
A seed-mediated approach to the general and mild synthesis of non-noble metal nanoparticles stabilized by a metal–organic framework for highly efficient catalysis†
Yu-Zhen
Chen‡
a,
Linfeng
Liang‡
ab,
Qihao
Yang
a,
Maochun
Hong
b,
Qiang
Xu
c,
Shu-Hong
Yu
a and
Hai-Long
Jiang
*ab
aHefei National Laboratory for Physical Sciences at the Microscale, Key Laboratory of Soft Matter Chemistry, Chinese Academy of Sciences, Collaborative Innovation Center of Suzhou Nano Science and Technology, Department of Chemistry, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: jianglab@ustc.edu.cn
bState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, P. R. China
cNational Institute of Advanced Industrial Science and Technology (AIST), Ikeda, Osaka 563-8577, Japan
First published on 22nd July 2015
Abstract
A novel and facile in situ seed-mediated synthetic approach has been developed to reduce non-noble metal precursors (Ni2+, Co2+, Fe2+, etc.) at room temperature, by taking advantage of a trace amount of noble metal cations (Ag+, Pd2+, Pt2+, Au3+, etc.) as a seed/initiator and NH3BH3 as a moderate reducing agent. The obtained noble metal-seed-mediated (NMSM) non-noble metal nanoparticles (NPs), stabilized by a metal–organic framework, are low-cost and display superior catalytic activity in the hydrolytic dehydrogenation of NH3BH3 under ambient conditions. As a representative catalyst, Ag-doped Ni/MIL-101 (with a Ag/Ni molar ratio as low as 1/200), exhibited a much higher activity than any of the other corresponding counterparts.
Conceptual insightsAlthough noble metal nanoparticles (NPs) exhibit excellent catalytic behavior in various reactions, the associated high cost and limited storage restrict their application. In reverse, the price of non-noble metal NPs is acceptable while their activity is usually inferior to noble metal NPs. More importantly, the reduction of non-noble metal precursors is generally carried out with a strong reductant or under harsh conditions that some supports, employed for stabilizing metal NPs, can hardly sustain. Therefore, it is highly desired to obtain highly active non-noble metal nanocatalysts with a moderate reducing agent under very mild conditions. In this work, for the first time, we develop a novel, general and facile one-step in situ noble metal seed-mediated (NMSM) synthetic approach to reduce non-noble metal cations under ambient conditions, by taking advantage of a trace amount of noble metal cations as an initiator and NH3BH3 as a moderate reducing agent. The obtained non-noble NPs, stabilized by a metal–organic framework (MOF), are low-cost and display superior catalytic activity in the hydrolytic dehydrogenation of NH3BH3 under ambient conditions. This general and facile approach paves a way to the synthesis of non-noble metal NPs under ambient conditions with a moderate reductant. |
Introduction
Metal nanoparticles (NPs) have attracted a great amount of attention because of their unique catalytic properties.1 Although noble metal NPs exhibit excellent catalytic behavior in various reactions, the associated high cost and limited storage restrict their application on an industrial scale. In contrast, the price of non-noble metal NPs is desirable while the activity is usually inferior to noble metal NPs. More importantly, the non-noble metal precursors have low reduction potentials and thus their reduction is generally carried out under harsh conditions, such as NaBH4 with strong basicity or H2 reduction under a high temperature or/and pressure.2 Given that tiny metal NPs with a high catalytic activity possess high surface energies, they are prone to aggregate and grow larger. One of the most effective solutions to avoid their aggregation is to stabilize them with supports/porous host materials, unfortunately, some of these structures can hardly sustain the harsh reaction conditions. Moreover, the strong reductants are detrimental to the control over the narrow size distribution of the metal NPs, which can be facilely generated by using moderate reducing agents.3 Therefore, it is highly desired and imperative to obtain highly active non-noble metal nanocatalysts under very mild conditions, which would guarantee the integrity of the NP supports/hosts during the synthesis and a low cost for the catalysts.On the other hand, studies have shown that noble metal precursors are readily reduced with moderate reductants.3–5 Therefore, it is possible to prepare non-noble metal NPs under ambient conditions by judiciously selecting a trace amount of noble metal as an initiator and choosing a suitable reductant, such as ammonia borane (NH3BH3, AB), which is not only a good/moderate reducing agent but also a promising hydrogen storage material with a high hydrogen capacity up to 19.6 wt%, exceeding that of gasoline.6 Hydrogen would be released from NH3BH3 over newly generated noble metal NPs to form intermediate M–H species with strong reducibility, which are able to further reduce the non-noble metal precursors at room temperature.5d The basic concept of such a noble metal-seed-mediated (NMSM) synthesis of non-noble metal NPs under mild conditions is to take advantage of the difference in the reduction potentials of the two soluble metal salts and a suitable reducing agent (NH3BH3).
Bearing this in mind, a trace amount (1/100–1/200, molar ratio of noble metal/non-noble metal) of noble metal precursors were introduced to non-noble metal precursors in a NH3BH3 hydrolysis system containing a metal–organic framework (MOF),7 which is a class of crystalline porous materials well demonstrated to be ideal supports/hosts providing a confinement effect to limit the aggregation and growth of the metal NPs.8 Strikingly, a trace amount of the different noble metal precursors were able to rapidly trigger the reduction of the non-noble metal cations at room temperature and the resultant in situ formed NPs exhibit an excellent catalytic performance in the hydrolytic dehydrogenation of NH3BH3. It is noteworthy that no non-noble metal NPs can be obtained without introducing noble metals, unless very harsh reduction conditions are used (Scheme 1). The current one-step in situ seed-mediated synthetic approach was conducted at room temperature under ambient atmosphere with the moderate reductant NH3BH3, which is very gentle and thus acceptable for diverse NP support/host materials. It is worthy to note that although bimetallic Au@Co and Au@Ni NPs have been reported based on a similar Au-induced reduction method,5a,b the amount of the noble metals used in this work is dramatically lower. To the best of our knowledge, this is the first general one-pot synthesis of non-noble metal NPs with a moderate reductant and a trace amount of noble metal precursors as the seed/initiator under ambient conditions.
