
Synthesis of tumor-associated MUC1-glycopeptides and their multivalent presentation by functionalized gold colloids†
Isabella
Tavernaro‡
a,
Sebastian
Hartmann‡
b,
Laura
Sommer
c,
Heike
Hausmann
d,
Christian
Rohner
a,
Martin
Ruehl
c,
Anja
Hoffmann-Roeder
e and
Sabine
Schlecht
*a
aInstitute of Inorganic and Analytical Chemistry, Justus-Liebig-University Giessen, Heinrich-Buff-Ring 58, 35392 Giessen, Germany. E-mail: Sabine.Schlecht@anorg.chemie.uni-giessen.de
bInstitute of Organic Chemistry, Johannes Gutenberg-University Mainz, Duesbergweg 10-14, 55128 Mainz, Germany
cInstitute of Food Chemistry and Food Biotechnology, Justus-Liebig-University Giessen, Heinrich-Buff-Ring 58, 35392 Giessen, Germany
dInstitute of Organic Chemistry, Justus-Liebig-University Giessen, Heinrich-Buff-Ring 58, 35392 Giessen, Germany
eInstitute of Organic Chemistry, Ludwig-Maximilian-University Munich, Butenandtstr. 5-13, Haus F, 81377 Munich, Germany
First published on 29th July 2014
Abstract
The mucin MUC1 is a glycoprotein involved in fundamental biological processes, which can be found over-expressed and with a distinctly altered glycan pattern on epithelial tumor cells; thus it is a promising target structure in the quest for effective carbohydrate-based cancer vaccines and immunotherapeutics. Natural glycopeptide antigens indicate only a low immunogenicity and a T-cell independent immune response; however, this major drawback can be overcome by coupling of glycopeptide antigens multivalently to immunostimulating carrier platforms. In particular, gold nanoparticles are well suited as templates for the multivalent presentation of glycopeptide antigens, due to their remarkably high surface-to-volume ratio in combination with their high biostability. In this work the synthesis of novel MUC1-glycopeptide antigens and their coupling to gold nanoparticles of different sizes are presented. In addition, the development of a new dot-blot immunoassay to test the potential antigen–antibody binding is introduced.
Introduction
Immunotherapy, a potential alternative to the established cancer therapy, is based on the possibility of activating the human immune system to recognize and effectively kill tumor cells.1,2 In comparison with standard cancer therapies, such as surgical removal of tumors, radiation and chemotherapy, this approach has the advantage of high selectivity and the delocalization of the immune reaction.3 With the help of synthetic vaccines the immune system would not only be activated to attack malignant cells specifically with minimal detriment to healthy cells, but also provide an opportunity to target primary and secondary metastases for the first time. Furthermore, an immunological memory that would provide long-term protection against new cancer diseases could be possible.4 In contrast to bacteria, viruses or other pathogens, tumor cells are endogenous materials against which the immune system necessarily shows tolerance. Therefore the synthetic vaccines also need to trigger a specific immune response, leading to the formation of antibodies that differentiate between normal and cancer cells.To distinguish between healthy and tumor cells, cell surface glycoproteins have proven to be potential target structures.5 These so-called mucins6 are a heterogeneous family of large and high molecular weight O-glycoproteins; the most studied member is the membrane-bound glycoprotein MUC1, found ubiquitously on the apical surface of epithelia and consists of numerous 20-mer-tandem repeats of the sequence HGVTSAPDTRPAPGSTAPPA which embodies five potential glycosylation sites.7 However, due to variations in enzyme activities, which lead to altered glycosylation patterns, these surface glycoproteins are modified on epithelial tumour cells. For example, MUC1 is overexpressed, distributed over the entire cell surface and bears smaller less branched tumor-associated carbohydrate antigens (TACA) in its tumor-associated form.8 Therefore, tandem repeat MUC1-glycopeptides with TACA side chains are of particular interest for antitumor vaccines.9
Although various mucin-type glycopeptides decorated with different TACA side chains have been successfully investigated as molecularly defined vaccine prototypes for triggering strong humoral immunity over the last few years,10 their targeting is often constricted by a limited metabolic stability, a weak immunogenicity and a T-cell-independent immune reaction.11 For that reason various strategies for vaccine design have focused on the enhancement of the immune response by the conjugation of TACAs and tumor-associated MUC1-glycopeptide fragments to immunogenic carriers like keyhole limpet hemocyanin, tetanus toxoid or BSA.12 For example, synthetic vaccines composed of tumor-associated MUC1 sequences, T-cell epitopes and Toll-like receptor 2 (TLR2) ligands elicited a strong immune response.13 Also, highly fluorinated MUC1-glycopeptide antigens have been developed.14 Another promising approach to enhance antigen density and therefore to overcome the mentioned obstacles is a multivalent antigen presentation by different templates such as immunogenic peptides,15 dendrimers,16 liposomes17 or bioactive nanoparticles.18 Although still at the beginning of its development, the first successful results indicate the great potential of MUC1-functionalized nanoparticles. The concept of multivalency describes the chemical interaction of multiple ligands of a biological unit with multiple receptors of another.19 The resulting bonds are, in many cases, significantly stronger than the mere multiplication of the individual amounts. In previous work we could indicate that the multivalent presentation of potential binding epitopes on the surface of nanoparticles can lead to higher binding affinities20 and so it is interesting to investigate their possible use in anti-cancer vaccines presentation.21 Recently, for example, the synthesis of nanosized polymer-based glycopeptide vaccines, which induce significant immune reactions in mice, was described.22 Due to their lack of immunogenicity, their remarkably high surface-to-volume ratio in combination with their high stability, and low toxicity23 gold nanoparticles are also well suited as templates for the multivalent presentation of antigens.
Herein, we present the synthesis and immobilization of novel MUC1-glycopeptide antigens by functionalized gold colloids and the detection of their selectively antigen–antibody binding by quartz crystal microbalance and a novel dot-blot immunoassay for the first time.
Results and discussion
Synthesis of the MUC1 glycopeptide antigen analogues
Following known strategies in the literature, the required building blocks such as the TN-antigen 5 and the spacer 10 for the synthesis of glycopeptides were prepared individually in multi-step procedures and then assembled using solid-phase peptide synthesis.24The synthesis of the TN-antigen block 5 was carried out by a previously prepared N-acetylgalactosamine–threonine conjugate,25 which reacted with a glycosyl donor through a variation of the Koenigs–Knorr glycosylation (Scheme 1a).26 Then the N-acetylgalactosamine–threonine conjugate 4 was produced by the conversion of the azide function into an acetamido group through reductive acetylation.27 In the terminal step the C-terminal tert-butylester protecting group was removed to achieve the desired glycosyl-aminoacid building block 5. The spacer 10 is used to guarantee flexibility and distance between the nanoparticle and glycopeptide and because it can be easily coupled to the N-terminal amino acid of the peptide chain. Because of its primary amino function the spacer also allows another easy access to the coupling to the nanoparticles. The synthesis of the spacer 10 follows the method described by Keil et al.16 (shown in Scheme 1b) and is described in detail in the Experimental section. The MUC1-glycopeptide antigens were assembled in an automated synthesizer by solid-phase peptide synthesis (SPPS) Fmoc-protocol employing trityl-TentaGel resin preloaded with Fmoc-Pro-OH, as previously described24 (Scheme 1c). Low loaded resins were used to avoid interfering interactions of the growing peptide chain and all the functional groups of the amino acid side chains were protected by acid-labile protecting groups.
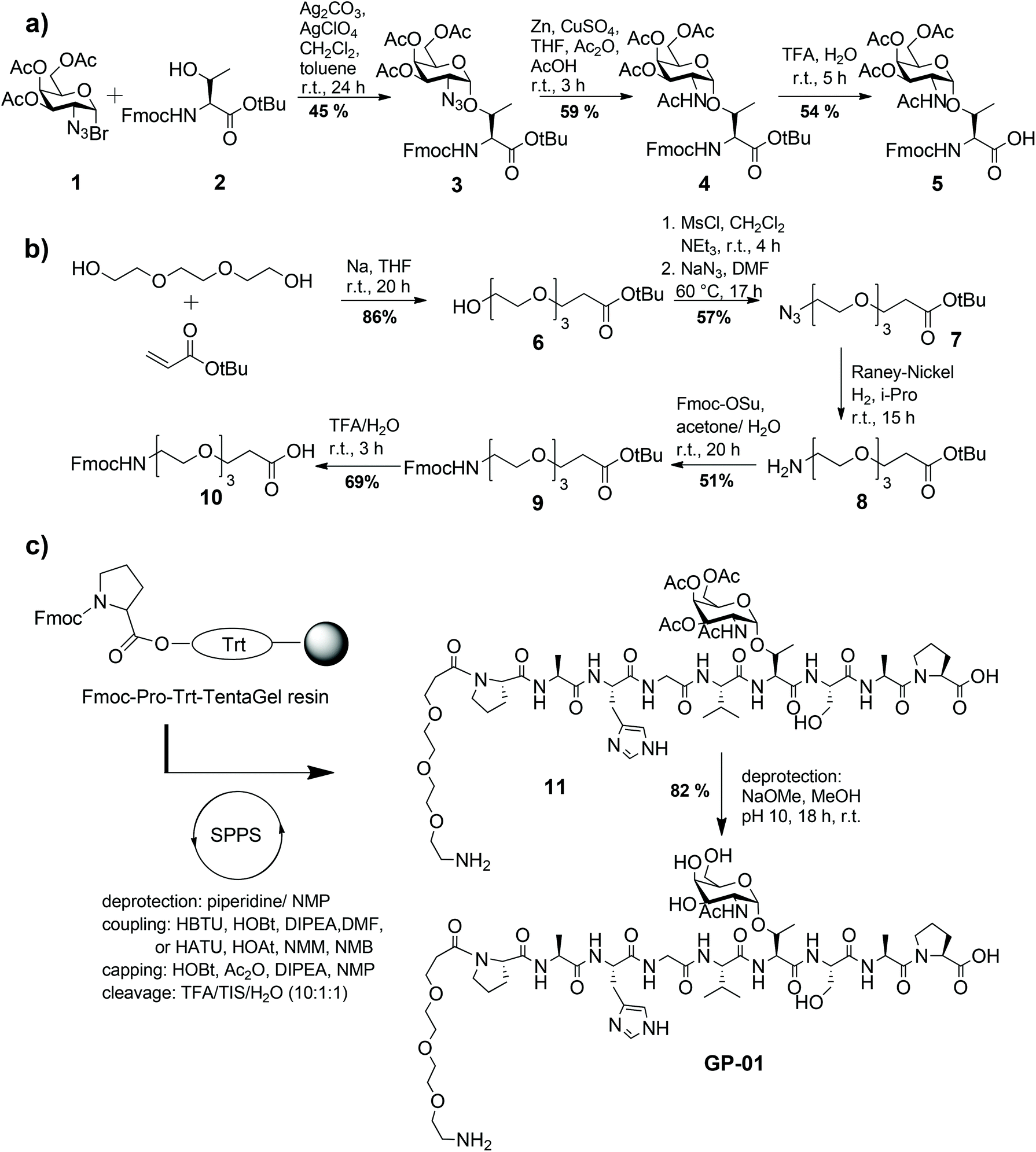 | ||
| Scheme 1 Synthesis routes of the TN-antigen (a) and the bi-functionalized triethylene glycol spacer (b). (c) Solid-phase peptide synthesis (SPPS) of the MUC1 like glycopeptides. Fmoc-Osu = N-(9H-Fluoren-9-ylmethoxycarbonyl)-succinimidylcarbonate NMP = N-methylpyrrolidone, HBTU = O-(1H-benzotriazole-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate, HOBt = N-hydroxy-benzotriazole, DIPEA = diisopropylethylamine, HATU = O-(7-aza-benzotriazole-1-yl)-N,N,N′,N′-tetramethyl-uronium hexafluoro-phosphate, HOAt = N-hydroxy-7-azabenzotriazole, NMM = N-methylmorpholine, TIS = triisopropylsilane. | ||
The coupling of the protected amino acids was performed with HBTU/HOBt and diisopropylethylamine (DIPEA) in DMF, while the coupling of the TN-antigen block 5 was performed using the more reactive HATU/HOAt mix and N-methyl-morpholine (NMM) in N-methyl-pyrrolidine (NMP). The last step of the solid-phase synthesis was the attachment of the synthesized triethylene glycol spacer 10, followed by the release of the glycopeptide 11 from the resin with simultaneous cleavage of the side chain amino acid protecting groups using an acidic mix of TFA–triisopropylsilane (TIS)–H2O (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1). Finally the glycopeptides were deacetylated with NaOMe in MeOH at pH 10 under Zemplén conditions.
