
Singlet fission induced giant optical limiting responses of pentacene derivatives†
Min
Zhao
a,
Ke
Liu
a,
You-Dan
Zhang
a,
Qiang
Wang
a,
Zhong-Guo
Li
b,
Ying-Lin
Song
bc and
Hao-Li
Zhang
*a
aState Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Key Laboratory of Special Function Materials and Structure Design, Ministry of Education, Lanzhou University, Lanzhou 730000, P. R. China. E-mail: haoli.zhang@lzu.edu.cn
bSchool of Physical Science and Technology, Soochow University, Suzhou 215006, P. R. China
cDepartment of Physics, Harbin Institute of Technology, Harbin 150001, P. R. China
First published on 4th September 2015
Abstract
Most organic molecules exhibit photo-induced bleaching under intense laser irradiation, while fewer materials show reversed saturable absorption, i.e. optical limiting properties. Here, we report that pentacene derivatives exhibit a giant nonlinear optical response at 532 nm. Upon investigation by the Z-scan technique, the pentacene derivatives demonstrated strong reverse saturable absorption and self-focusing effects using 532 nm pulsed lasers on a broad timescale from picosecond to nanosecond, with excellent output in solvents and polymer film matrices. Phase object (PO)-pump–probe and Z-scan measurements reveal that the superior optical limiting behaviors can be attributed to an excited state absorption mechanism associated with large absorption cross sections. We obtained a new benchmark of optical limiting onset of 2 mJ cm−2 with a concentrated solution, stemming from triplet state absorption due to the singlet fission process. This novel singlet fission induced nonlinear optical mechanism opens a new venue for the future design of organic optical limiting materials.
Introduction
Nonlinear optical (NLO) materials have found applications in a broad range of fields, such as optical switches,1,2 optical limiting,3–6 mode-locking7,8 and three-dimensional memory.9 Therefore, developing novel NLO materials is of both fundamental and practical importance, especially those based on π-conjugated organic molecules, which are particularly interesting due to their large third hyperpolarizabilities (γ) and fast response time.10,11 In the past two decades, a large number of organic and organometallic compounds with superior NLO performance have been discovered, such as fullerene derivatives,3,12 phthalocyanines13,14 and metalloporphyrins.15,16 However, reports on pure organic molecules exhibiting large nonlinear optical responses with desirable absorption wavelength and broad timescales are still scant.Pentacene has stimulated considerable interest since it was first synthesized in 1929.17–19 There have been more than 7000 articles published on pentacene and its derivatives by 2015, based on Web of Knowledge (ISI). The majority of these vast research works are focused on using pentacene as an organic semiconductor for various electronic applications. However, the NLO properties of this “star” molecule have not been thoroughly investigated,20,21 which is mainly hindered by the practical difficulties associated with its very low solubility in most solvents and poor photo/thermal stability.22
Recently, several groups including ours have developed various soluble pentacene derivatives for organic electronic applications.22–24 Introduction of appropriate substitutes groups into the pentacene framework25,26 could enhance their solubility and at the same time maintain their rigid and conjugated framework. Certain substitutes, such as halogen atoms, have been reported to significantly tune the optical properties of pentacene.24 We have reported various soluble heteropentacene derivatives exhibiting much improved solubility and photo/thermal stability,23,27,28 whose optical properties can be conveniently investigated in both solution and the solid state.
The most recent employment of pentacene and its derivatives in the organic electronic field is inspired by their unique singlet exciton fission effects, i.e., a singlet excited state decaying to form two triplet excitons on neighbouring molecules.29–31 The phenomenon can circumvent the Shockley–Queisser limit and has been utilized to enhance the external quantum yields in solar cells31 and photodetectors.32 The singlet exciton fission phenomenon of pentacene can even be observed in highly concentrated solutions due to their extremely high fission efficiency.30 We have recently proved that nitrogen-doping can further speed up the singlet fission process.33,34 These results prompted us to explore the NLO properties of pentacene and its derivatives, as these properties are closely related to the excited state dynamics of the materials.
In this paper, we present an experimental investigation on the NLO response of a series of pentacene derivatives in both solutions and thin films. We obtained remarkably low optical energy-limiting onset from the solutions of these materials on a broad timescale from picosecond to nanosecond, which is believed to be associated with the triplet state dynamics. We have found that singlet fission of pentacene derivatives at a high concentration can further enhance the optical limiting effect. Furthermore, thin films of these materials also show strong optical limiting properties with high stability, indicating their promising potential applications in NLO devices. To our knowledge, this is the first report that singlet fission can dramatically enhance the optical limiting activity of organic semiconductors.
Results and discussion
The chemical structures of the three pentacene derivatives are shown in Fig. 1a which are 6,13-bis(triisopropylsilylethynyl) pentacene (TP), 6,13-bis(triisopropyl silylethynyl)-1-azapentacene (1NTP) and 8,9,10,11-tetrachloro- 6,13-bis(triisopropyl silylethynyl)-1-azapentacene (4Cl1NTP). The synthesis of these molecules has been reported before.23,27 The normalized absorption spectra of TP and 1NTP are nearly overlapped (Fig. 1b.). In comparison, the absorption spectra of 4Cl1NTP are slightly red-shifted, likely due to the heavy atom effect of Cl.35 All the three pentacene derivatives exhibit weak absorption at 532 nm, favorable for excited state absorption at the wavelength.14 They give very weak fluorescence with their emission maximum centering around 650 nm.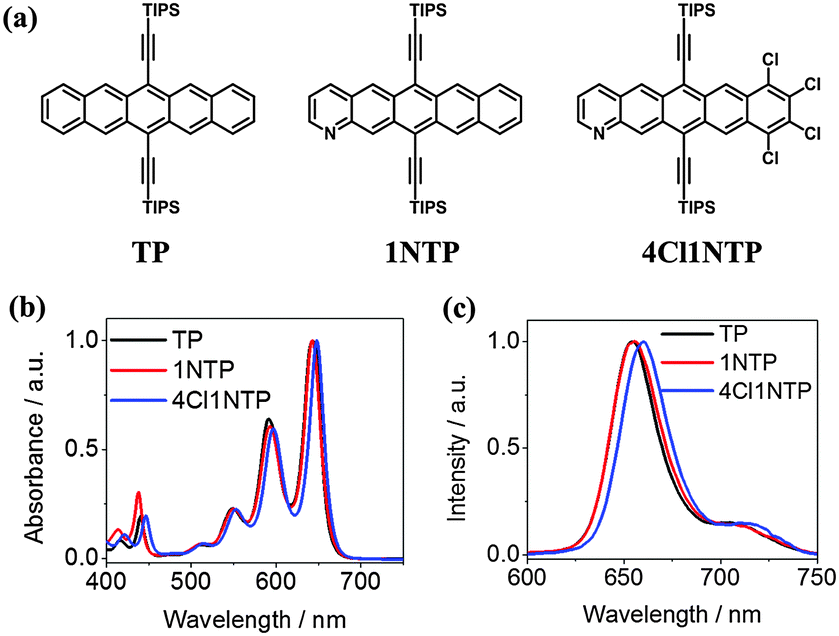 | ||
| Fig. 1 Structure of TP, 1NTP and 4Cl1NTP (a) and their normalized UV/Vis spectra (b) and fluorescence spectra (c) in toluene. The concentrations of all the solutions are 0.1 mM. | ||
Optical limiting characteristics of the pentacene derivatives were first measured using the nanosecond Z-scan technique. Laser pulses at wavelength of 532 nm with pulse width of 4 ns (FWHM) were generated from a frequency doubled and Q-switched Nd:YAG laser.36 The repetition rate was set to be 10 Hz in order to avoid the accumulated thermal effect. The transmittance of the sample was measured as a function of the incident laser fluence, which was varied by translating the sample through the focal plane along its propagation (Z) axis. In this measurement, normalized transmittance of 1.0 indicates that the material exhibits no NLO behavior. When the sample exhibits saturable absorption, normalized transmittance above 1.0 will be observed. In contrast, the normalized transmittance below 1.0 indicates that the sample exhibits reverse saturable absorption. For optical limiters, the measured curves would exhibit valleys at the focus and the deeper the valley, the stronger the optical limiting performance of the material.
Fig. 2a. displays the open-aperture Z-scan data of C60 (0.5 mM), TP (0.7 mM), 1NTP (0.2 mM) and 4Cl1NTP (0.3 mM) in toluene with input laser pulse energy of 2.7 μJ. C60 is shown here as a reference as it is a well-established benchmark with superior optical limiting properties.12,37–39 It can be seen that the optical limiting effects of TP, 1NTP and 4Cl1NTP are significantly stronger than that of C60. In addition, a plot of normalized transmittance versus input influence is presented in Fig. 2b for a quantitative comparison of these molecules, from which the optical energy-limiting onset fluence (Fon), defined as fluence where transmittance starts to fall to 95% of its original value can be extracted.40 Remarkably, the Fon values of all of the pentacene derivatives are much lower than that of C60 under the same experimental conditions (20, 20, 10 and 180 mJ cm−2 for TP, 1NTP, 4Cl1NTP and C60, respectively). 4Cl1NTP, in particular, demonstrates the lowest Fon of 10 mJ cm−2, one order of magnitude lower than that of C60, implying a new benchmark for optical limiting performance.40
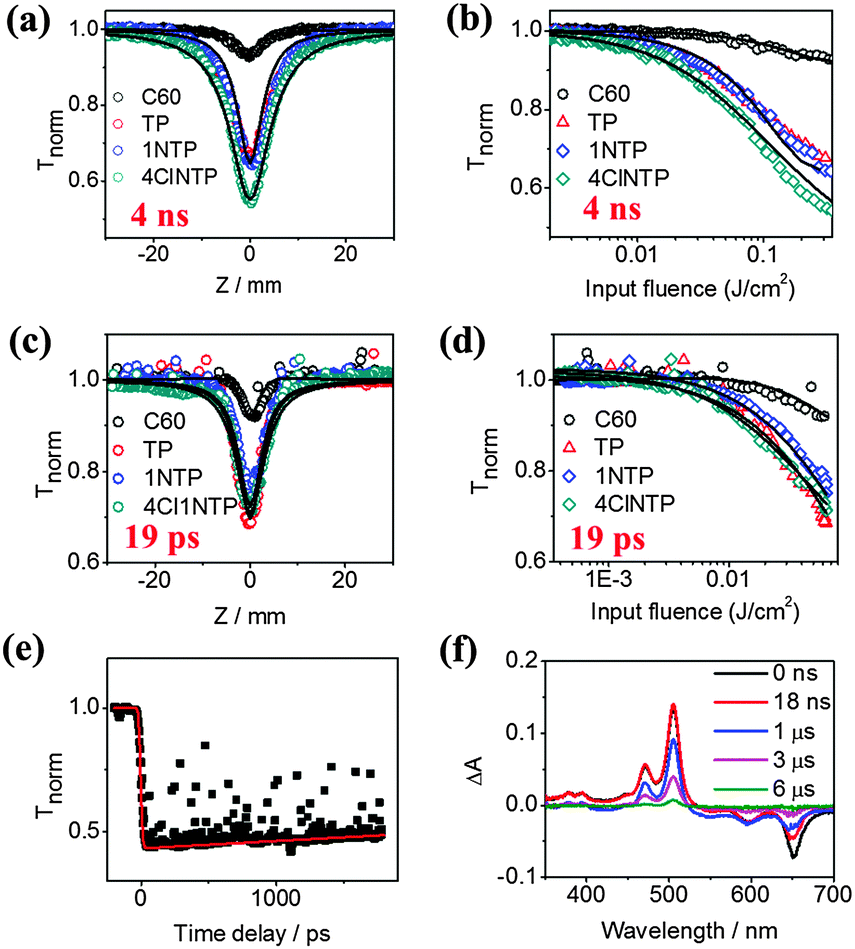 | ||
| Fig. 2 (a) Open aperture Z-scan data and theoretically fitted curves, and (b) corresponding plot of transmittance versus input fluence and theoretically fitted curves (solid curves) using 4 ns pulses at 532 nm for all the samples with same linear transmittance of 0.81. (c) Open aperture Z-scan data and theoretically fitted curves, and (d) corresponding plot of transmittance versus input fluence and theoretically fitted curves (solid curves) using 19 ps pulses at 532 nm for all the samples with the same linear transmittance of 0.70. Tnorm is the measured transmittance normalized by the linear transmittance of the sample. (e) Representative PO-pump–probe results for TP (black dots: open aperture pump–probe results, red line: theoretically fitted curves). The linear transmittance of all the samples is 0.57. Both pump and probe wavelength are 532 nm. (f) Representative transient absorption spectra of TP, on the timescale up to 6 μs at a pump wavelength of 532 nm. | ||