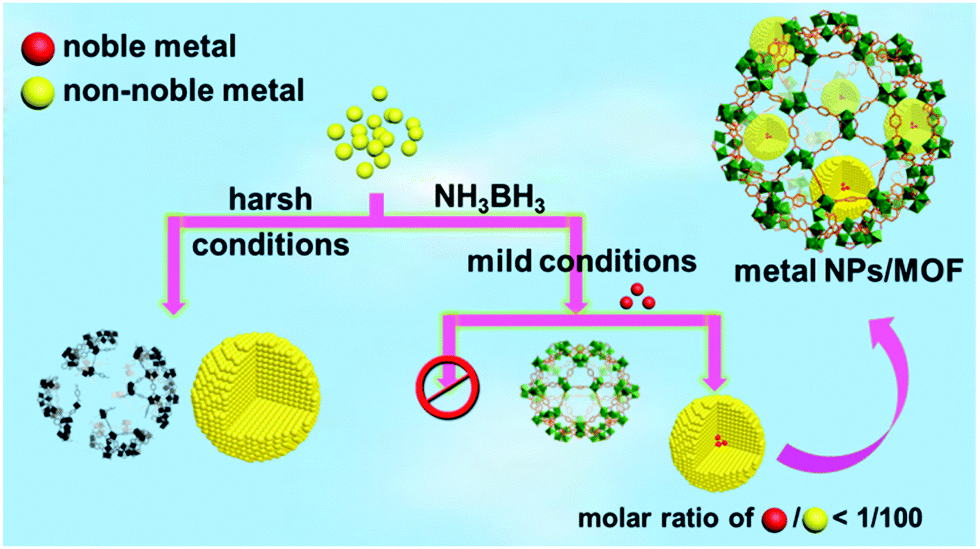 | ||
| Scheme 1 Schematic illustration showing the preparation of non-noble metal NPs under harsh conditions or via a NMSM synthetic approach with the moderate reductant NH3BH3 under ambient conditions. | ||
Experimental
General
All chemicals were from commercial sources and were used without further purification. De-ionized water with the specific resistance of 18.25 MΩ cm was obtained by reverse osmosis followed by ion-exchange and filtration (Cleaned Water Treatment Co., Ltd, Hefei). Powder X-ray diffraction (PXRD) data were collected on a Japan RigakuSmartLab™ rotation anode X-ray diffractometer equipped with graphite monochromatized Cu Kα radiation (λ = 1.54 Å). Transmission electron microscopy (TEM), high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and energy-dispersive X-ray spectroscopy (EDS) were performed on a JEOL-2010 and a JEOL-2100F with an electron acceleration energy of 200 kV. The nitrogen sorption isotherms were measured by using automatic volumetric adsorption equipment (Micrometritics ASAP 2020). Prior to the nitrogen adsorption/desorption measurements, the samples were dried overnight at 160 °C under vacuum. The content of the metals in the samples or solutions was quantified by an Optima 7300 DV inductively coupled plasma atomic emission spectrometer (ICP-AES).Preparation of MIL-101
MIL-101 was synthesized according to the previous report with modifications.9 Typically, a mixture of terephthalic acid (332 mg, 2.0 mmol) with Cr(NO3)3·9H2O (800 mg, 2.0 mmol) in the presence of aqueous HF (0.1 mL, 0.5 mmol) and de-ionized water (9.5 mL) was reacted at 200 °C for 8 h. The reaction produced a microcrystalline green powder of MIL-101 with the formula Cr3X(H2O)2O[(O2C)C6H4(CO2)]3·nH2O (X = F or OH, n ≤ 25). MIL-101 was purified in water at the reflux temperature for 12 h, and was then twice purified in ethanol at 100 °C for 24 h, before being washed with hot ethanol, and further purified by a NH4F solution. The resultant green solid was finally dried overnight at 150 °C under vacuum prior to further use.Preparation of Ag/MIL-101, Ag-doped Ni/MIL-101, Au-doped Ni/MIL-101, Pd-doped Co/MIL-101, Pd-doped Ni/MIL-101, Pd-doped Fe/MIL-101, Pt-doped Co/MIL-101 and Pt-doped Ni/MIL-101
The aqueous solutions AgNO3 (0.24 mol L−1), Pd(NO3)2 (0.25 mol L−1) or H2PtCl6·6H2O (0.02 mol L−1) and Ni(NO3)2·6H2O (0.5 mol L−1), FeSO4·7H2O (0.5 mol L−1) or Co(NO3)2·6H2O (0.5 mol L−1) with the desired contents as well as 20 mL of pure water were charged to a flask with 40 mg MIL-101, which was dried overnight at 160 °C under vacuum prior to use. The reaction proceeded when ammonia borane was added into the flask. The noble metal ions were first reduced by the ammonia borane to behave as in situ seeds for the subsequent reduction of the non-noble metal ions to produce non-noble based NPs stabilized by MIL-101. It should be noted that magnetic stirring was not preferred and that shaking was employed for the reaction in order to avoid the attachment of Ni or Co species onto the magnetic stirring bar. In this work, the molar quantities of (Au + Ni), (Pd + Ni), (Pd + Co), (Pd + Fe), (Pt + Ni), (Pt + Co) and (Ag + Ni) were 0.02, 0.015, 0.015, 0.02, 0.02, 0.02 and 0.015 mmol, respectively. The theoretical molar ratios of Au/Ni, Pd/Ni, Pd/Co, Pd/Fe, Pt/Ni, Pt/Co and Ag/Ni were 1/100, 1/200, 1/150, 1/100, 1/100, 1/100 and 1/200, respectively. In addition, pure Ag-doped Ni NPs with a Ag/Ni molar ratio of 1/200 were also obtained in the absence of MIL-101 or another support/stabilizer.Preparation of Ni/MIL-101 and PVP-stabilized Ni NPs
The aqueous solution of Ni(NO3)2·6H2O (29.8 μL, 0.5 mol L−1) and 20 mL of pure water were introduced to 40 mg of MIL-101 or PVP placed at the bottom of a flask, then NaBH4 (10 mg) was added into the flask to afford PVP-protected Ni NPs. It should be noted that magnetic stirring was not preferred and that shaking was employed for the reaction in order to avoid the attachment of Ni species onto the magnetic stirring bar.Attempt to prepare Ag-doped Ni/MIL-101 reduced by NaBH4
The aqueous solutions AgNO3 (0.24 mol L−1) and Ni(NO3)2·6H2O (0.5 mol L−1) with desired contents as well as 20 mL of pure water were charged to a flask with 40 mg of MIL-101, then a fresh 0.6 M NaBH4 aqueous solution was added into the flask for the reduction of the metal cations. Similarly, magnetic stirring was not preferred and shaking was employed for the reaction in order to avoid the attachment of Ni species onto the magnetic stirring bar.Preparation of PVP-stabilized Ag@Ni core–shell NPs
The aqueous solutions of AgNO3 (31 μL, 0.24 mol L−1), Ni(NO3)2·6H2O (15 μL, 0.50 mol L−1) and 20 mL of pure water were introduced to 40 mg of PVP that was placed at the bottom of a flask, then NH3BH3 (30 mg) was added into the flask with vigorous shaking to afford the PVP stabilized Ag@Ni core–shell NPs. It should be noted that magnetic stirring was not preferred and that shaking was employed for the reaction in order to avoid the attachment of Ni species onto the magnetic stirring bar.Catalytic activity characterization
In general, a two-necked round-bottomed flask (50 mL) with a mixture of 40 mg of MIL-101, the metal precursors of desired concentration and 20 mL of pure water was placed in a mechanical oscillator in order to avoid the attachment of Ni or Co NPs onto the magnetic stirring bar. A gas burette filled with water was connected to the reaction flask to measure the volume of hydrogen evolved. The reaction started when 30 mg of NH3BH3 was added into the flask. The volume of the evolved hydrogen gas was monitored by recording the displacement of the water in the gas burette. The reaction was completed when there was no more gas generated. For the catalytic recyclability/durability experiments, the same amount of NH3BH3 (30 mg) was added into the flask to initiate the reaction.Results and discussion
A representative mesoporous MOF, MIL-101 with the molecular formula Cr3F(H2O)2O[(O2C)C6H4(CO2)]3·nH2O (n = ∼25),9 was selected as a host matrix based on its high specific surface area (BET, >3000 m2 g−1) and large pore size (2.9 to 3.4 nm), in order to avoid the aggregation and growth of resultant metal NPs. No reaction occurred when the NH3BH3 aqueous solution involved only the non-noble metal precursor and MIL-101 in the flask. Upon the introduction of a trace amount of noble metal precursor (1/100–1/200, molar ratio of noble metal/non-noble metal), the reaction was initiated immediately, accompanied by a color evolution of the reaction solution (Fig. S1, ESI†). Based on this NMSM synthetic approach, diverse non-noble metal NPs can be obtained by using different kinds of noble metal precursors as reduction initiators. Typically, Pd-doped Ni, Pd-doped Co, Pt-doped Ni, Pt-doped Co, Ag-doped Ni and Au-doped Ni NPs stabilized by MIL-101 have been successfully synthesized involving only trace amounts of noble metal species. Due to the high reduction potentials of the soluble noble metal salts (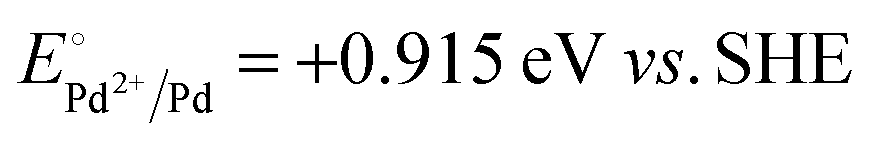 ,
, 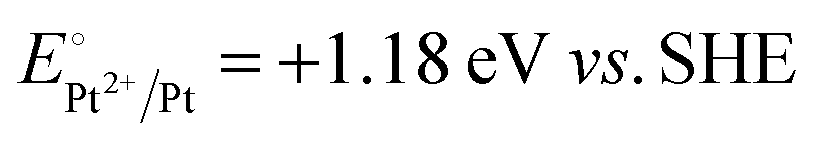 ,
, 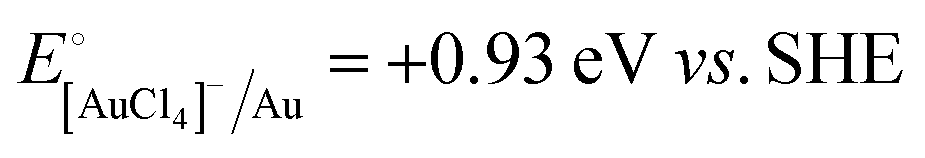 and
and 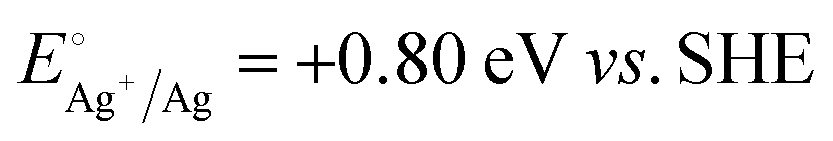 ), the noble metal ions were first reduced to NPs by NH3BH3 and were then used as the initiator for the subsequent reduction of the non-noble metal precursors.
), the noble metal ions were first reduced to NPs by NH3BH3 and were then used as the initiator for the subsequent reduction of the non-noble metal precursors.
The high dispersion of Pd-doped Ni, Pd-doped Co, Au-doped Ni, Pt-doped Ni, Pt-doped Co and Ag-doped Ni NPs can be evidenced by transmission electron microscopy (TEM) (Fig. 1a–f), which shows that the nanoparticle average size is 2–3 nm and indicates the universality of this noble metal-triggered synthetic approach (Fig. 1f). To gain further structural information on these metal NPs, high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM), high-resolution TEM (HRTEM) and energy-dispersive X-ray spectroscopy (EDS) have been employed to investigate the Ag-doped Ni/MIL-101 catalyst as a representative nanocomposite (Fig. 1g and h). The lattice fringes with a spacing of 0.206 nm are assigned to the (111) planes of the face-centered cubic (fcc) Ni, which are clearly observable from the HRTEM image of Ag-doped Ni/MIL-101 (Fig. 1h). The SAED pattern (Fig. 1h, inset) shows diffuse diffraction rings, indicating that the Ni is mostly in an amorphous phase.2a,6i The EDS spectrum of the Ag-doped Ni NPs identifies Ni species only, revealing a very low content of Ag doping in the Ni NPs (Fig. S2, ESI†). For comparison, PVP-protected Ni NPs with much larger particle sizes were obtained by the reduction of NaBH4 (Fig. S3b, ESI†), the lattice fringes demonstrate that the NPs are composed of Ni.