:1:1). Finally the glycopeptides were deacetylated with NaOMe in MeOH at pH 10 under Zemplén conditions.
According to the described synthesis a short glycosylated sequence consisting of nine amino acids and the spacer was prepared initially GP-01. This compound was used to test the immobilization on the particle surface and the resulting multivalent effect. Finally, a single glycosylated MUC1 partial structure with a full, 20 amino acids containing TR domain and the spacer was made GP-02 and used for binding studies of the functionalized Au-NPs (Fig. 1).
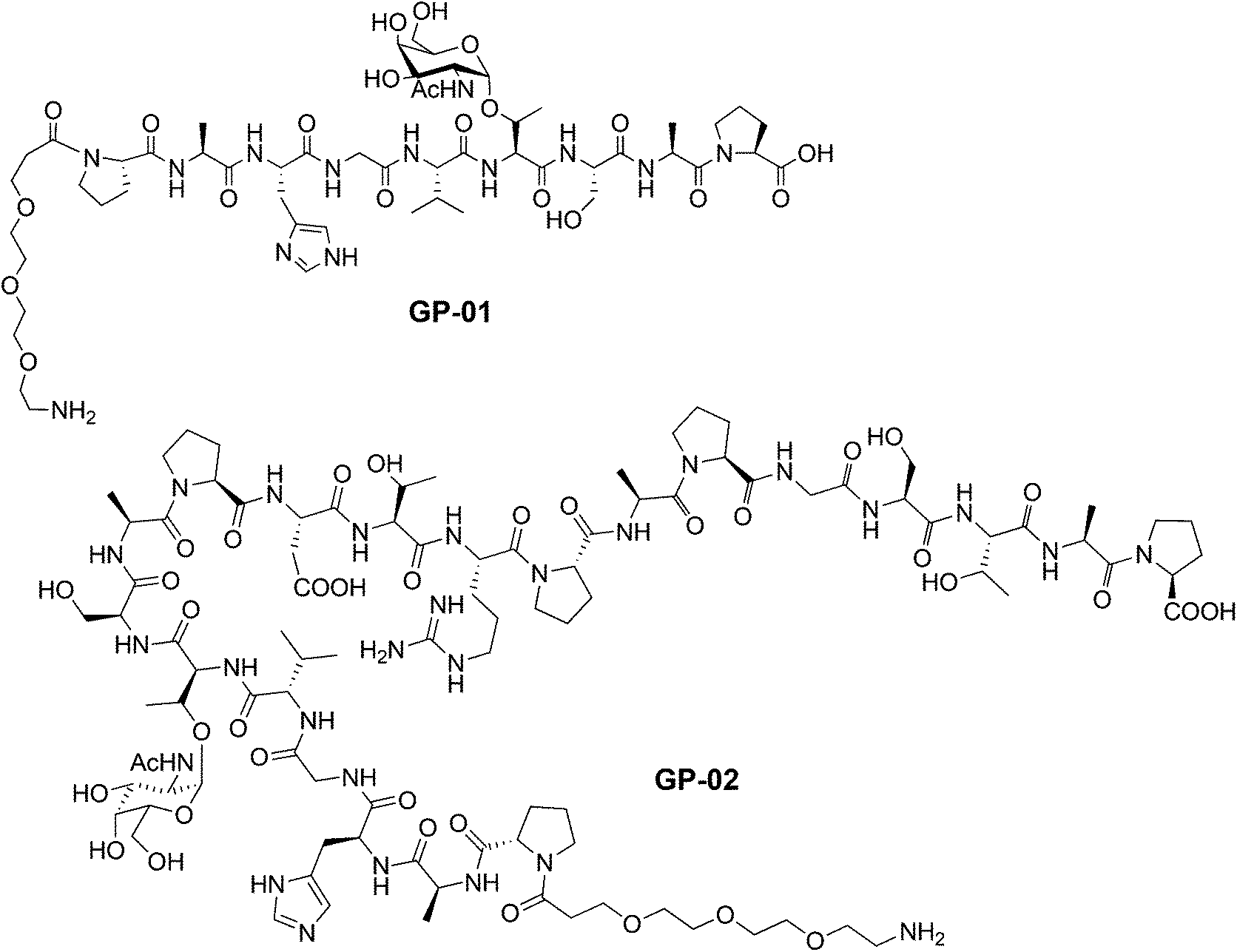 | ||
| Fig. 1 Glycopeptides GP-01 and GP-02 with a PEG-spacer and a TN antigen synthesized by SPPS. | ||
Preparation of surface-modified gold nanoparticles and the immobilization of the glycopeptides by peptide coupling reaction
For a multivalent presentation of the prepared potential MUC1-glycopeptide antigen analogues GP-01 and GP-02 the synthesis of nearly monodispersed nanoparticles is necessary. In this work we focused on gold nanoparticles, due to their mentioned unique properties.23 The synthesis of gold nanoparticles is known in the literature28 and based on the reduction of a gold salt in the presence of a stabilizer. By variation of the reaction conditions functionalized gold nanoparticles with a diameter less than 10 nm and a narrow size distribution were obtained. Although with a multidentate thiolinker, like for example the bidentate lipoic acid, functionalized gold nanoparticles are nearly resistant against ligand exchange reactions and have a considerably higher stability in water,29 we decided to bind the MUC1-glycopeptide antigen analogues covalently to the particle surface by a monodentate thiolinker based on 11-mercaptoundecanoic acid, which was used in our group previously.20 Due to its limited required space on the particle surface the density of ligands should be higher.30 The synthesis of N-hydroxysuccinimide-11-mercaptoundecanoate (MUDHSE) coordinated gold nanoparticles 14 with a diameter of 7 nm was carried out by a two-step route described in Scheme 2. In the first step dodecanethiol (DT) coordinated gold nanoparticles 12 were prepared by a one-pot synthesis, which was established by Stucky et al.28 In the second step the dodecanethiol ligand was exchanged for the previously prepared N-hydroxysuccinimide-11-mercaptoundecanoate2413 by a one step ligand exchange reaction in absolute DMF. To present the MUC1-glycopeptide antigen analogues multivalently, they were coupled to the prepared MUDHSE functionalized nanoparticles. Although the water solubility of the particles was a first hint of the successful immobilization, the characterization of the glycopeptide-functionalized nanoparticles was carried out by various methods. TEM images, UV/Vis spectra and dynamic light scattering (DLS) measurements were applied to determine the average diameter, shape and the size distribution of the nanoparticles. The TEM image shown in Fig. 2 indicates spherical particles with a narrow size distribution and an average diameter of 7 ± 1 nm. Also the UV/Vis spectra, which show only one maximum at 523 nm (Fig. 3), and the DLS measurements confirmed the total size including the shell of the particles with its adequate hydrodynamic diameter. In addition, the potential influence of the ligand exchange on the agglomeration state of the nanoparticles could be checked by these characterisation methods. There is no evidence that the ligand exchange reaction influences the agglomeration state. The successful change of the ligand shell was controlled directly by NMR spectroscopy. For this, the characteristic signals of the NMR spectra of the free glycopeptides were compared with those of the glycopeptide-coordinated gold nanoparticles. On the basis of 2D DQF-COSY and HSQC spectra, the newly formed amide bond could be clearly identified (Fig. 4 and 5). A differentiation between immobilized and free ligands is also possible through typical effects in nanoparticle NMR spectra. These effects are the significant line broadening and downfield shifts of the resonances of the immobilized ligands.31 Unbound impurities and ligands retain their sharp resonances and can be distinguished from the bound ones. Moreover, IR spectra were used to prove the immobilization by identifying the amide bond.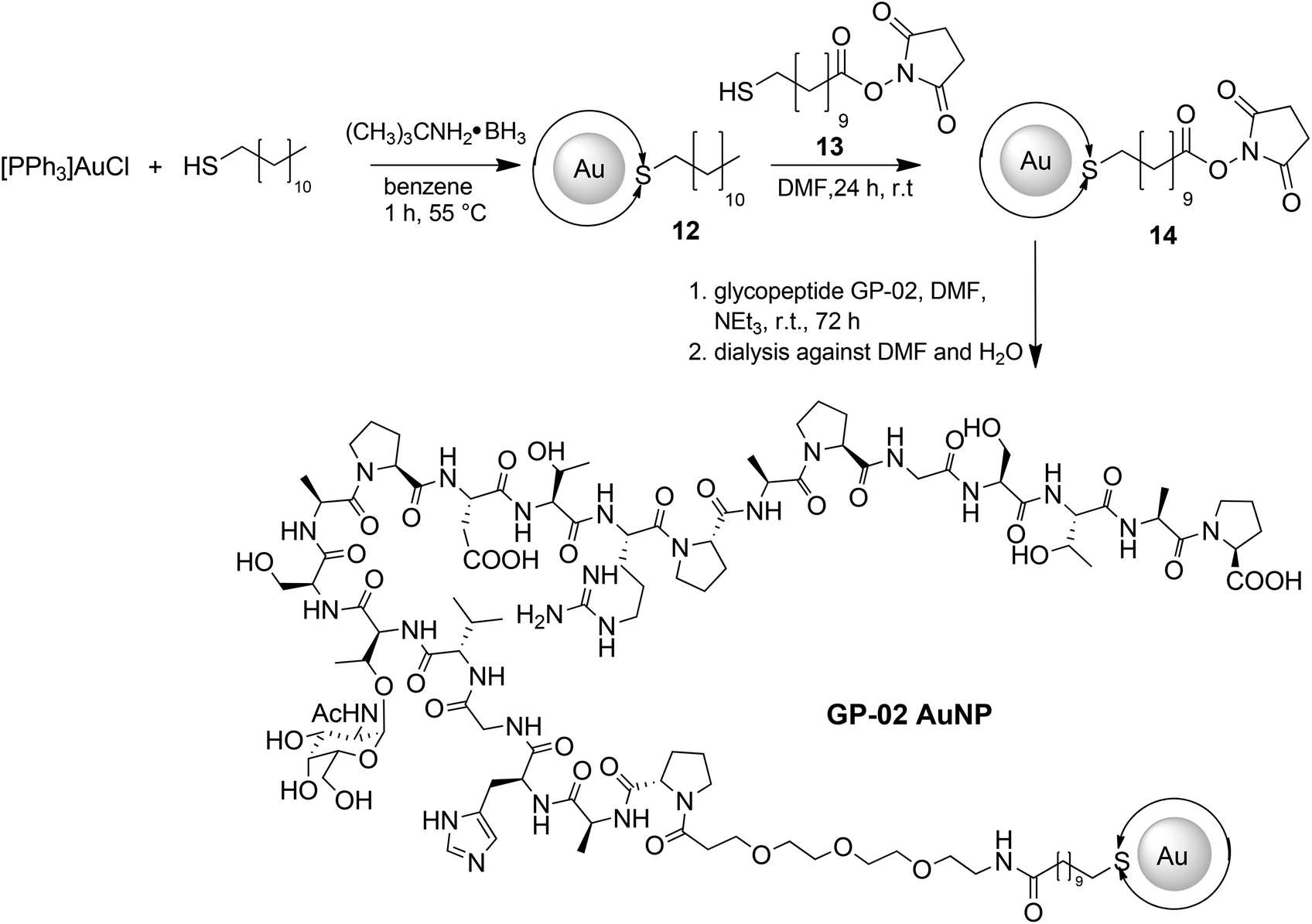 | ||