As the laser pulse width frequently influences the optical limiting behaviors of molecules due to induction of different nonlinear optical processes, we also performed Z-scan measurements with picosecond laser durations.41 Similarly, all of the pentacene derivatives demonstrated superior optical limiting effects to C60 (Fig. 2c and d). In addition, ESI† Fig. S1 shows the closed aperture Z-scan data of TP, 1NTP, 4Cl1NTP in toluene solutions in the picosecond region. It shows that the sign of nonlinear refractive index of all the compounds is positive due to the self-focusing effect, indicative of potential applications for optical switch.2
How does a small organic molecule like the pentacene derivative could exhibit such strong optical limiting effects in both nanosecond and picosecond timescales, even significantly surpassing traditional fullerene? To unravel the mechanism of the nonlinear optical process, we carried out a modified time-resolved pump–probe measurement using a phase object (PO-pump–probe), a third-order nonlinear optical time domain technique capable of simultaneously measuring the dynamics of nonlinear absorption and refraction, which we have previously reported.42–44 The representative PO-pump–probe curves of TP in toluene under open conditions are shown in Fig. 2e (the data of 1NTP and 4Cl1NTP are shown in the ESI,† Fig. S2). A control experiment with neat toluene verified that the nonlinear optical response of sample solution originated solely from the solute. For all of the three molecules, no sharp valley or peak-indicator of two-photon absorption and instantaneous Kerr refraction nonlinearity appeared near the zero-delay time, which suggested herein that other nonlinear optical processes like excited state absorption (ESA) account for the giant optical limiting effects of the samples.42 Within the time window (∼4 ns) of the current setup, none of the molecules showed any significant decay at the probe wavelength of 532 nm. Therefore a long pulse duration measurement is needed to extract the lifetime of the excited state for the samples.
We then performed a laser flash photolysis experiment with time window extending up to the millisecond timescale. According to the literature,30,45 the strongest transient absorption at 504 nm as shown in Fig. 2f was attributed to the triplet state absorption of TP, and the corresponding ones for 1NTP (Fig. S3a, ESI†) and 4Cl1NTP (Fig. S3b, ESI†) red shifted to 511 and 525 nm, respectively. A long lifetime of ∼1.7 μs can be extracted for all of the samples in the concentration of 10−4 M (Fig. S4, ESI†), corroborating the existence of the triplet state.30 Whether the elongated lifetime components compared with previously reported values of pentacene46 or TP films45 contribute to the outstanding optical limiting performance of the pentacene derivatives needs further exploration. Meanwhile, the negative peaks at ∼650 nm for all of the three molecules are expected to arise from ground-state bleaching because they absorb strongly around this wavelength (Fig. 1b). We also carried out fluorescence lifetime measurement to identify the short-lived singlet state, where a short lifetime of ∼13 ns was obtained for all of the three molecules at the concentration of 1.0 × 10−4 M (ref. 47) (Fig. S5, ESI†).
The above PO-pump–probe and laser photolysis measurements imply an ESA mechanism for the excellent optical limiting performance of the pentacene derivatives. Therefore, any effects that can modulate the population of the triplet states are expected to have significant impacts on the optical limiting performance.40,48 Given that singlet exciton fission could dramatically enhance triplet state formation,30 we studied the optical limiting properties of highly concentrated TP solution. Fig. 3 illustrates the Z-scan measurements of TP with concentration varied from 1.0 × 10−4 M to 1.0 × 10−2 M using a picosecond laser. Clearly, with the same input laser pulse energy of 0.4 μJ, the higher the concentration of TP, the lower the Fon values. Fon dropped down to a remarkable 2 mJ cm−2 at the concentration of 0.01 M, one order of magnitude lower than that of dilute solution of 1.0 × 10−4 M. As a reference, Fon of C60, which has hitherto shown no singlet fission even at high concentration, is independent on the solution concentration (shown in Fig. S6, ESI†). Nanosecond Z-scan measurements gave similar results (Fig. S7, ESI†). Corresponding PO-pump–probe experiments were also carried out (Fig. S8, ESI†) in order to combine Z-scan data for a simultaneous fit for the TP molecules, to extract parameters such as absorption cross sections and excited state lifetimes associated with different energy levels (see the ESI†). It is found that the absorption cross sections for the ground state (σS0), first singlet state (σS1) and first triplet state (σT1) of TP are 1.1 × 10−21 m2, 1.2 × 10−20 m2, and 3.1 × 10−21 m2, respectively. Clearly, both types of excited states show much larger absorption cross sections than that of the ground state of TP, leading to strong reverse saturable absorption behaviors of TP molecules. Meanwhile, σS1 and σT1 values of TP are also significantly larger than their counterparts for C60 (1.6 × 10−21 m2 and 0.95 × 10−21 m2, respectively),44 explaining the origin of the superior optical limiting performance of TP to C60 as observed in this work.
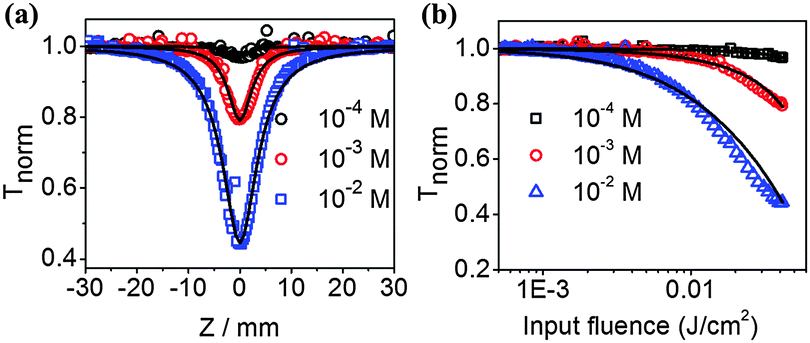 | ||
| Fig. 3 (a) Open aperture Z-scan data and theoretically fitted curves, and (b) corresponding plot of transmittance versus input fluence and theoretically fitted curves (solid curves) using 19 ps pulses at 532 nm for TP at different concentrations. | ||
Friend and co-workers have found that the quantitative singlet-fission yield for TP solution at a concentration of 0.075 mol l−1 could reach as large as 200%.30 Hence, in our case, we attribute the significant drop of the Fon value of TP with the concentration increase to the effect of singlet exciton fission. We took the absorption spectra of TP in different concentrations (Fig. S9, ESI†), and found that the absorption peaks have no obvious shift at any concentration from 1.0 × 10−4 to 1.0 × 10−2 mol L−1. We conclude that the TP molecules get no significant aggregates at high concentrations and are electronically decoupled in the ground state.30 To prove the occurrence of singlet fission in highly concentrated pentacene derivative solutions, we performed steady-state and time-resolved spectroscopic measurements. The photoluminescence spectra of TP showed a substantial change as the concentration increased (Fig. S10, ESI†). The emission wavelength shifts to red gradually along with concentration increase, which is due to the self-absorption presented in the sample. In accordance with previous literature,30 the redshifted feature at 710 nm stemmed from a TP excimer of [S1–S1]* (shown in Fig. 4) in the excited state. Corresponding fluorescence lifetime measurement indicated a significantly reduced lifetime from ∼13 ns to <1 ns (Fig. S11, ESI†), with the latter attributed to the time constant for the singlet fission process.49 We then performed broadband femtosecond pump–probe measurement in order to observe direct singlet fission dynamics. As shown in Fig. S12 (ESI†), for concentrated TP solution (0.01 M), the triplet state absorption band at ∼520 nm emerged on an ultrafast timescale of several picoseconds and exhibited a long lifetime over 2 ns (the time window of the setup). Whereas for very diluted TP solution (0.0001 M) no obvious triplet state absorption band was detected on the picosecond timescale, probably due to the absence of the singlet fission process. Femtosecond transient absorption spectroscopy was used to probe the photogeneration and population of the involved excited states (Fig. S13, ESI†). Fig. S13a (ESI†) shows that the positive absorption at ∼460 nm is attributed to the Sn ← S1 transition and the positive absorption at ∼505 nm is attributed to the Tn ← T1 transition. It also shows that as the singlets decay, the triplets rise, demonstrating the singlet fission process in TP solutions. The kinetics of singlet and triplet photoinduced absorption in Fig. S13b (ESI†) prove the occurrence of singlet fission. The calculated SF efficiency is 112 ± 10% for TP.33 These observations further evidenced the occurrence of singlet fission in the high concentration TP solutions used in our experiment.
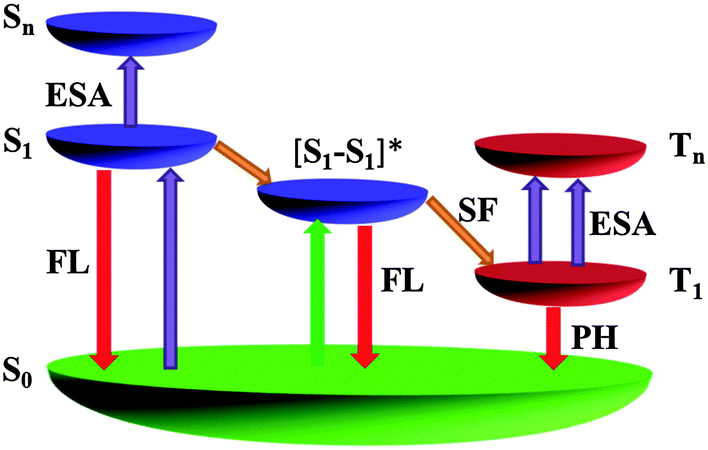 | ||
| Fig. 4 An energy level model demonstrating a singlet fission induced optical response of the pentacene derivatives on both fast and slow timescales (FL: fluorescence; PH: phosphorescence; ESA: excited state absorption; SF: singlet exciton fission; [S1–S1]*: TP excimer). | ||
Based on above observations, we proposed an energy level diagram in Fig. 4 to elucidate the origin of the giant nonlinear optical responses of the pentacene derivatives,14,21,50 which showed the possibility of both excited singlet and excited triplet state absorption. Initially, in dilute solution (<10−3 M) the molecules in the ground state (S0) absorb optical energy weakly at 532 nm and are excited to the vibrational manifold of the first singlet state (S1). Thereafter, dependent on the laser pulse width, these molecules may undergo two different pathways: with picosecond or femtosecond input pulses shorter than the time required to populate the triplet state, these molecules can absorb optical energy strongly and hop to the higher singlet state (Sn) to induce the optical limiting effect. If the width of a laser pulse is long enough (like nanosecond), the molecules would instead undergo relatively fast intersystem crossing to the first triplet state (T1), and subsequently absorb optical energy to reach the higher triplet state (Tn) (Fig. S14, ESI†). By contrast, in highly concentrated (0.01 M) solution, the singlet fission provides alternative channels for the generation of triplet states instead of intersystem crossing under the same incident light energy. As the singlet fission occurs on an ultrafast timescale ranging from sub-picosecond to sub-nanosecond,33,49 concentrated pentacene derivatives exhibit optical limiting effects upon excitation by both short and long pulse durations. Considering the large σT1 of TP (3.1 × 10−21 m2 measured in this work, or the reported extinction coefficient as large as 64![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 for the T1 → Tn absorption51), the molecule will exhibit much stronger triplet state absorption with regard to ground state absorption and thus result into giant optical limiting effects. In summary, either way the mechanism for nonlinear optical responses of these molecules is dominated by excited state absorption. Thus, pentacene derivatives are able to respond on both fast and slow timescales, making them particularly attractive for optical limiting applications.
000 M−1 cm−1 for the T1 → Tn absorption51), the molecule will exhibit much stronger triplet state absorption with regard to ground state absorption and thus result into giant optical limiting effects. In summary, either way the mechanism for nonlinear optical responses of these molecules is dominated by excited state absorption. Thus, pentacene derivatives are able to respond on both fast and slow timescales, making them particularly attractive for optical limiting applications.
The pentacene derivatives can be conveniently prepared into polystyrene thin films by mixing them in polystyrene solution. Fig. 5 clearly indicates that the thin film of TP showed strong optical limiting properties and good transparency (inset of Fig. 5 at the bottom). The absorption spectra in ESI† Fig. S15 and the fluorescence spectra in Fig. S16 (ESI†) show that TP in the polystyrene film exhibits similar optical properties as that in toluene. The results prove that TP has no significant aggregates in the polystyrene film. The nonlinear scattering from heat induced micro bubbles could be largely excluded in the solid state, which further verified that the superior optical limiting behaviors of the pentacene derivatives are stemmed from an ESA mechanism. The results from the same Z-scan measurement for 1NTP and 4Cl1NTP films are provided in ESI,† Fig. S17. Additional investigations have been performed to confirm the stability of the films. After the film was irradiated by 4 ns pulses at 532 nm for 20 minutes, no noticeable change was observed for the Z-scan measurement, suggesting the pentacene derivatives in the form of solid films are of great value for practical applications.
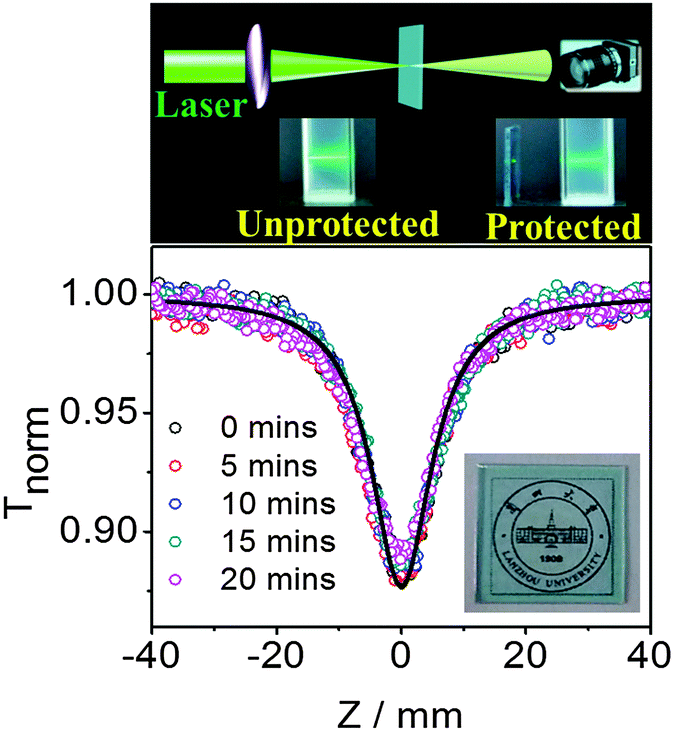 | ||
| Fig. 5 Schematic illustration of the optical limiting effect of pentacene derivatives (top) and open aperture Z-scan data for a neat film of TP in polystyrene irradiated by 4 ns pulses at 532 nm in different time (down). The solid curve is numerical fit for data. The linear transmittance of the sample given by the Fout/Fin ratio in the limit of zero fluence is 0.85. Inset: photograph of the transparent film cast on fused silica. | ||
Conclusions
Compared to previous reports, our pentacene derivatives of TP, 1NTP and 4Cl1NTP exhibited much superior optical limiting performance. All of the three compounds exhibit strong reverse saturable absorption and self-focusing effects on a 532 nm pulsed laser. The optical-limiting threshold achieved herein is 2 mJ cm−2 at the concentration of 0.01 M, which thus sets a new performance benchmark in small organic molecules. The result is even better than the previous record obtained on graphene single sheets.40 In addition, these materials can be conveniently prepared into thin films for future practical applications. PO-pump–probe and Z-scan experimental results reveal that the superior optical limiting behaviors of these molecules arise from a singlet fission induced excited state absorption mechanism, capable of responding on both fast and slow timescales. The study provided insight into the new tricks of the pentacene derivatives with giant optical limiting effects and their potential applications in nonlinear optics. More important, the singlet fission induced nonlinear optical mechanism uncovered in this work opens a new venue for the design of high performance organic nonlinear optical materials.Experimental section
The collection of the UV/Vis absorption spectra was performed on a TU-1810 spectrophotometer (Beijing Purkinje General Instrument, China).Fluorescence measurement
The steady-state fluorescence spectra were collected on a FLS 920 fluorescence spectrometer (Edinburgh Instrument, UK).Fluorescence lifetime measurement
The fluorescence lifetime measurements were carried out on a FluoTime 200 (PicoQuant GmbH, Germany) TCSPC time-resolved fluorescence platform, equipped with a nanosecond pulsed LED (Pulse width <750 ps) at 464 nm as the excitation source.Laser flash photolysis experiment
The laser flash photolysis experiment was carried on a LP920 laser flash photolysis instrument (Edinburgh Instruments Ltd) equipped with a pulsed OPO laser (Opolette HE355 LD-UVDM, OPOTEK Inc., USA), with tunable wavelength from 220 nm to 2400 nm and pulse duration of 5–7 ns as the pump light source. The probe source is a 450 W ozone-free pulsed Xe arc lamp.Broadband fs-pump–probe spectroscopy
Broadband fs-pump–probe measurements were carried out using an optical parametric amplifier (Light Conversion ORPHEUS) pumped with a mode-locked Yb:KGW-based femtosecond laser (1.20 eV, 190 fs, 6 kHz) as the light source. The whitelight probe pulses were produced by focusing a 1.2 eV laser pulse onto a sapphire plate. Time resolution of the measurement system is ∼280 fs.Z-scan setup
We refer the readers to the ESI† for the detailed schematic setup (Fig. S18, ESI†) and explanations therein. Briefly, for a nanosecond Z-scan measurement, 4 ns (FWHM), 532 nm laser pulses with a repetition rate of 10 Hz from a frequency-double Q-switched Nd:YAG laser (Continuum, Model Surelite SL-I-10) were used as the light sources. Two corresponding pyroelectric detectors (Laser Probe, RJ-735; with RJ7620 dual channel power meter) were used to measure changes in laser transmission. The samples were placed in quartz cells with thicknesses of 2 mm and 0.2 mm. For a picosecond Z-scan measurement, the laser was simply replaced by a mode locked Nd:YAG laser (EKSPLA PL2143B) with pulse width of 19 ps (FWHM) as the light source while all of the other parts remain unchanged.PO-pump–probe measurement
We refer the readers to the ESI† for the detailed schematic setup (Fig. S19, ESI†) and explanations therein. Briefly, the laser source was a mode locked Nd:YAG laser (EKSPLA PL2143B) with pulsewidth of 19 ps (FWHM). The signals were recorded by two silicon detectors (Laser Probe, Rjp-765a) simultaneously.Preparation of pentacene derivative films in polystyrene
Initially, pentacene derivatives were dissolved in toluene with a concentration of 10−4 M and a linear transmittance of 80% at 532 nm at room temperature. Thereafter, 0.3 g of polystyrene was added to 5.0 ml of this solution under vigorous stirring for 24 hours. The mixed solution was then drop-cast on fused silica, baked for 1 h at 50 °C.Acknowledgements
This work is supported by National Basic Research Program of China (973 Program) No. 2012CB933102, National Natural Science Foundation of China (NSFC. 21233001, 21190034, 21073079), Specialized Research Fund for the Doctoral Program of Higher Education (SRFDP. 20110211130001), the Fundamental Research Funds for the Central Universities and 111 Project.References
- M. Hochberg, T. Baehr-Jones, G. Wang, M. Shearn, K. Harvard, J. Luo, B. Chen, Z. Shi, R. Lawson, P. Sullivan, A. K. Jen, L. Dalton and A. Scherer, Nat. Mater., 2006, 5, 703–709 CrossRef CAS PubMed.
- R. L. Gieseking, S. Mukhopadhyay, C. Risko and J.-L. Brédas, ACS Photonics, 2014, 1, 261–269 CrossRef CAS.
- L. W. Tutt and A. Kost, Nature, 1992, 356, 225–226 CrossRef CAS PubMed.
- G. J. Zhou and W. Y. Wong, Chem. Soc. Rev., 2011, 40, 2541–2566 RSC.
- L. W. Tutt and T. F. Boggess, Prog. Quantum Electron., 1993, 17, 299–338 CrossRef CAS.
- T. M. Wang, B. Gao, Q. Wang, M. Zhao, K. B. Kang, Z. G. Xu and H. L. Zhang, Chem. – Asian J., 2013, 8, 912–918 CrossRef CAS PubMed.
- Q. L. Bao, H. Zhang, Z. H. Ni, Y. Wang, L. Polavarapu, Z. X. Shen, Q. H. Xu, D. Y. Tang and K. P. Loh, Nano Res., 2011, 4, 297–307 CrossRef CAS.
- K. Wang, J. Wang, J. Fan, M. Lotya, A. O'Neill, D. Fox, Y. Feng, X. Zhang, B. Jiang, Q. Zhao, H. Zhang, J. N. Coleman, L. Zhang and W. J. Blau, ACS Nano, 2013, 7, 9260–9267 CrossRef CAS PubMed.
- W. Zhou, S. M. Kuebler, K. L. Braun, T. Yu, J. K. Cammack, C. K. Ober, J. W. Perry and S. R. Marder, Science, 2002, 296, 1106–1109 CrossRef CAS PubMed.
- H. S. Nalwa, Adv. Mater., 1993, 5, 341–358 CrossRef CAS PubMed.
- J. L. Bredas, C. Adant, P. Tackx, A. Persoons and B. M. Pierce, Chem. Rev., 1994, 94, 243–278 CrossRef CAS.
- S. V. Rao, D. N. Rao, J. A. Akkara, B. S. DeCristofano and D. V. G. L. N. Rao, Chem. Phys. Lett., 1998, 297, 491–498 CrossRef CAS.
- J. W. Perry, K. Mansour, I. Y. S. Lee, X. L. Wu, P. V. Bedworth, C. T. Chen, D. Ng, S. R. Marder, P. Miles, T. Wada, M. Tian and H. Sasabe, Science, 1996, 273, 1533–1536 CAS.
- T. H. Wei, D. J. Hagan, M. J. Sence, E. W. Vanstryland, J. W. Perry and D. R. Coulter, Appl. Phys. B: Photophys. Laser Chem., 1992, 54, 46–51 CrossRef.
- Y. F. Xu, Z. B. Liu, X. L. Zhang, Y. Wang, J. G. Tian, Y. Huang, Y. F. Ma, X. Y. Zhang and Y. S. Chen, Adv. Mater., 2009, 21, 1275–1279 CrossRef CAS PubMed.
- M. O. Senge, M. Fazekas, E. G. A. Notaras, W. J. Blau, M. Zawadzka, O. B. Locos and E. M. N. Mhuircheartaigh, Adv. Mater., 2007, 19, 2737–2774 CrossRef CAS PubMed.
- E. Clar and F. John, Ber. Dtsch. Chem. Ges. A, 1929, 62, 3021–3029 CrossRef PubMed.
- E. Clar and F. John, Ber. Dtsch. Chem. Ges. A, 1930, 63, 2967–2977 CrossRef PubMed.
- E. Clar and F. John, Ber. Dtsch. Chem. Ges. A, 1931, 64, 981–988 CrossRef PubMed.
- R. A. Ganeev, A. I. Ryasnyansky, R. I. Tugushev and T. Usmanov, in Organic Nanophotonics, ed. F. Charra, V. Agranovich and F. Kajzar, Springer, Netherlands, 2003, vol. 100, pp. 367–384 Search PubMed.
- K. Kamada, K. Ohta, T. Kubo, A. Shimizu, Y. Morita, K. Nakasuji, R. Kishi, S. Ohta, S.-i. Furukawa, H. Takahashi and M. Nakano, Angew. Chem., Int. Ed., 2007, 119, 3614–3616 CrossRef PubMed.
- U. H. Bunz, J. U. Engelhart, B. D. Lindner and M. Schaffroth, Angew. Chem., Int. Ed., 2013, 52, 3810–3821 CrossRef CAS PubMed.
- Y. Y. Liu, C. L. Song, W. J. Zeng, K. G. Zhou, Z. F. Shi, C. B. Ma, F. Yang, H. L. Zhang and X. Gong, J. Am. Chem. Soc., 2010, 132, 16349–16351 CrossRef CAS PubMed.
- A. L. Appleton, S. M. Brombosz, S. Barlow, J. S. Sears, J. L. Bredas, S. R. Marder and U. H. Bunz, Nat. Commun., 2010, 1, 91 CrossRef PubMed.
- J. E. Anthony, J. S. Brooks, D. L. Eaton and S. R. Parkin, J. Am. Chem. Soc., 2001, 123, 9482–9483 CrossRef CAS.
- J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS PubMed.
- C. L. Song, C. B. Ma, F. Yang, W. J. Zeng, H. L. Zhang and X. Gong, Org. Lett., 2011, 13, 2880–2883 CrossRef CAS PubMed.
- J. Wang, K. Liu, Y.-Y. Liu, C.-L. Song, Z.-F. Shi, J.-B. Peng, H.-L. Zhang and X.-P. Cao, Org. Lett., 2009, 11, 2563–2566 CrossRef CAS PubMed.
- M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891–6936 CrossRef CAS PubMed.
- B. J. Walker, A. J. Musser, D. Beljonne and R. H. Friend, Nat. Chem., 2013, 5, 1019–1024 CrossRef CAS PubMed.
- D. N. Congreve, J. Lee, N. J. Thompson, E. Hontz, S. R. Yost, P. D. Reusswig, M. E. Bahlke, S. Reineke, T. Van Voorhis and M. A. Baldo, Science, 2013, 340, 334–337 CrossRef CAS PubMed.
- J. Lee, P. Jadhav and M. A. Baldo, Appl. Phys. Lett., 2009, 95, 033301 CrossRef PubMed.
- Y. Wu, K. Liu, H. Liu, Y. Zhang, H. Zhang, J. Yao and H. Fu, J. Phys. Chem. Lett., 2014, 5, 3451–3455 CrossRef CAS PubMed.
- J. Herz, T. Buckup, F. Paulus, J. Engelhart, U. H. F. Bunz and M. Motzkus, J. Phys. Chem. Lett., 2014, 5, 2425–2430 CrossRef CAS PubMed.
- C.-L. Sun, J. Li, H.-W. Geng, H. Li, Y. Ai, Q. Wang, S.-L. Pan and H.-L. Zhang, Chem. – Asian J., 2013, 8, 3091–3100 CrossRef CAS PubMed.
- J. T. Seo, Q. Yang, W.-J. Kim, J. Heo, S.-M. Ma, J. Austin, W. S. Yon, S. S. Jung, S. W. Han, B. Tabibi and D. Temple, Opt. Lett., 2009, 34, 307–309 CrossRef CAS.
- B. Gao, M. Zhao, Q. Wang, K.-B. Kang, Z.-G. Xu and H.-L. Zhang, New J. Chem., 2013, 37, 1692–1695 RSC.
- G. Mountrichas, S. Pispas, E. Xenogiannopoulou, P. Aloukos and S. Couris, J. Phys. Chem. B, 2007, 111, 4315–4319 CrossRef CAS PubMed.
- K.-S. Liao, J. Wang, D. Früchtl, N. J. Alley, E. Andreoli, E. P. Dillon, A. R. Barron, H. Kim, H. J. Byrne, W. J. Blau and S. A. Curran, Chem. Phys. Lett., 2010, 489, 207–211 CrossRef CAS PubMed.
- G. K. Lim, Z. L. Chen, J. Clark, R. G. S. Goh, W. H. Ng, H. W. Tan, R. H. Friend, P. K. H. Ho and L. L. Chua, Nat. Photonics, 2011, 5, 554–560 CrossRef CAS PubMed.
- W. Sun, B. Zhang, Y. Li, T. M. Pritchett, Z. Li and J. E. Haley, Chem. Mater., 2010, 22, 6384–6392 CrossRef CAS.
- Z.-G. Li, Y.-T. Lu, J.-Y. Yang, Z.-Q. Nie, M. Shui, X. Jin, J.-F. Ge and Y.-L. Song, Mater. Chem. Phys., 2013, 139, 975–978 CrossRef CAS PubMed.
- J. Yang, Y. Song, Y. Wang, C. Li, X. Jin and M. Shui, Opt. Express, 2009, 17, 7110–7116 CrossRef CAS.
- G. Shi, C. He, Y. Li, R. Zou, X. Zhang, Y. Wang, K. Yang, Y. Song and C. H. Wang, J. Opt. Soc. Am. B, 2009, 26, 754–761 CrossRef CAS.
- C. Ramanan, A. L. Smeigh, J. E. Anthony, T. J. Marks and M. R. Wasielewski, J. Am. Chem. Soc., 2012, 134, 386–397 CrossRef CAS PubMed.
- A. Rao, M. W. Wilson, J. M. Hodgkiss, S. Albert-Seifried, H. Bassler and R. H. Friend, J. Am. Chem. Soc., 2010, 132, 12698–12703 CrossRef CAS PubMed.
- A. D. Platt, J. Day, S. Subramanian, J. E. Anthony and O. Ostroverkhova, J. Phys. Chem. C, 2009, 113, 14006–14014 CAS.
- S. Hirata, K. Totani, T. Yamashita, C. Adachi and M. Vacha, Nat. Mater., 2014, 13, 938–946 CrossRef CAS PubMed.
- B. J. Walker, A. J. Musser, D. Beljonne and R. H. Friend, Nat. Chem., 2013, 5, 1019–1024 CrossRef CAS PubMed.
- C. W. Spangler, J. Mater. Chem., 1999, 9, 2013–2020 RSC.
- A. Scarpaci, A. Nantalaksaku, J. M. Hales, J. D. Matichak, S. Barlow, M. Rumi, J. W. Perry and S. R. Marder, Chem. Mater., 2012, 24, 1606–1618 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c5mh00120j |
| This journal is © The Royal Society of Chemistry 2015 |