 | ||
| Fig. 1 TEM images of (a) Pd-doped Ni, (b) Pd-doped Co, (c) Au-doped Ni, (d) Pt-doped Ni, (e) Pt-doped Co, and (f) Ag-doped Ni NPs stabilized by MIL-101 (inset: size distribution). (g) HAADF-STEM image, and (h) HRTEM image of the Ag-doped Ni/MIL-101 catalyst (inset: SAED pattern). | ||
For further comparison, TEM observation for Ni/MIL-101 (reduced by NaBH4), Ag/MIL-101 (reduced by NH3BH3), Ni2+/MIL-101, pure Ag-doped Ni (1/200) NPs without stabilizer and PVP-stabilized Ag@Ni core–shell NPs (reduced by NH3BH3) with a Ag/Ni molar ratio of 1/1 was also performed (Fig. S3, ESI†). The Ni/MIL-101 reduced with NaBH4 shows relatively larger Ni NPs, possibly because the structure of MIL-101 was partly damaged by the strong reductant NaBH4 (Fig. S3c, ESI†). In contrast, Ag/MIL-101 shows an average size of ∼2 nm with confinement by MIL-101 (Fig. S3d, ESI†). However, the Ag-doped Ni NPs present an apparent aggregation to ∼20 nm in the absence of a support (Fig. S3e, ESI†). Ni2+/MIL-101 presents almost no particles (Fig. S3g, ESI†). In order to validate the seed role of noble metals for inducing the reduction of the non-noble metals, as a representative nanocomposite, the PVP stabilized Ag@Ni core–shell NPs with increasing Ag content were synthesized with NH3BH3. The TEM image clearly shows the core–shell structure for Ag@Ni and the HRTEM further confirms the Ag core (Fig. S3h, ESI†), suggesting that the in situ formation of the Ni NPs is triggered by the Ag seed.
An attempt to prepare Ag-doped Ni/MIL-101 reduced by the strong reductant NaBH4 even under ambient conditions is demonstrated to have failed, as the powder X-ray diffraction (PXRD) peak intensity for the resultant composite is very weak, suggesting a partly collapsed MIL-101 structure (Fig. S4a, ESI†). In contrast, the PXRD profiles show that there is no apparent loss of crystallinity and no identifiable peaks for metal NPs after the reduction with NH3BH3, indicating retained integrity of the MIL-101 framework and tiny metal NPs (Fig. S4b and c, ESI†). The impact of the metal NP loading on the porosity of MIL-101 was established by studying the N2 adsorption/desorption at 77 K, which revealed that the highly porous structure of MIL-101 remained. The appreciable decrease in the amount of N2 sorption and surface area indicates that the cavities of the host framework could be occupied by the dispersed metal NPs or blocked by the NPs located on the surface (Fig. S5, ESI†).
To investigate the catalytic performance of the resultant noble metal-doped non-noble metal NPs, the hydrolytic dehydrogenation of NH3BH3 was employed. The very high hydrogen capacity of 19.6 wt%, excellent solubility, nontoxicity, stability in water and easy storage of NH3BH3 make it one of the most attractive candidates for chemical hydrogen storage. Among various catalysts for the hydrolytic dehydrogenation of NH3BH3,6g–i,10 platinum possesses the highest activity.10a It is clearly a desired goal to prepare low-cost catalysts with a high catalytic performance and rapid kinetics of hydrogen release under ambient conditions for the ultimate practical application of reaction systems in fuel cells. In this work, the reaction was initiated by adding NH3BH3 to a flask containing MIL-101, noble metal (Ag+, Pd2+, Pt2+, Au3+, etc.) and non-noble metal (Ni2+, Co2+, etc.) precursors of desired concentrations under vigorous shaking and ambient conditions. Fig. 2 shows plots of the H2 generation versus time during the catalytic hydrolysis of NH3BH3 over different NMSM non-noble metal-based catalysts with a noble metal/non-noble metal molar ratio as low as 1/100–1/200. The results indicate that the catalytic activity of the in situ formed non-noble metal NPs is significant and that all hydrogen can be released from NH3BH3 in less than 10 min (H2/NH3BH3 ≈ 3). Among these noble metals, Ag is the most inexpensive and is a good initiator to trigger the reduction of non-noble metals. Meanwhile, Ni is one of the most intensively studied non-noble metals with a high catalytic activity toward the hydrolytic dehydrogenation of NH3BH3.2c,6i,10b,i Therefore, the Ag-doped Ni/MIL-101 catalyst, as a representative nanocomposite, was further investigated in detail. Fig. 3 shows the catalytic activity for the hydrolysis of NH3BH3 over Ag-doped Ni/MIL-101 with Ag/Ni molar ratios varying from 10/1 to 1/200, while the molar ratio of total metal/NH3BH3 was fixed at a constant of 0.017. The results revealed that the catalytic activity did not significantly change even when the molar ratio of Ag/Ni was decreased sharply. Remarkably, a trace amount of Ag dopant was able to trigger the reduction of Ni2+ (Ag/Ni molar ratio = 1/200) and the resultant Ag-doped Ni/MIL-101 catalyst presents superior catalytic activity with completion of the hydrolysis in 12 min (containing ∼2 min of the initiation/induction period). The same parallel tests with various noble/non-noble metal molar ratios for other samples such as Pd-doped Ni/MIL-101, Pd-doped Co/MIL-101, Au-doped Ni/MIL-101, Pt-doped Ni/MIL-101 and Pt-doped Co/MIL-101 were also conducted for a better comparison (Fig. S6, ESI†).
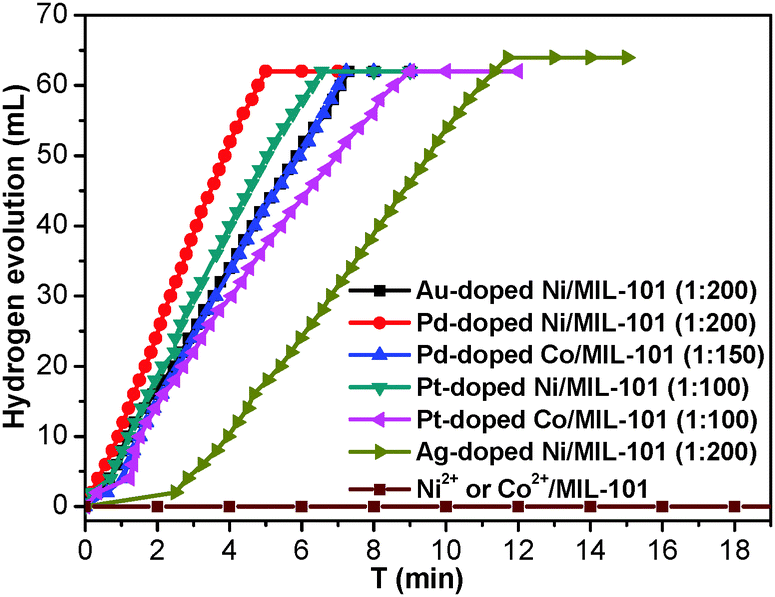 | ||
| Fig. 2 Plots of time vs. volume of hydrogen generated from the catalytic hydrolysis of NH3BH3 (0.875 mmol in 20 mL water) over Pd-doped Ni/MIL-101, Pd-doped Co/MIL-101, Au-doped Ni/MIL-101, Pt-doped Ni/MIL-101, Pt-doped Co/MIL-101 and Ag-doped Ni/MIL-101 catalysts at 25 °C. The data in parentheses indicate the molar ratios of noble metal/non-noble metal in the catalysts. | ||
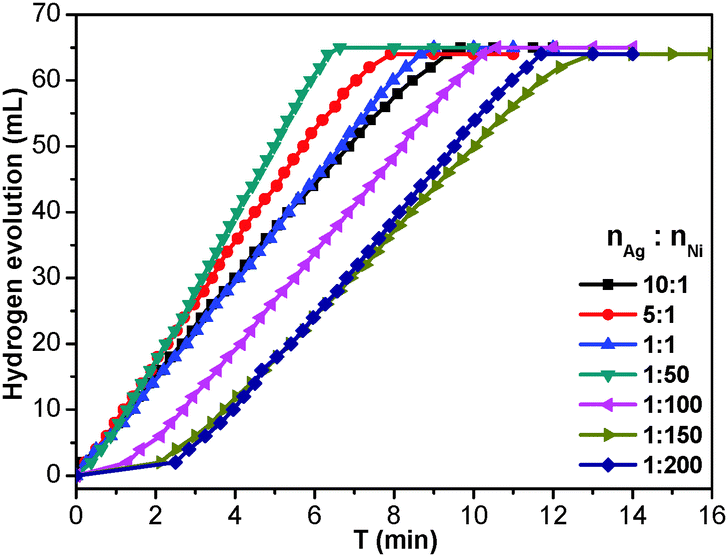 | ||
| Fig. 3 Plots of time vs. volume of hydrogen generated from the catalytic hydrolysis of NH3BH3 over Ag-doped Ni/MIL-101 with different Ag/Ni molar ratios at 25 °C. The molar ratio of (Ag + Ni)/NH3BH3 is fixed at a constant of 0.017 for all catalysts. | ||
When the small amount of noble metal precursors was introduced, an induction time was observed to initiate the reaction (Fig. 2 and 3). Interestingly, the induction time is not only dependent on the content of Ag+, but is also related to the reaction temperature as well as the concentration of NH3BH3.6i A higher reaction temperature or concentration of NH3BH3 leads to a shortened or even no induction period. The induction process was attributed to the slow generation of the Ag–H reductive species from trace amounts of the Ag precursor or a low temperature or a low concentration of NH3BH3, and thus the Ni2+ would be reduced slowly. Detectable levels of hydrogen were released once a certain amount of Ni NPs, the active species, was produced and following that, a nearly linear rate of hydrogen was rapidly generated and all the remaining Ni2+ cations were fully reduced to further efficiently catalyze the hydrolytic reaction. Although the Ag amount is very low, the limited Ag–H is able to induce the reduction of a small amount of Ni2+, during which the formation of Ni–H as new reductant occurs.10e,11 Subsequently, more Ni2+ is reduced and more Ni–H can be successively formed until all the Ni2+ ions are reduced.
Importantly, although the Ag doping is negligible in the resultant Ag-doped Ni NPs, the catalytic activity is much higher than that of the monometallic counterparts Ag and Ni NPs (Fig. 4). Moreover, the Ni/MIL-101 obtained by NaBH4 reduction also takes a longer time (over 22 min) to complete the reaction. The Ag/MIL-101 catalyst obtained by the reduction of NH3BH3 also showed a low activity performance and the hydrolysis of NH3BH3 was not completed even after 60 min. In addition, no H2 was generated without the introduction of a Ag+ salt because the Ni2+ cannot be directly reduced by NH3BH3 under ambient conditions. For comparison, Ag-doped Ni NPs with a Ag/Ni molar ratio of 1/200 reduced by NH3BH3 in the absence of a MIL-101 support and MIL-101/PVP-protected Ni NPs reduced by NaBH4 were also applied as catalysts in the hydrolysis of NH3BH3 (Fig. 4). The incomplete dehydrogenation of NH3BH3 over PVP-protected Ni NPs was observed and only ∼40 mL of hydrogen was released after 45 min, which presented much slower catalytic kinetics than that of the in situ formed Ag-doped Ni/MIL-101. The catalytic dehydrogenation of NH3BH3 over Ag-doped Ni NPs without a MIL-101 support was much slower and complete hydrogen release took more than 60 min under identical reaction conditions, possibly due to the agglomeration of the Ag-doped Ni NPs in the absence of the confinement effect of MIL-101. The above results indicate the electronic structure modification-induced synergistic effects between Ag and Ni species in the Ag-doped Ni NPs, which have been documented in previous reports.1d,12 The trace amount of Ag triggered the in situ formation of a Ni nanocatalyst which is very efficient for the hydrolytic dehydrogenation of NH3BH3 and presents superior activity to other corresponding catalysts. Evidently, each of the Ag, Ni and MIL-101 components in the Ag-doped Ni/MIL-101 catalyst plays a respective critical role towards its excellent catalytic performance.
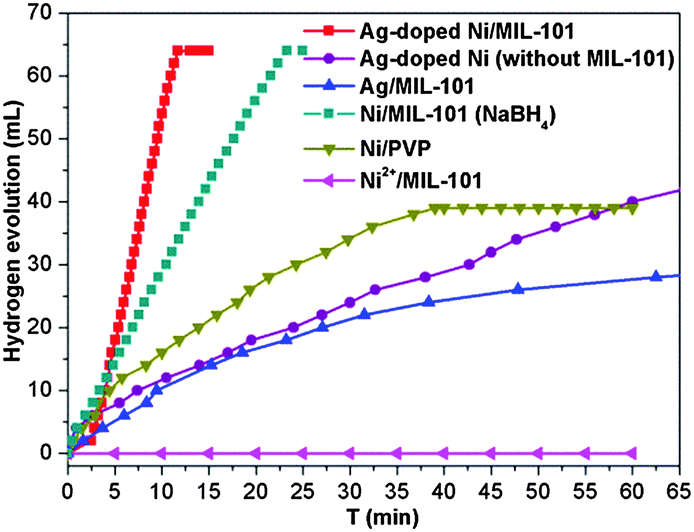 | ||
| Fig. 4 Plots of time vs. volume of hydrogen generated from the catalytic hydrolysis of NH3BH3 over Ag-doped Ni/MIL-101 (Ag/Ni molar ratio of 1/200), Ag-doped Ni NPs (Ag/Ni molar ratio of 1/200) in the absence of MIL-101, Ag/MIL-101, Ni/MIL-101 and PVP-protected Ni NPs. The total amount of Ag + Ni is fixed to be (Ag + Ni)/NH3BH3 = 0.017 (molar ratio) for all the catalysts. | ||
The catalytic reaction rate constant (k) and the total turnover frequency (TOF) values of the Ag-doped Ni/MIL-101 catalysts with different Ag content for the hydrolytic dehydrogenation of NH3BH3 are shown in Table S1 (ESI†). It is noteworthy that the catalyst with the lowest Ag/Ni molar ratio of 1/200 in Ag-doped Ni/MIL-101 has an excellent catalytic activity with a high TOF value of 20.2 molH2 molmetal−1 min−1, which is higher than most of the non-noble metals such as Ni catalysts toward the hydrolysis of NH3BH3 (Table S2, ESI†). Inductively coupled plasma atomic emission spectrometry (ICP-AES) has confirmed that the actual Ag/Ni molar ratio (1/170) is very close to the nominal value (1/200) (Table S3, ESI†). From the Arrhenius plots of the reaction over Ag-doped Ni/MIL-101 (Ag/Ni molar ratio of 1/200) obtained at different temperatures ranging from 25 to 40 °C (Fig. S8, ESI†), the activation energy (Ea) was calculated to be 25 kJ mol−1 (Fig. S9, ESI†), which is lower than most of the reported catalysts (Table S2, ESI†), indicating the superior catalytic performance of the catalyst.
Another remarkable benefit of the Ag-doped Ni/MIL-101 catalyst for the hydrolysis of NH3BH3 is its good stability and reusability. It can be seen that there is no significant decrease in the activity during five consecutive usages without any treatment or activation of the catalyst (Fig. 5a). The Ag-doped Ni NPs have well retained sizes (about 2.5 nm) even after several runs, showing a good confinement effect of MIL-101 due to its porous structure (Fig. 5b). The powder XRD profiles show that there is no apparent loss of crystallinity and no identifiable peaks for the metal NPs after five cycles (Fig. S11, ESI†), suggesting the good recyclability and stability of the catalyst.
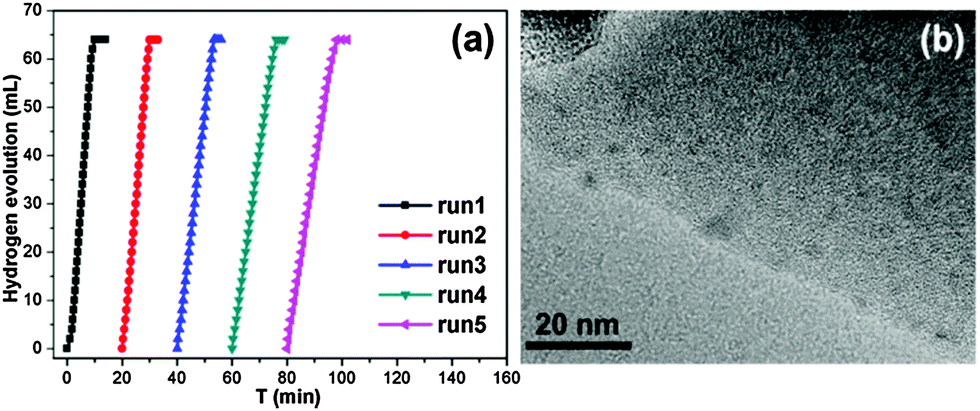 | ||
| Fig. 5 (a) Recyclability test for the hydrogen generation from an aqueous NH3BH3 solution (0.875 mmol in 20 mL H2O, T = 30 °C) over the Ag-doped Ni/MIL-101 catalyst. (b) TEM image for the Ag-doped Ni/MIL-101 catalyst with a Ag/Ni molar ratio of 1/200 after several cycles of the NH3BH3 hydrolysis reaction. | ||
Conclusions
In summary, for the first time, we have successfully developed a novel, general and simple one-step in situ NMSM synthetic approach to reduce non-noble metal precursors under very mild conditions by taking advantage of noble metals, even in a trace amount, as the seed and initiator. The obtained catalysts are low-cost and have superior catalytic activity in the hydrolytic dehydrogenation of ammonia borane compared to the corresponding monometallic counterparts. Nevertheless, these non-noble metal NPs cannot be obtained in the absence of a noble metal as the seed/initiator unless with harsh reduction conditions. As a representative example, the Ag-doped Ni/MIL-101 catalyst (Ag/Ni molar ratio of 1/200) has a superb catalytic activity in the hydrolysis of NH3BH3. Furthermore, the catalyst also exhibits a very low activation energy of 25 kJ mol−1 and good recyclability for the hydrolytic dehydrogenation of NH3BH3. This general and facile approach paves a way to the synthesis of non-noble metal NPs under ambient conditions with a moderate reductant, such as NH3BH3. In addition, the outstanding catalytic performance of such non-noble metal nanocatalysts via a NMSM synthetic approach presents a promising step for the application of ammonia borane as a feasible chemical hydrogen storage material.Acknowledgements