| Scheme 2 Synthetic route for the preparation of nearly monodisperse gold nanoparticles with terminally functionalized thiol shell and the immobilization of the glycopeptide GP-02via postsynthetic shell modifications. Double arrows at the Au core represent the ligand shell completely surrounding the gold nanoparticles. | ||
 | ||
| Fig. 2 TEM images of the synthesized gold nanoparticles with an average diameter of 7 nm. (a) DT coord. AuNP; (b) MUDHSE coord. AuNP; (c) GP-01 coord. AuNP; (d) GP-02 coord. AuNP. | ||
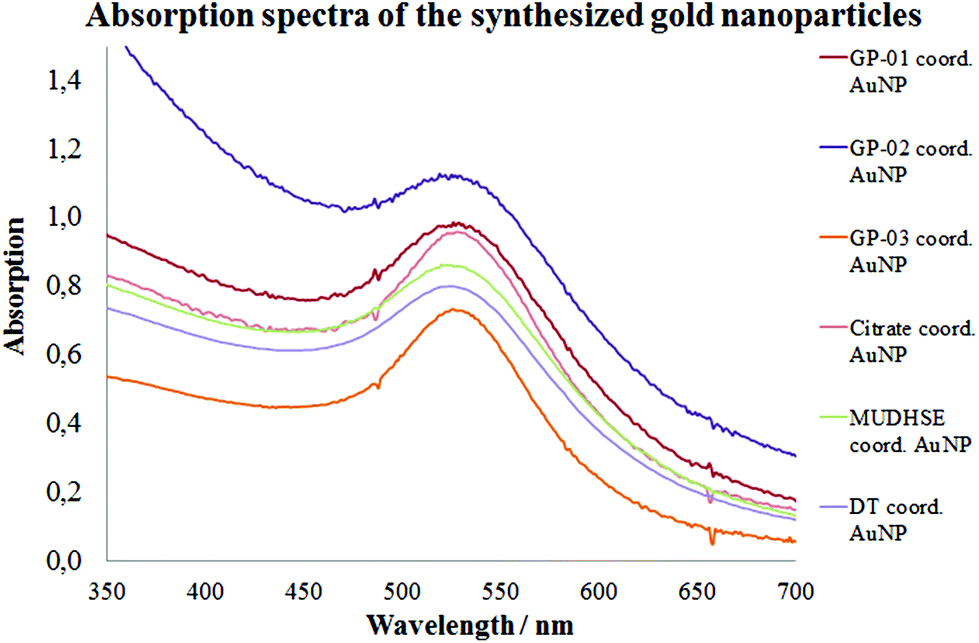 | ||
| Fig. 3 UV/Vis spectra of the synthesized nanoparticles. | ||
 | ||
| Fig. 4 HSQC-spectra of the GP-01 coordinated AuNP; red = coordinated ligand, black = free glycopeptides. | ||
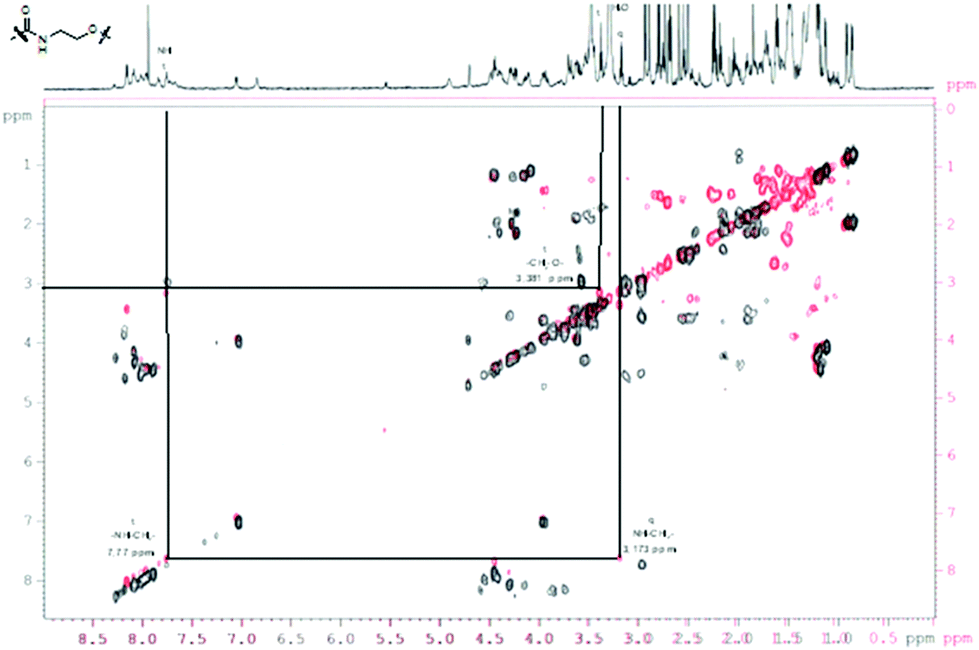 | ||
| Fig. 5 COSY-DQF-spectrum of the GP-01 coordinated AuNP; red = coordinated ligand, black = free ligand. | ||
Due to the fact that repulsive interactions of ligand molecules at less curved surfaces are weaker, a higher density of ligand molecules and thus a higher multivalent presentation could be possible on nanoparticles with a larger diameter.23,32 To investigate this potential effect of the nanoparticle size on chemical and biological properties like stability, antigen presentation and bioavailability, we also synthesized and used gold nanoparticles with a larger diameter.
The large gold nanoparticles with a diameter of 14 nm were prepared by the well-known citrate method, which was first mentioned by Turkevich et al.28b The reaction is based on the reduction of the tetrachloroauric acid (HAuCl4) with sodium citrate in water. Depending on the amount of citrate, it is possible to generate nanoparticles with a modest monodispersity and a size of around 10–20 nm. The citrate ligands of the large gold nanoparticles are only bound weakly via van der Waals interactions to the particle surface and so their replacement by water-soluble thiol-functionalized ligands is relatively facile. A ligand exchange with the N-hydroxysuccinimide-11-mercaptoundecanoate 13 is not realizable because of the insolubility of the active ester in water. For this reason, the coupling between the glycopeptides and the thiolinkers was carried out in the final step of the solid phase peptide synthesis. To avoid side reactions in the synthesis, the reactive thiol group had to be protected, which was done by a trityl protecting group which could be removed like the other acid-labile side chain protecting groups during the cleavage from the resin. So, the linker 15 was prepared by the reaction of 11-mercaptoundecanoic acid with trityl chloride (Scheme 3a). After the solid phase peptide synthesis and the cleavage of the protecting groups the glycopeptide GP-03 was immobilized by a ligand exchange reaction in water (Scheme 3b), followed by dialysis against water. The high affinity of the thiol linker to the gold nanoparticles and the resulting covalent bond lead to a full replacement of the citrate ligand.
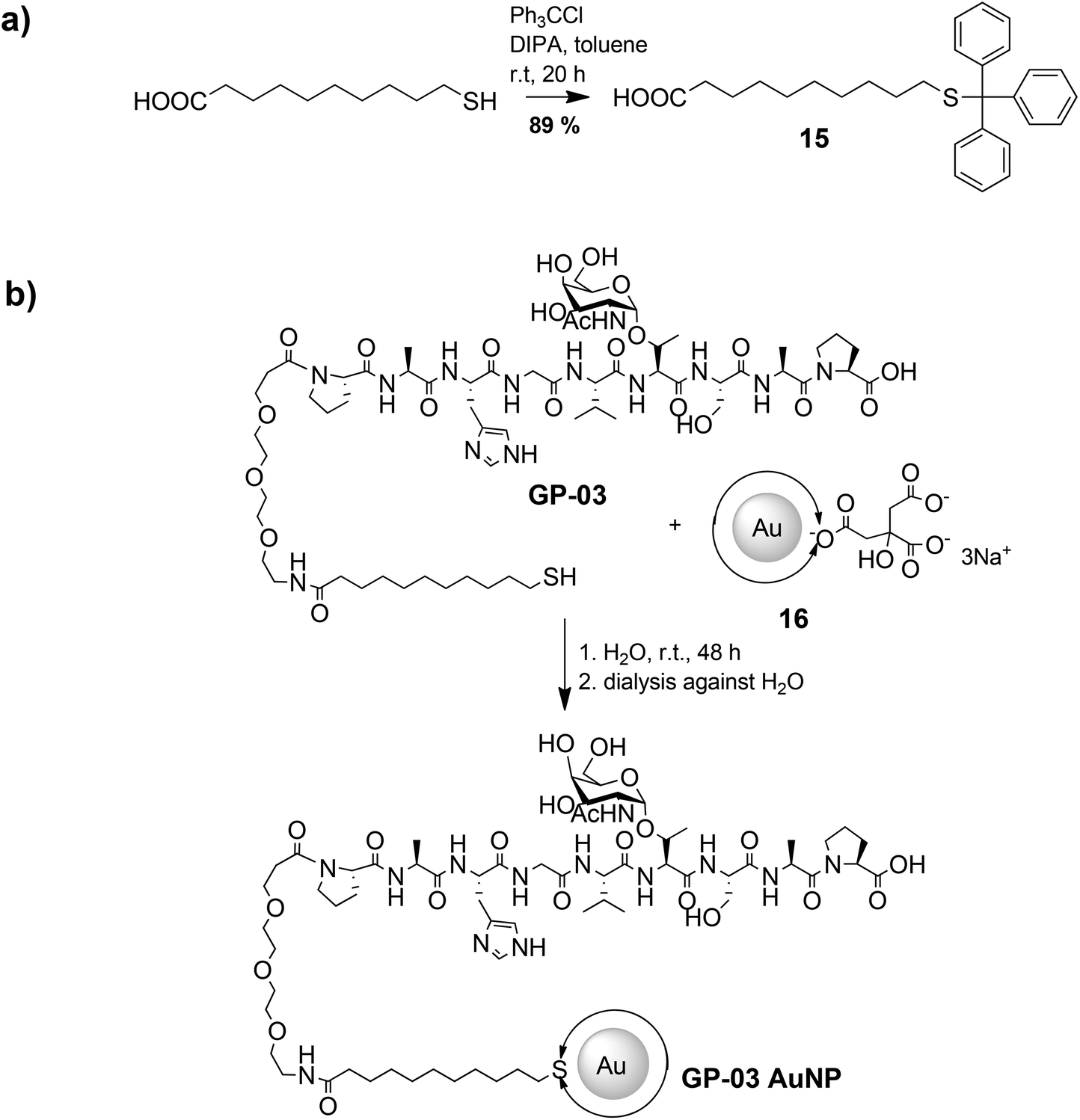 | ||
| Scheme 3 Synthetic procedure of the immobilization of glycopeptides on the surface of large gold nanoparticles. | ||
The TEM images shown in Fig. 6 indicate that the received citrate coordinated gold nanoparticles have mostly a spherical shape, and only a small percentage of triangular nanoparticles deviate from it. The average diameter d = 14 ± 1 nm of the citrate coordinated gold nanoparticles was determined by TEM images and UV/Vis spectra. The hydrodynamic diameter received by DLS measurements is dh = 18 ± 4 nm. The immobilization of the glycopeptides was checked by NMR and IR spectra. The aggregation state of the particles and the size and shape of the coordinated gold nanoparticles were also investigated like before by TEM, DLS measurements and absorption spectra. The obtained data indicated that the average diameter of the particle core remained 14 ± 1 nm and the maxima of the absorption spectra (Fig. 3) did not change (λmax = 531 nm) either. This result confirms that the ligand exchange reaction had no effect on the particle and the agglomeration state.
