
Artificial photosynthesis on tree trunk derived alkaline tantalates with hierarchical anatomy: towards CO2 photo-fixation into CO and CH4†
Han
Zhou
ab,
Peng
Li
ac,
Jianjun
Guo
ac,
Runyu
Yan
b,
Tongxiang
Fan
b,
Di
Zhang
b and
Jinhua
Ye
*acd
aInternational Center for Materials Nanoarchitectonics (WPI-MANA) and Environmental Remediation Materials Unit, National Institute for Materials Science (NIMS), 1-1, Namiki, Tsukuba, Ibaraki 305-0044, Japan. E-mail: Jinhua.YE@nims.go.jp
bState Key Lab of Metal Matrix Composites, Shanghai Jiaotong University, Shanghai, 200240, China
cGraduate School of Chemical Sciences and Engineering, Hokkaido University, Sapporo, Japan
dTU–NIMS Joint Research Center, School of Materials Science and Engineering, Tianjin University, 92 Weijin Road, Nankai District, Tianjin 300072, P. R. China
First published on 16th September 2014
Abstract
Artificial photosynthesis, the photochemical fixation and recycling of CO2 back to hydrocarbon fuels using sunlight and water, is both a significant challenge and an opportunity that, if realized, could have a revolutionary impact on our energy system. Herein, we demonstrate one of the first examples using biomass derived hierarchical porous photocatalysts for CO2 photo-fixation into sustainable hydrocarbon fuels. A generic method is proposed to build a series of alkaline tantalates MTaO3 (M = Li, Na, K) with hierarchical anatomy from macro- to nanoscales using activated carbonized tree trunks as templates. Artificial photosynthesis is carried out on MTaO3 series using only artificial sunlight, water, and carbon dioxide as inputs to produce carbon monoxide and methane as the main outputs. The CO2 photo-fixation performance can be enhanced by introducing a macropore network, which mainly enhances light transfer and accelerates gas diffusion. The research provides prototype models that integrate individual nanoscale components into higher level macroscopic artificial photosynthetic systems for better solar-to-fuel conversion efficiencies. This work would have potential significance for the ultimate construction of “artificial trees” and provide envisions creating “forests” of these CO2-capturing artificial trees to remove carbon dioxide from the atmosphere and convert it into sustainable fuels.
Introduction
The significant rise in atmospheric CO2 levels due to the combustion of hydrocarbon fuels has generated worldwide concern. Among various CO2 sequestration technologies, a compelling approach, normally known as artificial photosynthesis, is photochemical fixation and recycling of CO2 back to hydrocarbon fuels (e.g. CO, CH4, CH3OH, etc.) using sunlight and water. The realization of artificial photosynthesis is a significant challenge and could have a revolutionary impact on our energy system.1–5 Since the first study on the photo-fixation of CO2 was reported in 1978,6 tremendous efforts have been devoted to develop new promising photocatalysts and to modify the photocatalysts for enhanced efficiency.7–9 In particular, some multi-metallic catalysts, such as Zn2GeO4,10,11 ZnGa2O4,12,13 NaNbO3,14 Zn2SnO4,15 and Bi2WO6,16,17 have become very attractive in recent years due to their wide band gaps with high reduction potentials. However, up to now materials that are reported to be active for CO2 photo-fixation are still very limited. Tantalates exhibit high photocatalytic performance because they possess conduction bands at a more negative position than that of titanates, and thus a variety of mixed metal oxides containing closed-shell Ta5+ transition-metal ions have been studied recently.18,19 As a typical example, alkaline tantalates MTaO3 (M = Li, Na, K) show high activities for water splitting.18,19 Therefore, they are likely to be promising for CO2 photoreduction. However, in general, these tantalates are synthesized via a conventional solid-state reaction or polymerized via a complex method at high temperatures, leading to products with rather low porosity and low surface area, which restricts their performance, especially for reactions involving gases.Morphology control strategy has been an effective and versatile approach for promoting CO2 photo-fixation activities,20 involving the construction of nanostructures,10,21,22 micro/mesoporous structures,11,12 hollow structures,23–25 metal–organic frameworks (MOFs)26 with highly porous cavities, etc. In recent years, materials with hierarchical porous anatomy have been extensively researched and are widely used in catalysis.27–29 This is because materials with hierarchical porosity can provide macro to nanolength sizes of pores for reaction, interfacial transport, or dispersion of active sites and reduce diffusion paths or reduce diffusion.27,28 However, to date there are only few reports on the oriented design, controlled synthesis and ultimate application of hierarchically porous materials for CO2 photo-fixation. Furthermore, the general synthesis of alkaline tantalates with a well-defined hierarchical porous anatomy has not been reported yet.
In green plants, leaves are well-known as the main photosynthetic organs. Tree trunks, a main part of trees, function as water/mineral transportation pathways for the photosynthesis of leaves. They exhibit a hierarchically built anatomy which act as the central “plumbing system” in a tree, forming a network of tubes as transportation paths with three-dimensional (3D), highly hierarchical, porosity and high system connectivity that carries water and minerals up from the roots to the leaves, and sugar from the leaves down to the branches, trunk, and roots. The most distinct morphological feature of tree trunks is the open porous system of the tracheidal cells, which provide the transportation path for water in the living wood and yields an uniaxial pore structure with anisotropic mechanical properties.30 Furthermore, tree trunks have a rather high surface area of up to hundreds m2 g−1. Thus, such a hierarchical anatomy would be a potential candidate for the directed synthesis of hierarchical systems.
Herein, we demonstrate one of the first examples using biomass derived hierarchical porous catalysts for CO2 photo-fixation into sustainable hydrocarbon fuels. A generic method is proposed to build a series of alkaline tantalates MTaO3 (M = Li, Na, K) with hierarchical anatomy using activated carbonized tree trunks as templates. Artificial photosynthesis is carried out on MTaO3 series using only artificial sunlight, water, and carbon dioxide as inputs to produce carbon monoxide and methane as the main outputs. The CO2 photo-fixation performance can be enhanced by introducing a macropore network because of the enhanced light transfer and faster gas diffusion in hierarchical meso/macroporous systems. The research provides prototype models to integrate individual functional components into higher level macroscopic artificial photosynthetic systems for better solar-to-fuel conversion efficiencies. This work would have potential significance for ultimate construction toward “artificial trees” or “artificial plants” and one could envision creating “forests” of these CO2-capturing artificial trees to remove carbon dioxide from the atmosphere and convert it into sustainable fuels.
Experimental section
Pretreatment/activation of wood template
First, the specimens (10 × 10 × 10 mm3) of White Pine wood were boiled in 5% ammonia solution overnight to remove the wood extractive compounds, such as organic acids and lipids, so that the connectivity of the wood's pores can be increased for better soaking with precursor. The extracted wood templates were completely washed with deionized water. The wood was calcined at 400 °C for 2 h under N2 atmosphere at a heating rate of 1 °C min−1. The obtained carbonized wood was infiltrated in NaOH solution (mass ratio of carbonized wood: NaOH = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) at 80 °C until the aqueous solution was evaporated. For the synthesis of KTaO3 and LiTaO3, KOH and LiOH solutions were used, respectively. Then, the carbonized wood was calcined at 600 °C for 2 h under N2 atmosphere at a heating rate of 1 °C min−1. The obtained activated carbonized wood was dispersed in DI water and then concentrated HNO3 (70%) was added dropwise until the solution was acidic. Then, the wood was washed with DI water and ethanol completely and dried in a vacuum oven at 60 °C overnight.
:4) at 80 °C until the aqueous solution was evaporated. For the synthesis of KTaO3 and LiTaO3, KOH and LiOH solutions were used, respectively. Then, the carbonized wood was calcined at 600 °C for 2 h under N2 atmosphere at a heating rate of 1 °C min−1. The obtained activated carbonized wood was dispersed in DI water and then concentrated HNO3 (70%) was added dropwise until the solution was acidic. Then, the wood was washed with DI water and ethanol completely and dried in a vacuum oven at 60 °C overnight.
Synthesis of MTaO3 (M = Li, Na, K) from the activated carbonized wood
Typically, for the synthesis of NaTaO3, 10 mmol sodium acetate was first dissolved in 100 mL anhydrous ethanol and stirred for 30 min. Then, stoichiometric amounts of tantalum ethoxide dissolved in 100 mL 2-methoxyethanol solution was added into the abovementioned solution and stirred for 2 h. 1.0 g activated carbonized wood was immersed into the precursor solution and infiltrated under vacuum at 50 °C for 8 h. Excess precursor solution was washed away by anhydrous ethanol four times. The as-obtained infiltrated carbonized wood was dried at room temperature overnight and then calcined at 550 °C for 10 h under O2 atmosphere at a heating rate of 1 °C min−1. For the synthesis of KTaO3 and LiTaO3, potassium acetate and lithium acetate were used instead of sodium acetate, respectively.Loading the co-catalysts
The loading of Au was also performed by a precipitation method. The precipitation procedure was performed at 343 K and pH 9 for 4 h with 1 wt% HAuCl4·3H2O as the Au source using (0.2 M) NaOH to maintain the pH constant. The catalyst was then recovered, filtered, washed with deionized water, and dried at 373 K overnight. Finally, the powder was calcined at 473 K in air for 4 h.CO2 photoreduction measurements
CO2 photoreduction experiments were carried out in a gas closed circulation system with an upside window (ESI, Fig. S1†). The catalyst (50 mg) was dispersed on a small glass cell with a base area of 8.1 cm2 and then loaded into a Pyrex reaction cell. After that, 2 mL of distilled water was added into the gas closed reaction system. The volume of the reaction system was around 390 mL. The entire system was then evacuated and filled with 80 kPa of pure CO2 gas. The light source was a 200 W Hg–Xe arc lamp (ILC Technology, CERMAX LX-300). Organic products were sampled and measured with a gas chromatograph (GC-14B, Shimadzu) equipped with a flame ionization detector (FID) according to the standard curves. For CH4 detection, a PEG-1000 chromos column was used. For CO detection, a Propark Q column was used. H2 and O2 evolution was measured with an online gas chromatograph (GC-8A, Shimadzu) with a TCD detector according to the standard curve.Characterization
Crystal structures of samples were determined with an X-ray diffractometer (Rint-2000, Rigaku Co., Japan) with Cu Kα radiation. The diffuse reflection spectra were measured with an integrating sphere equipped with a UV-visible recording spectrophotometer (UV-2600, Shimadzu Co., Japan) using BaSO4 as a reference, and the optical absorptions were converted from the reflection spectra according to the Kubelka–Munk equation. Scanning electron microscopy (SEM) images were recorded to observe the morphology with a scanning electron microscope (JEOL 6700F field emission scanning electron microscope). Transmission electron microscopy and high-resolution images were recorded with a field emission transmission electron microscope (2100F, JEOL Co., Japan) operated at 200 kV. The specific surface areas were determined with a surface-area analyzer (BEL Sorp-II mini, BEL Japan Co., Japan) by the Brunauer–Emmett–Teller (BET) method. Mercury porosimetry measurements were performed using an Autopore IV 9500 (Micromertitics Company).Results and discussion
Water is essential to photosynthesis. Large amounts of water are transpired by a tree on a daily basis. As shown in Fig. 1a, water is absorbed by the root system and is transported up the tree trunks that act like pipes to the leaves for photosynthesis. Food prepared in the leaves is then transported down to the roots and other parts of the tree for growth. The trunks contain a network of tubes (Fig. 1b) that run between the roots and the leaves and act as the central plumbing system for the tree. Tree trunk, generally referred to as wood, is a natural composite and exhibits an anisotropic, 3D porous morphology. Macroscopically, wood is characterized by the formation of growth ring structures. The microstructural hierarchical features of wood range from the millimeter (growth ring structures) through the micrometer (tracheidal cell patterns, macro- and microfibril cell-wall textures) down to the nanometer scale (molecular fiber and membrane structures of cell walls (Fig. 1b–1d).30 Wood tissue is characterized by a hierarchical anatomy of unidirectionally oriented cells. The morphology and arrangement of the cells vary between deciduous and coniferous wood. Coniferous wood has a very uniform structure (Fig. 1b) and consists of 90–95% tracheids, which are long and slender cells tapered at the ends. Deciduous wood is less homogeneous (ESI, Fig. S2†). Tracheids are oriented in the direction of the trunk axis. They form transportation paths as long continuous tubes for water and minerals within the living tree (Fig. 1b).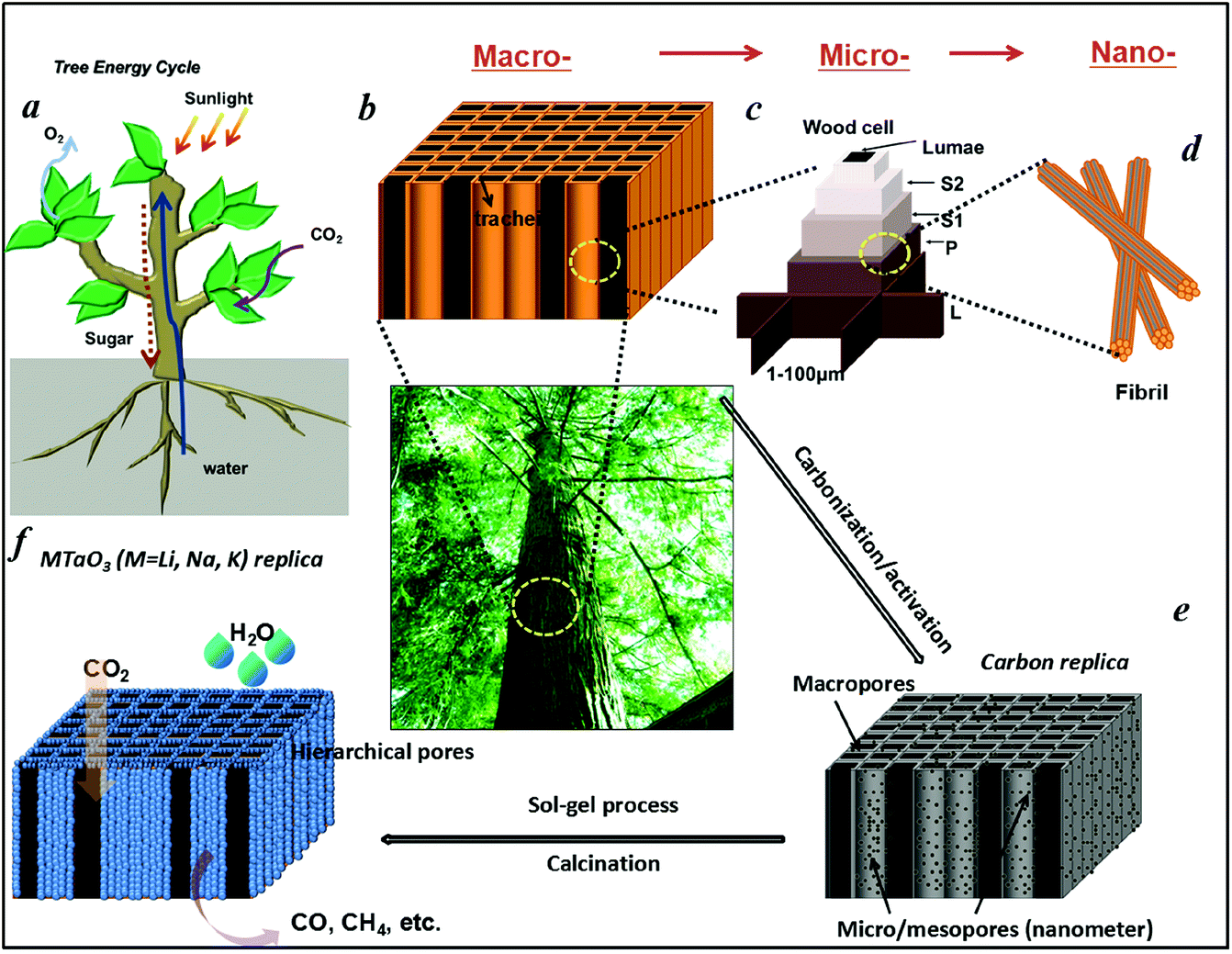 | ||
| Fig. 1 Schematic illustration of (a) Tree energy cycle. (b) Hierarchical macro- and (c) microscopic cell structures of tree trunks. A single longitudinal tracheidal cell exhibits a layered wall structure, a thin primary wall (P), and a thicker secondary wall composed of sublayers (S1, S2), which vary in cellulose microfibril orientation and play a key role in the mechanical behavior of cellular tissue. Adjacent tracheids are joined together by a highly lignified layer (middle lamella L). (d) Fibril structure of a cell wall at the nanoscale. (e) Carbonization/activation process for the production of activated carbonized tree trunk templates with hierarchical pores. (f) Hierarchical porous MTaO3 (M = Li, Na, K) replica obtained via sol–gel/calcination process with the conceptual drawing of an artificial system that uses only light, water, and carbon dioxide as inputs and produces sustainable fuels. | ||
In this work, a typical coniferous wood White pine was chosen as a template. As shown in Fig. 2a and 2b, white pine has a nearly monomodal microsized pore distribution. The mean pore diameter of the tracheids is 10 μm. The longitudinal section is composed of oriented long microtubes with a diameter of 10 μm (Fig. 2c and 2d). Cells arranged in a radial direction (rays) and pores in the cell walls create a 3D macropore network for transportation. Furthermore, cell walls are composed of numerous micropores according to the N2 adsorption measurement (Fig. 3a), which are from the nanosized molecular fibers. Such hierarchical porous anatomy endows the wood a high surface area of up to 345 m2 g−1 (Fig. 3a). Here, an activation process was applied on the carbonized wood template to significantly increase its surface area, approaching ∼1757 m2 g−1 (Fig. 1e and 3a). An activation process causes more micropores to form and thereby causing a major relative increase in pore volume in the micropore range (Fig. 3a). Obviously, main micropores are formed during partial oxidation and removal of carbon in hot air, which leads to the increase in specific surface area and porosity. Activated carbonized wood with 3D hierarchical micro/meso/macroporous anatomy and high surface area was a promising template for the synthesis of the corresponding alkaline tantalates replica. Under vacuum infiltration conditions, it was easier for the precursor solution to infiltrate into the micro/mesopores of the carbonized samples. Because there are functional groups on the surfaces of the carbonized samples, the precursors could interact with the surfaces, which facilitated targeted materials assembly. The major biopolymeric constituents in wood are cellulose, hemicellulose, and lignin with some additional macromolecular compounds, such as different kinds of fat, oil, wax, minerals, alcaloides, etc., as minor constituents.31 According to TGA data (ESI, Fig. S3a†), the organics could be decomposed completely at around 450 °C. Hence, after calcination at 550 °C in O2 atmosphere for 10 h, the organics would be removed completely (ESI, Fig. S2†) leaving crystalline tantalates MTaO3 (M = Li, Na, K), as demonstrated by XRD (Fig. 3b). FESEM and TEM images show the hierarchical porous anatomy of the products. Considering NaTaO3 as an example, the product has a very loose feature (ESI, Fig. S4†). It can be observed from the cross-section that the products have highly porous structures with pores in the range of several micrometers (Fig. 2e). Longitudinally, it has tube-like structures (Fig. 2f). A TEM image (inset of Fig. 2e) shows that the macropore networks are composed of mesoporous nanocrystalline building blocks.
 | ||
| Fig. 2 FESEM images of (a) the cross-section of carbonized white pine; inset: 3D schematic illustration. (b) Magnified image of the cross-section. (c) Longitudinal section of carbonized white pine. (d) Magnified image of the longitudinal section. (e) Porous NaTaO3 replica derived from the activated carbon templates; inset: corresponding TEM image indicating the mesoporous feature. (f) FESEM image of the tubular structure of NaTaO3; inset: schematic illustration. | ||
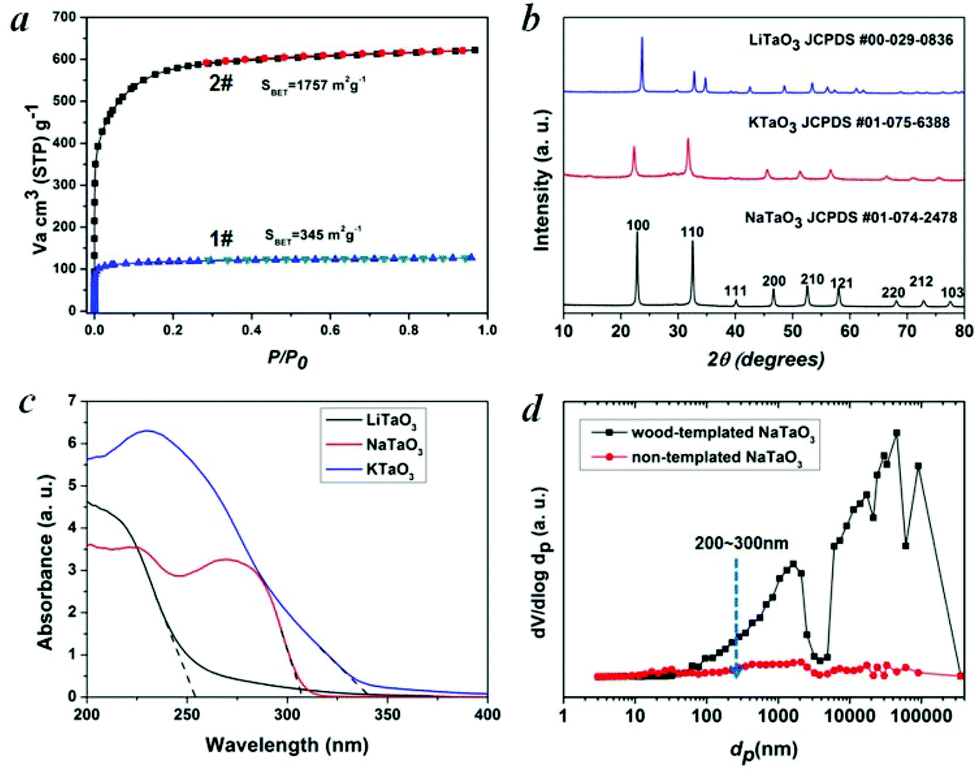 | ||
| Fig. 3 (a) N2 adsorption isotherms of wood templates: 1# carbonized wood and 2# activated carbonized wood. (b) PXRD patterns of wood-templated MTaO3 (M = Li, Na, K) series. (c) UV-vis absorption spectra of wood-templated MTaO3 (M = Li, Na, K) series. (d) Mercury intrusion porosimetry of NaTaO3 for the characterization of hierarchical pore size distributions. | ||
In general, CO2 in the presence of water vapor can be photoreduced to hydrocarbon fuels using a wide-band-gap semiconductor as a photocatalyst.32 Because of the irradiation of full arc Xe lamp, the existing form of water for the reaction is vapor. Theoretically, alcohol or methanol could serve as electron donors to increase the activities. However, because of the well-known process of photocatalytic reforming of ethanol, in the presence of alcohol the production of CO and CH4 could increase significantly, which is derived from the photocatalytic reforming of ethanol. Thus, it is not easy to analyze the carbon sources of CO and CH4. According to the diffuse reflection spectra of alkali tantalates (Fig. 3c), the onsets of the absorption of LiTaO3, NaTaO3 and KTaO3 were 256, 310 and 344 nm, respectively. Thus, the band gaps of LiTaO3, NaTaO3 and KTaO3 were estimated to be 4.8, 4.0 and 3.6 eV, respectively. Here, we performed artificial photosynthesis of CO2 + gaseous H2O reaction in a gas–solid system over the MTaO3 series. Considering NaTaO3 as an example, bare NaTaO3 evolves CO and CH4 as the main products in the absence of any sacrificial agents (Fig. 4a and 4b). It is well-known that CO2 photoreduction mainly undergoes two courses, including oxidation and reduction processes. In the oxidation process, the photogenerated holes in the valence band oxidize water to generate hydrogen ions via the half-reaction (2H2O + 4h+ → O2 + 4H+). In the reduction course, there is a chain reaction to reduce CO2 to CH4 (CO2 + 2H+ + 2e− → CO + H2O, CO2 + 4H+ + 4e− → HCHO + H2O, CO2 + 6H+ + 6e− → CH3OH + H2O, CO2 + 8H+ + 8e− → CH4 + 2H2O).32 The edges of the valence band (EVB) and conduction band (ECB) of NaTaO3 are determined to be 2.993 and −1.007 V (vs. normal hydrogen electrode, NHE) via the Mulliken electronegativity method, respectively.33EVB of NaTaO3 is more positive than that of E°(H2O/H+) (H2O → 1/2O2 + 2H+ + 2e−, E° redox = 0.82 V vs. NHE), and ECB is more negative than that of E° (CO2/CO) (CO2 + 2e− + 2H+ → CO + H2O, E° redox = −0.53 V vs. NHE) and E° (CO2/CH4) (CO2 + 8e− + 8H+ → CH4 + 2H2O, E° redox = −0.24 V vs. NHE). This indicates that the photogenerated electrons and holes on the irradiated NaTaO3 can react with the adsorbed CO2 and H2O to produce CO and CH4. Other products, such as HCOOH, HCHO, CH3OH, were not detected probably because of the gas-phase CO2 photoreduction with low proton concentrations; CO is the primary first step product instead of HCOOH.34–36 The intermediates produced during the reduction tend to favor the formation of CH4 instead of CH3OH, HCHO and HCOOH.34–36 Furthermore, the strong oxidation power of photogenerated holes (or OH radicals) that can react with intermediates and products of CO2 conversion in reactions34–36 make the net yield negligible. It is also worth mentioning that in our semiconductor systems, H2 was not detected probably because in the absence of sacrificial agents in our system, the produced H2 served as reductant for CO2 photoreduction.
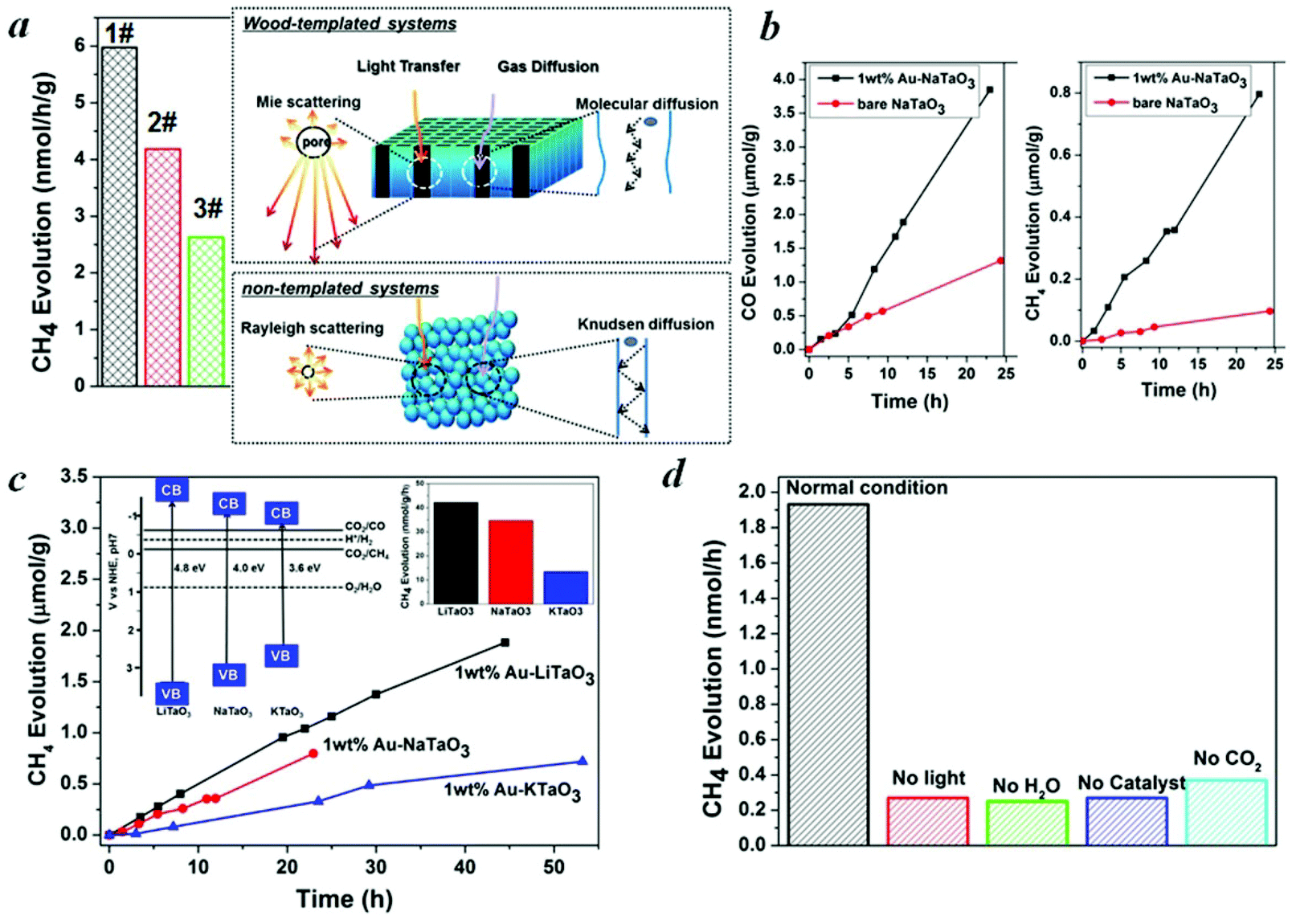 | ||
| Fig. 4 Photocatalytic CO2 reduction activities. (a) Left part: CH4 evolution on a bare NaTaO3 series, right part: schematic illustration of light transfer and gas diffusion in wood-templated hierarchical porous systems and non-templated systems, respectively. 1# NaTaO3 derived from activated carbonized wood, 2# NaTaO3 derived from carbonized wood, 3# non-templated NaTaO3. (b) CO and CH4 evolution on NaTaO3 with and without loading with Au cocatalysts. (c) Photocatalytic formation of CH4 on MTaO3 (M = Li, Na, K) series derived from activated carbonized wood after loading with 1 wt% Au as a cocatalyst, with the inset of the energy band structure of MTaO3 (M = Li, Na, K). (d) CH4 evolution in reference experiments in the conditions without H2O, CO2, light irradiation, and catalyst compared with that in normal conditions. Au (1 wt%)-NaTaO3 derived from activated carbonized wood was measured as the normal condition. | ||
Hierarchical porous anatomy could influence their activity. Considering NaTaO3 as an example, activated carbonized wood-templated NaTaO3 exhibits about a 2.23-fold improvement in activities than non-templated NaTaO3 based on the same amount (Fig. 4a) mainly due to enhanced light transfer and faster gas diffusion. Hendricks and Howell37 indicated that the porous structure of ceramics creates complex electromagnetic scattering and interference patterns within the structure. Particularly, when the size of the voids is comparable to the wavelength of the incident light, Mie's scattering occurs, in which light scatters strongly along the forward direction. This scattering produces a pattern similar to an antenna lobe with a sharper and more intense forward lobe (Fig. 4a, right part).38 According to the diffuse reflection spectra and the spectrum of Hg–Xe lamp (ESI, Fig. S5†), the effective incident light wavelength is between 200–310 nm. According to the mercury porosimeter (Fig. 3d), the quantity of macropores around 200–310 nm comparable with the incident light wavelength in wood-templated NaTaO3 are obviously much more than that of the non-templated NaTaO3. Therefore, the path length of the incident light increases,39 leading to an enhanced light utilization efficiency. The scattered light then transfers to and is absorbed by the neighboring pores, thus inducing more photo-excited electrons for the reduction (Fig. 4a, right part).
On the other hand, gas diffusivity is another key factor. When the diameter of the pore is considerably greater than the mean free path of the gas molecules, the diffusing molecules interact with each other more than with the pore walls, thereby minimizing the wall effects on transport.40 In our case, the mean free paths of CO2, CH4, CO and H2O were calculated to be about 74, 70, 72 and 139 nm according to eqn (1), respectively. Where K is 1.38 × 10−23, T is assumed to be 323 K in our experiment and D0 refers to the diameter of gas molecules (nm).
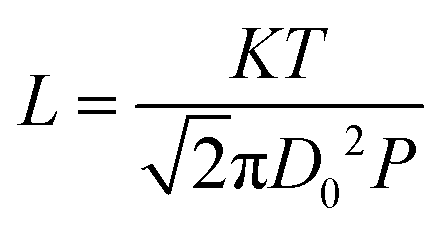 | (1) |
According to the SEM and mercury porosimeter, a number of macropores are in the range of several to hundreds of micrometers, and thus the pore diffusivity is essentially the same as the molecular diffusivity, in which the diffusion coefficient is considerably larger than that in mesopores (Fig. 4a, right part). In contrast, gas diffusion in non-templated system with much fewer and smaller pores is mainly Knudsen diffusion, where the gas molecules frequently collide with the pore walls (Fig. 4a, right part). Thus, the gas diffusion rate is smaller and a longer time is required for reactants to move into the deeper layers as well as for products to move from the deeper layers into the atmosphere. The higher activity of the activated carbonized wood-templated system (Fig. 4a, 1#) than the carbonized wood-templated system (Fig. 4a, 2#) is mainly because of higher surface areas and higher volume of mesopores derived from the porous activated carbon templates, (ESI, Table S1†) and provide more reaction sites, thus enhancing the overall performance.
The evolution rates of CO and CH4 over NaTaO3 could be significantly enhanced by a factor of 3.1 and 8.4, respectively, after loading Au cocatalyst to facilitate the proton-coupled multielectron transfer (Fig. 4b). A precipitation method was used for the deposition of Au nanoparticles. XPS spectrum (ESI, Fig. S6†) demonstrates the formation of metallic Au. The size of Au is only about 3 nm (ESI, Fig. S7†) with good homogeneity, which coincides with a literature report.41 After loading with about 1 wt% Au onto MTaO3 series, the activities were in the order of LiTaO3 > NaTaO3 > KTaO3, as compared in Fig. 4c. According to the Mulliken electronegativity method,33 the edges of the conduction band of LiTaO3, NaTaO3, KTaO3 were estimated to be −1.302 V, −1.007 V and −0.98 V (vs. NHE), respectively (inset of Fig. 4c). The order of the photocatalytic activities was consistent with that of the conduction band levels, indicating that wide band gap semiconductors with higher reduction potentials are more favorable for CO2 photo-fixation. We compared our results with other photocatalytic systems with respect to efficiency and selectivity, and the data is shown in Table 1. However, because the experimental conditions (light intensity, irradiation distance, catalysts amount, and others) are different, it may not be suitable comparison. Here, we also compared our photocatalytic systems with some typical semiconductors (published by our group) measured with the same reactor system (Table S2†).
| Light source | Catalyst | Co-catalyst | Reaction medium | Products | SelCH4a/% | Ref. |
|---|---|---|---|---|---|---|
| a SelCH4/% = mol (CH4)/mol (CH4 + CO). | ||||||
| 300 W Xe lamp | Leaf-architectured SrTiO3 | 1 wt% Au | CO2 and H2O vapor | CO (350 nmol h−1 g−1) | 44.1 | 46 |
| CH4 (275 nmol h−1 g−1) | ||||||
| UV Xe lamp | TiO2 rods with {010} facets | 1 wt% Pt | CO2 and H2O vapor | CH4 (5.7 nmol h−1 g−1) | 100 | 47 |
| 300 W Xe lamp UV | hollow anatase TiO2 single crystals with {101} facets | 1 wt% RuO2 | CO2 and H2O vapor | CH4 (1.725 nmol h−1 g−1) | 100 | 48 |
| 350 W Xe lamp UV | SiO2-Pillared HNb3O8 | 0.4 wt% Pt | CO2 and H2O vapor | CH4 (2.9 nmol h−1 g−1) | 100 | 49 |
| 300 W Xe arc lamp visible light λ > 420 nm | Na2V6O16 nanoribbons | 1 wt% Pt and 1 wt% RuO2 | CO2 and H2O vapor | CH4 (190 nmol h−1 g−1) | 100 | 50 |
| Xe arc lamp visible light λ > 420 nm | W18O49 nanowires | — | CO2 and H2O vapor | CH4 (666 ppm h−1 g−1) | 100 | 21 |
| visible light λ > 420 nm | ZnAl2O4-modified mesoporous ZnGaNO | 0.5 wt% Pt | CO2 and H2O vapor | CH4 (9.2 nmol h−1 g−1) | 100 | 51 |
| Xe lamp UV | Zn2GeO4 nanobelts | 1 wt% Pt and 1 wt% RuO2 | CO2 and H2O vapor | CH4 (25 nmol h−1 g−1) | 100 | 10 |
| 300 W Xe arc lamp visible light λ > 420 nm | sheaf-like, hyperbranched Zn1.7GeN1.8O | 1 wt% Pt and 1 wt% RuO2 | CO2 and H2O vapor | CH4 (9 nmol h−1 g−1) | 100 | 52 |
| 200 W Hg-Xe arc lamp | NaTaO3 | 1 wt% Au | CO2 and H2O vapor | CO (173 nmol h−1 g−1) | 17.3 | This work |
| CH4 (36 nmol h−1 g−1) | ||||||
For all the samples, oxygen is not detected. Water oxidation is much more difficult kinetically and energetically than H2 evolution, requiring a large overpotential. Oxygen generated by water oxidation is partially used for the oxidation of the evolved products (HCOOH, HCHO, CO, etc.)42 On the other hand, O2 evolution is controlled by the interfacial reaction such that the semiconductor should provide a favorable reaction site for the O2 formation process. Otherwise, O2 can be absorbed on surface oxygen vacancies43 and undesirably absorbs electrons to form O2− or other species, such as H2O2, that chemisorbs on the surfaces of catalysts and can increase the redox energy of the half-reaction, making the necessary overvoltage still larger,44 thereby further inhibiting O2 production. We checked the XRD patterns before and after irradiation on wood-templated NaTaO3 (Fig. S8†) to test the chemical stability. Furthermore, Fig. 4b and 4c show that after long term irradiation (24 h–54 h), the activities of MTaO3 (M = Li, Na, K) series remained almost linear, which further demonstrates the stability of these catalysts. Finally, to confirm that the hydrocarbon fuels were generated from the reduction of CO2via the protons released from H2O oxidization and photogenerated electrons, control experiments were carried out, as shown in Fig. 4d. When the experiment was carried out in the absence of H2O, catalyst or light irradiation, very little CH4 was detected due to only 1 ppm of CH4 contamination from air during samples. When the CO2 gas was replaced by Ar gas, a small amount of CH4 was found which should be generated from the photoreduction of the remaining CO2 on the sample surface.45
Conclusions
To conclude, we have successfully synthesized alkaline tantalates MTaO3 (M = Li, Na, K) with hierarchical porous anatomy via the development of a general approach using activated carbonized tree trunks as templates. CO2 photo-fixation processes indicate that H2O supplies protons, CO2 offers a carbon source, and the photocatalyst gives redox potentials for the entire reaction to finally produce CO and CH4. The research opens a new pathway for oriented design, controlled synthesis and application of 3D hierarchical porous systems for the realization of artificial photosynthesis, especially for gas–solid reactions. From this work, it is predicted that activated carbonized biomass with rather high surface area and high hierarchical pore volume could be promising models for the design and fabrication of a new class of hierarchical catalysts for improved performance. A similar strategy could be extended to a wide range of multi-metallic catalysts. Because natural patterns and shapes arise in innumerable ways on a range of scales, our results suggest that the discovery of topological morphologies associated with photosynthesis is of great significance and the unique morphology models are promising for the biomimetic synthesis of artificial analogues. Moreover, the research would provide a conceptual blueprint for the ultimate construction of “artificial trees” and we envision creating “forests” of these carbon-capturing artificial trees for large scale CO2 photo-fixation into sustainable fuels. Finally, we anticipate that this concept may be of interest to a variety of energy capture, conversion and storage areas, and may also result in significant improvements in various applications, ranging from solar cells, fuels cells and lithium-based battery.Acknowledgements
We are grateful for financial support by the World Premier International Research Center Initiative on Materials Nanoarchitectonics, MEXT, Japan, and National Basic Research Program of China (973 Program, 2014CB239301) and the National Natural Science Foundation of China (51102163).Notes and references
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- J. Michl, Nat. Chem., 2011, 3, 268–269 CrossRef CAS PubMed.
- S. Bensaid, G. Centi, E. Garrone, S. Perathoner and G. Saracco, ChemSusChem, 2012, 5, 500–521 CrossRef CAS PubMed.
- M. Antonietti, Angew. Chem., Int. Ed., 2013, 52, 1086–1087 CrossRef CAS PubMed.
- J. Liu and M. Antonietti, Energy Environ. Sci., 2013, 6, 1486–1493 CAS.
- M. Halmann, Nature, 1978, 275, 115–116 CrossRef CAS.
- S. C. Roy, O. K. Varghese, M. Paulose and G. A. Grimes, ACS Nano, 2010, 4, 1259–1278 CrossRef PubMed.
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, ChemSusChem, 2013, 6, 562–577 CrossRef CAS PubMed.
- M. R. Hoffmann, J. A. Moss and M. M. Baum, Dalton Trans., 2011, 40, 5151 RSC.
- Q. Liu, Y. Zhou, J. Kou, X. Chen, Z. Tian, J. Gao, S. Yan and Z. G. Zou, J. Am. Chem. Soc., 2010, 132, 14385–14387 CrossRef CAS PubMed.
- N. Zhang, S. X. Ouyang, P. Li, Y. J. Zhang, G. C. Xi, T. Kako and J. H. Ye, Chem. Commun., 2011, 47, 2041–2043 RSC.
- S. C. Yan, S. X. Ouyang, J. Gao, M. Yang, J. Y. Feng, X. X. Fan, L. J. Wan, Z. S. Li, J. H. Ye, Y. Zhou and Z. G. Zou, Angew. Chem., Int. Ed., 2010, 49, 6400–6404 CrossRef CAS PubMed.
- S. C. Yan, J. J. Wang, H. Gao, N. Wang, H. Yu, Z. Li, Y. Zhou and Z. Zou, Adv. Funct. Mater., 2013, 23, 758–763 CrossRef CAS.
- P. Li, S. Ouyang, G. Xi, T. Kako and J. Ye, J. Phys. Chem. C, 2012, 116, 65–70 Search PubMed.
- Z. Li, Y. Zhou, J. Zhang, W. Tu, Q. Liu, T. Yu and Z. Zou, Cryst. Growth Des., 2012, 12, 1476–1481 CAS.
- H. Cheng, B. Huang, Y. Liu, Z. Wang, X. Qin, X. Zhang and Y. Dai, Chem. Commun., 2012, 48, 9729–9731 RSC.
- Y. Zhou, Z. Tian, Z. Zhao, Q. Liu, J. Kou, X. Chen, J. Gao, S. Yan and Z. Zou, ACS Appl. Mater. Interfaces, 2011, 3, 3594–3601 CAS.
- F. E. Osterloh, Chem. Mater., 2008, 20, 35–54 CrossRef CAS.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- H. Tong, S. X. Ouyang, Y. P. Bi, N. Umezawa, M. Oshikiri and J. H. Ye, Adv. Mater., 2012, 24, 229–251 CrossRef CAS PubMed.
- G. C. Xi, S. X. Ouyang, P. Li, J. H. Ye, Q. Ma, N. Su, H. Bai and C. Wang, Angew. Chem., Int. Ed., 2012, 51, 2395–2399 CrossRef CAS PubMed.
- J. Liu, R. Cazelles, Z. P. Chen, H. Zhou, A. Galarneau and M. Antonietti, Phys. Chem. Chem. Phys., 2014, 16, 14699–14705 RSC.
- W. Tu, Y. Zhou, Q. Liu, Z. Tian, J. Gao, X. Chen, H. Zhang, J. Liu and Z. Zou, Adv. Funct. Mater., 2012, 22, 1215–1221 CrossRef CAS.
- S. In, D. D. Vaughn and R. E. Schaak, Angew. Chem., Int. Ed., 2012, 51, 3915–3918 CrossRef CAS PubMed.
- O. K. Varghese, M. Paulose, T. J. Latempa and C. A. Grimes, Nano Lett., 2009, 9, 731–737 CrossRef CAS PubMed.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS PubMed.
- S. Mitchell, N. L. Michels, K. Kunze and J. P. Ramirez, Nat. Chem., 2012, 4, 825–831 CrossRef CAS PubMed.
- Y. Li, Z. Y. Fu and B.-L. Su, Adv. Funct. Mater., 2012, 22, 4634–4667 CrossRef CAS.
- H. Zhou, X. F. Li, T. X. Fan, F. E. Osterloh, J. Ding, E. M. Sabio, D. Zhang and Q. X. Guo, Adv. Mater., 2010, 22, 951–956 CrossRef PubMed.
- H. Sieber, C. Hoffmann, A. Kaindl and P. Greil, Adv. Eng. Mater., 2000, 2, 105–109 CrossRef CAS.
- P. Greil, J. Eur. Ceram. Soc., 2001, 21, 105–118 CrossRef CAS.
- I. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef.
- M. A. Butler and D. S. Ginley, J. Electrochem. Soc., 1978, 125, 228 CrossRef CAS PubMed.
- B. Kumar, M. Llorente, J. Froehlich, T. Dang, A. Sathrum and C. P. Kubiak, Annu. Rev. Phys. Chem., 2012, 63, 541–569 CrossRef PubMed.
- A. A. Peterson, F. A. Pedersen, F. Studt, J. Rossmeisl and J. K. Norskov, Energy Environ. Sci., 2010, 3, 1311–1315 Search PubMed.
- G. Halasi, G. Schubert and F. Solymosi, J. Phys. Chem. C, 2012, 116, 15396–15405 CAS.
- T. J. Hendricks and J. R. Howell, J. Heat Transfer, 1996, 118, 911–917 CrossRef CAS.
- G. Mie, Ann. Phys., 1908, 330, 377–445 CrossRef.
- H. J. Koo, J. Park, B. Yoo, K. Yoo, K. Kim and N. G. Park, Inorg. Chim. Acta, 2008, 361, 677–683 CrossRef CAS PubMed.
- L. Cindrella, A. M. Kannan, J. F. Lin, K. Saminathan, Y. Ho, C. W. Lin and J. Wertz, J. Power Sources, 2009, 194, 146–160 CrossRef CAS PubMed.
- C. G. Silva, R. Juarez, T. Marino, R. Molinari and H. Garcia, J. Am. Chem. Soc., 2011, 133, 595–602 CrossRef PubMed.
- Z. Gorem, I. Willner, A. J. Nelson and A. J. Frank, J. Phys. Chem., 1990, 94, 3784–3790 CrossRef.
- E. Lira, S. Wendt, P. Huo, J. Hansen, R. Streber, S. Porsgaard, Y. Wei, R. Bechstein, E. Lagsgaard and F. Besenbacher, J. Am. Chem. Soc., 2011, 133, 6529–6532 CrossRef CAS PubMed.
- P. Salvador and C. Gutierrez, Surf. Sci., 1983, 124, 398–406 CrossRef.
- N. M. Dimitrijevic, B. K. Vijayan, O. G. Poluektov, T. Rajh, K. A. Gray, H. He and P. Zapol, J. Am. Chem. Soc., 2011, 133, 3964–3971 CrossRef CAS PubMed.
- H. Zhou, J. Guo, P. Li, T. X. Fan, D. Zhang and J. H. Ye, Sci. Rep., 2013, 3, 1667 Search PubMed.
- J. Pan, X. Wu, L. Wang, G. Liu, G. Q. Lu and H. M. Cheng, Chem. Commun., 2011, 47(29), 8361–8363 RSC.
- W. Jiao, L. Wang, G. Liu, G. Q. Lu and H. M. Cheng, ACS Catal., 2012, 2(9), 1854–1859 CrossRef CAS.
- X. Li, W. Li, Z. Zhuang, Y. Zhong, Q. Li and L. Wang, J. Phys. Chem. C, 2012, 116(30), 16047–16053 CAS.
- S. Feng, X. Chen, Y. Zhou, W. Tu, P. Li, H. Li and Z. Zou, Nanoscale, 2014, 6(3), 1896–1900 RSC.
- S. Yan, H. Yu, N. Wang, Z. Li and Z. Zou, Chem. Commun., 2012, 48(7), 1048–1050 RSC.
- Q. Liu, Y. Zhou, Z. Tian, X. Chen, J. Gao and Z. Zou, J. Mater. Chem., 2012, 22(5), 2033–2038 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: FESEM images, TGA curves, XPS spectrum, TEM image, the spectrum of the lamp, reaction setup. See DOI: 10.1039/c4nr03019b |
| This journal is © The Royal Society of Chemistry 2015 |