We gratefully thank the reviewers for their valuable suggestions. This work is supported by the NSFC (21371162 and 51301159), the 973 program (2014CB931803), NSF of Anhui Province (1408085MB23), Research Fund for the Doctoral Program of Higher Education of China (20133402120020), the Recruitment Program of Global Youth Experts and the Fundamental Research Funds for the Central Universities (WK2060190026).References
- (a) A. Cao, R. Lu and G. Veser, Phys. Chem. Chem. Phys., 2010, 12, 13499–13510 RSC; (b) X.-H. Li and M. Antonietti, Chem. Soc. Rev., 2013, 42, 6593–6604 RSC; (c) A. Dhakshinamoorthy and H. Garcia, Chem. Soc. Rev., 2012, 41, 5262–5284 RSC; (d) S. Alayoglu, A. U. Nilekar, M. Mavrikakis and B. Eichhorn, Nat. Mater., 2008, 7, 333–338 CrossRef CAS PubMed; (e) Y. Sun and Y. Xia, Science, 2002, 298, 2176–2179 CrossRef CAS PubMed; (f) Metal Nanoparticles for Catalysis: Advances and Applications, ed. F. Tao, Royal Society of Chemistry, Cambridge, 2014 Search PubMed; (g) D. Astruc, Nanoparticles and Catalysis, Wiley-VCH, Weinheim, 2008, vol. 2, pp. 1–640 Search PubMed.
- (a) J.-M. Yan, X.-B. Zhang, S. Han, H. Shioyama and Q. Xu, Angew. Chem., Int. Ed., 2008, 47, 2287–2289 CrossRef CAS PubMed; (b) Ö. Metin and S. Özkar, Energy Fuels, 2009, 23, 3517–3526 CrossRef; (c) P.-Z. Li, A. Aijaz and Q. Xu, Angew. Chem., Int. Ed., 2012, 51, 6753–6756 CrossRef CAS PubMed; (d) C.-Y. Cao, C.-Q. Chen, W. Li, W.-G. Song and W. Cai, ChemSusChem, 2010, 3, 1241–1244 CrossRef CAS PubMed; (e) W. Lin, H. Cheng, J. Ming, Y. Yu and F. Zhao, J. Catal., 2012, 291, 149–154 CrossRef CAS; (f) K.-I. Shimizu, N. Imaiida, K. Kon, S. M. A. H. Siddiki and A. Satsuma, ACS Catal., 2013, 3, 998–1005 CrossRef CAS; (g) C. Li, Y. Chen, S. Zhang, S. Xu, J. Zhou, F. Wang, M. Wei, D. G. Evans and X. Duan, Chem. Mater., 2013, 25, 3888–3896 CrossRef CAS; (h) J. Hannauer, O. Akdim, U. B. Demirci, C. Geantet, J.-M. Herrmann, P. Miele and Q. Xu, Energy Environ. Sci., 2011, 4, 3355–3358 RSC.
- (a) N. Zheng and G. D. Stucky, J. Am. Chem. Soc., 2006, 128, 14278–14280 CrossRef CAS PubMed; (b) N. Zheng, J. Fan and G. D. Stucky, J. Am. Chem. Soc., 2006, 128, 6550–6551 CrossRef CAS PubMed.
- (a) Y. W. Lee, N. H. Kim, K. Y. Lee, K. Kwon, M. Kim and S. W. Han, J. Phys. Chem. C, 2008, 112, 6717–6722 CrossRef CAS; (b) M. Zahmakıran and S. Özkar, Appl. Catal., B, 2009, 89, 104–110 CrossRef.
- (a) J.-M. Yan, X.-B. Zhang, T. Akita, M. Haruta and Q. Xu, J. Am. Chem. Soc., 2010, 132, 5326–5327 CrossRef CAS PubMed; (b) D. Wang and Y. Li, J. Am. Chem. Soc., 2010, 132, 6280–6281 CrossRef CAS PubMed; (c) L. Yang, W. Luo and G. Cheng, ACS Appl. Mater. Interfaces, 2013, 5, 8231–8240 CrossRef CAS PubMed; (d) Y.-Z. Chen, Q. Xu, S.-H. Yu and H.-L. Jiang, Small, 2015, 11, 71–76 CrossRef CAS PubMed.
- (a) P. Chen, Z. Xiong, J. Luo, J. Lin and K. L. Tan, Nature, 2002, 420, 302–304 CrossRef CAS PubMed; (b) W. Grochala and P. P. Edwards, Chem. Rev., 2004, 104, 1283–1316 CrossRef CAS PubMed; (c) A. Gutowska, L. Li, Y. Shin, C. M. Wang, X. S. Li, J. C. Linehan, R. S. Smith, B. D. Kay, B. Schmid, W. Shaw, M. Gutowski and T. Autrey, Angew. Chem., Int. Ed., 2005, 44, 3578–3582 CrossRef CAS PubMed; (d) M. E. Bluhm, M. G. Bradley, R. Butterick III, U. Kusari and L. G. Sneddon, J. Am. Chem. Soc., 2006, 128, 7748–7749 CrossRef CAS PubMed; (e) C. W. Hamilton, R. T. Baker, A. Staubitz and I. Manners, Chem. Soc. Rev., 2009, 38, 279–293 RSC; (f) R. J. Keaton, J. M. Blacquiere and R. T. Baker, J. Am. Chem. Soc., 2007, 129, 1844–1845 CrossRef CAS PubMed; (g) H.-L. Jiang, S. K. Singh, J.-M. Yan, X.-B. Zhang and Q. Xu, ChemSusChem, 2010, 3, 541–549 CrossRef CAS PubMed; (h) D. Sun, V. Mazumder, Ö. Metin and S. Sun, ACS Nano, 2011, 5, 6458–6464 CrossRef CAS PubMed; (i) Ö. Metin, V. Mazumder, S. Özkar and S. Sun, J. Am. Chem. Soc., 2010, 132, 1468–1469 CrossRef PubMed.
- (a) H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed; (b) H.-C. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC; (c) Z. Wang and S. M. Cohen, Chem. Soc. Rev., 2009, 38, 1315–1329 RSC; (d) D. Farrusseng, S. Aguado and C. Pinel, Angew. Chem., Int. Ed., 2009, 48, 7502–7513 CrossRef CAS PubMed; (e) B. Chen, S. Xiang and G. Qian, Acc. Chem. Res., 2010, 43, 1115–1124 CrossRef CAS PubMed; (f) T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC; (g) P. Hu, J. V. Morabito and C.-K. Tsung, ACS Catal., 2014, 4, 4409–4419 CrossRef CAS; (h) J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed; (i) Z. Hu, K. Zhang, M. Zhang, Z. Guo, J. Jiang and D. Zhao, ChemSusChem, 2014, 7, 2791–2795 CrossRef CAS PubMed; (j) H.-R. Fu, Z.-X. Xu and J. Zhang, Chem. Mater., 2015, 27, 205–210 CrossRef CAS.
- (a) S. Hermes, M.-K. Schröter, R. Schmid, L. Khodeir, M. Muhler, A. Tissler, R. W. Fischer and R. A. Fischer, Angew. Chem., Int. Ed., 2005, 44, 6237–6241 CrossRef CAS PubMed; (b) Y. K. Hwang, D. Y. Hong, J. S. Chang, S. H. Jhung, Y. K. Seo, J. Kim, A. Vimont, M. Daturi, C. Serre and G. Férey, Angew. Chem., Int. Ed., 2008, 47, 4144–4148 CrossRef CAS PubMed; (c) T. Ishida, M. Nagaoka, T. Akita and M. Haruta, Chem. – Eur. J., 2008, 14, 8456–8460 CrossRef CAS PubMed; (d) H.-L. Jiang, B. Liu, T. Akita, M. Haruta, H. Sakurai and Q. Xu, J. Am. Chem. Soc., 2009, 131, 11302–11303 CrossRef CAS PubMed; (e) B. Yuan, Y. Pan, Y. Li, B. Yin and H. Jiang, Angew. Chem., Int. Ed., 2010, 49, 4054–4058 CrossRef CAS PubMed; (f) H.-L. Jiang and Q. Xu, Chem. Commun., 2011, 47, 3351–3370 RSC; (g) J. Hermannsdörfer, M. Friedrich, N. Miyajima, R. Q. Albuquerque, S. Kümmel and R. Kempe, Angew. Chem., Int. Ed., 2012, 51, 11473–11477 CrossRef PubMed; (h) C. Wang, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2012, 134, 7211–7214 CrossRef CAS PubMed; (i) G. Lu, S. Li, Z. Guo, O. K. Farha, B. G. Hauser, X. Qi, Y. Wang, X. Wang, S. Han, X. Liu, J. S. DuChene, H. Zhang, Q. Zhang, X. Chen, J. Ma, S. C. J. Loo, W. D. Wei, Y. Yang, J. T. Hupp and F. Huo, Nat. Chem., 2012, 4, 310–316 CrossRef CAS PubMed; (j) H. R. Moon, D.-W. Lim and M. P. Suh, Chem. Soc. Rev., 2013, 42, 1807–1824 RSC; (k) M. Zhao, K. Deng, L. He, Y. Liu, G. Li, H. Zhao and Z. Tang, J. Am. Chem. Soc., 2014, 136, 1738–1741 CrossRef CAS PubMed; (l) X.-H. Liu, J.-G. Ma, Z. Niu, G.-M. Yang and P. Cheng, Angew. Chem., Int. Ed., 2015, 54, 988–991 CrossRef CAS PubMed; (m) Y.-Z. Chen, Y.-X. Zhou, H. Wang, J. Lu, T. Uchida, Q. Xu, S.-H. Yu and H.-L. Jiang, ACS Catal., 2015, 5, 2062–2069 CrossRef CAS.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef PubMed.
- (a) M. Chandra and Q. Xu, J. Power Sources, 2006, 156, 190–202 CrossRef CAS; (b) F. Cheng, H. Ma, Y. Li and J. Chen, Inorg. Chem., 2007, 46, 788–794 CrossRef CAS PubMed; (c) S. Karahan, M. Zahmakıran and S. Özkar, Chem. Commun., 2012, 48, 1180–1182 RSC; (d) S. B. Kalidindi, U. Sanyal and B. R. Jagirdar, Phys. Chem. Chem. Phys., 2008, 10, 5870–5874 RSC; (e) H.-L. Jiang and Q. Xu, Catal. Today, 2011, 170, 56–63 CrossRef CAS; (f) U. B. Demirci and P. Miele, J. Power Sources, 2010, 195, 4030–4035 CrossRef CAS; (g) J.-M. Yan, Z.-L. Wang, H.-L. Wang and Q. Jiang, J. Mater. Chem., 2012, 22, 10990–10993 RSC; (h) Y. Yamada, K. Yano and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 5356–5363 RSC; (i) M. Yadav and Q. Xu, Energy Environ. Sci., 2012, 5, 9698–9725 RSC; (j) Ö. Metin and S. Özkar, Int. J. Hydrogen Energy, 2011, 36, 1424–1432 CrossRef; (k) L.-T. Guo, Y.-Y. Cai, J.-M. Ge, Y.-N. Zhang, L.-H. Gong, X.-H. Li, K.-X. Wang, Q.-Z. Ren, J. Su and J.-S. Chen, ACS Catal., 2015, 5, 388–392 CrossRef CAS.
- H.-L. Jiang, T. Akita and Q. Xu, Chem. Commun., 2011, 47, 10999–11001 RSC.
- J. R. Kitchin, J. K. Norskov, M. A. Barteau and J. G. Chen, Phys. Rev. Lett., 2004, 93, 156801–156804 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, and figures referred in the text. See DOI: 10.1039/c5mh00125k |
| ‡ Yu-Zhen Chen and Linfeng Liang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |