
Amino acid-linked porphyrin-nitroimidazole antibiotics targeting Porphyromonas gingivalis†
Simon A.
Dingsdag‡
ab,
Benjamin C-M.
Yap‡
ac,
Neil
Hunter
ab and
Maxwell J.
Crossley
*c
aInstitute of Dental Research, Westmead Millennium Institute and Centre for Oral Health, Westmead, NSW 2145, Australia
bFaculty of Dentistry, The University of Sydney, NSW 2006, Australia
cSchool of Chemistry, The University of Sydney, NSW 2006, Australia. E-mail: maxwell.crossley@sydney.edu.au
First published on 22nd October 2014
Abstract
The periodontal pathogen Porphyromonas gingivalis requires porphyrin supplementation for growth. Previously, in order to inhibit P. gingivalis growth, we synthesised very effective ‘Trojan horse’ ester and amide-linked deuterporphyrin-nitroimidazole (DPIX-Nim) adducts that exploited this requirement to transport metronidazole-derived antibiotics with excellent antimicrobial selectivity and recognition by the HA2 porphyrin binding site. Herein, in the context of developing topical agents to target P. gingivalis, L-amino acids are incorporated into adducts as linkers to improve uptake. Ten 13- and 17-propionic amide regioisomers of L-amino acid-linked deuterporphyrin-nitroimidazole adducts were synthesised using a peptide coupling approach. DPIX-Lys regioisomers without attached nitroimidazole were also synthesised as comparison compounds. All the porphyrin adducts bound (Kd50 7 to 20 nM) to a recombinant HA2 receptor with similar binding affinity to haem, except the lysine-proline linked DPIX-Lys(Boc)Pro-Nim adducts (Kd50 300 nM) and the DPIX-Lys(Nim)-Nim adducts (Kd50 200 nM), both of which have large appended groups. DPIX-Lys(Boc)-Nim, DPIX-Lys(OH)-Nim, and DPIX-Pro-Nim adducts were shown to be very effective against P. gingivalis. DPIX-Lys(Boc)Pro-Nim adducts and DPIX-Lys(Nim)-Nim adducts showed weak activity. Importantly, DPIX-Lys(Boc)-Nim adducts were selective for P. gingivalis and, unlike metronidazole, did not kill a range of other anaerobic bacteria isolated from the human gastrointestinal tract.
Introduction
The anaerobic bacterium Porphyromonas gingivalis, implicated in periodontal disease, is unable to synthesise porphyrin. The requirement for external porphyrin (for growth) is satisfied in vitro by haem 1. Unlike haem 1, deuteroporphyrin IX (DPIX 2) does not feature in the bacterial biosynthetic pathway.DPIX 2 has no central metal iron, and does not contain vinyl groups on the northern face (positions 3 and 8 of the porphyrin macrocycle). Our group previously demonstrated DPIX 2 supports growth of P. gingivalis.1 DPIX 2 is recognised in part by a recombinant preparation of HA2, an outer membrane sub-domain protein, located within the gingipains of P. gingivalis.2,3
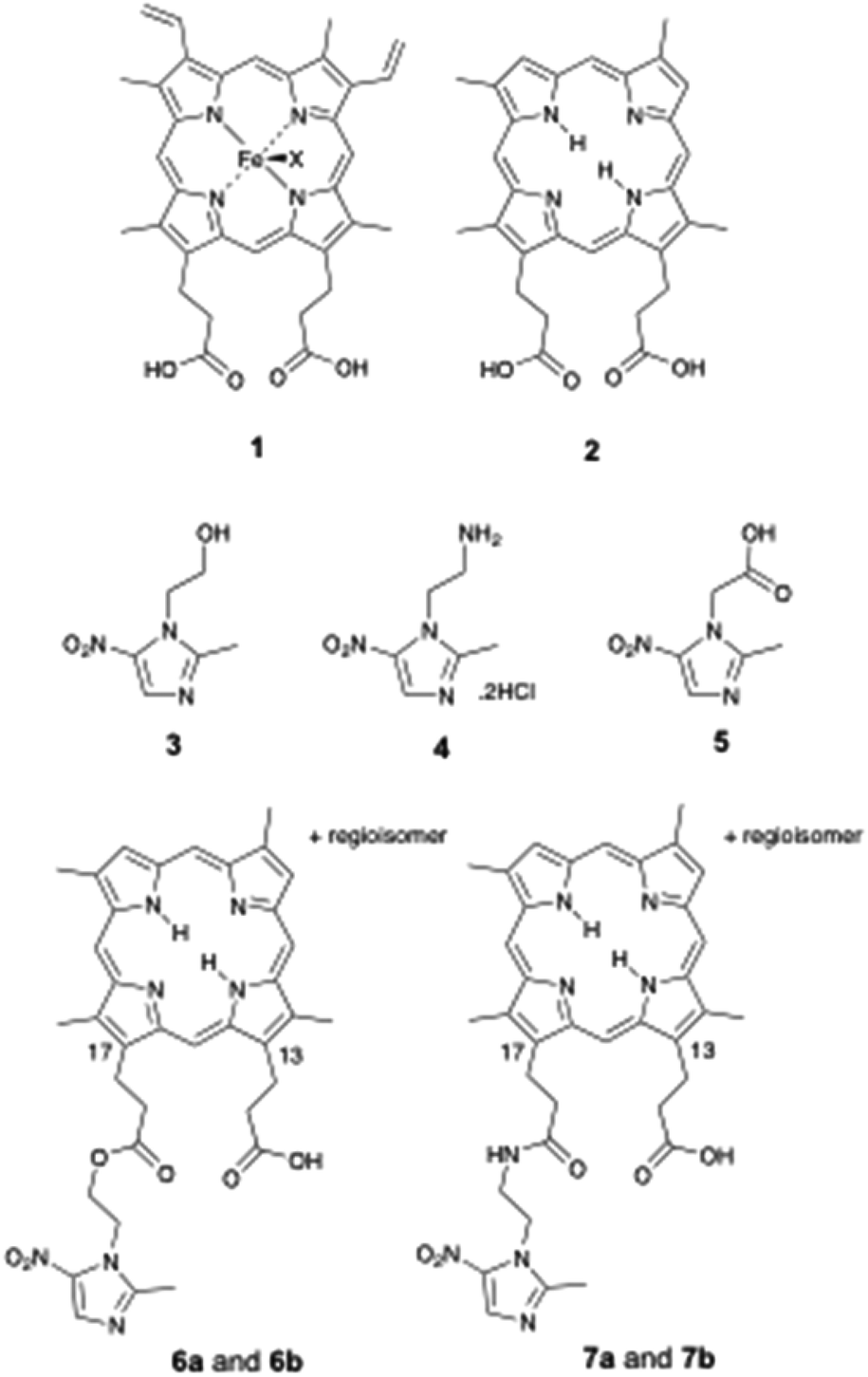
Broad-spectrum antibiotics, such as metronidazole (Mtz) 3, are commonly used to treat bacterial infections such as during periodontitis. In this work, we also make use of the derivatives Mtz-NH24 and Mtz-CO2H 5. The nitroimidazole (Nim) antibiotics are pro-drugs, inactive until taken up by anaerobic bacteria and ‘parasites’ (Entamoeba and Giardia), and activated by nitro group reduction. The human gastrointestinal tract contains extremely complex bacterial populations. Unfortunately, broad-spectrum antibiotics perturb these complex microbial ecosystems within humans and promote drug resistance in bacteria.4,5