 | ||
| Fig. 6 TEM images of the synthesized large gold nanoparticles; (a + b) citrate coordinated gold nanoparticles with an average diameter of 14 nm; (c + d) TEM images of GP-03 coordinated gold nanoparticles. | ||
Binding and immune tests of the glycopeptides coordinated gold nanoparticles
In order to investigate the biological activity of the glycopeptide nanoparticle conjugates, the GP-02 coordinated AuNPs were tested in quartz crystal microbalance (QCM) experiments for their recognition by specific serum antibodies.33 For that reason the commercially available anti-MUC1 antibody [SM3],34 was used for detection of the antigen–antibody-binding. The QCM experiments were performed using commercially available quartz crystals coated with 100 nm gold layers. The binding of the SM3 antibody to the gold layer occurs through peripheral methionine binding sites, like Met18, Met34, Met82 and Met13535 and because of this the methionine binding sites of the antibody do not interfere with the antigen binding site. During the QCM measurement the SM3 antibody was immobilized on the crystal, SM3 injection was stopped after no further increase in weight (Δf = const.) was achieved. After several washing steps with PBS buffer (Fig. 7), the GP-02-AuNP was added and washed again with PBS buffer.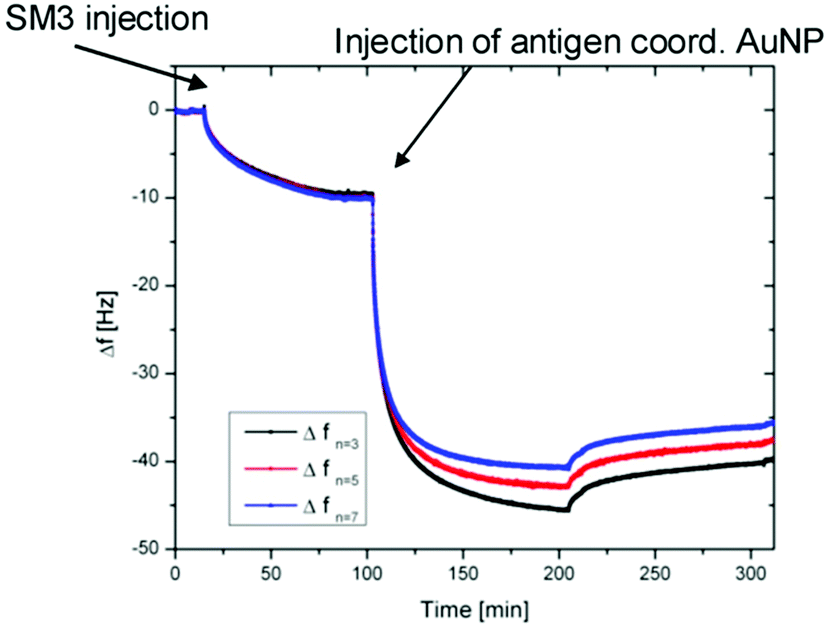 | ||
| Fig. 7 Results of QCM measurements: QCM frequency shift vs. time for the binding of the GP-02 coordinated AuNP to the preadsorbed SM3 antibody. | ||
After addition of the potential antigenic GP-02 coordinated AuNP, a typical Langmuir adsorption isotherm was obtained. Due to the fact that the nanoparticle addition leads to a significant decrease of the Δf value, a binding of the GP-AuNP antigen colloids to the previously immobilized SM3 antibodies is indicated. At a certain point no further increase in weight can be recognized; this is the point where all available antibodies are bound to the antigen coated nanoparticles.
The obtained result of the QCM experiment only indicates a binding of the GP-AuNPs to the SM3 antibody. To obtain information about the nature of the binding, the undiluted GP-01 (c = 0.13 μM) and GP-02 (0.32 μM) coordinated gold nanoparticle conjugates were tested in a simple and fast dot-blot immunoassay experiment. An undiluted citrate coordinated AuNP solution (c = 2.6 nM) was used as a negative control sample to preclude unspecific bindings of the gold particles. Also a serial dilution of GP-02 coordinated AuNP was blotted to find the qualitative detection limits of the dot-blot immunoassay experiment.
The principle of the developed dot-blot immunoassay is shown in Fig. 8. Firstly the undiluted GP-01 and GP-02 coordinated AuNP, the serial dilution of GP-02 coordinated AuNP and the negative control (AuCitrate) were immobilized onto the nitrocellulose membrane and incubated overnight (Fig. 9a). The colored dots of the undiluted gold nanoparticles were still visible after several washing steps with PBS, which indicates that the immobilization of the gold nanoparticles onto the nitrocellulose membrane was still intact. In the second step the membrane was blocked by milk powder in PBS buffer for 2 h at room temperature to block the remaining sites of the nitrocellulose membrane, and then the primary antibody (monoclonal anti-MUC1 antibody) was added and attached to the antigen. In the next step the enzyme-labeled secondary antibody was added and incubated for 1 h at room temperature, before the substrate solution was finally added.
 | ||
| Fig. 8 Schematic process of the dot-blot immunoassay. | ||
 | ||
| Fig. 9 Qualitative results of the dot-blot immunoassay (a) dot-blot immunoassay before the staining with the color reaction; (b) comparison of the binding affinities of GP-02, citrate and GP-01 coord., AuNP after the staining reaction; (c) comparison of diluted and undiluted GP-02 coord., AuNP after the staining reaction. | ||
After adding the substrate solution, the horseradish peroxidase conjugated to the second antibody oxidized 3,3′,5,5′-tetramethylbenzidine (TMB) in the presence of H2O2 into a radical cation that forms a charged transfer complex with the unoxidized TMB. This blue complex led to dark stains in the spots containing the GP-02 coordinated AuNP (Fig. 9b). These dark spots, visible to the naked eye, developed within minutes and indicated an antigen–antibody binding. The absence of color or change in color in the reaction of the negative control and the GP-01 experiment revealed that this antigen–antibody binding is selective and specific for the binding of the glycosylated MUC1 partial structures with the full 20 amino acids containing the TR domain.
A qualitative detection of the antigen–antibody binding through a pale dot was still possible for the 1:50 dilution of GP-02 coordinated AuNP (Fig. 9c). As the concentration of the serial dilution increases, the color of the blotted dots changes from pale to strong. Therefore, we can speculate that this method is sensitive even at low concentrations of the glycopeptide nanoparticle conjugates.
Conclusion
In this work we have successfully synthesized three different MUC1-glycopeptides using solid-phase peptide synthesis to assemble the individual components, such as the spacer or the TN-antigen. These MUC1-glycopeptides were combined into potential vaccine candidates with gold colloids of different sizes in different synthetic ways, in order to override the natural tolerance of the immune system by multivalent presentation. The MUC1-glycopeptide immobilization on the gold colloid surface was characterized by TEM, UV/Vis spectra, DLS measurements, IR- and NMR-spectroscopy, which all indicated a high stability and solubility in water of the particles.In addition, a novel dot-blot immunoassay was developed to analyze the nature of the antigen–antibody-binding of the MUC1-glycopeptide coord. gold colloids. Due to the strong affinity between the MUC1-glycopeptide and the SM3 antibody, blue dots can be visible to the naked eye without any instrument. The experimental results demonstrate that this new assay indicates a high selectivity, specificity and sensitivity for MUC1-glycopeptides containing the TR domain. These promising results suggest that gold colloids are robust and interesting platforms for presenting MUC1-glycopeptide antigens multivalently to their corresponding antibodies, which may lead to new potential developments in the construction of efficient immunotherapeutics, for example, antitumor vaccines.
Experimental section
Materials
All reactions of nanoparticles in aqueous solution were conducted in purified Millipore water. Organic solvents were distilled before use with the colloids. All other chemicals were purchased from commercial sources and used as received. The ligand exchange and coupling reactions were prepared under argon conditions. For peptide synthesis, all preloaded resins were obtained from Rapp Polymers GmbH (Tuebingen, Germany), the protected Fmoc-aminoacids were obtained from Orpengen Pharma (Heidelberg, Germany) and peptide grade solvents were purchased from Iris Biotech (Marktredwitz, Germany).Instrumentation
The glycopeptides were synthesized on an automated ABI 433A peptide synthesizer (Applied Biosystems, Carlsbad, USA), which was equipped with an external 200 UV/Vis detector (Perkin Elmer) to control the synthesis process. Purification of the peptides was realized by the semi-preparative HPLC system (Jasco, Gross-Umstadt, Germany) consisting of two HPLC-pumps (type:PU-2087Plus), a degasser and a dynamic mixer. Detection was applied with a Jasco UV/Vis-detector at 214 nm. A semi-preparative column purchased from Phenomenex (Torrance, USA) with the dimension 250 nm × 30 mm filled with Luna RP-C18(2), 100 A, Axia, 10 μm was used. All runs were performed at flow rates of 20.00 mL min−1. As an eluent a mixture of acetonitrile and Millipore water both acidified with 0.1% TFA was used. Two different water–acetonitrile gradients were used: -gradient A: 95:5 at 0 min, 60:40 at 30 min, 0:100 at 40 min-gradient B: 75:25 at 0 min, 80:20 at 30 min, 0:100 at 40 min. All NMR spectra were recorded with a Bruker WT-300, a Bruker AM-400 and a Bruker Avance III 600 MHz spectrometer. Chemical shifts (δ) are given in ppm relative to TMS. Samples were prepared in deuterated solvents and their signals referenced to residual non-deuterated solvent signals. Because of the rigidity of the alkyl chains in the thiol shells and the resulting large differences in the relaxation times of different protons, no integrals are given for the proton NMR data of the gold colloids.
IR spectra were obtained on a Bruker IFS48 spectrometer in ATR mode. ESI-MS analyses were performed using a navigator instrument from Thermoelectronics with a sample concentration of 0.1 mg mL−1, 0.75 mL min−1 flow rate, cone voltage 70 V, 45 V or 35 V and nitrogen flow rate 300 L min−1. TEM measurements were performed on a Philips CM30 STEM (300 kV, LaB6-cathode) equipped with a GATAN digital camera. TEM images were recorded using a digital micrograph. The average particle core sizes were determined by measuring at least 100 individual particles from recorded TEM images. The UV/Vis spectra were plotted with an Agilent 8453 spectrophotometer (Agilent Technologies Inc., Santa Clara, CA, USA). Dynamic light scattering measurements were performed with a StabiSizer PMX 200C from Particlemetrix (Meerbusch, Germany).
000-fold in a wash buffer containing 0.5% BSA, was added and incubated for 1 h. For the substrate solution 1 mL of a TMB stock solution (1.2 mg 3,3′,5,5′-tetramethylbenzidine per mL of dimethyl sulfoxide) is filled up to 10 mL with sodium acetate buffer (18 mM, pH 4.5) and 0.05% (0.017 mL of a 30% solution) H2O2 was added. The membrane was covered with the substrate solution and incubated in the dark for 15 min.
:1); yield: 20.42 g (52.00 mmol, 81%); Rf = 0.58 (cHex–EtOAc, 1:1). C12H16BrN3O7 (M = 394.18 g mol−1) [393.02].
1H-NMR (300 MHz, CDCl3): δ (ppm) = 6.47 (d, 1H, JH1,H2 = 3.9 Hz, 1-H), 5.50–4.48 (m, 1H, 4-H), 5.33 (dd, 1H, JH3,H4 = 3.6 Hz, JH3,H2 = 10.8 Hz, 3-H), 4.50–4.45 (m, 1H, 5-H), 4.20–4.06 (m, 2H, 6a,b-H), 3.98 (dd, 1H, JH2,H1 = 3.8 Hz, JH2,H3 = 10.7 Hz, 2-H), 2.15, 2.06, 2.05 (3 × s, 9H, 3 × CH3 (Ac)).
:1 → 2:1); yield: 16.38 g (41.00 mmol, 56%); Rf = 0.61 (cHex–EtOAc, 2:1). C23H27NO5 (M = 397.46 g mol−1) [397.19].
1H-NMR (300 MHz, CDCl3): δ (ppm) = 7.77 (d, 2H, JH4,H3 = JH5,H6 = 7.2 Hz, 4-H-, 5-H-Fmoc), 7.63–7.60 (m, 2H, 1-H-, 8-H-Fmoc), 7.40 (t, 2H, JH3,H2/H4 = JH6,H5/H7 = 7.4 Hz, 3-H-, 6-HFmoc),7.34–7.28 (m, 2H, 2-H-, 7-H-Fmoc), 5.59 (d, 1H, JNH,Tα = 8.7 Hz, NH), 4.41 (d, 2H, JCH,CH2 = 7.2 Hz, CH2 (Fmoc)), 4.33 (m, 1H, 9-H-Fmoc), 4.31–4.22 (m, 2H, Tα, Tβ), 2.08 (sb,1H, OH), 1.43 (s, 9H, CH3 (tBu)), 1.24 (d, 3H, JTγ,Tβ = 6.3 Hz, Tγ).
:1) was stirred for 1 h with activated powdered molecular sieves (4 Å) under an argon atmosphere and exclusion of light. Then the mixture was cooled to 0 °C and 7.74 g (28.07 mmol) Ag2CO3 and 1.22 g (5.43 mmol) AgClO4 in 8 mL of toluene were added. The mixture was stirred for 30 min at 0 °C before 10.00 g (0.025 mol) αAc3N3Gal-Br 1 in toluene (35 mL) and dichloromethane (35 mL) was added dropwise to the mixture over 90 min. After stirring overnight at room temperature, the mixture was diluted with 100 mL dichloromethane and filtered through Hyflo®. Then the filtrate was extracted twice with 150 mL of a saturated sodium hydrogen carbonate solution, followed by washing twice with 200 mL of brine solution. The aqueous layer was separated from the organic layer, which was dried with magnesium sulfate and filtered. The solvent was removed in vacuo and the crude product was purified by flash chromatography with silica (CH2Cl2–EtOAc, 10:1). Yield: 7.92 g (11.00 mmol, 45%), Rf = 0.74 (CH2Cl2–EtOAc, 10:1).
C35H42N4O12 (M = 710.73 g mol−1) [710.28].
1H-NMR (300 MHz, CDCl3): δ (ppm) = 7.75 (d, 2H, JH4,H3 = JH5,H6 = 7.5 Hz, 4-H-, 5-H-Fmoc), 7.64 (d, 2H, JH1,H2 = JH7,H8 = 7.3 Hz, 1-H-, 8-H-Fmoc), 7.40 (t, 2H, JH3,H2/H4 = JH6,H5/H7 = 7.44 Hz, 3-H-, 6-H-Fmoc), 7.34–7.28 (m, 2H, 2-H-, 7-H-Fmoc), 5.66 (d, 1H, JNH,Tα = 9.3 Hz, NH (Fmoc)), 5.47 (d, 1H, JH4,H3 = 2.7 Hz, 4-H), 5.35 (dd, 1H, JH3,H4 = 3.0 Hz, JH3,H2 = 11.2 Hz, 3-H), 5.11 (d, 1H, JH1,H2 = 3.7 Hz, 1-H), 4.46–4.43 (m, 1H, Tβ), 4.40–4.36 (m, 2H, 6a,b-H), 4.32–4.25 (m, 3H, Tα, 5-H, 9-H-Fmoc), 4.10 (d, 1H, JCH2,CH = 6.3 Hz, CH2 (Fmoc)), 3.64 (dd,1H, JH2,H1 = 3.4 Hz, JH2,H3 = 11.0 Hz, 2-H), 2.15, 2.08, 2.04 (3 × s, 9H, 3 × CH3 (Ac)), 1.51 (s, 9H, CH3 (tBu)), 1.36 (d, 3H, JTγ,Tβ = 6.5 Hz, Tγ).
:2:1). The mixture was then stirred at room temperature for 3 h. After completion of the reaction, the mixture was diluted with 500 mL, filtered through Hyflo®, concentrated and purified by flash chromatography with silica (cHex–EtOAc, 1:2). Yield: 1.63 g (2.24 mmol, 59%); Rf = 0.24 (cHex–EtOAc, 1:2). C37H46N2O13 (M = 726.77 g mol−1) [726.30].
1H-NMR (300 MHz, CDCl3), δ (ppm) = 7.73 (d, 2H, JH4,H3 = JH5,H6 = 7.3 Hz, 4-H-, 5-H-Fmoc), 7.62 (d, 2H, JH1,H2 = JH7,H8 = 7.1 Hz, 1-H-, 8-H-Fmoc), 7.39 (t, 2H, JH3,H2/H4 = JH6,H5/H7 = 7.4 Hz, 3-H-, 6-H-Fmoc), 7.33–7.30 (m, 2H, 2-H-, 7-H-Fmoc), 5.99 (d, 1H, JNH,Tα = 9.9 Hz, NH (Fmoc)), 5.55 (d, 1H, JNH,H2 = 8.9 Hz, NH (GalNAc)), 5.38 (d, 1H, JH4,H3 = 2.4 Hz, 4-H), 5.07 (dd, 1H, JH3,H4 = 2.4 Hz, JH3,H2 = 10.7 Hz, 3-H), 4.86 (d, 1H, JH1,H2 = 2.7 Hz, 1-H), 4.62–4.57 (m, 1H, 2-H), 4.46–4.35 (m, 2H, CH2 (Fmoc)), 4.27–4.04 (m, 6H, 9-H-Fmoc, 5-H, 6a,b-H, Tα, Tβ), 2.14 (s, 3H, CH3 (Ac)), 2.02 (s, 3H, CH3 (AcNH)), 1.98 (2 × s, 6H, 2 × CH3 (Ac)), 1.44 (s,9H, CH3 (tBu)), 1.30 (d, 3H, JTγ,Tβ = 6.0 Hz, Tγ).
:5:1) to yield compound 5 (618 mg, 0.92 mmol, 54%); Rf = 0.10 (CH2Cl2–MeOH–AcOH, 95:5:1).
C33H38N2O13 (M = 670.66 g mol−1) [670.24].
1H-NMR (300 MHz, CDCl3), δ (ppm) = 7.76 (d, 2H, JH4,H3 = JH5,H6 = 7.2 Hz, 4-H-, 5-H-Fmoc), 7.62 (d, 2H, JH1,H2 = JH7,H8 = 7.3 Hz, 1-H-, 8-H-Fmoc), 7.39 (t, 2H, JH3,H2/H4 = JH6,H5/H7 = 7.2 Hz, 3-H-, 6-H-Fmoc), 7.33–7.30 (m, 2H, 2-H-, 7-H-Fmoc), 6.05 (d, 1H, JNH,Tα = 8.9 Hz, NH(Fmoc)), 5.91 (d, 1H, JNH,H2 = 9.3 Hz, NH (GalNAc)), 5.38 (d, 1H, JH4,H3 = 2.9 Hz, 4-H), 5.14 (dd, 1H, JH3,H4 = 2.7 Hz, JH3,H2 = 11.2 Hz, 3-H), 4.98 (d, 1H, JH1,H2 = 3.3 Hz, 1-H), 4.66–4.58 (m, 1H, 2-H), 4.52–4.38 (m, 2H, CH2 (Fmoc)), 4.29–4.04 (m, 6H, 9-H-Fmoc, 5-H, 6a,b-H, Tα,Tβ), 2.17 (s, 3H, CH3 (Ac)), 2.04 (s, 3H, CH3 (AcNH)), 1.98 (2× s, 6H, 2× CH3 (Ac)), 1.29 (d, 3H, JTγ,Tβ = 6.2 Hz, Tγ).
1H-NMR (300 MHz, CDCl3), δ (ppm) = 3.72–3.69 (m, 4H, 3-CH2, 14-CH2), 3.67–3.58 (m, 10H, 5 × OCH2), 2.50 (t, 2H, JCH2, CH2 = 6.7 Hz, 2-CH2), 1.43 (s, 9H, tBu).
:1) to yield 9.63 g (31.00 mmol, 57%) of a yellow oil. Rf = 0.22 (cHex–EtOAc, 3:1).
C13H25N3O5 (M = 303.35 g mol−1) [303.18].
1H-NMR (300 MHz, CDCl3): δ (ppm) = 3.73–3.68 (m, 4H, 3-CH2, 14-CH2), 3.67–3.58 (m, 10H, 5 × OCH2), 2.49 (t, 2H, JCH2,CH2 = 6.9 Hz, 2-CH2), 1.44 (s, 9H, tBu).
:1).
C13H27NO5 (M = 277.36 g mol−1) [277.19].
:1) and then 2.53 g (30.80 mmol) sodium hydrogen carbonate was added. After adding 10.30 g (30.60 mmol) Fmoc-Os slowly to the reaction mixture, it was then vigorously stirred at room temperature for 90 h. HCl was added until pH 6 was reached, before acetone was evaporated. After phase separation, the aqueous layer was extracted four times with 100 mL of dichloromethane. The combined organic layers were washed with 250 mL of 1 M HCl, 250 mL of H2O and dried with magnesium sulfate. The solvent was removed in vacuo and the crude product was purified by flash chromatography with silica (cHex–EtOAc, 2:1 → 1:1 → 1:2). Yield: 7.62 g (15.20 mmol, 51%); Rf = 0.22 (cHex–EtOAc, 2:1).
C28H37NO7 (M = 499.60 g mol−1) [499.26].
1H-NMR (300 MHz, CDCl3): δ (ppm) = 7.76 (d, 2H, JH4,H3 = JH5,H6 = 7.9 Hz, 4-H-, 5-H-Fmoc), 7.60 (d, 2H, JH1,H2 = JH8,H7 = 7.4 Hz, 1-H-, 8-H-Fmoc), 7.42–7.14 (m, 4H, 2-H-, 3-H-, 6-H-, 7-H-Fmoc), 4.38 (d, 2H, JCH2,CH = 6.9 Hz, CH2-Fmoc), 4.20 (t, 1H, 9-H-Fmoc, JH9,CH2 = 6.8 Hz),3.76–3.49 (m, 12H, 6 × OCH2), 3.41 (t, 2H, JCH2,CH2 = 5.4 Hz, 12-CH2), 2.60 (t, 2H, JCH2,CH2 = 6.7 Hz, 2-CH2), 1.43 (s, 9H, CH3-tBu).
:1:0.5). Yield: 4.61 g (10.40 mmol, 69%); Rf = 0.32 (CH2Cl2–MeOH–EtOAc, 19:1:0.5). C24H29NO7 (M = 443.49 g mol−1) [443.19].
1H-NMR (300 MHz, CDCl3): δ (ppm) = 7.77 (d, 2H, JH4,H3 = JH5,H6 = 7.9 Hz, 4-H-, 5-H-Fmoc), 7.60 (d, 2H, JH1,H2 = JH8,H7 = 7.4 Hz, H-1-, H-8-Fmoc), 7.42–7.14 (m, 4H, 2-H-, 3-H-, 6-H-, 7-H-Fmoc), 6.40 (sb, 1H, NH-Fmoc), 4.39 (d, 2H, JCH2,CH = 6.7 Hz, CH2-Fmoc), 4.20 (t, 1H, 9-H-Fmoc, JH9,CH2 = 6.7 Hz), 3.71 (t, 2H, JCH2,CH2 = 5.8 Hz, 11-CH2), 3.68–3.49 (m, 10H, 5 × OCH2), 3.47–3.36 (m, 2H, 12-CH2), 2.60 (t, 2H, JCH2,CH2 = 6.7 Hz, 2-CH2).
Standard protocol of the glycopeptide synthesis
The glycopeptides were synthesized using a standard fluorenyl-methoxycarbonyl (Fmoc) chemistry protocol with 454.5 mg Fmoc-Pro-O-Trt Tentagel resin (Rapp Polymere, 0.22 mmol g−1). The standard coupling of an amino acid was performed in a three-part cycle of coupling. In the first step the Fmoc protecting group of the starting amino acid was cleaved with 20% solution of piperidine in NMP and then in the next step, 1.00 mmol (10 eq.) of the Fmoc-amino acid was activated with a solution of 1.00 mmol HBTU, 1.00 mmol HOBt and 2.00 mmol DIPEA in NMP. The coupling time was 20 min at room temperature. The third step, the capping, was carried out by acetic anhydride, while all free amino groups were acetylated. After the capping step, the cycle began with the removal of the terminal Fmoc protecting group again. After the last amino acid the Fmoc group was removed again and the resin was thoroughly washed with NMP and dichloromethane. Finally the glycopeptides were cleaved from the resin with 10 mL of trifluoroacetic acid, 1 mL of triisopropylsilane and 1 mL of water and the solution was washed three times with 3 mL of trifluoroacetic acid.The entire filtrate was treated with 20 mL of toluene and concentrated in vacuo by evaporating the solvent. The residue was co-distilled five-times with toluene and then dissolved in 10 mL of water and lyophilized.
:1:1) was added. The vessel was shaken for 3 h. The solution was filtered, and the resin was washed two times with 2 mL trifluoroacetic acid. Toluene (20 mL) was added to the combined solutions, the solvent was removed in vacuo, and the residue was co-distilled two times with toluene (20 mL) and lyophilized. The lyophilisate was directly used in the next step, without further purification.
Yield: 59 mg (0.043 mmol, 43%), colorless lyophilisate. Rt = 16.14 min (Phenomenex Luna, gradient see Instrumentation) C59H93N13O24 (M = 1368.44 g mol−1) [1367.65].
ESI-MS (positive), m/z: 1368.66 ([M + H]+, ber.: 1368.65), 684.84 ([M + 2H]2+, ber.: 684.83).
Yield: 44 mg (0.035 mmol, 82%) colorless lyophilisate; Rt = 10.34 min (Phenomenex Luna, gradient A see Instrumentation).
C53H87N13O21 (M = 1242.33 g mol−1) [1241.61].
ESI-MS (positive), m/z: 1242.61 ([M + H]+, ber.: 1242,62), 621.79 ([M + 2H]2+, ber.: 621.81).
1H-NMR (400 MHz, D2O, COSY, HSQC): δ (ppm) = 8.52 (d, 1H, JHε,Hδ = 1.25 Hz, Hε), 7.22 (, 1H, JHδ,Hε = 1.43 Hz, Hδ), 4.83 (d, 1H, JH1,H2 = 3.91 Hz, H1), 4.85–4.11 (m, 9H, Hα{4.60}, Tα{4.52}, A1α {4.39}, Sα{4.38}, P1α{4.31}, P2α{4.26}, Tβ{4.21}, Vα{4.20}, A2α{4.12}), 3.98 (dd, 1H, JH2, H1 = 3.95 Hz, JH2,H3 = 10.58 Hz, H2), 3.93–3.33 (m, 27H, H5 {3.89}, Gαa{3.85}, Gαb{3.84}, H4 {3.83}, H3 {3.74}, H6a,b{3.68}, 3-CH2-Spacer {3.67}, Sβ{3.65}, 5 × CH2O-Spacer {3.62}, P1δ{3.55}, P2δ{3.53}, 2-CH2-Spacer {3.45}), 3.20 (dd, 1H, JHβ,Hα = 5.64 Hz, Hβ), 3.08 (m, 2H, CH2-Spacer), 2.70–2.52 (m, 4H, 12-CH2-Spacer {2.65}, 2-CH2-Spacer {2.57}), 2.31–2.07 (m, 4H, P1β{2.22}, P2β{2.15}), 2.05–1.70 (m, 5H, Vβ{1.99}, P1γ{1.91}, P2γ{1.89}), 1.32–1.19 (m, 6H, A1β{1.28}, A2β{1.23}), 1.16 (d, 3H, JTγ,Hβ = 6.41 Hz, Tγ), 0.86 (t, 6 H, JVγ,Vβ = 6.34 Hz, Vγ).
13C-NMR (100.6 MHz, D2O, HSQC): δ (ppm) = 175.9, 174.9, 174.4, 173.7, 172.9, 172.4, 172.0, 171.5, 171.1, 170.9, 170.6 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CO–acetyl), 163.1 (CNH), 133.4 (Hε), 128.4 (Hγ), 117.2 (Hδ), 99.0 (C1), 76.8 (Tβ), 71.3 (C5), 69.6, 69.5, 69.5, 69.4 (CH2O-Spacer), 68.5 (C4), 68.2 (C3), 66.3 (S), 61.3 (11-CH2-Spacer), 60.0 (P2α), 59.5 (P1α), 59.4 (Vα), 57.0 (Tα), 54.9 (Sα), 52.3 (Hα), 49.6 (C2), 49.6 (A2α), 48.7, 48.0 (2-CH2-Spacer), 47.7 (P2δ), 47.6 (A1α), 47.5 (P1δ), 47.4 (C6), 42.3 (Gα), 39.0, 34.0 (12-CH2-Spacer), 30.1 (Vβ), 29.6 (P2β), 28.7 (P1β), 28.7 (P2γ), 28.6 (P1γ), 26.2 (Hβ), 22.2 (CH3-AcNH), 18.4 (Tγ), 18.24 (Vγb), 17.7 (Vγa), 16.3 (A2β), 15.0 (A1β).
O, CO–acetyl), 163.1 (CNH), 133.4 (Hε), 128.4 (Hγ), 117.2 (Hδ), 99.0 (C1), 76.8 (Tβ), 71.3 (C5), 69.6, 69.5, 69.5, 69.4 (CH2O-Spacer), 68.5 (C4), 68.2 (C3), 66.3 (S), 61.3 (11-CH2-Spacer), 60.0 (P2α), 59.5 (P1α), 59.4 (Vα), 57.0 (Tα), 54.9 (Sα), 52.3 (Hα), 49.6 (C2), 49.6 (A2α), 48.7, 48.0 (2-CH2-Spacer), 47.7 (P2δ), 47.6 (A1α), 47.5 (P1δ), 47.4 (C6), 42.3 (Gα), 39.0, 34.0 (12-CH2-Spacer), 30.1 (Vβ), 29.6 (P2β), 28.7 (P1β), 28.7 (P2γ), 28.6 (P1γ), 26.2 (Hβ), 22.2 (CH3-AcNH), 18.4 (Tγ), 18.24 (Vγb), 17.7 (Vγa), 16.3 (A2β), 15.0 (A1β).
:1:1) was added. The vessel was shaken for 3 h. The solution was filtered, and the resin was washed two times with 2 mL trifluoroacetic acid. Toluene (20 mL) was added to the combined solutions, the solvent was removed in vacuo, and the residue was co-distilled two times with toluene (20 mL) and lyophilized. The lyophilisate was directly used in the next step without further purification.
Yield: 128 mg (0.053 mmol, 53%), colorless lyophilisate. Rt = 13.64 min (Phenomenex Luna, gradient see Instrumentation). C103H163N27O40 (M = 2419.55 g mol−1) [2418.16].
ESI-MS (positive), m/z: 1210.56 ([M + 2H]2+, ber.: 1210.09).
To remove the carbohydrate protecting groups the glycopeptide was dissolved in 25 mL methanol and a solution of 0.5 g sodium in 25 mL methanol was added dropwise until the pH reached 10.5. After stirring for 18 h at room temperature the mixture was neutralized with three drops of concentrated acetic acid, before the solvent was evaporated in vacuo. The crude product was purified by a semi-preparative RP-HPLC.
Yield: 45 mg (0.020 mmol, 20%) colorless lyophilisate. Rt = 8.67 min (Phenomenex Luna, gradient A see Instrumentation).
C97H157N27O37 (M = 2293.44 g mol−1) [2292.12].
ESI-MS (positive), m/z: 1147.65 ([M + 2H]2+ cal.: 1147.07), 765.11 ([M + 3H]3+, cal.: 765.05), 1529.92 ([2M + 3H]3+, cal.: 1529.10), 2294.35 ([M + H]+, cal.: 2293.13).
HR-MS (positive), m/z: 1147.0693 ([M + 2H]2+, cal.: 1147.0695).
1H-NMR (400 MHz, MeOH-d4, COSY, HSQC): δ (ppm) = 8.79 (d, 1H, JHε,Hδ = 1.49 Hz, Hε), 7.43 (d, 1H, JHδ, Hε = 1.43 Hz, Hδ), 5.02 (d, 1H, JH1,H2 = 3.63 Hz, H1), 4.70–4.21 (m, 21H, Dα {4.68}, Hα {4.66}, Rα {4.64}, A3α {4.63}, A2α {4.62}, S2α {4.59}, A4α {4.57}, S1α {4.54}, P1-5α {4.51, 4.50, 4.46, 4.45, 4.41}, Vα {4.39}, TTnα {4.36}, TTnβ {4.35}, A1α {4.32}, T1α {4.29}, T2α {4.27}, T2β {4.26}, T1β {4.24}), 4.17 (dd, 1H, JH2,H1 = 3.57 Hz, JH2,H3 = 10.80 Hz, H2), 4.10–3.60 (m, 31H, G1αa {4.08}, G1αb {4.05}, H5 {4.04}, S2β {4.01}, P1δ {3.95}, G2α {3.93}, S1β {3.85}, H3 {3.81}, H6a,b {3.78}, P1δ {3.76}, P2δ {3.75}, H4 {3.69}, 3-CH2-Spacer {3.67}, 2-CH2-Spacer {3.67}, 5 × CH2O-Spacer {3.66}, P2-5δ {3.64, 3.63, 3.62}), 3.40 (d, 1H, JHβ,Hα = 5.65 Hz, Hβa), 3.24–3.12 (m, 5H, CH2-Spacer {3.22}, Hβb {3.20}, Dβa {3.17}, Rδ {3.12}), 2.92 (m, 1H, Dβb), 2.73–2.65 (m, 2H, 12-CH2-Spacer), 2.31–2.20 (m, 4H, P1,2βa {2.27, 2.25}, CH2-Spacer {2.24}), 2.18–2.09 (m, 4H, CH2-Spacer), 2.08–1.84 (m, 21H, P3-5βa {2.07, 2.05, 2.04}, Vβ {2.03}, P1-5γ {2.02, 2.01}, P1-5βb {2.00}, Rβa {2.15}, AcNH {s, 1.86}), 1.74–1.64 (m, 3H, Rβb {1.70}, Rγ {1.68}), 1.43–1.29 (m, 12H, A2β {1.40}, A4β {1.37}, A3β {1.36}, A1β {1.35}), 1.26 (d, 3H, JTγ,Tβ = 6.35 Hz, TTnγ), 1.22–1.14 (m, 6H, JTγ,Tβ = 6.57 Hz, T1γ {1.20}, T2γ {1.18}), 0.99 (t, 6H, JVγ,Vβ = 5.18 Hz, Vγ).
13C-NMR (100.6 MHz, MeOH-d4, HSQC): δ (ppm) = 173.8, 173.7, 173.7, 173.0, 173.0, 172.8, 172.6, 172.6, 172.3, 172.2, 173.1, 171.6, 171.5, 171.4, 171.2, 171.0, 170.9, 170.8, 170.55, 170.5, 170.4, 170.3 (CO, CO–acetyl), 157.0 (CNH), 133.5 (Hε), 129.2 (Hγ), 117.4 (Hδ), 99.0 (C1), 76.4 (TTnβ), 70.0 (CH2-Spacer), 69.9 (C3), 69.7 (C4), 68.9 (T1β), 66.8 (C5), 66.4 (T2β), 66.2 (C6), 61.7 (S1β), 61.5 (S2β), 61.3, 61.1, 60.3, 60.2, 60.1 (P1-5α), 59.8 (T2α), 58.9 (T1α), 58.7 (Vα), 58.4 (TTnα), 56.4 (S2α), 55.6 (S1α), 55.1 (Hα), 52.2 (Rα), 50.6 (Dα), 49.6 (C2), 48.3 (A3α), 48.1 (A1α), 47.9 (A4α), 47.7, 47.5, 47.2, 47.0, 46.9, 46.7 (P1-5δ), 42.3 (G2α), 41.9 (G1α), 40.6 (CH2-Spacer), 39.1 (Rδ), 34.3 (Dβ), 30.1 (Vβ), 29.3, 29.1, 29.0, 28.8, 28.6 (P1-5β), 27.8 (Rβ), 26.4 (Hβ), 24.8, 24.6, 24.5, 24.4, 24.3 (P1-5γ), 24.1 (Rγ), 21.8 (CH3-AcNH), 18.8 (T1γ), 18.7 (T2γ), 18.4 (Vγa), 17.8 (TTnγ), 17.5 (Vγb), 15.7, 15.5, 15.3, 15.0 (A1-4β).
Synthesis of the gold nanoparticles
DLS: dh = 7 ± 1 nm; TEM: d = 7 ± 1 nm; UV/Vis: λmax = 524 nm; 1H-NMR (400 MHz, CDCl3): δ = 0.88 (t, 3J = 7,2 Hz, 3H, CH3); 1.26–1.38 (bs, 20H, CH2); 1.59 (bs, 2H, CH2) ppm.
ATR-IR: 2916.1 cm−1 (s, νC–H)); 2849.0 cm−1 (s, νC–H)); 1455.7 cm−1 (m, δ(C–H)); 1433.8 cm−1 (m, δ(C–H)); 1404.8 cm−1 (m, δ(C–H)); 1378.4 cm−1 (m, δ(CH3)); 720.5 cm−1 (w, CH2-rocking).
1H-NMR (400 MHz, DMSO-d6): δ = 1.26–1.35 (m, CH2); 1.49–1.64 (m, CH2); 2.23 (m, SH); 2.46 (t, CH2); 2.66 (t, CH2); 2.81 (s, CH2) ppm.; 13C-NMR (100 MHz, DMSO-d6): δ = 23.72 (CH2); 24.25 (CH2); 25.41 (CH2); 27.71 (CH2); 27.95 (CH2); 28.43 (CH2); 28.73 (CH2); 28.81 (CH2); 30.15 (CH2); 30.74 (CH2); 33.36 (CH2); 35.75 (CH2); 162.27 (CO); 168.97 (CO); 170.23 (CO) ppm.; ATR-IR: 2917.0 cm−1 (s,ν(C–H)); 2849.1 cm−1(s,ν(C–H)); 1812.0 cm−1 (m,ν(CO)); 1783.8 cm−1 (m,ν(CO)); 1727.5 cm−1 (m,ν(CO)); 1651.1 cm−1 (s,ν(CO)); 1525.4 cm−1 (w, ν(OC–N)); 1371.6 cm−1 (w, δ(C–H)); 1309.6 cm−1 (w, δ(C–H)); 1201.4 cm−1 (s,ν(C–O)); 1070.2 cm−1 (s, ν(C–O)).
O)); 1726.4 cm−1 (s, ν(CO)); 1518.4 cm−1 (w, ν(OC–N)); 1441.8 cm−1 (s, δ(C–H)); 1406.3 cm−1 (s, δ(C–H)); 1366.2 cm−1 (m, δs(CH3)); 1201.4 cm−1 (s,ν(C–O)); 721.0 cm−1 (w, CH2-rocking).
:1); Rf = 0.16 (cHex–EtOAc, 6:1)). Yield: 5.60 g (12.16 mmol, 89%) C30H36O2S (M = 460.67 g mol−1) [460.24]. ESI-MS (positive), m/z: 483.24 ([M + Na]+, cal.: 483.23), 499.22 ([M + K]+, cal.: 499.20).
1H-NMR (300 MHz, CDCl3), δ (ppm) = 7.41–7.37 (m, 6H, H2ar, H6ar), 7.28–7.23 (m, 6H, H3ar, H5ar), 7.21–7.17 (m, 3H, H4ar), 2.32 (t, 2H, J = 7.4 Hz, CH2–S), 2.12 (t, 2H, J = 7.3 Hz, CH2–COOH), 1.63–1.58 (m, 2H, CH2-CH2–S), 1.37–1.17 (m, 14H, CH2).
13C-NMR (75.5 MHz, CDCl3), δ (ppm): 179.5 (COOH), 145.1 (3 × C1ar), 129.7 (3 × C3ar, 3 × C5ar), 127.8 (3 × C2ar, 3 × C6ar), 126.5 (3 × C4ar), 66.3 (S-C-Ph3), 33.8 (CH2–COOH), 32.0 (CH2-S-C-Ph3), 29.3, 29.16, 29.1, 29.0, 28.9, 28.5, 26.9, 24.7 (8 × CH2).
The lyophilisate was dissolved in 1 mL of methanol–toluene (4:1) and added slowly into 50 mL cooled Et2O. The glycopeptide precipitated immediately and was obtained after ultracentrifugation, decantation, redissolvation in water and lyophilization.
Yield: 35.00 mg (0.023 mmol, 23%) colorless lyophilisate.
R t = 16.14 min (Phenomenex Luna, gradient B see Instrumentation).
ESI-MS (positive), m/z: 1568.89 ([M + H]+, ber.: 1568.78), 784.97 ([M + 2H]2+, ber.: 784.89).
1H-NMR (400 MHz, MeOH-d4, COSY, HSQC), δ (ppm) = 8.54 (d, 1H, JHε,Hδ = 1.02 Hz, Hε), 7.31 (d, 1H, JHδ,Hε = 0.66 Hz, Hδ), 5.12 (d, 1H, JH1,H2 = 3.22 Hz, H1), 4.68–4.17 (m, 10H, Hα {4.65}, Tα {4.60}, A1α {4.52}, Sα {4.45}, P1α {4.40}, 4.33 (dd, 1H, JH2,H1 = 5.24 Hz, JH2,H3 = 12.07 Hz, H2), P2α {4.31}, Tβ {4.27}, Vα {4.22}, A2α {4.20}), 3.82–3.55 (m, 27H, H5 {3.82}, Gαa {3.78}, Gαb {3.76}, H4 {3.74}, H3 {3.64}, H6a,b {3.64}, 3-CH2-Spacer {3.63}, Sβ {3.62}, 5 × CH2O-Spacer {3.62}, P1δ {3.60}, P2δ {3.59}, 2-CH2-Spacer {3.55}, 3.33 (dd, 1H, JHβ,Hα = 5.54 Hz, Hβ), 3.18–3.09 (m, 2H, CH2-Spacer), 2.91–2.80 (m, 4H, 12-CH2-Spacer {2.89}, 2-CH2-Spacer {2.83}), 2.53–2.42 (m, 4H, P1β {2.49}, P2β {2.41}, 2.12, 1.98, 1.97 (s, 9H, 3 × Ac), 2.10–1.90 (m, 9H, Vβ {2.06 }, P1γ {2.04}, P2γ {2.03}, 2 × CH2-linker {1.91}, 1.37–1.18 (m, 20H, A1β {1.36}, A2β {1.34}, 7 × CH2-linker {1.29}), 1.21 (d, 3H, JTγ,Hβ = 6.93 Hz, Tγ), 0.97 (t, 6H, JVγ,Vβ = 6.21 Hz, Vγ).
13C-NMR (100.6 MHz, MeOH-d4, HSQC), δ (ppm) = 172.2, 172.1, 172.1, 172.1, 172.0, 171.9, 171.7, 171.4, 171.2, 171.0, 170.8, 170.5, 170.3, 170.1, 169.9 (CO, CO–acetyl), 160.9 (CNH), 130.5 (Hε), 129.5 (Hγ), 128.4 (Hδ), 99.1 (C1), 77.1 (Tβ), 72.7 (C5), 71.6, 71.4, 71.3, 71.2, 71.1 (CH2O-Spacer) 70.6 (C4), 69.0 (C3), 67.9 (Sβ), 60.6 (11-CH2-Spacer), 59.9 (P2α), 59.5 (P1α), 59.2 (Vα), 57.88 (Tα), 56.7 (Sα), 53.8 (Hα), 51.3 (C2), 49.8 (A2α), 49.0, 48.9 (2-CH2-Spacer), 48.1 (P2δ), 47.6 (A1α), 47.6 (P1δ), 47.5 (C6), 42.4 (Gα), 40.2, 37.1 (12-CH2-Spacer), 36.0 (CH2–CO–N-Linker), 31.4 (CH2-S-C-Ph3), 31.0 (Vβ), 30.8, 30.6, 30.5, 30.5, 30.4 (5 × CH2-Linker), 30.3 (P2β), 30.2 (CH2-Linker), 28.9 (P1β), 28.8 (P2γ), 28.8 (P1γ), 28.5, 28.4 (2 × CH2-Linker), 27.0 (Hβ), 23.8, 23.2 (2 × CO–CH3), 22.0 (CH3–AcNH), 20.7 (CO–CH3), 19.9 (Tγ), 19.2 (Vγb), 19.1 (Vγa), 16.7 (A2β), 14.7 (A1β).
To remove the carbohydrate protecting groups the glycopeptide was dissolved in 25 mL of methanol and a solution of 0.5 g sodium in 25 mL of methanol was added dropwise until the pH reached 10.5. After stirring for 18 h at room temperature the mixture was neutralized with three drops of concentrated acetic acid, before the solvent was evaporated in vacuo. The crude product was purified by a semi-preparative RP-HPLC.
Yield: 20.21 mg (0.0140 mmol, 62.2%) colorless lyophilisate.
R t = 14.88 min (Phenomenex Luna, gradient B see Instrumentation).
C64H107N13O22S (M = 1442.67 g mol−1) [1441.74].
ESI-MS (positive), m/z: 1442.75 ([M + H]+, ber.: 1442.75), 721.89 ([M + 2H]2+, ber.: 721.87), 732.87 ([M + H + Na]2+, ber.: 732.87).
Immobilization of the glycopeptides
0.017 g (0.013 mmol) glycopeptide GP-01 was dissolved in 2 mL of anhydrous DMF, and 1 nmol of MUDHSE coordinated gold nanoparticles solution in 5 mL of anhydrous DMF was added dropwise to the stirring solution. After 30 min 15 μL (0.11 mmol) of triethylamine was added and the solution was stirred at room temperature for another 72 h. Finally the nanoparticle solution was dialysed three times against 400 mL of DMF and 300 mL of triple-distilled water. A red colloid solution with a particle concentration of 0.13 μM was obtained and stored at 4 °C.
DLS: dh = 17 ± 3 nm; TEM: d = 7 ± 1 nm; UV/Vis: λmax = 524 nm.
1H-NMR (600 MHz, DMSO-d6, COSY-DQF, HSQC): d = 1.18 (d, 6H, CH3); 1.25 (d, 3H, CH3); 1.32–1.41 (m, 12H, CH2); 1.46 (d, 6H, CH3); 1.55 (quin, 4H, CH2); 1.74 (m, 4H, CH2); 1.98 (m, 1H, CH); 2.01 (m, 4H, CH2); 2.17 (t, 2H, CH2); 2.26 (t, 2H, CH2); 2.45 (m, 1H, CH); 2.55 (t, 2H, CH2); 2.94 (d, 2H, CH2); 3.18 (q, 2H, CH2); 3.38 (t, 2H CH2); 3.45 (t, 4H, CH2); 3.50 (bm, 8H, CH2); 3.65 (t, 2H, CH2); 3.76 (s, 3H, CH3); 3.91 (q, 1H, CH); 3.95 (d, 2H, CH2); 4.12 (d, 1H, CH); 4.15 (t, 2H, CH2); 4.24 (q, 1H, CH); 4.27 (d, 1H, CH); 4.29 (d, 2H, CH); 4.38 (m, 2H, CH); 4.43–4.48 (m, 4H, CH); 4.51 (m, 2H, CH); 4.72 (d, 1H, CH); 4.92 (bs, 5H, OH); 7.77 (t, 1H, NH); 7.85 (d, 1H, CH); 7.98 (d, 1H, CH); 8.17 (d, 7H, NH); 8.33 (d, 1H, NH) ppm; ATR-IR: 3273.0 cm−1 (m; ν(N–H)); 2925.6 cm−1 (s; ν(C–H)); 2853.4 cm−1 (st; ν(C–H)); 2324.5 cm−1 (w); 2164.8 cm−1 (w); 1782.6 cm−1 (w, ν(CO)); 1626.1 cm−1 (s, ν(CO)); 1527.1 cm−1 (s, ν(CC)); 1450.8 cm−1 (s, δ(C–H)); 1398.8 cm−1 (m, δ(C–H)); 1339.4 cm−1 (s, δ(C–H)); 1201.2 cm−1 (s, ν(C–O)); 1125.3 cm−1 (w); 1076.5 cm−1 (w); 1044.5 cm−1 (w); 718.2 cm−1 (w, CH2-rocking).
DLS: dh = 34 ± 7 nm; TEM: d = 7 ± 1 nm; UV/Vis: λmax = 524 nm.
DLS: dh = 19 ± 4 nm; TEM: d = 14 ± 1 nm; UV/Vis: λmax = 531 nm.
Acknowledgements
The authors thank Timo Schueler for the QCM measurements.References
- (a) J. N. Blattman and P. D. Greenberg, Science, 2004, 305, 200 CrossRef CAS PubMed; (b) C. J. Melief and S. H. van der Burg, Nat. Rev. Cancer, 2008, 8, 351 CrossRef CAS PubMed.
- (a) J. A. Berzofsky, M. Terabe, S. Oh, I. M. Belyakov, J. D. Ahlers, J. E. Janik and J. C. Morris, J. Clin. Invest., 2004, 113, 1515–1525 CrossRef CAS; (b) I. Espinoza-Delgado, Oncologist, 2002, 7(Suppl 3), 20 CrossRef CAS PubMed; (c) E. Jager, D. Jager and A. Knuth, Int. J. Cancer, 2003, 106, 817–820 CrossRef PubMed.
- M. S. Mitchell, Int. J. Immunopharmacol., 2003, 3, 1051 CrossRef CAS.
- Y. Krishnamachari, S. M. Geary, C. D. Lemke and A. K. Salem, Pharm. Res., 2011, 28, 215–236 CrossRef CAS PubMed.
- (a) P. E. Constantinou, B. P. Danysh, N. Dharmaraj and D. D. Carson, Expert Rev. Endocrinol. Metab., 2011, 6, 835 CrossRef CAS PubMed; (b) A. Hoffmann-Röder, A. Kaiser, S. Wagner, N. Gaidzik, D. Kowalczyk, U. Westerlind, B. Gerlitzki, E. Schmitt and H. Kunz, Angew. Chem., Int. Ed., 2010, 122, 1 CrossRef; (c) T. Renno, S. Lebecque, N. Renard, S. Saeland and A. Vicari, Cell. Mol. Life Sci., 2003, 60, 1296 CrossRef CAS; (d) M. L. Disis, K. H. Grabstein, P. R. Sleath and M. A. Cheever, Clin. Cancer Res., 1999, 5, 1289 CAS.
- (a) J. R. Gum Jr., Biochem. Soc. Trans., 1995, 23, 795 CAS; (b) I. Carlsted and J. R. Davies, Biochem. Soc. Trans., 1997, 25, 214 Search PubMed; (c) A. Girling, J. Bartkova, J. Burchell, S. Gendler, C. Gillet and J. Taylor-Papadimiriou, Int. J. Cancer, 1989, 43, 1072 CrossRef CAS; (d) M. A. Tarp and H. Clausen, Biochim. Biophys. Acta Gen. Subj., 2008, 1780, 546 CrossRef CAS PubMed.
- (a) U. Westerlind, H. Schröder, A. Hobel, N. Gaidzik, A. Kaiser, C. M. Niemeyer, E. Schmitt, H. Waldmann and H. Kunz, Angew. Chem., Int. Ed., 2009, 48, 8263 CrossRef CAS PubMed; (b) F. Carzana, J. M. Busto, F. Marcelo, M. G. de Luis, J. C. Abensio, S. Martín-Santamaría, J. Jiménez-Barbero, A. Avenoza and J. M. Peregrina, Chem. – Eur. J., 2011, 17, 3105 CrossRef PubMed.
- (a) A. Kaiser, N. Gaidzik, U. Westerlind, D. Kowalczyk, A. Hobel, E. Schmitt and H. Kunz, Angew. Chem., Int. Ed., 2009, 121, 7688 CrossRef; (b) D. H. Dube and C. R. Bertozzi, Nat. Rev. Drug Discovery, 2005, 4, 477 CrossRef CAS PubMed.
- (a) N. Gaidzik, U. Westerlind and H. Kunz, Chem. Soc. Rev., 2013, 42, 4421 RSC; (b) T. Buskas, P. Thompson and G. J. Boons, Chem. Commun., 2009, 5335 RSC; (c) R. D. Astronomo and D. R. Burton, Nat. Rev. Drug Discovery, 2009, 9, 308 CrossRef PubMed; (d) S. F. Slovin, S. J. Keding and G. Ragupathi, Immunol. Cell Biol., 2005, 83, 418 CrossRef CAS PubMed; (e) R. E. Beatson, J. Taylor-Papadimitriou and J. M. Burchell, Immunotherapy, 2010, 2, 305 CrossRef CAS PubMed; (f) S. H. Itzkowitz, M. Yuan, C. K. Montgomery, T. Kjeldsen, H. K. Takahashi, W. L. Bigbee and Y. S. Kim, Cancer Res., 1989, 49, 197–204 CAS.
- Y. S. Kim, J. Gum Jr. and I. Brockhausen, Glycoconjugate. J., 1996, 13, 693 CrossRef CAS.
- F. Berti and R. Adamo, Chem. Biol., 2013, 8, 1653 CAS.
- (a) S. J. Danishefsky and J. A. Allen, Angew. Chem., Int. Ed., 2000, 112, 882 CrossRef; (b) G. D. MacLean, M. A. Reddish, R. R. Koganty and B. M. Longenecker, J. Immunother., 1996, 19, 59 CrossRef CAS PubMed; (c) B. M. Longenecker, M. Reddish, R. Koganty and G. D. MacLean, Adv. Exp. Med. Biol., 1994, 353, 105 CrossRef CAS; (d) N. Gaidzik, A. Kaiser, D. Kowalczyk, U. Westerlind, B. Gerlitzki, H. P. Sinn, E. Schmitt and H. Kunz, Angew. Chem., Int. Ed., 2011, 123, 10153 CrossRef; (e) T. Nolan, S. Lambert, D. Roberton, H. Marshall, P. Richmond, C. Streeton, J. Poolman and D. Boutriau, Vaccine, 2007, 25, 8487 CrossRef CAS PubMed.
- P. Spohn, U. Buwitt-Beckmann, R. Bock, G. Jung, A. J. Ulmer and K.-H. Wiesmueller, Vaccine, 2004, 22, 2494 CrossRef PubMed.
- A. Hoffmann-Roeder and M. Johannes, Chem. Commun., 2011, 47, 9903 RSC.
- O. Renaudet and P. Dumy, Org. Lett., 2003, 5, 243 CrossRef CAS PubMed.
- S. Keil, A. Kaiser, F. Syed and H. Kunz, Synthesis, 2009, 1355 CAS.
- S. Ingale, T. Buskas and G.-J. Boons, Org. Lett., 2006, 8, 5785 CrossRef CAS PubMed.
- (a) J. M. de la Fuente and S. Penadés, Biochim. Biophys. Acta, 2006, 1760, 636 CrossRef CAS PubMed; (b) R. P. Brinas, A. Sundgren, P. Sahoo, S. Morey, K. Rittenhouse-Olson, G. E. Wilding, W. Deng and J. J. Barchi Jr., Bioconjugate Chem., 2012, 23, 1513 CrossRef CAS PubMed; (c) B. P. Danysh, P. E. Constantinou, E. Y. Lukianova-Hleb, D. O. Lapotko and D. D. Carson, Theranostics, 2012, 2, 777 CrossRef CAS PubMed; (d) R. Savla, O. Taratula, O. Garbuzenko and T. Minko, J. Controlled Release, 2011, 153, 16 CrossRef CAS PubMed; (e) E. S. Glazer, C. Zhu, K. L. Massey, C. S. Thompson, W. D. Kaluarachchi and A. N. Hamir, Clin. Cancer Res., 2010, 16, 5712 CrossRef CAS PubMed.
- (a) G. B. Sigal, M. Mammen, G. Dahmann and G. M. Whitesides, J. Am. Chem. Soc., 1996, 118, 3789 CrossRef CAS; (b) M. Mammen, S.-K. Choi and G. M. Whitesides, Angew. Chem., 1998, 110, 2908 CrossRef.
- (a) J. Dernedde, S. Enders, H.-U. Reissig, M. Roskamp, S. Schlecht and S. Yekta, Chem. Commun., 2009, 8, 932 RSC; (b) M. Roskamp, S. Enders, F. Pfrengle, S. Yekta, V. Dekaris, J. Dernedde, H.-U. Reissig and S. Schlecht, Org. Biomol. Chem., 2011, 9, 7448 RSC.
- (a) C. M. Niemeyer, Angew. Chem., Int. Ed., 2001, 113, 4254 CrossRef; (b) C. Buzea, I. I. Pacheco and K. Robbie, Biointerphases, 2007, 4, 17 Search PubMed; (c) C. M. Niemeyer, Angew. Chem., Int. Ed., 2001, 40, 4128 CrossRef CAS; (d) R. Ojeda, J. L. de Paz, A. G. Barrientos, M. M. Lomas and S. Penadés, Carbohydr. Res., 2007, 342, 448 CrossRef CAS PubMed; (e) A. Sundgren and J. J. Barchi Jr., Carbohydr. Res., 2008, 343, 1594 CrossRef CAS PubMed.
- L. Nuhn, S. Hartmann, B. Palitzsch, B. Gerlitzki, E. Schmitt, R. Zentel and H. Kunz, Angew. Chem., Int. Ed., 2013, 52, 10652 CrossRef CAS PubMed.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293 CrossRef CAS PubMed.
- A. Kaiser, N. Gaidzik, U. Westerlind, D. Kowakczyk, A. Hobel, E. Schmitt and H. Kunz, Angew. Chem., Int. Ed., 2009, 48, 7551 CrossRef CAS PubMed.
- H. Paulsen and K. Adermann, Liebigs Ann. Chem., 1989, 751 CrossRef CAS.
- (a) H. Paulsen and J. P. Hoelck, Carbohydr. Res., 1982, 109, 89 CrossRef CAS; (b) W. Koenigs and E. Knorr, Chem. Ber., 1901, 34, 957 CrossRef.
- E. Meinjohanns, M. Meldal, A. Schleyer, H. Paulsen and K. Bock, J. Chem. Soc., Perkin Trans. 1, 1996, 985 RSC.
- (a) D. Stucky, et al. , J. Am. Chem. Soc., 2006, 128, 6550 CrossRef PubMed; (b) J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55 RSC; (c) J. Turkevich, Gold Bull., 1985, 18, 86 CrossRef CAS; (d) G. Frens, Nat. Phys. Sci., 1973, 241, 20 CrossRef CAS.
- E. Oh, K. Susumu, A. J. Makinen, J. R. Deschamps, A. L. Huston and I. L. Medintz, J. Phys. Chem. C, 2013, 117, 18947 CAS.
- B. C. Mei, E. Oh, K. Susumu, D. Farrell, T. J. Mountziaris and H. Mattoussi, Langmuir, 2009, 25, 10604 CrossRef CAS PubMed.
- A. Dass, R. Guo, J. B. Tracy, R. Balasubramanian, A. D. Douglas and R. W. Murray, Langmuir, 2008, 24, 310 CrossRef CAS PubMed.
- K. B. Cederquistm and C. D. Keating, Nano, 2009, 3, 256 Search PubMed.
- T. Platen, T. Schueler, W. Tremel and A. Hoffmann-Roeder, Eur. J. Org. Chem., 2011, 3878 CrossRef CAS.
- (a) J. Burchell, S. Gendler, J. Taylor-Papadimitriou, A. Girling, A. Lewis, R. Millis and D. Lamport, Cancer Res., 1987, 47, 5476–5482 CAS; (b) J. Burchell, J. Taylorpapadimitriou, M. Boshell, S. Gendler and T. Duhig, Int. J. Cancer, 1989, 44, 691–696 CrossRef CAS.
- H. Moeller, N. Serttas, H. Paulsen, J. M. Burchell, J. Taylor-Papadimitriou and B. Meyer, Eur. J. Biochem., 2002, 269, 1444 CrossRef.
- R. U. Lemieux and R. M. Ratcliffe, Can. J. Chem., 1979, 57, 1244 CrossRef CAS PubMed.
- A. Paquet, Can. J. Chem., 1982, 60, 976 CrossRef CAS.
- B. Liebe and H. Kunz, Helv. Chim. Acta, 1997, 80, 1473 CrossRef CAS.
- S. Keil, C. Claus, W. Dippold and H. Kunz, Angew. Chem., 2001, 113, 379 CrossRef.
- O. Seitz and H. Kunz, J. Org. Chem., 1997, 62, 813 CrossRef CAS.
- D. Liu, H. Shao and X. Jiang, Angew. Chem., Int. Ed., 2009, 48, 4406 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ob01339e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |