
Electrochemical wastewater treatment with carbon nanotube filters coupled with in situ generated H2O2†
Yanbiao
Liu
a,
Jianping
Xie
b,
Choon Nam
Ong
a,
Chad D.
Vecitis
c and
Zhi
Zhou
*d
aNUS Environmental Research Institute, National University of Singapore, 117411, Singapore
bDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 117585, Singapore
cSchool of Engineering and Applied Sciences, Harvard University, Cambridge, Massachusetts 02138, USA
dDivision of Environmental and Ecological Engineering and School of Civil Engineering, Purdue University, West Lafayette, Indiana 47907, USA. E-mail: zhizhou@purdue.edu
First published on 23rd July 2015
Abstract
Electrochemically active carbon nanotube (CNT) filters can effectively adsorb and oxidize chemical compounds in the anode, but the role of a cathode in electrochemical filters beyond a counter electrode has not been thoroughly investigated. In this study, a novel wastewater treatment system was developed to combine both adsorption and oxidation in the CNT anode and additional oxidation with in situ generated hydrogen peroxide (H2O2) in the CNT cathode. The impacting factors, treatment efficiency, and oxidation mechanism of the system were systematically studied. The results demonstrated that H2O2 flux could be affected by the electrode material, cathode potential, pH, flow rate, and dissolved oxygen (DO). The maximum H2O2 flux of 1.38 mol L−1 m−2 was achieved with C-grade CNT at an applied cathode potential of −0.4 V (vs. Ag/AgCl), a pH of 6.46, a flow rate of 1.5 mL min−1, and an influent DO flux of 1.95 mol L−1 m−2. Additionally, phenol was used as a model aromatic compound to evaluate the removal efficiency of the system and its oxidation rate was directly correlated with H2O2 flux. H2O2 was likely reacting with a phenol species that was anodically activated to a radical form, since H2O2 alone cannot remove phenol efficiently. Furthermore, electrochemical polymer formation via phenolic radical chain reactions may also contribute to 13% of phenol removal. A stable phenol removal efficiency of 87.0 ± 1.8% within 4 h of continuous operation was achieved with an average oxidation rate of 0.059 ± 0.001 mol h−1 m−2. The developed electrochemical CNT filtration system coupled with in situ generated H2O2 is a new application of carbon nanotube filters and can be used as an effective wastewater treatment system to remove organic pollutants or as a promising point-of-use wastewater treatment system.
Water impactThe presence of organic contaminants in natural surface water and ground water can lead to severe human health effects. In this study, we developed a novel wastewater treatment system that combines both adsorption and oxidation in the carbon nanotube (CNT) anode and additional oxidation with in situ generated hydrogen peroxide (H2O2) in the CNT cathode. Compared with existing electrochemical systems, this system exhibited excellent removal efficiencies. The impacting factors, treatment efficiency, and oxidation mechanism of the system were systematically studied using phenol as a model compound. Overall, these results highlight the potential for the developed CNT filters coupled with in situ produced H2O2 to serve as an effective wastewater treatment system to remove organic pollutants. |
Introduction
With rapid population growth and fast industrial development in recent decades, large amounts of organic pollutants have been discharged into water bodies and substantially damaged aquatic environments.1 Therefore, there is a critical need to develop effective wastewater treatment technologies to remove these organic pollutants. Recent advances in membrane technology have led to an increased use of synthetic organic/inorganic membranes to remove viruses and hazardous chemicals from contaminated sources of water.2 Among all the membrane materials, carbon nanotubes (CNTs) have been considered as a good option due to their mechanical stability, flexibility, and chemical resistivity.3 CNTs can be easily formed into porous 3D networks and used as filters for contaminant sorption and electrochemical degradation due to their high specific surface area (30–650 m2 g−1)4 and high conductivity (104–106 S m−1).5 An electrochemical CNT filter brings this concept one step further by not only adsorptively trapping, but also electrochemically oxidizing the target compounds. Previous results have demonstrated that CNT-based electrochemical filters were effective in removing aqueous organic pollutants, such as salts,6,7 proteins,8 viruses,9 azo dyes,10 pharmaceuticals,11,12 perfluorinated chemicals,13 and phenol.14 Within the electrochemical CNT filtration system, organic pollutants were adsorbed and oxidized via a direct/indirect oxidation process on the anodic CNT filters,15 and a titanium ring or CNT filters were used as counter cathodes to provide the required potential.10,16 However, the role of a cathode in electrochemical filters beyond a counter electrode has not been thoroughly investigated, and previous studies mainly focused on the anodic oxidation of organic pollutants.10,14A cathode provides electrons and only supports reduction instead of oxidation, and therefore it cannot be directly used to oxidize organic pollutants in wastewater, which are mostly reduced hydrocarbons. As CNTs were regarded as a new generation of oxygen reduction reaction (ORR) catalysts,17–19 a CNT cathode could be used to reduce oxygen to generate hydrogen peroxide (H2O2, eqn (1)) with the counter electrode serving as a functional cathode.20–23
| O2 + 2H+ + 2e− → H2O2 | (1) |
As a strong oxidant (E0 = 1.763 V vs. SHE), H2O2 can oxidize various organic pollutants via Fenton reactions,21,24,25 and produce oxygen and water as by-products after oxidation.26–29 However, the high cost and potential hazards associated with the transport and handling of commercial concentrated H2O2 as well as its limited working pH range greatly restrained the practical application of classical Fenton reactions (Fe2+/H2O2).21 To overcome these obstacles, electro-Fenton technology has attracted extensive attention due to the in situ production of H2O2via ORR, which can avoid the addition of expensive and hazardous H2O2. The carbon-based materials have been proven to be effective cathode materials for electro-Fenton systems because of their large surface area, excellent stability and conductivity. For instance, Plakas et al. developed an electrochemical filtration system for in situ generation of H2O2 by employing three different carbonaceous cathode materials (woven carbon fibers, loose carbon fibers, and powdered carbon) in a continuous flow-through mode, and their results demonstrated that the electrode materials, applied potential, pH, and ionic strength were key factors affecting H2O2 yield.30
The objective of this study was to develop a novel wastewater treatment system combining adsorption and oxidation in the CNT anode and additional oxidation with in situ generated H2O2 in the CNT cathode. The impacting factors, treatment efficiency, and oxidation mechanism were systematically studied with phenol as a model aromatic compound because of its toxicity and frequent detection in industrial and municipal sewage.31,32 Finally, the efficacy and efficiency of organic pollutant removal with the proposed filtration system were evaluated using three additional organic compounds: tetracycline (a common antibiotic released from wastewater treatment plants), methyl orange (a common azo dye compound found in industrial wastewater), and geosmin (a common taste & odour compound present at parts per trillion level in water). To the best of our knowledge, the combination of electrochemical CNT filtration and oxidation with in situ generated H2O2 has not been reported previously.
Materials and methods
Chemicals and materials
Sodium sulphate (Na2SO4, ≥99.0%), hydrochloric acid (HCl, ACS reagent, 37%), potassium iodide (KI, ACS reagent, ≥99.0%), ammonium molybdate ((NH4)2MoO4, 99.98% trace metal basis), potassium hydrogen phthalate (C8H5KO4, ≥99.95%), sodium chloride (NaCl, ACS reagent, ≥99%), sodium hydroxide (NaOH, ACS reagent, ≥97.0%), tetracycline (C22H24N2O8·xH2O, ≥98.0%), and methyl orange (C14H14N3NaO3S, ≥85.0%) were purchased from Sigma-Aldrich (St. Louis, MO). Ethanol (EtOH) and n-methyl-2-pyrrolidone (NMP, ACS grade, >99.0%) were purchased from VWR (Singapore). Geosmin standard solution (C12H22O, >98.0%) was purchased from Wako Pure Chemical Industries, Ltd. (Japan). Suwannee River natural organic matter (NOM), extracted from soil and other solid phase sources using a strong base (e.g. NaOH or KOH), was purchased from the International Humic Substances Society (St. Paul, MN). Aqueous solutions were prepared with deionized water (DI-H2O) from an ELGA PURELAB Option system (Singapore) with a resistivity of ≥18.2 MΩ cm−1. CNTs (<d> = 15 nm and <l> = 100 μm, as provided by the manufacturer; BET surface area = 88.5 m2 g−1) were purchased from NanoTechLabs (Yadkinville, NC).Preparation of electrochemical CNT filters
Three types of CNTs with the same length (<l> = 100 μm, as provided by the manufacturer) were used: C-grade (<d> = 15 nm), M-grade (<d> = 40 nm), and J-grade (<d> = 100 nm). All the raw CNTs were calcined at 350 °C for 1 h before use to remove amorphous carbon. Some C-grade CNTs were further treated with HCl at 70 °C for 12 h to remove the remaining iron/iron oxide catalyst. The cathodic and anodic CNT filters were produced by dispersing 15 mg of CNT into 0.5 mg mL−1 NMP and probe-sonicating for 15 min (100 W, LABSONIC® M, Sartorius). The post-sonicated homogeneous dispersion of CNT and NMP was then vacuum-filtered onto a 5 μm Millipore JMWP PTFE membrane (Billerica, MA) and washed sequentially with 100 mL of ethanol, 100 mL of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DI-H2O:EtOH, and 250 mL of DI-H2O before use.
:1 DI-H2O:EtOH, and 250 mL of DI-H2O before use.
Electrochemical filtration apparatus and characterization
All filtration experiments were conducted with an electrochemically modified Whatman filtration casing (Piscataway, NJ) as described previously.16 Two PTFE-supported CNT networks served as the cathode and anode (Fig. S1†) and the inter-electrode distance was twice the thickness of the PTFE membrane (0.11 mm). Both CNT networks were mechanically brought into contact with a titanium current collector and the electrochemistry was driven with a CHI 604E electrochemical analyzer (Austin, TX). A 1 mol L−1 Ag/AgCl reference electrode (Model CHI 111P, CH Instruments) was applied in all cases and located ~2 mm away from the working electrode in a flow cell configuration. Additionally, a perforated titanium cathode was also utilized to compare the effect of cathode materials on both H2O2 flux and phenol removal. After sealing the filtration casing and priming with DI-H2O, a Masterflex L/S digital peristaltic pump (Vernon Hills, IL) was used to pump DI-H2O through the filter for rinsing and calibration. Then, phenol solutions containing different DO fluxes were pumped into the filter. All aqueous concentrations were normalized by the active cross-sectional area of the filters (706 mm2). The electrochemical filtration system was operated at a flow rate of 1.5 mL min−1 unless otherwise noted and 10 mmol L−1 Na2SO4 or NaCl was used as a background electrolyte to normalize the ionic strength and conductivity. The chronoamperometric curves and cyclic voltammograms (CV) were measured with a CHI 604E electrochemical analyzer using a three-electrode system: a cathodic CNT working electrode, an Ag/AgCl reference electrode, and an anodic CNT electrode. Unless otherwise stated, liquid samples flowed through the anodic CNT filter first, then through the cathodic CNT filter.Material characterization and effluent analysis
Liquid chromatography-mass spectrometry (LC-MS) analysis of phenol was performed on an Agilent 1290 UHPLC system (Waldbronn, Germany) coupled to a 6540 quadrupole-time-of-flight (Q-TOF) mass detector. The concentration of H2O2 was measured by the potassium iodide method. Concentrations of DO and pH levels were measured using an Agilent 3200M multi-parameter analyzer (Singapore). The details of CNT network characterization, and other analytical methods are available in the ESI.†Results and discussion
Cyclic voltammetry
The fundamental oxygen electrochemical characteristics of the CNT cathode within the electrochemical filter were examined by cyclic voltammetry (Fig. 1). The CV curves under saturated O2 flux (1.95 mol L−1 m−2) remain flat between −0.1 and 0 V (vs. Ag/AgCl) and exhibited a steep decrease in the reduction current at more negative potentials (≤−0.1 V vs. Ag/AgCl), indicating that O2 reduction dynamics increases significantly with overpotential. Lower currents were observed with higher initial DO flux, which indicated the excellent electro-activity of the CNT cathode for O2 reduction. The reduction peak at −0.32 V (vs. Ag/AgCl) can be ascribed to the generation of H2O2via an O2 reduction process (eqn (1), E = +0.07 V vs. Ag/AgCl at pH 6.46). However, there was no obvious current reduction peak observed under deoxygenated conditions. It is noteworthy that the CV curves with increasing influent O2 also became steeper and this can be explained by the increase of the reduction current with the oxidant (i.e. O2) and the rate-limiting mass transfer of O2.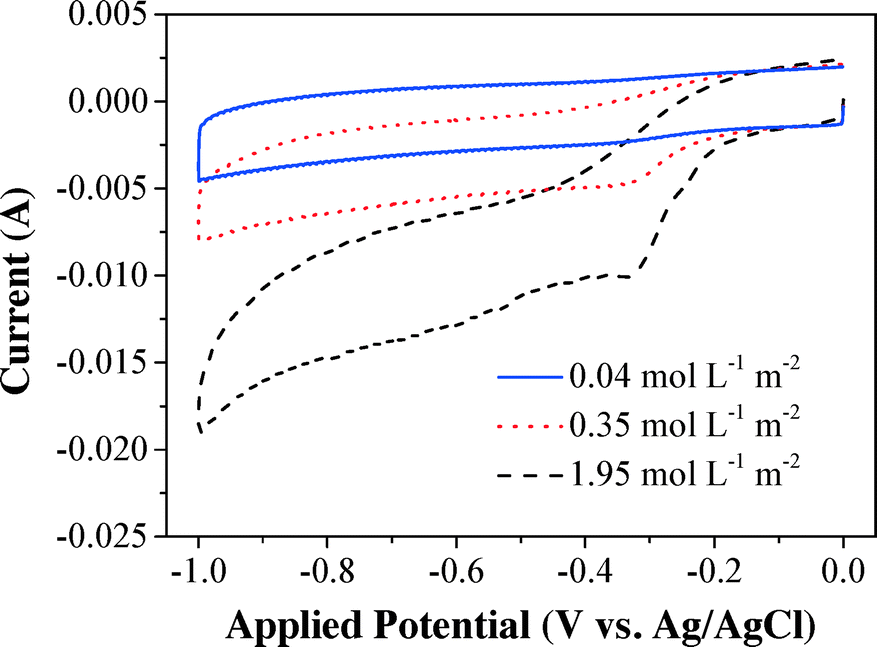 | ||
| Fig. 1 Cyclic voltammetry curves of the CNT electrochemical filter as a functional of applied cathode potential and DO flux levels. Experimental conditions: [Na2SO4] = 10 mmol L−1, flow rate = 1.5 mL min−1, and scan rate = 10 mV s−1. | ||
Effects of the cathode material, cathode potential, pH, DO, and flow rate on H2O2 flux
The performance of electrochemical CNT filters in the degradation of organic pollutants was directly correlated with the amount of H2O2 and/or other reactive oxygen species produced in the medium, which is affected by the cathode materials (Fig. S2†), cathode potential, pH, DO flux, cathode position (Fig. S3†), electrolyte (Fig. S4†), and flow rate (Fig. S5†).The cathode materials significantly affected the system performance. The comparison of perforated titanium and CNT cathodes showed that H2O2 flux and phenol removal rates were improved significantly using the CNT cathode (Fig. S2†). For instance, at an applied cathodic potential of −0.4 V (vs. Ag/AgCl), the H2O2 flux and phenol oxidation rates for the CNT cathode were 140-fold and 41-fold higher than those for the titanium cathode, respectively. The titanium cathode had a total surface area less than 15 cm2 (current density of 0.05–0.50 mA cm−2), while the CNT cathode had a surface area around 5000 cm2 (current density of 0.001–0.010 mA cm−2). The different specific surface areas of the cathodes led to different distributions of the total cell potential. The increased surface area of the CNT cathode increased the fraction of potential going towards the cathodes for O2 reduction,33 and greatly reduced resistance to electron transfer, resulting in increased current efficiency, H2O2 flux, and phenol removal kinetics.
H2O2 flux was also affected by functional CNT cathodes of different dimensions (Fig. 2a). The C-grade CNT with the smallest nanotube diameter (<d> = 15 nm) showed the highest H2O2 flux of 1.04 mol L−1 m−2 (cathode area), which can be ascribed to its largest specific surface area (BET = 88.5 m2 g−1). The C-grade CNT after surface treatment by refluxing in HCl further improved the H2O2 production rate to 1.38 mol L−1 m−2, which occurred because most internal or external Fe2O3 catalysts were removed during acid treatment and more surficial sp2-bonded carbon atoms favored oxygen adsorption and electron transfer.14 H2O2 was constantly produced within a residence time of τ = 1.2 s and H2O2 flux was maintained after 5 min and 30 min. The H2O2 flux in our system (1.38 mol L−1 m−2 or 4.70 mg L−1 cm−2) was significantly improved in reference to previously reported values produced in batch systems, demonstrating that the proposed flow-through filtration system evidently enhanced the mass transfer and reaction kinetics. For example, Isarain-Chávez et al. reported an H2O2 flux of 1.25 mg L−1 cm−2 with a constant current density of 31 mA cm−2 and a pH of 3.0 in a BDD-based electrochemical flow-by system.27 In another separate study, Zhou et al. observed an average H2O2 flux of 0.75 mg L−1 cm−2 min−1 at a cathodic potential of −0.55 V (vs. SCE) in a graphite–PTFE cathode batch system.34
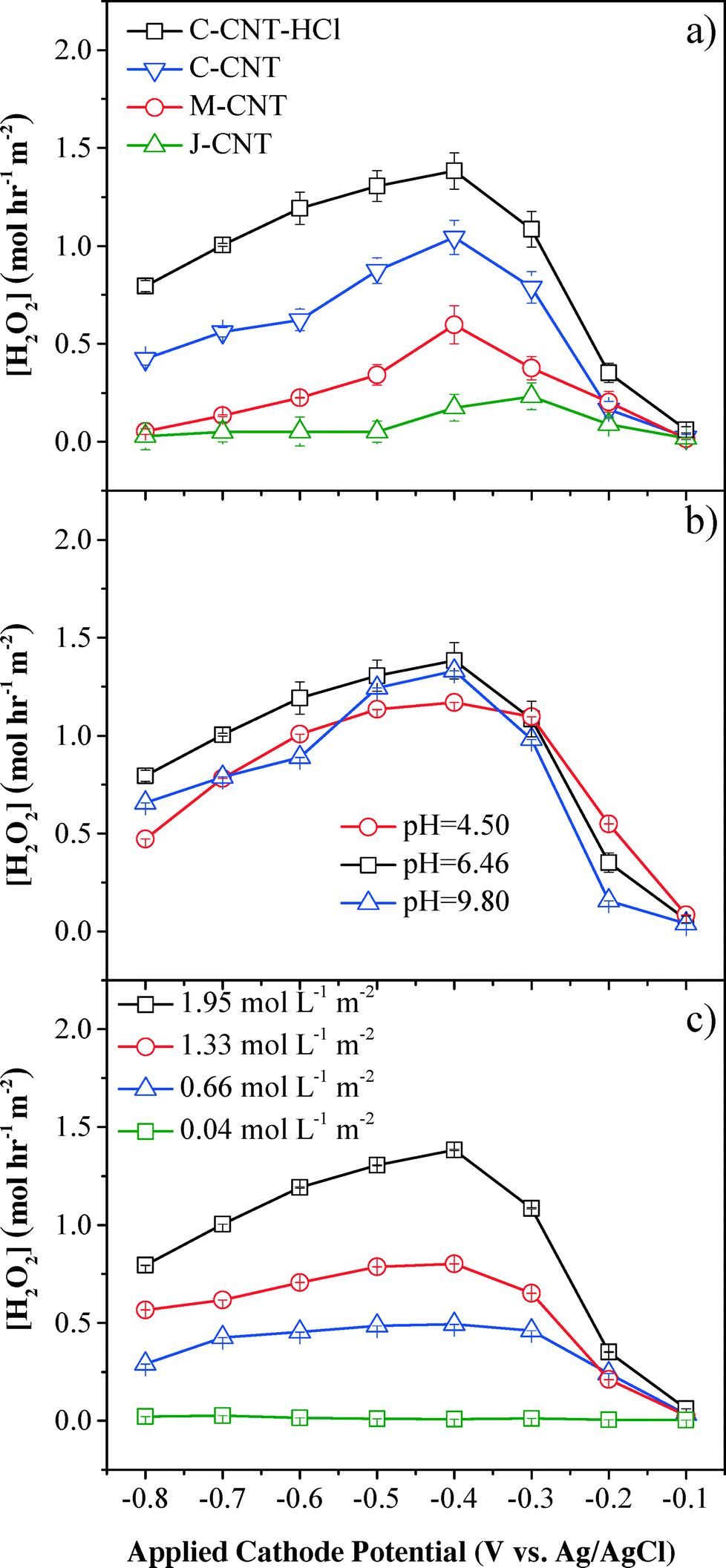 | ||
| Fig. 2 Electrochemical H2O2 flux as a function of (a) CNT dimensions and treatment method, (b) pH, and (c) influent DO flux. Experimental conditions: DO flux = 1.95 mol L−1 m−2 (except in (c)), [Na2SO4] = 10 mmol L−1, and flow rate = 1.5 mL min−1. | ||
The results also demonstrated that H2O2 flux was related to the cathode potential, and the maximum H2O2 flux was observed at −0.4 V (vs. Ag/AgCl), which was consistent with previous results in a batch system.35,36 The initial increase of H2O2 flux between −0.1 V and −0.4 V (vs. Ag/AgCl) was due to the enhanced kinetics, as indicated by the Butler–Volmer equation37 for the reduction of oxygen to produce H2O2 (eqn (1)). When the cathode potential was below −0.4 V (vs. Ag/AgCl), H2O2 flux gradually dropped because of side reactions, e.g., H2O2 was decomposed to O2 (eqn (2), E = +0.49 V vs. Ag/AgCl at pH 6.46), O2 was reduced to water (eqn (3), E = +0.62 V vs. Ag/AgCl at pH 6.46) and H2O2 was directly reduced to H2O (eqn (4), E = +1.15 V vs. Ag/AgCl at pH 6.46). Additionally, [H+] reduction to H2 at more negative cathode potentials may also contribute to the current efficiency decrement (eqn (5), E = −0.60 V vs. Ag/AgCl at pH 6.46).
| 2H2O2 → 2H2O + O2 | (2) |
| O2 + 4H+ + 4e− → 2H2O | (3) |
| H2O2 + 2H+ + 2e− → 2H2O | (4) |
| 2H+ + 2e− → H2 | (5) |
Influent pH was another important impacting factor for the production of H2O2 (Fig. 2b). Compared with basic and acidic conditions, neutral pH conditions presented the highest H2O2 flux at an applied cathode potential of −0.4 V (vs. Ag/AgCl), although the difference with other pH conditions was not significant (<18%). However, it has been extensively reported that acidic conditions (pH ~3) were optimal for H2O2 production from O2 reduction in a conventional batch system.21 This difference could be attributed to the convection-enhanced transfer of the target O2 molecules (τ = 1.2 s) to the CNT surface, which significantly weakened the pH effect on H2O2 yield in this convective flow-through system. A recent study by Agladze et al. also indicated that near neutral pH conditions favored H2O2 production by utilizing a gas diffusion electrode and an undivided reactor.38 Under acidic conditions, the competing reactions, such as eqn (4), consumed more electrons and reduced the current efficiency for H2O2 production (eqn (1)). Under basic conditions (pH = 9.8) with applied potentials below −0.4 V (vs. Ag/AgCl), H2O2 production was lower than that under neutral conditions, which could be caused by the decomposition reaction of H2O2 to O2:
| H2O2 + 2OH− → O2 + 2H2O + 2e− | (6) |
However, further experiments indicated that H2O2 was stable under strongly basic conditions (pH = 12) in the absence of applied potentials, suggesting that other reactions may be involved in H2O2 decomposition under basic conditions with applied potentials. As other factors, such as the reactor configuration, electrode materials, electrolyte, and operation conditions, may also affect H2O2 production, the pH effect on H2O2 production deserves further investigation.
As the main reactant to produce H2O2, influent O2 flux affected H2O2 flux both directly and indirectly. A control experiment carried out by flowing 10 mmol L−1 Na2SO4 solution with saturated O2 at 1.95 mol L−1 m−2 through the filtration system in the absence of an applied potential demonstrated that the O2 loss was negligible (<1%). However, the maximum initial DO flux was limited to 1.95 mol L−1 m−2 due to the low saturated concentration of O2 in water. H2O2 flux increased with higher initial DO flux (Fig. 2c) and the maximum H2O2 flux was obtained at an applied cathode potential of −0.4 V (vs. Ag/AgCl) and an O2 flux of 1.95 mol L−1 m−2, which was 1.7-, 2.8- and 46.8-fold higher than those at DO flux of 1.33, 0.66, and 0.04 mol L−1 m−2, respectively. The maximum current efficiency for H2O2 generation was only 52.9 ± 1.2% (Fig. S6†), suggesting the existence of H2O2 decomposition processes or other electron-consuming reactions. For example, H2O2 could undergo chemical decomposition to O2 either on the anode (heterogeneous process) or in the medium (homogeneous process, eqn (2)). Reduction of H2O2 to OH− on the CNT cathodes could also consume H2O2 and electrons (eqn (7)).34,39 Additionally, H2O2 could be consumed indirectly by other reactive species, such as OH˙ to produce HO2˙ at the anode (eqn (8))40 and eventually decomposed to O2 (eqn (9)).21,39
| H2O2 + 2e− → 2OH− | (7) |
| H2O2 + OH. → HO·2 + H2O | (8) |
| HO·2 → O2 + H+ + e− | (9) |
Effects of the cathode potential on electrochemical and effluent characteristics
Steady-state currents increased rapidly with applied cathode potential, becoming more negative till −0.4 V (vs. Ag/AgCl), then increased slowly between −0.4 to −0.6 V (vs. Ag/AgCl) because of the potential-independent mass transfer limit (Fig. 3a). The steady-state currents continued to increase when the applied cathode potential was below −0.6 V (vs. Ag/AgCl), which suggests that other side reactions, such as eqn (4) and (5), could have contributed to the continuously increased currents and overcome mass transfer limits. The steady-state currents were almost the same under neutral and basic pH conditions, but slightly lower under acidic conditions.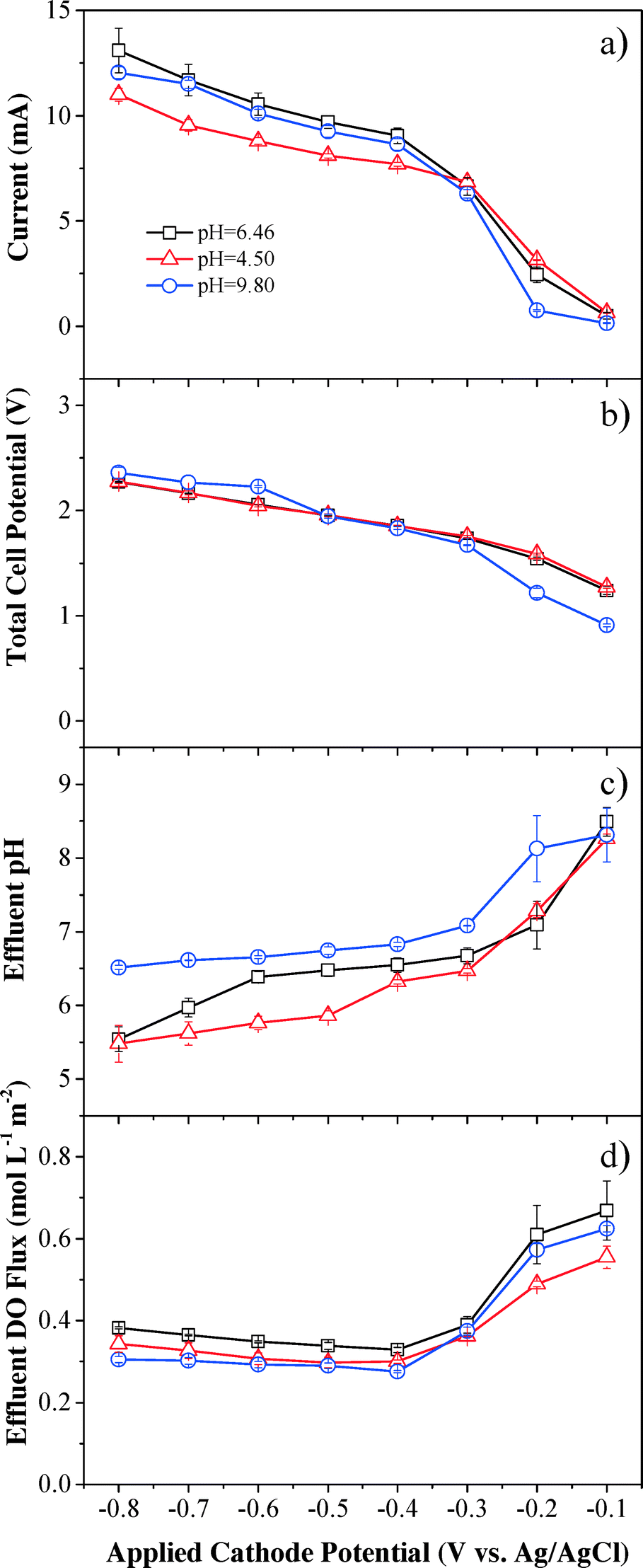 | ||
| Fig. 3 Effects of pH and applied cathode potential on (a) steady-state current, (b) total cell potential, (c) effluent pH, and (d) effluent DO concentration. Experimental conditions: DO flux = 1.95 mol L−1 m−2, [Na2SO4] = 10 mmol L−1, and flow rate = 1.5 mL min−1. | ||
The applied voltage increased monotonically from 0.8 to 2.3 V with decreasing applied cathode potential (Fig. 3b). Almost identical voltages were observed under neutral and acidic pH conditions and the change in voltage was similar to the trend of the steady-state current. The current was higher than 1 mA at a cell voltage higher than 1.2 V and increased monotonically with increasing voltage, indicating that oxidation reactions such as water splitting to O2 at the anode contributed to the current.
Effluent pH was affected by both the applied cathode potential and influent pH (Fig. 3c). Effluent pH values decreased when lower cathode potentials were applied, and effluent pH values were higher when influent pH values were higher. Upon application of −0.1 V (vs. Ag/AgCl) cathode potential, the effluent pH values under neutral and acidic conditions increased in reference to their corresponding influent pH values, indicating that hydrogen ions readily participated in H2O2 production and were removed from the solution, and therefore pH was increased. The effluent pH slightly decreased to 8.31 at −0.1 V (vs. Ag/AgCl) cathode potential under basic conditions with an initial influent pH of 9.80, indicating that electrogenerated H2O2 decomposed to O2 and H2O rapidly in basic media viaeqn (6) and consumption of OH− decreased the effluent pH. The effluent pH then gradually decreased to nearly neutral (pH = 6.56 ± 0.25) at the applied cathode potential of −0.4 V (vs. Ag/AgCl) for all conditions, indicating that the cathodic and anodic processes neutralize each other.
Effluent DO flux showed an inverse trend in comparison to H2O2 flux under all pH conditions (Fig. 3d), since effluent DO was the remnant O2 in solution after O2 reduction reaction (eqn (1)) and the reactants included the influent DO and the O2 produced from water oxidation at the anode. DO flux under acidic and basic conditions was similar and slightly lower than that under neutral pH conditions. The maximum DO efficiency was achieved at an applied cathode potential of −0.4 V (vs. Ag/AgCl) (Fig. S7†). A gap was observed between the saturated influent DO flux (1.95 mol L−1 m−2) and the sum of effluent DO flux and DO utilized for H2O2 production. For example, a DO efficiency of 71.0% under neutral solution conditions at −0.4 V (vs. Ag/AgCl) indicated that 71.0% of DO in the influent (1.38 mol L−1 m−2) was converted to H2O2 through reduction reaction at the cathode. In addition to a DO flux of 0.33 mol L−1 m−2 in the effluent, there was still a gap of 0.24 mol L−1 m−2 DO that disappeared in the system. The potential gas exchange between the reaction system and ambient air may release some DO and other side reactions may also consume DO during the filtration process.
Mechanism of phenol removal
To further unveil the underlying chemistry, various factors were evaluated for their effects on phenol removal. All experiments were conducted under the following optimized conditions for H2O2 production: influent phenol of 0.53 mmol L−1, applied cathode potential of −0.4 V (vs. Ag/AgCl), pH of 6.46, flow rate of 1.5 mL min−1 and Na2SO4 of 10 mmol L−1.Both system setup and electrochemistry were important for effective phenol removal. To evaluate whether phenol can be efficiently oxidized by H2O2 alone without using the CNT filtration system, a batch system was used to mix 0.53 mmol L−1 phenol solution with 1 mmol L−1 H2O2, which was similar to the typical H2O2 concentration produced by the electrochemical filter system. However, no significant change in phenol concentration was observed after 2 h of mixing, indicating that H2O2 alone cannot oxidize phenol efficiently in a conventional batch system (Fig. S8†). Additionally, phenol was not effectively removed by physical adsorption using the CNT filtration system. Breakthrough of 0.53 mmol L−1 phenol in the filtration system with only CNT adsorption occurred in less than 30 min (Fig. S9†), indicating that phenol molecules consumed all reactive surface sites and therefore physical adsorption using CNT filters alone is not sustainable for wastewater treatment.
Although phenol was poorly removed by H2O2 in a batch system, the convection-enhanced transfer of phenol molecules could greatly enhance mass transfer and may result in high phenol removal efficiency in the electrochemical system. Phenol removal rates under different initial phenol flux are shown in Fig. 4a. The HPLC-MS/Q-TOF results (Fig. S10†) indicate that the characteristic phenol peak (m/z = 93.0346) was observed in the influent sample and was significantly decreased by 16.8–92.1% at applied cathodic potentials of −0.1 to −0.8 V (vs. Ag/AgCl), indicating that the parent phenol molecule had been removed or oxidized (Fig. S11†). Maximum phenol removal rates were achieved at an applied cathode potential of −0.4 V (vs. Ag/AgCl), which was consistent with the H2O2 flux results as shown in Fig. 2. The maximum removal kinetics of phenol increased with the initial phenol flux and reached 0.069, 0.024 and 0.007 mol h−1 m−2 for influent phenol flux of 2.20, 0.88 and 0.44 mol L−1 m−2, respectively, which can be ascribed to increased diffusion rates and enhanced mass transfer within the convective flow-through system at high concentrations of phenol. Phenol was efficiently removed when passing through the filters. For example, at an influent phenol flux of 0.44 mol L−1 m−2 (Phenolin = 0.53 mmol L−1 and J = 1.5 mL min−1), the phenol conversion efficiency increased significantly from 17.3% (−0.1 V vs. Ag/AgCl) to 92.1% (−0.4 V vs. Ag/AgCl) and then slightly decreased to 81.3% with further decrement of the applied cathode potential to −0.8 V vs. Ag/AgCl. At lower influent phenol flux (e.g. 0.44 mol L−1 m−2), a potential-independent mass transfer-limited plateau was obtained when the cathode potential was below −0.4 V. However, no plateau was observed for higher phenol flux due to increased diffusion rates at higher concentrations that overcome the mass transfer limitations, indicating mixed control of advection and diffusion. The applied voltage and steady-state current were 1.7–38.5% and 28.0–41.4% lower than those without phenol under the same cathode potentials, respectively, indicating that system resistance was increased in the presence of phenol. One possible mechanism for phenol removal is that the phenol species may be anodically activated to the radical form when passing through the anodic CNT filter which can be efficiently oxidized by H2O2 produced at the cathodic CNT, since H2O2 alone cannot remove phenol efficiently (Fig. S8†). This assumption can be verified through a control experiment by sparging N2 at the CNT cathode to inhibit H2O2 production and mixing the effluent with H2O2 directly. The results show that the phenol species after activation by the anode can be efficiently removed (from 1.2–3.4% to 81.8–82.6%) by the subsequent addition of H2O2 (Fig. S12†), which is in well accordance with our assumption.
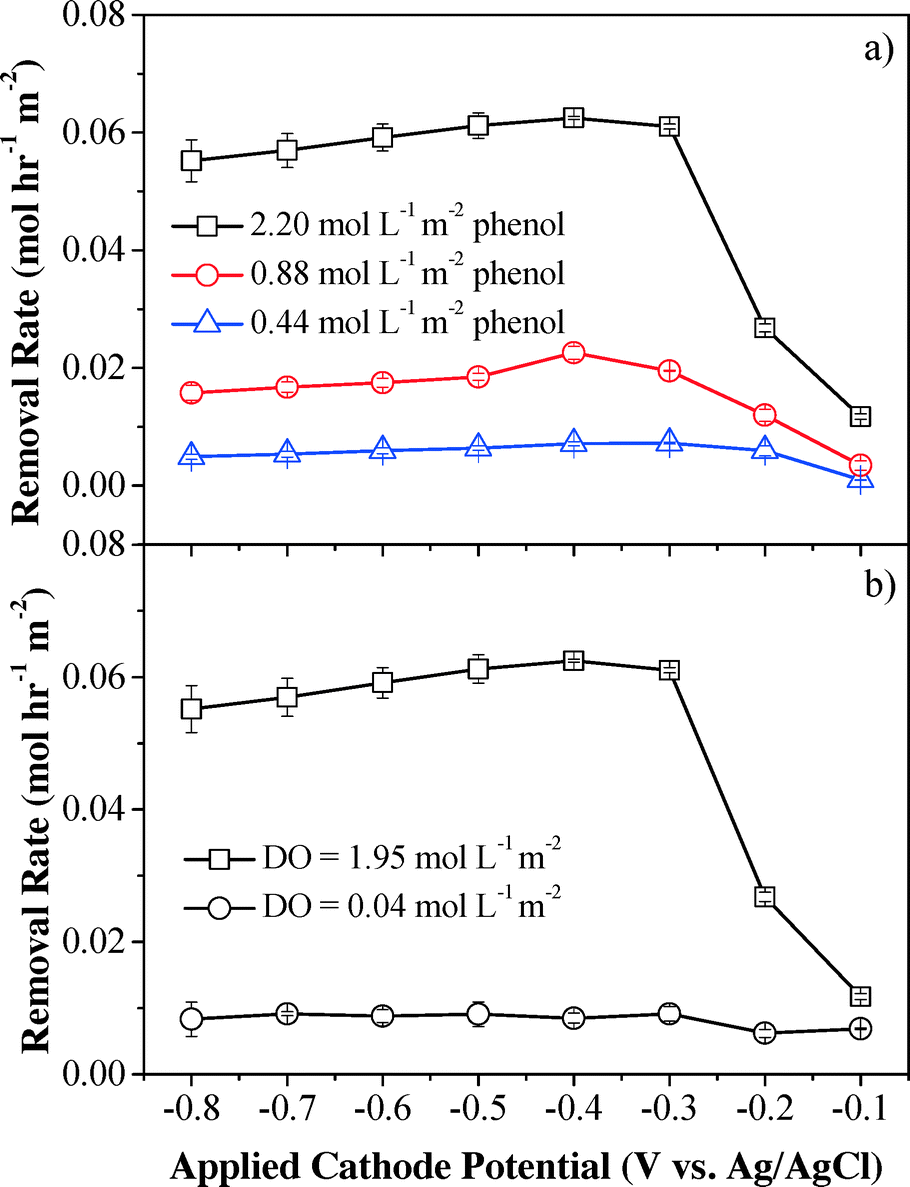 | ||
| Fig. 4 Effect of influent (a) phenol flux and (b) DO flux on phenol removal kinetics. Experimental conditions: [Na2SO4] = 10 mmol L−1, pH = 6.46, and flow rate = 1.5 mL min−1. | ||
The electrochemical polymer formation via phenolic radical chain reactions may also contribute to phenol removal as previously reported.41 Phenol electropolymerization could start with one-electron oxidation of phenol (eqn (10)) and the generated phenolic radical could react with another phenol molecule or phenol oxidation product to initiate a chain polymerization process (eqn (11)).
| C6H5OH + h+ → C6H5O. | (10) |
| C6H5O. + C6H5OH → C6H5OC6H4O | (11) |
The polymerization process was supported by the increased weight in the anodic CNT filter network (+56.9%), decreased steady-state current (−42.4%) after 5 h of continuous operation (Fig. S13†), and SEM images of the anodic CNT filters (Fig. S14†). The detection of filtration by-products also supports this polymerization mechanism. A few anionic products were detected in all effluent samples (Fig. S15 & S16†), such as C6H4O (m/z = 91.0189, 7.4% of influent phenol area), C5H4O2 (m/z = 95.0137, 40.3% of influent phenol area), C6H6O2 (m/z = 109.0294, 1026.4% of influent phenol area), C7H6O2 (m/z = 121.0294, 114.0% of influent phenol area), and C7H6O3 (m/z = 137.0243, 69.1% of influent phenol area), suggesting that polymers were formed during the filtration process. Hydroquinone (C6H6O2) was found to be a dominant by-product in this study. It was reported that hydroquinone could be transformed into benzoquinone via redox reactions,42 and quinone compounds could activate persulfate and induce the production of sulfate radicals (SO4˙−),43 which may further contribute to phenol oxidation and removal.
To further illustrate the phenol removal mechanism, the contribution from the anodic CNT filter alone was quantified. The CNT cathode was sparged with pure N2 instead of O2 to inhibit H2O2 production from the initial dissolved O2, and therefore removal of the remaining phenol can be mainly ascribed to direct anodic oxidation, and anodically induced polymerization. The results demonstrated that the average phenol removal rate sharply decreased by 87% to 0.0082 ± 0.0011 mol h−1 m−2 when the influent DO flux was around zero and only 13% phenol removal was derived from the anode part (Fig. 4b). This result is consistent with the previous assumption that H2O2 is likely reacting with a phenol species that is anodically activated to a radical form, since both H2O2 alone and anodic oxidation/polymerization alone cannot remove phenol efficiently.
In addition to electrogenerated H2O2, other reactive oxygen species (e.g., O2˙−, HO2˙ and OH˙) could be produced within the CNT filtration system and may be involved in phenol removal. Superoxide radicals, O2˙−, are reactive compounds produced when oxygen is reduced by electrons (eqn (12), E = −0.79 V vs. Ag/AgCl at pH 6.46) and occur widely in nature.44 O2˙− is in equilibrium in aqueous solution with the hydroperoxyl radical, HO2˙ (eqn (13), E = +0.18 V vs. Ag/AgCl at pH 6.46).45 The low pKa (4.8) of HO2˙ suggests that most HO2˙ radicals are converted to O2˙− under neutral conditions. Meanwhile, HO2˙ can also convert to H2O2 (eqn (14), E = +1.21 V vs. Ag/AgCl at pH 6.46) and H2O (eqn (15), E = +1.42 V vs. Ag/AgCl at pH 6.46) under more negative cathode potentials.
| O2 + e− → O˙−2 | (12) |
| O˙−2 + H2O → HO˙2 + OH− | (13) |
| HO˙2 + H+ + e− → H2O2 | (14) |
| HO˙2 + 3H+ + 3e− → 2H2O | (15) |
To evaluate the contribution of reactive oxygen species to phenol oxidation, benzoquinone (O2˙− scavenger,46,47k = 109 M−1 s−1) and tert-butanol (OH˙ scavenger,48,49k = 6 × 108 M−1 s−1) were spiked into phenol solution before phenol was pumped into the electrochemical filter. Scavenger tests showed that phenol oxidation was mainly due to H2O2. Compared with the control without scavengers, the phenol removal rate decreased by 24% after benzoquinone was introduced at the start of filtration (0 s) or after the system was operated for 540 s, suggesting that O2˙− contributed to 20.9% of phenol removal in the electrochemical CNT filter (Fig. 5a). Conversely, no significant change in the phenol oxidation rate was observed when tert-butanol was spiked, indicating that phenol removal by OH˙ was negligible or did not occur within the electrochemical CNT filter (Fig. 5b). The presence of high concentrations of by-products and limited cell potential also supported this assumption. Therefore, the remaining 66.1% of phenol removal can be attributed to the synergistic effect of anodic oxidation/polymerization and H2O2 reacting with the anodically activated phenol species during filtration. Contrarily, Zaky and Chaplin previously studied phenolic compound oxidation at a reactive electrochemical Ti4O7 membrane and proposed that the produced OH˙ species were the main contributor.50
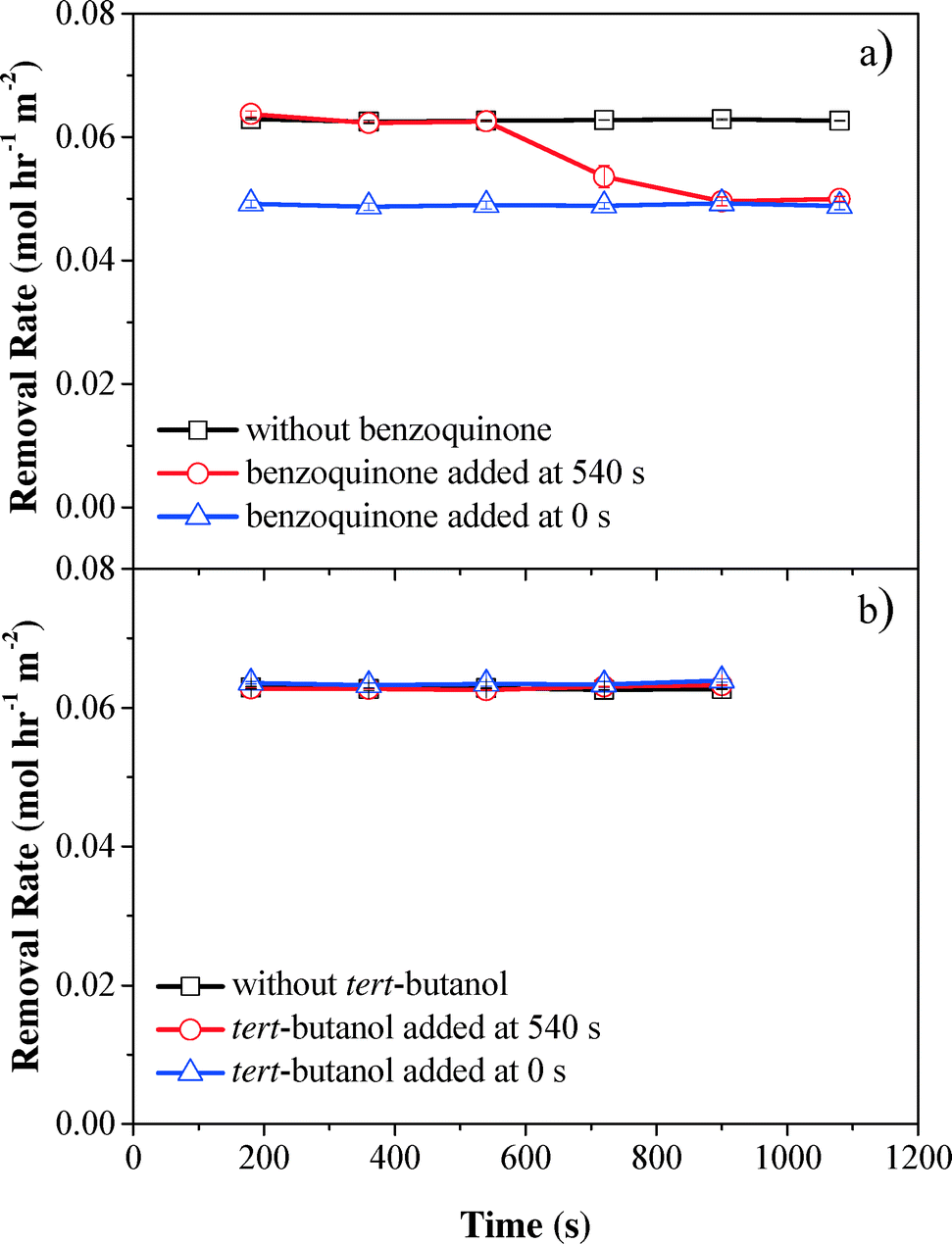 | ||
| Fig. 5 Effect of (a) benzoquinone and (b) tert-butanol on phenol removal kinetics. Experimental conditions: applied cathode potential = −0.4 V vs. Ag/AgCl, [Na2SO4] = 10 mmol L−1, pH = 6.46, and flow rate = 1.5 mL min−1. | ||
The efficient removal rates of phenol revealed that H2O2 production coupled with electrochemical CNT filters could be used to efficiently remove phenolic compounds in wastewater. As the electrochemical removal of phenol may decrease due to reduced reactive CNT surface sites that were consumed by adsorbed compounds or oxidation by-products, continuous operation for 4 h was conducted to evaluate phenol removal and the short-term stability of the system. Although a low degree of polymerization was observed on the anodic CNT surface (Fig. S14†), an average phenol oxidation rate of 0.059 ± 0.001 mol h−1 m−2 and a phenol removal efficiency of 87.0 ± 1.8% were achieved after 4 h of continuous operation under an applied cathode potential of −0.4 V (vs. Ag/AgCl), and no significant efficiency deterioration was observed (Fig. S17†). The high oxidation rates and absence of complete breakthrough indicated that the primary removal mechanism of phenol was anodic polymerization and H2O2 oxidation, rather than physical adsorption.
Other applications for organic contaminant removal and energy consumption
The effective removal of phenol suggested that such an electrochemical filter could be used for water purification. To further explore the potential of using this electrochemical system for contaminant removal, the filter was tested with three additional organic compounds: tetracycline, methyl orange, and geosmin. All three organic compounds were efficiently removed under optimized conditions (i.e., applied cathode potential = −0.4 V vs. Ag/AgCl, DO flux = 1.95 mol L−1 m−2, pH = 6.46) and removal efficiencies for 0.1 mmol L−1 tetracycline, 0.1 mmol L−1 methyl orange, and 0.55 nmol L−1 geosmin were 90.3%, 96.0%, and 87.2%, respectively (Fig. S18†). The results suggest that such an electrochemical filter can be widely used to treat a variety of different organic contaminants for water purification.Energy consumption is an important aspect for any new wastewater treatment system. The energy consumption for electrochemical phenol filtration was calculated to be 0.19 kW h m−3 at a total cell potential of 1.85 V (corresponding to a cathode potential of −0.4 V vs. Ag/AgCl) and a current of 9.05 mA. Additionally, the pumping energy was considered to pump the liquid solution through the filter. At a common backpressure of 15 kPa,14 a flow rate of 1.5 mL min−1, and a pumping efficiency of 75%, the total energy cost for pumping was calculated to be 1.35 J,15 which is only 2.2% of the energy used for electrochemical H2O2 production. These values are similar or lower than state-of-the-art electrochemical oxidation processes with energy consumptions in the range of 0.1–10 kW h m−3.51–53
There are several limitations on our proposed filtration system. For instance, both previous studies54,55 and current work have demonstrated that anodic CNT electrode passivation and clogging caused by electrochemical filtration of phenolic compounds may significantly limit the practical applications towards real contaminated water. Therefore, further systematic studies will be required towards the electrode passivation mechanism, electrode regeneration technologies, and passivation prevention techniques. Additionally, the presence of dissolved natural organic matter (NOM) may significantly decrease (−17%) the removal efficiency of the filtration system (Fig. S19†). The potential formation of chlorinated by-products during treatment of real wastewater samples deserves further investigation (Fig. S20†).
Conclusions
In summary, an effective and novel wastewater treatment system was developed by combining oxidation & adsorption at the CNT anode and additional oxidation with in situ generated H2O2 at the CNT cathode. The electrode materials, applied potential, pH, electrolyte, and DO were found to be important parameters influencing the H2O2 yield and pollutant oxidation performance. Although there is additional electrical energy input in the treatment setup, the benefits of high H2O2 yield, high oxidation kinetics, short residence time, ease of regeneration, and in situ H2O2 production can compensate for the additional energy input. Furthermore, a solar panel can be employed to provide the low cell potential, and the proposed wastewater treatment system can be widely used as efficient and cost-effective end-treatment equipment for point-of-use applications.Acknowledgements
This work is supported by the Singapore National Research Foundation under its Environment and Water Technologies Strategic Research Programme and administered by the Environment and Water Industry Programme Office (EWI) of the PUB under project 1102-IRIS-14-03. Y. Liu, J. Xie and Z. Zhou also thank research support from NERI-GE (GE-NERI 2014, grant number R-706-005-004-592) and a start-up grant from Purdue University. We thank Ms. Qiaoying Zhang and Mr. Han Liu at Harvard University, Prof. Guandao Gao at Nankai University and Dr. Hui Zhang at NUS Environmental Research Institute for their valuable technical discussions. We thank Mr. Qing Xia and Mr. Yi Kong for performing preliminary experiments and Dr. Ying Ma at the National University of Singapore for the FESEM analysis.Notes and references
- Y. Liu, J. Li, B. Zhou, X. Li, H. Chen, Q. Chen, Z. Wang, L. Li, J. Wang and W. Cai, Water Res., 2011, 45, 3991–3998 CrossRef CAS PubMed.
- S. C. Lewis, S. Datta, M. H. Gui, E. L. Coker, F. E. Huggins, S. Dauner, L. Bachas and D. Bhattacharyya, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8577–8582 CrossRef PubMed.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS PubMed.
- S. M. Lee, S. C. Lee, J. H. Jung and H. J. Kim, Chem. Phys. Lett., 2005, 416, 251–255 CrossRef CAS PubMed.
- R. Z. Ma, C. L. Xu, B. Q. Wei, J. Liang, D. H. Wu and D. J. Li, Mater. Res. Bull., 1999, 34, 741–747 CrossRef CAS.
- C. F. De Lannoy, D. Jassby, K. Gloe, A. D. Gordon and M. R. Wiesner, Environ. Sci. Technol., 2013, 47, 2760–2768 CrossRef CAS PubMed.
- Y. Liu, J. H. Dustin Lee, Q. Xia, Y. Ma, Y. Yu, L. Y. Lanry Yung, J. Xie, C. N. Ong, C. D. Vecitis and Z. Zhou, J. Mater. Chem. A, 2014, 2, 16554–16562 CAS.
- X. Sun, J. Wu, Z. Chen, X. Su and B. J. Hinds, Adv. Funct. Mater., 2013, 23, 1500–1506 CrossRef CAS PubMed.
- M. S. Rahaman, C. D. Vecitis and M. Elimelech, Environ. Sci. Technol., 2012, 46, 1556–1564 CrossRef CAS PubMed.
- C. D. Vecitis, G. Gao and H. Liu, J. Phys. Chem. C, 2011, 115, 3621–3629 CAS.
- H. Li, D. Zhang, X. Han and B. Xing, Chemosphere, 2014, 95, 150–155 CrossRef CAS PubMed.
- Y. Liu, H. Liu, Z. Zhou, T. Wang, C. N. Ong and C. D. Vecitis, Environ. Sci. Technol., 2015, 49, 7974–7980 CrossRef CAS PubMed.
- S. Deng, Q. Zhang, Y. Nie, H. Wei, B. Wang, J. Huang, G. Yu and B. Xing, Environ. Pollut., 2012, 168, 138–144 CrossRef CAS PubMed.
- G. D. Gao and C. D. Vecitis, Environ. Sci. Technol., 2011, 45, 9726–9734 CrossRef CAS PubMed.
- H. Liu and C. D. Vecitis, J. Phys. Chem. C, 2012, 116, 374–383 CAS.
- M. H. Schnoor and C. D. Vecitis, J. Phys. Chem. C, 2013, 117, 2855–2867 CAS.
- H. Li, H. Liu, Z. Jong, W. Qu, D. Geng, X. Sun and H. Wang, Int. J. Hydrogen Energy, 2011, 36, 2258–2265 CrossRef CAS PubMed.
- X. Zhang, J. Fu, Y. Zhang and L. Lei, Sep. Purif. Technol., 2008, 64, 116–123 CrossRef CAS PubMed.
- M. Zhang, Y. Yan, K. Gong, L. Mao, Z. Guo and Y. Chen, Langmuir, 2004, 20, 8781–8785 CrossRef CAS PubMed.
- Y. B. Xie and X. Z. Li, J. Hazard. Mater., 2006, 138, 526–533 CrossRef CAS PubMed.
- E. Brillas, I. Sires and M. A. Oturan, Chem. Rev., 2009, 109, 6570–6631 CrossRef CAS PubMed.
- J. J. Pignatello, E. Oliveros and A. MacKay, Crit. Rev. Environ. Sci. Technol., 2006, 36, 1–84 CrossRef CAS PubMed.
- Y. L. Hsiao and K. Nobe, Chem. Eng. Commun., 1993, 126, 97–110 CrossRef CAS PubMed.
- X. Z. Li and H. S. Liu, Environ. Sci. Technol., 2005, 39, 4614–4620 CrossRef CAS.
- C. H. Feng, F. B. Li, H. J. Mai and X. Z. Li, Environ. Sci. Technol., 2010, 44, 1875–1880 CrossRef CAS PubMed.
- S. Yuan, Y. Fan, Y. Zhang, M. Tong and P. Liao, Environ. Sci. Technol., 2011, 45, 8514–8520 CrossRef CAS PubMed.
- E. Isarain-Chávez, C. d. l. Rosa, C. A. Martínez-Huitle and J. M. Peralta-Hernández, Int. J. Electrochem. Sci., 2013, 8, 3084–3094 Search PubMed.
- J. C. Forti, J. A. Nunes, M. R. V. Lanza and R. Bertazzoli, J. Appl. Electrochem., 2007, 37, 527–532 CrossRef CAS.
- N. Guillet, L. Roué, S. Marcotte, D. Villers, J. P. Dodelet, N. Chhim and S. T. Vin, J. Appl. Electrochem., 2006, 36, 863–870 CrossRef CAS.
- K. V. Plakas, A. J. Karabelas, S. D. Sklari and V. T. Zaspalis, Ind. Eng. Chem. Res., 2013, 52, 13948–13956 CrossRef CAS.
- J. Xiao and L. Qi, Nanoscale, 2011, 3, 1383–1396 RSC.
- A. O. Olaniran and E. O. Igbinosa, Chemosphere, 2011, 83, 1297–1306 CrossRef CAS PubMed.
- Q. Zhang and C. D. Vecitis, J. Membr. Sci., 2014, 459, 143–156 CrossRef CAS PubMed.
- M. Zhou, Q. Yu and L. Lei, Dyes Pigm., 2008, 77, 129–136 CrossRef CAS PubMed.
- J. S. Do and C. P. Chen, J. Electrochem. Soc., 1993, 140, 1632–1637 CrossRef CAS PubMed.
- G. V. Kornienko, N. V. Chaenko, I. S. Vasil'eva and V. L. Kornienko, Russ. J. Electrochem., 2004, 40, 148–152 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods. Fundamentals and Applications, Wiley, New York, 2nd edn, 2001 Search PubMed.
- G. R. Agladze, G. S. Tsurtsumia, B. I. Jung, J. S. Kim and G. Gorelishvili, J. Appl. Electrochem., 2007, 37, 375–383 CrossRef CAS.
- E. Brillas, R. M. Bastida and E. Llosa, J. Electrochem. Soc., 1995, 142, 1733–1741 CrossRef CAS PubMed.
- W. R. Haag and C. C. David Yao, Environ. Sci. Technol., 1992, 26, 1005–1013 CrossRef CAS.
- G. Gao and C. D. Vecitis, ACS Appl. Mater. Interfaces, 2012, 4, 6096–6103 CAS.
- P. Petrangolini, A. Alessandrini and P. Facci, J. Phys. Chem. C, 2013, 117, 17451–17461 CAS.
- G. Fang, J. Gao, D. D. Dionysiou, C. Liu and D. Zhou, Environ. Sci. Technol., 2013, 47, 4605–4611 CrossRef CAS PubMed.
- G. W. Thorpe, M. Reodica, M. J. Davies, G. Heeren, S. Jarolim, B. Pillay, M. Breitenbach, V. J. Higgins and I. W. Dawes, Mol. Biol. Cell, 2013, 24, 2876–2884 CrossRef CAS PubMed.
- B. H. J. Bielski, D. E. Cabelli, R. L. Arudi and A. B. Ross, J. Phys. Chem. Ref. Data, 1985, 14, 1041–1100 CrossRef CAS PubMed.
- N. Oshitani, A. Kitano, H. Okabe, S. Nakamura, T. Matsumoto and K. Kobayashi, Gut, 1993, 34, 936–938 CrossRef CAS.
- Y. Sawada, T. Iyanagi and I. Yamazaki, Biochemistry, 1975, 14, 3761–3764 CrossRef CAS.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS PubMed.
- J. Ma, M. Sui, T. Zhang and C. Guan, Water Res., 2005, 39, 779–786 CrossRef CAS PubMed.
- A. M. Zaky and B. P. Chaplin, Environ. Sci. Technol., 2014, 48, 5857–5867 CrossRef CAS PubMed.
- C. Cameselle, M. Pazos and M. A. Sanromán, Chemosphere, 2005, 60, 1080–1086 CrossRef CAS PubMed.
- M. Panizza and G. Cerisola, Chem. Rev., 2009, 109, 6541–6569 CrossRef CAS PubMed.
- M. Panizza and G. Cerisola, Water Res., 2006, 40, 1179–1184 CrossRef CAS PubMed.
- G. Gao and C. D. Vecitis, ACS Appl. Mater. Interfaces, 2012, 4, 1478–1489 CAS.
- G. Gao and C. D. Vecitis, Electrochim. Acta, 2013, 98, 131–138 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ew00128e |
| This journal is © The Royal Society of Chemistry 2015 |