J. Ind. Eng. Chem., 2010, 16, 385–389 CrossRef CAS PubMed
- H. Qiu and F. Zou, ACS Appl. Mater. Interfaces, 2012, 4, 1404–1410 CAS
- S. H. Joo, S. J. Choi, I. Oh, J. Kwak, Z. Liu, O. Terasaki and R. Ryoo, Nature, 2001, 412, 169–172 CrossRef CAS PubMed
- T. Huang, J. Liu, R. Li, W. Cai and A. Yu, Electrochem. Commun., 2009, 11, 643–646 CrossRef CAS PubMed
- N. V. Long, Y. Yang, C. M. Thi, N. V. Minh, Y. Cao and M. Nogami, Nano Energy, 2013, 2, 636–676 CrossRef PubMed
- J. M. Sieben and M. M. E. Duarte, Int. J. Hydrogen Energy, 2012, 37, 9941–9947 CrossRef CAS PubMed
- J. L. Gómez de la Fuente, M. V. Martínez-Huerta, S. Rojas, P. Hernández-Fernández, P. Terreros, J. L. G. Fierro and M. A. Peña, Appl. Catal., B, 2009, 88, 505–514 CrossRef PubMed
- V. A. Ribeiro, O. V. Correa, A. O. Neto, M. Linardi and E. V. Spinacé, Appl. Catal., A, 2010, 372, 162–166 CrossRef CAS PubMed
- D. R. M. Godoi, J. Perez and H. M. Villullas, J. Phys. Chem. C, 2009, 113, 8518–8525 CAS
- H. Wang, L. R. Alden, F. J. DiSalvo and H. c. D. Abruña, Langmuir, 2009, 25, 7725–7735 CrossRef CAS PubMed
- T. Yajima, H. Uchida and M. Watanabe, J. Phys. Chem. B, 2004, 108, 2654–2659 CrossRef CAS
- A. Velázquez-Palenzuela, E. Brillas, C. Arias, F. Centellas, J. A. Garrido, R. M. Rodríguez and P.-L. Cabot, J. Power Sources, 2012, 208, 306–315 CrossRef PubMed
- F. Liu, J. Y. Lee and W. J. Zhou, Small, 2006, 2, 121–128 CrossRef CAS PubMed
- Z. B. Wang, G. P. Yin, P. F. Shi and Y. C. Sun, Electrochem. Solid-State Lett., 2006, 9, A13–A15 CrossRef CAS PubMed
- D.-Y. Wang, H.-L. Chou, Y.-C. Lin, F.-J. Lai, C.-H. Chen, J.-F. Lee, B.-J. Hwang and C.-C. Chen, J. Am. Chem. Soc., 2012, 134, 10011–10020 CrossRef CAS PubMed
- T. Huang, J. Liu, R. Li, W. Cai and A. Yu, Electrochem. Commun., 2009, 11, 643–646 CrossRef CAS PubMed
- P. Strasser, Q. Fan, M. Devenney, W. H. Weinberg, P. Liu and J. K. Nørskov, J. Phys. Chem. B, 2003, 107, 11013–11021 CrossRef CAS
- M. K. Jeon, J. Y. Won, K. R. Lee and S. I. Woo, Electrochem. Commun., 2007, 9, 2163–2166 CrossRef CAS PubMed
- T. Kawaguchi, Y. Rachi, W. Sugimoto, Y. Murakami and Y. Takasu, J. Appl. Electrochem., 2006, 36, 1117–1125 CrossRef CAS
- S. Kageyama, A. Murakami, S. Ichikawa, S. Seino, T. Nakagawa, H. Daimon, Y. Ohkubo, J. Kugai and T. A. Yamamoto, J. Mater. Res., 2012, 27, 1037–1045 CrossRef CAS
- S. Guo, S. Zhang, X. Sun and S. Sun, J. Am. Chem. Soc., 2011, 133, 15354–15357 CrossRef CAS PubMed
- C. Koenigsmann and S. Wong, Energy Environ. Sci., 2011, 4, 1045–1528 Search PubMed
- Q. Yuan, D.-B. Huang, H.-H. Wang and Z.-Y. Zhou, Langmuir, 2014, 30, 5711–5715 CrossRef CAS PubMed
- L. Cademartiri and G. A. Ozin, Adv. Mater., 2009, 21, 1013–1020 CrossRef CAS
- M. K. Jeon, K. R. Lee, H. Daimon, A. Nakahara and S. I. Woo, Catal. Today, 2008, 132, 123–126 CrossRef CAS PubMed
- C. Koenigsmann, M. Scofield, H. Liu and S. Wong, J. Phys. Chem. Lett., 2012, 3, 3385–3398 CrossRef CAS
- Y. Song, R. M. Garcia, R. M. Dorin, H. Wang, Y. Qiu, E. N. Coker, W. A. Steen, J. E. Miller and J. A. Shelnutt, Nano Lett., 2007, 7, 3650–3655 CrossRef CAS PubMed
- S. Yang, F. Hong, L. Wang, S. Guo, X. Song, B. Ding and Z. Yang, J. Phys. Chem. C, 2009, 114, 203–207 Search PubMed
- H. Liu, C. Koenigsmann, R. R. Adzic and S. S. Wong, ACS Catal., 2014, 4, 2544–2555 CrossRef CAS
- C. Koenigsmann, A. C. Santulli, K. Gong, M. B. Vukmirovic, W.-p. Zhou, E. Sutter, S. S. Wong and R. R. Adzic, J. Am. Chem. Soc., 2011, 133, 9783–9795 CrossRef CAS PubMed
- C. Koenigsmann, D. B. Semple, E. Sutter, S. E. Tobierre and S. S. Wong, ACS Appl. Mater. Interfaces, 2013, 5, 5518–5530 CAS
- E. Antolini, Mater. Chem. Phys., 2003, 78, 563–573 CrossRef CAS
- V. R. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross and N. M. Markovic, J. Am. Chem. Soc., 2006, 128, 8813–8819 CrossRef CAS PubMed
- T. Toda, H. Igarashi, H. Uchida and M. Watanabe, J. Electrochem. Soc., 1999, 146, 3750–3756 CrossRef CAS PubMed
- C. Xu, Q. Li, Y. Liu, J. Wang and H. Geng, Langmuir, 2012, 28, 1886–1892 CrossRef CAS PubMed
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Nat. Chem., 2010, 2, 454–460 CrossRef CAS PubMed
- T. Toda, H. Igarashi and M. Watanabe, J. Electrochem. Soc., 1998, 145, 4185–4188 CrossRef CAS PubMed
- S. Stolbov, M. A. Ortigoza, R. Adzic and T. S. Rahman, J. Chem. Phys., 2009, 130, 124714 CrossRef PubMed
- K. Sasaki, J. X. Wang, M. Balasubramanian, J. McBreen, F. Uribe and R. R. Adzic, Electrochim. Acta, 2004, 49, 3873–3877 CrossRef CAS PubMed
- X. Zhang, F. Zhang, R.-F. Guan and K.-Y. Chan, Mater. Res. Bull., 2007, 42, 327–333 CrossRef CAS PubMed
- S. M. M. Ehteshami, Q. Jia, A. Halder, S. H. Chan and S. Mukerjee, Electrochim. Acta, 2013, 107, 155–163 CrossRef CAS PubMed
- W. H. Zhong, Y. X. Liu and D. J. Zhang, J. Phys. Chem. C, 2012, 116, 2994–3000 CAS
- D. Yuan, X. Gong and R. Wu, J. Chem. Phys., 2008, 128, 064706 CrossRef PubMed
- J. Rossmeisl, P. Ferrin, G. A. Tritsaris, A. U. Nilekar, S. Koh, S. E. Bae, S. R. Brankovic, P. Strasser and M. Mavrikakis, Energy Environ. Sci., 2012, 5, 8335–8342 CAS
- H. Wang, T. Loffler and H. Baltruschat, J. Appl. Electrochem., 2001, 31, 759–765 CrossRef CAS
- W.-P. Zhou, M. Li, C. Koenigsmann, C. Ma, S. S. Wong and R. R. Adzic, Electrochim. Acta, 2011, 56, 9824–9830 CrossRef CAS PubMed
- S. Wang, S. P. Jiang, X. Wang and J. Guo, Electrochim. Acta, 2011, 56, 1563–1569 CrossRef CAS PubMed
- F. Si, L. Ma, C. Liu, X. Zhang and W. Xing, RSC Adv., 2012, 2, 401–403 RSC
- B. Y. Xia, H. B. Wu, Y. Yan, X. W. D. Lou and X. Wang, J. Am. Chem. Soc., 2013, 135, 9480–9485 CrossRef CAS PubMed
- S. C. S. Lai, N. P. Lebedeva, T. H. M. Housmans and M. T. M. Koper, Top. Catal., 2007, 46, 320–333 CrossRef CAS
- J. Xu, D. Yuan, F. Yang, D. Mei, Z. Zhang and Y.-X. Chen, Phys. Chem. Chem. Phys., 2013, 15, 4367–4376 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ee02162b |
| This journal is © The Royal Society of Chemistry 2015 |