The problems associated with the use of broad-spectrum antibiotics prompted our group to synthesise porphyrin-based inhibitors, designed to act as a “Trojan-horse”: being recognised and imported as an essential porphyrin nutrient, while also delivering metronidazole derivatives.6–8 Facilitated delivery of such nitroimidazole antibiotics was envisaged to overcome indiscriminate suppression of non-target bacteria within the polymicrobial ecosystem of the oral cavity thereby resulting in selectivity for P. gingivalis.
In the absence of a structure of the porphyrin binding site on rHA2, a model of the site (Fig. 1) was proposed on the basis of growth and recognition studies.
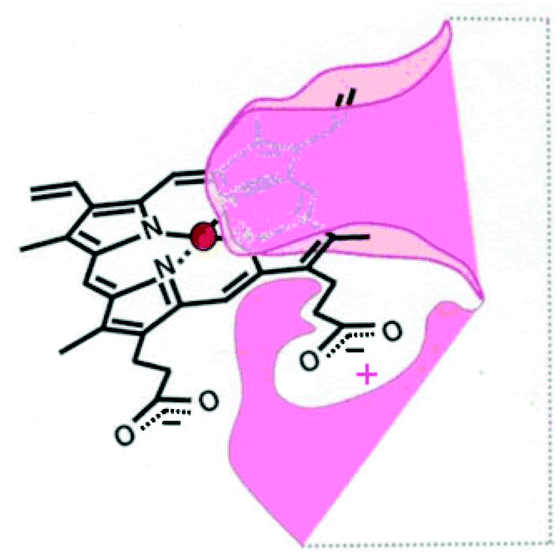 | ||
| Fig. 1 Proposed HA2 binding site.6 | ||
Based on the model (Fig. 1), we synthesised deuteroporphyrin-nitroimidazole ester adducts 6a and 6b and amide adducts 7a and 7b whereby the 17-propionate (a series) and 13-propionate (b series) of the porphyrin is linked directly to the antibiotic. These adducts are recognised by the HA2 receptor of P. gingivalis, as strongly as haem 1 to HA2 (Kd50 10 to 20 nM) and are highly potent towards P. gingivalis [Minimum Inhibitory Concentration (MIC) 0.625 to 2 μM].6 This activity matches that of metronidazole 3 (MIC 1 μM) and surpasses that of Mtz-NH24 (MIC 15 μM), suggesting that the pro-drug does not require cleavage from the porphyrin for activity.
Consistent with the binding site model (Fig. 1), the individual deuteroporphyrin-nitroimidazole esters, 6a and 6b, were found to bind to rHA2 equally well and they showed the same potency towards P. gingivalis. This indicates that either face of the porphyrin ring of deuteroporphyrin can be recognised by rHA2 by binding the 13-propionate or the 17-propionate if the ring is flipped over. Thus both 6a and 6b can bind into the rHA2 site by making use of the available propionate thereby leaving the 17-side-chain ester unimpaired in the case of 6a, and the 13-side-chain ester unimpaired in the case of 6b. A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 regioisomeric mixture 6a/6b also showed the same binding and inhibition properties as the individual isomers; thus separation of the regioisomers is not required.
:1 regioisomeric mixture 6a/6b also showed the same binding and inhibition properties as the individual isomers; thus separation of the regioisomers is not required.
The work herein involves the design, synthesis and evaluation of new porphyrin-based growth inhibitors for P. gingivalis. With consideration for topical therapy, amino acids lysine (Lys) and proline (Pro) were incorporated into porphyrin-nitroimidazole adducts as linkers, permitting derivatisation to increase potency and water solubility. Synthesis of amino acid-linked porphyrin-nitroimidazole adducts; DPIX-Lys(Boc)-Nim adducts 12a and 12b, DPIX-Lys(H)-Nim adducts 14a and 14b, DPIX-Lys(Boc)-Pro-Nim adducts 22a and 22b, and DPIX-Pro-Nim adducts 24a and 24b is reported herein. DPIX-Lys(Boc)-H adducts 29a and 29b, without attached nitroimidazoles function as controls. We also characterise the rate of kill and minimum inhibitory concentrations of 7a and 7b (synthesised previously)6 and 12a and 12b against a range of anaerobic bacteria known to be sensitive to metronidazole (Fusobacterium nucleatum, Bacteroides fragilis, Tannerella forsythia and Prevotella melaninogenica). Evaluation of rHA2 binding and growth inhibitory activities against anaerobic bacteria provides a framework for porphyrin-based antibiotics as alternatives to broad-spectrum antibiotics.
Results and discussion
Design of an inhibitor
A recombinant preparation of the HA2 surface receptor of P. gingivalis binds porphyrin independent of the central iron.1,2 On this basis, a free-base porphyrin was chosen, designed to render the porphyrin unpalatable to other bacteria with porphyrin scavengers such as the HasA hemophore in Serratia marcescens that recognise iron-metallated porphyrin.9 Also, rHA2 recognition is dependent on the structure of the porphyrin macrocyle. At least one free propionic acid side-chain is necessary and certain modifications of the vinyl aspect of these haem analogues are not tolerated.1,3The synthetic route involves elaboration of an amino acid-linked nitroimidazole derivative, coupled to the carboxylic acid group of DPIX mono-methyl esters 10 to give the desired mono-amino acid-linked porphyrin-antibiotic adducts. One propionic acid is then free for HA2 recognition. DPIX mono-methyl esters 10 were chosen as there is no central metal, envisaged thereby to minimize uptake by other bacteria.
Previous attempts to form a mono-adduct directly between DPIX 2 and nitroimidazoles resulted in a mixture of products that proved difficult to separate due to the polarity of free carboxylic acid groups.6 Further, by-products from the reaction could not be recycled easily. The route using 10 for coupling avoids these issues. Subsequent removal of protecting groups and hydrolysis of the methyl ester should be achieved selectively and in high yield to afford the desired amino acid-linked porphyrin-nitroimidazole adducts.
P. gingivalis Kgp gingipain is lysine-specific.6 Using lysine linkers also provides the opportunity to explore gingipain-specific cleavage by P. gingivalis.
Synthesis
Fmoc-Lys(Boc)-Nim 9 was prepared in 80% yield from Mtz-NH24 and Fmoc-Lys(Boc)-OH 8 using o-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU) (Scheme 1). Mtz-NH24 was prepared in 85% yield from 3 following the literature.8 DPIX mono-methyl esters 10 were prepared in a 49% yield by the controlled hydrolysis of DPIX dimethyl ester with 4 M hydrochloric acid in refluxing methanol.10 Fmoc deprotection of 9 by base in situ and attachment to the carboxylic acid group of 10 by reaction at its newly liberated amine gave the DPIX-Lys(Boc)-Nim mono-methyl ester adducts 11a and 11b in 90% yield (Scheme 1). Hydrolysis of adducts 11a and 11b by treatment with excess LiOH in a CH3OH–THF–H2O mixture overnight at RT gave the free acids 12a and 12b in quantitative yield.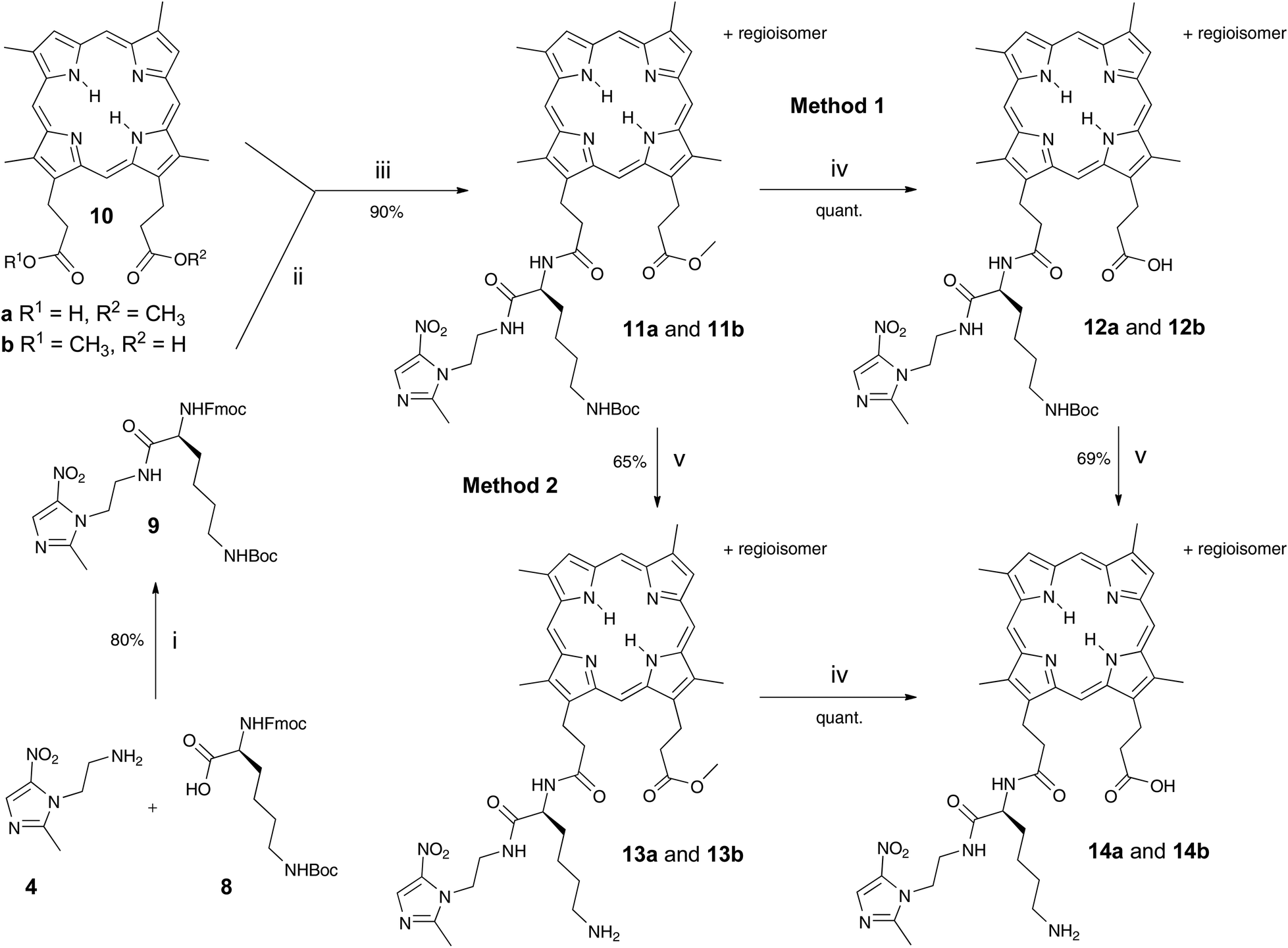 | ||
| Scheme 1 Reagents and conditions: i HBTU, DIPEA, stir under N2, 3 h, then HOBt; ii DBU, CH2Cl2, stir under N2, 30 min; iii HOBt, HBTU, DIPEA, stir under N2, overnight; iv LiOH in CH3OH–THF–H2O (3:4:1) stir, overnight; v TFA, CH2Cl2, stir under N2, 3 h. | ||
The fully deprotected adducts 14a and 14b were obtained by two routes The first route involved Boc deprotection of adducts 12a and 12b by treatment with CF3CO2H in CH2Cl2 overnight to give the desired products 14a and 14b in 69% yield. The second route involved reversal of the sequence. Removal of the Boc groups from adducts 11a and 11b and subsequent ester hydrolysis of the resultant adducts 13a and 13b gave the desired products 14a and 14b in 65% yield.
Adducts 17a and 17b, with two nitroimidazoles attached, were synthesised in a bid to improve potency and this was also achieved by two routes (Scheme 2). The first route involved prior preparation of the di-Nim-linked lysine 15. Mtz-CO2H 5 was prepared from Mtz 3 in 32% yield following the literature.11 The di-Nim-linked lysine 15 was prepared by initial Boc deprotection of 9 and subsequent coupling to 5 with HBTU (Scheme 2). Fmoc deprotection of 15 with DBU and coupling to the DPIX mono-methyl esters 10 gave adducts 16a and 16b in a 90% yield. Hydrolysis to the corresponding free acids 17a and 17b occurred quantitatively. The second route involved HBTU-induced coupling of conjugates 13a and 13b with 5, followed by quantitative hydrolysis.
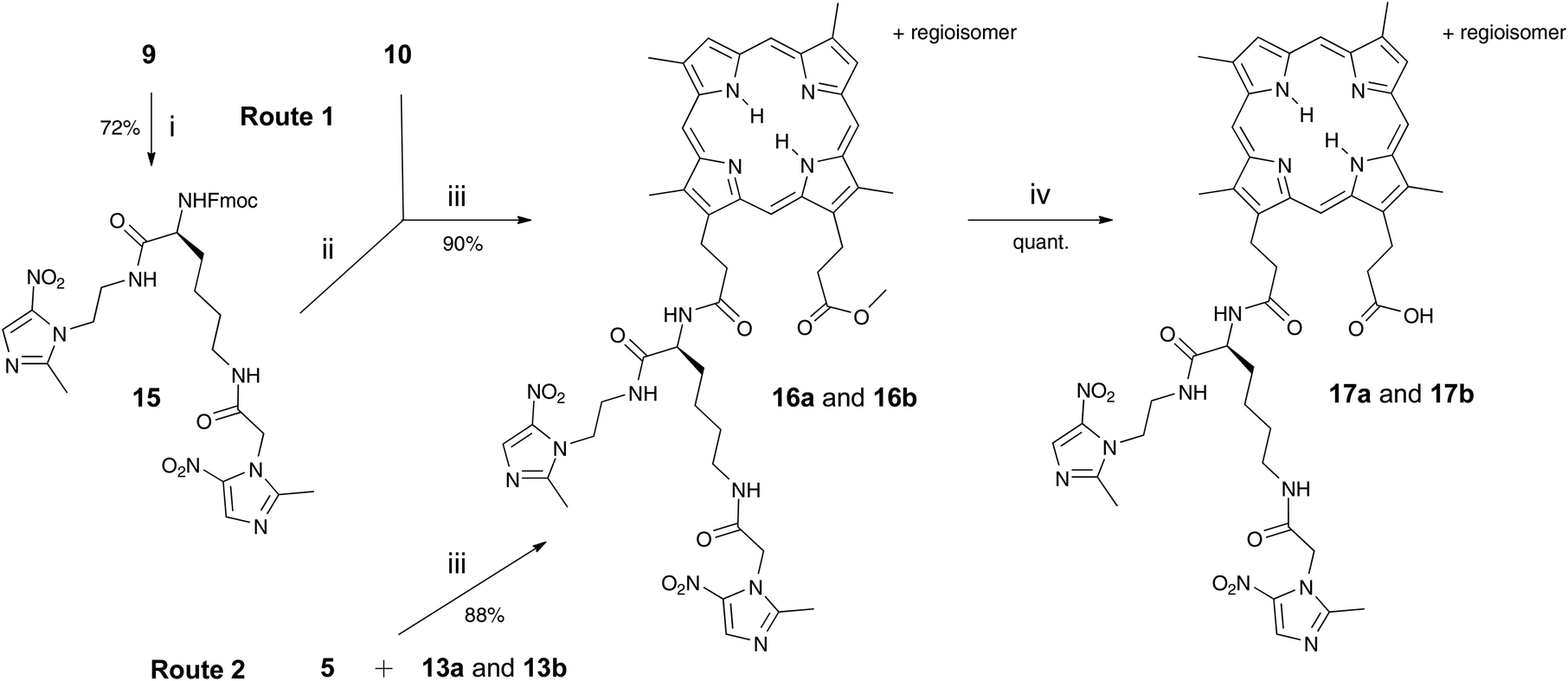 | ||
| Scheme 2 Reagents and conditions: i TFA, CH2Cl2, stir under N2, 3 h, then Mtz-CO2H 5, HBTU, DIPEA, stir under N2, overnight; ii DBU, CH2Cl2, stir under N2, 30 min then HOBt; iii HBTU, DIPEA, stir under N2, overnight; iv LiOH in CH3OH–THF–H2O (3:4:1) stir, overnight. | ||
DPIX-Lys(Boc)-Pro-Nim adducts 22a and 22b, whereby a proline-lysine linker was used to bridge the nitroimidazole and the porphyrin, were synthesised in good overall yield (Scheme 3). First, the Fmoc-Pro-Nim 19 was prepared in 85% yield from Mtz-NH24 and Fmoc-Pro-OH 18 with the use of peptide coupling reagent HBTU. Fmoc-Lys(Boc)-Pro-Nim 20 was then prepared in 81% yield by HBTU-induced coupling of Fmoc-Lys(Boc)-OH 8 and the amine derived by in situ base deprotection of the Fmoc-Pro-Nim derivative 19. In turn the Fmoc-Lys(Boc)-Pro-Nim 20 was Fmoc-deprotected and coupled in situ to the DPIX mono-methyl esters 10 which gave the methyl ester adducts 21a and 21b in a 81% yield. Hydrolysis of the methyl esters gave the DPIX-Lys(Boc)-Pro-Nim adducts 22a and 22b in quantitative yield.
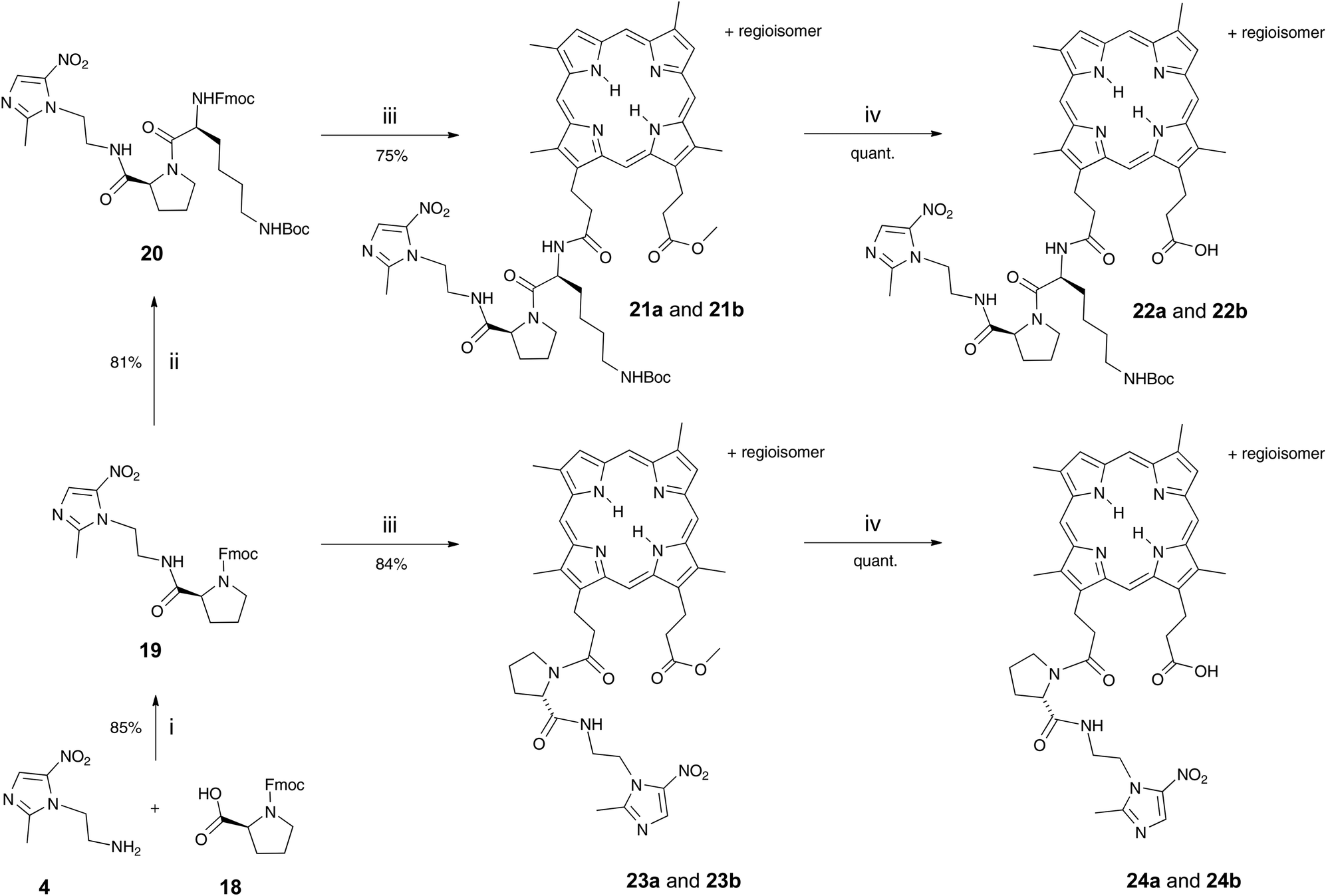 | ||
| Scheme 3 i HBTU, DIPEA, stir under N2, overnight; ii DBU, CH2Cl2, stir under N2, 30 min; then HOBt, Fmoc-Lys(Boc)-OH 8, HBTU, DIPEA, stir under N2, overnight; iii DBU, CH2Cl2, stir under N2, 30 min; then DPIX mono-methyl esters 10, HOBt, HBTU, DIPEA, stir under N2, overnight; iv LiOH in THF, CH3OH and water mixture, stir, overnight. | ||
DPIX-Pro-Nim adducts 24a and 24b, whereby a proline linker was used to bridge the nitroimidazole and the porphyrin, were synthesised as a control for the enzyme activity experiment. Fmoc-Pro-Nim derivative 19 was Fmoc deprotected by base in situ and attached at its newly liberated amine to the carboxylic acid group of 10 to give the DPIX-Pro-Nim mono-methyl ester adducts 23a and 23b (Scheme 3) in 84% yield. Subsequent hydrolysis with LiOH dissolved in a CH3OH–THF–H2O mixture at RT overnight gave the corresponding free acids 24a and 24b in quantitative yield. Controls 29a and 29b, essentially 12a and 12b without nitroimidazole attached were synthesised (Schemes 4 and 5). First, preparation of the triply-protected lysine 27 involved reacting prenyldimethylsulfonium tetrafluoroborate 26 with Fmoc-Lys(Boc)-OH 8 with catalytic CuBr. The sulfonium salt 26 was prepared from 3-methyl-2-buten-1-ol 25 in 60% yield following the literature (Scheme 4).12 Fmoc-Lys(Boc)-dimethylallyl ester 27 was Fmoc deprotected using DBU and then attached to the DPIX mono-methyl esters 10 by an amide linkage through one of the propionic acid side-chains of the porphyrin macrocycle to give diesters 28a and 28b in 82% yield (Scheme 5). Subsequent hydrolysis of the methyl and dimethylallyl esters to give adducts 29a and 29b occurred quantitatively on treatment over night under standard conditions.
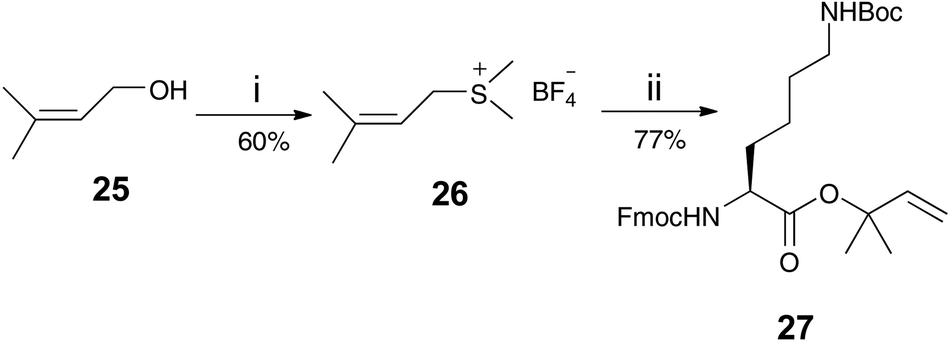 | ||
| Scheme 4 Reagents and conditions: i (CH3)2S, HBF4 (54 wt% in diethyl ether), CH2Cl2, stir under N2 at 0 °C, 6 h, then at room temperature, 14 h; ii Fmoc-Lys(Boc)-OH 8, K2CO3, CuBr, CH2Cl2, stir under N2 at 25 °C, overnight. | ||
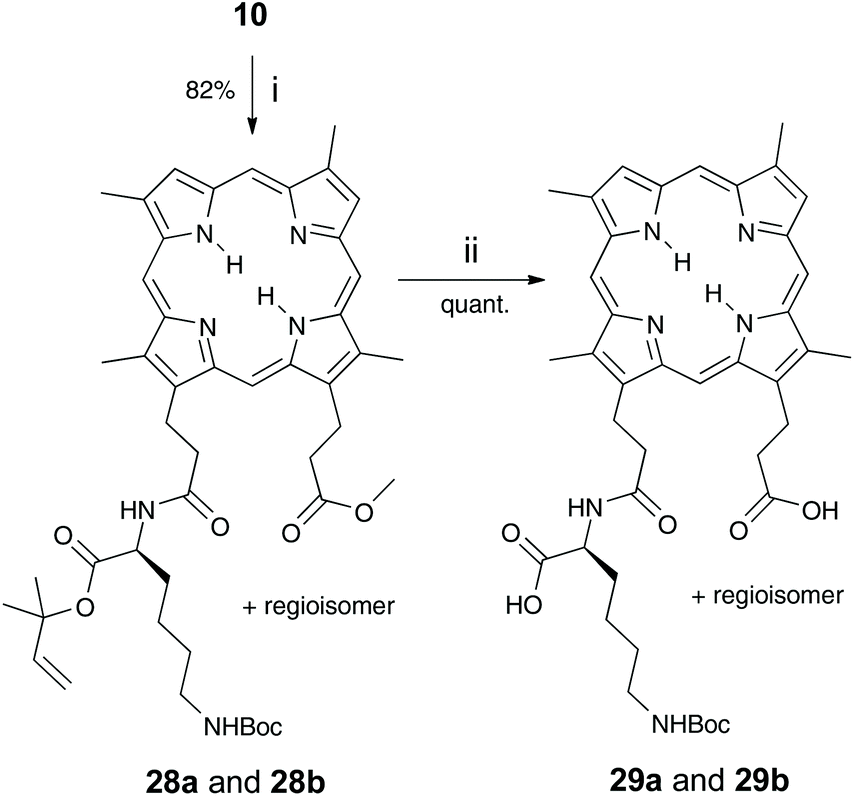 | ||
| Scheme 5 Reagents and conditions: i Fmoc-Lys(Boc)-dimethylallyl ester 27, DBU, CH2Cl2, stir under N2, 30 min; then HOBt, HBTU, DIPEA, stir under N2, overnight; ii LiOH in CH3OH–THF–H2O (3:4:1) stir, overnight. | ||
rHA2 binding
DPIX-Lys(Boc)-Nim adducts 12a and 12b, DPIX-Lys(H)-Nim adducts 14a and 14b, DPIX-Pro-Nim adducts 24a and 24b, and DPIX-Lys(Boc)-H adducts 29a and 29b bind rHA2 as measured by solid-phase ELISA (where binding affinity to rHA2 was defined as the Kd50) with similar binding affinities (Kd50 7 to 20 nM) to that of haem 1 (Fig. 2).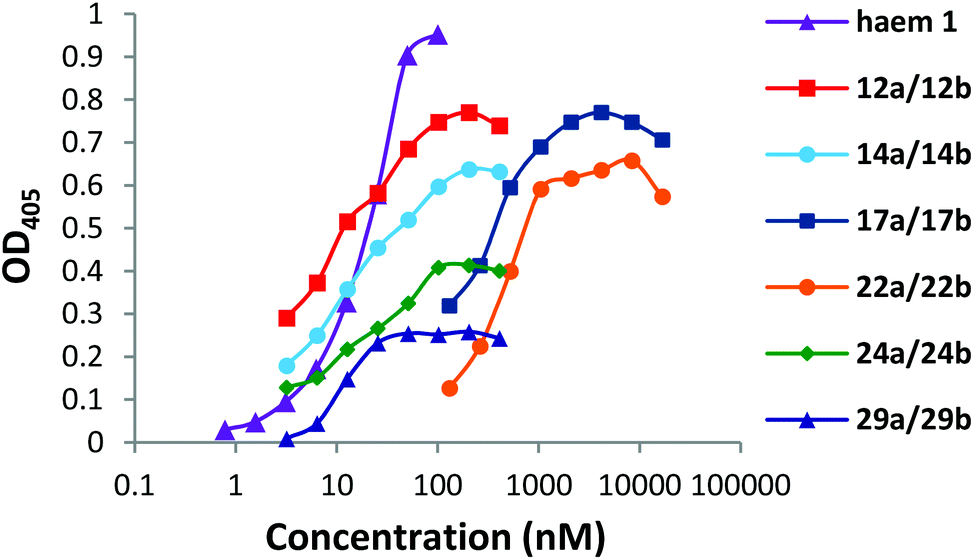 | ||
| Fig. 2 Binding curves of haem 1 and porphyrin-nitroimidazole adducts 12a/12b, 14a/14b, 22a/22b, 24a/24b, porphyrin-bis(nitroimidazole) adducts 17a/17b and the porphyrin-lysine controls 29a/29b to rHA2. | ||
DPIX-Lys-diNim adducts 17a and 17b (Kd50 200 nM) and DPIX-Lys(Boc)-Pro-Nim adducts 22a and 22b (Kd50 300 nM), with larger appended groups, had a significantly reduced affinity for rHA2, probably because of some size restrictions around the second propionate group. This might also have resulted from π–π interaction between the two nitroimidazoles or the nitroimidazole and the porphyrin ring leading to a greater three-dimensional bulk, thereby interfering with HA2 recognition. NMR spectroscopy rules this out, however, as the chemical shifts of the nitroimidazole protons are essentially the same across the range of adducts prepared in this study, even when such a π–π interaction cannot occur.
Bacteriology
Growth inhibition profiles in CDC media of P. gingivalis and a range of other anaerobic bacteria isolated from the human gastrointestinal tract, were determined in order to understand the behaviour of porphyrin nitroimidazole adducts (Fig. 3 and Table 1). To accommodate growth requirements of all nominated bacteria it was necessary to use enriched media (eBHI). On eBHI media, rate of kill comparisons were made to assess effectiveness of uptake and processing.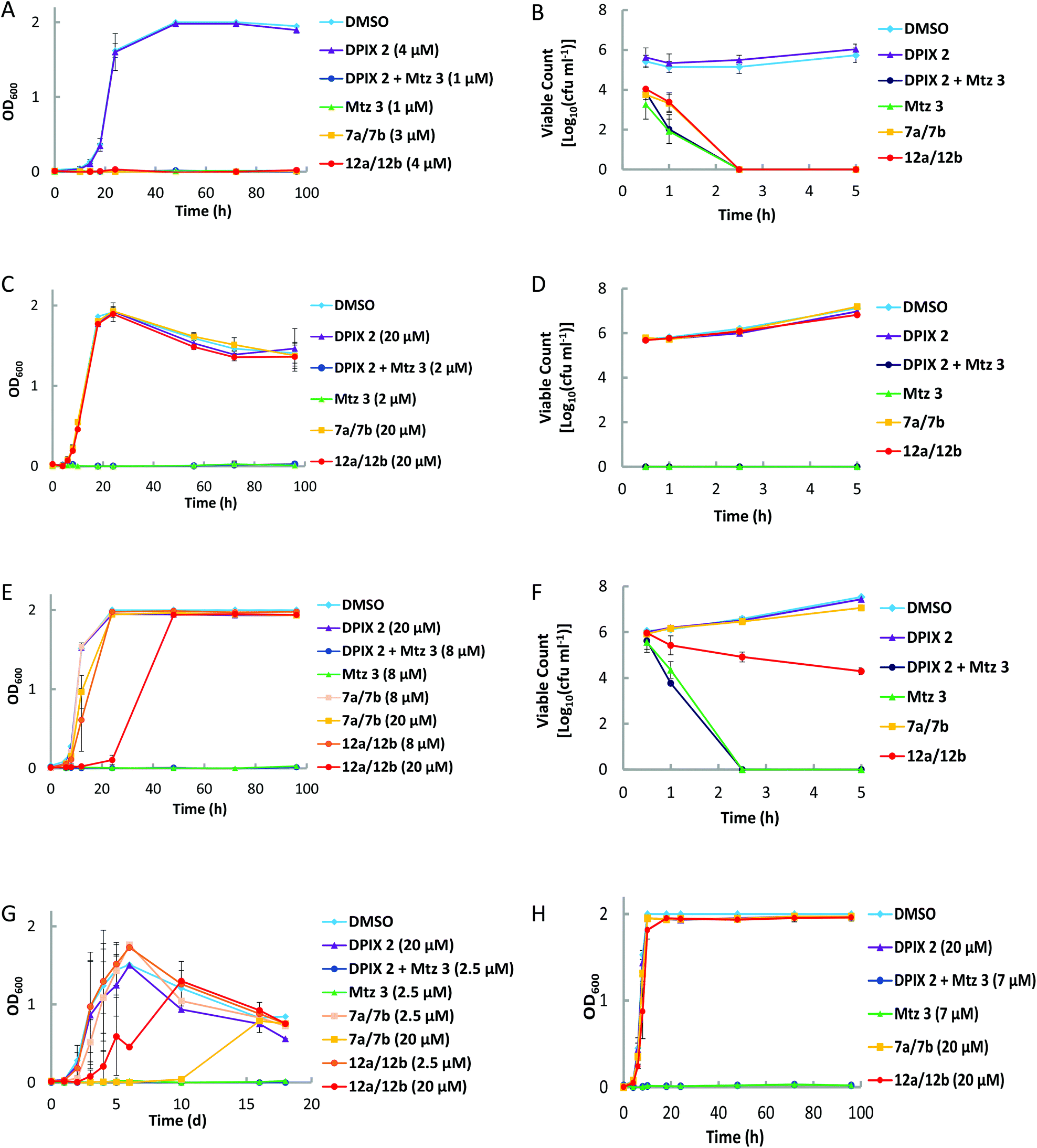 | ||
| Fig. 3 Growth curves or rate of kill assays for bacterial strains. (A) Growth of P. gingivalis in eBHI media; (B) viable count of P. gingivalis on solid eBHI media with 5 μM treatments; (C) growth of F. nucleatum in eBHI media; (D) viable count of F. nucleatum on solid eBHI media with 20 μM treatments; (E) growth of P. melaninogenica in eBHI media; (F) viable count of P. melaninogenica on solid eBHI media with 20 μM treatments; (G) growth of T. forsythia in eBHI media; (H) growth of B. fragilis in eBHI media. | ||
| Adduct | MIC (μM) |
|---|---|
| a See Fig. S1–S6 for growth curves. | |
| Mtz 3 | 1–2 |
| Mtz-NH24 | 10–20 |
| 12a and 12b | 1–2 |
| 14a and 14b | 10–20 |
| 17a and 17b | Not killed (at 20 μM) |
| 22a and 22b | Not killed (at 20 μM) |
| 24a and 24b | 1 |
| 29a and 29b | Growth supported |
DPIX-Lys(Boc)-Nim adducts 12a and 12b, whereby Mtz-NH24 is attached to DPIX 2via a Boc-protected lysine, have an MIC of <4 μM for P. gingivalis (in eBHI, Fig. 3A). In eBHI Mtz 3 has a lower MIC (<1 μM, Fig. 3A). It was found that 5 μM Mtz 3 killed P. gingivalis within 2.5 hours, as did equimolar 12a and 12b and 7a and 7b (in eBHI, Fig. 3B). The MIC for 12a and 12b in CDC was 1 μM (Table 1, Fig. S1†). In previous experiments (using CDC media), no growth of P. gingivalis was observed in Mtz 3 at <2 μM and in DPIX-Nim 7a and 7b at <1.25 μM.6
In eBHI 20 μM Mtz 3 rapidly killed F. nucleatum (<30 minutes), whilst 12a and 12b and 7a and 7b did not affect F. nucleatum (Fig. 3C). The MIC of Mtz 3 for F. nucleatum was 2 μM (Fig. 3D). Like F. nucleatum, B. fragilis was unaffected by adducts in eBHI media, but no growth was detected in the presence of 7 μM Mtz 3 (Fig. 3H).
It was found that 20 μM Mtz 3 killed P. melaninogenica, within 2.5 hours (Fig. 3F) and the MIC was 8 μM (Fig. 3E). DPIX-Lys(Boc)-Nim adducts 12a and 12b caused a dose-dependent initial reduction in P. melaninogenica (Fig. 3E and F) with recovery between 24–48 hours similar to the biomass of untreated controls (Fig. 3F). Adducts 7a and 7b (20 μM) affected the growth of P. melaninogenica slightly (Fig. 3E and F). The growth rate of T. forsythia was slowed by 12a and 12b as well as 7a and 7b in a dose-dependent manner (Fig. 3G). Mtz 3 at 2.5 μM prevented growth of T. forsythia. Indeed, P. melaninogenica and T. forsythia grew in 50 μM 7a and 7b and 12a and 12b, respectively.
DPIX-Lys(H)-Nim adducts 14a and 14b, essentially adducts 12a and 12b without a Boc group, are active against P. gingivalis at 20 μM (Table 1, Fig. S2†).
In an attempt to increase the potency of adducts against P. gingivalis, the DPIX-Lys(Nim)-Nim adducts 17a and 17b, with two nitroimidazoles attached, were synthesised. However, adducts 17a and 17b are inactive against P. gingivalis at 20 μM (Table 1, Fig. S3†).
DPIX-Lys(Boc)-Pro-Nim adducts 22a and 22b, with a proline-lysine linker bridging Mtz 3 to DPIX 2, retarded growth, but did not kill P. gingivalis at 20 μM. This could be due to weaker binding affinity for rHA2 (Fig. S4†).
DPIX-Pro-Nim adducts 24a and 24b, whereby Mtz-NH24 is attached to DPIX 2via a proline, are as active as the DPIX-Lys(Boc)-Min adducts 12a and 12b, with an MIC less than 1 μM (Table 1, Fig. S5†).
DPIX-Lys(Boc)-OH adducts 29a and 29b, essentially 12a and 12b without nitroimidazole attached, were synthesised as a control. Controls 29a and 29b supported growth of P. gingivalis at 20 μM (Table 1, Fig. S6†). This was expected as no antibiotic is attached, so these controls behave like DPIX 2 (support growth). Controls 29a and 29b were shown to bind rHA2 with a similar binding affinity to that of haem 1 (Fig. 2).
Gingipain activity studies
The P. gingivalis Kgp gingipain is lysine-specific and is associated with porphyrin accumulation at the surface of the bacterium. Preliminary work demonstrated that a peptide-biotin compound with a lysine-proline sequence was preferentially cleaved by P. gingivalis Kgp whereas this linkage is inefficiently cleaved by trypsin.6,13In vitro studies demonstrated efficient cleavage of DPIX-Lys(Boc)-Pro-Nim adducts 22a and 22b by purified Kgp (see Fig. S7a–S7c†). A kgp− mutant of P. gingivalis 33277 (KDP 129) and wild type were grown in the presence of adducts 22a and 22b with similar growth inhibitory activities observed for both adducts. In contrast the proline-linked DPIX-Pro-Nim adducts 24a and 24b were not cleaved by Kgp but were as active as metronidazole 3 for P. gingivalis (see Table 1). These results suggest that Kgp catalytic activity is not required for uptake and processing by P. gingivalis (see Fig. S7a–S7c†).14
Confocal studies
H413 are a human oral epithelial cell line. H413 readily take up 12a and 12b (Fig. 4). Concentrated depots of 12a and 12b were observed in the cytoplasm of the epithelial cells. Amide-linked adducts 6a and 6b and 7a and 7b were not taken up by epithelial cells (results not shown). The lysine-linked adducts 12a and 12b may therefore be effective against intracellular P. gingivalis.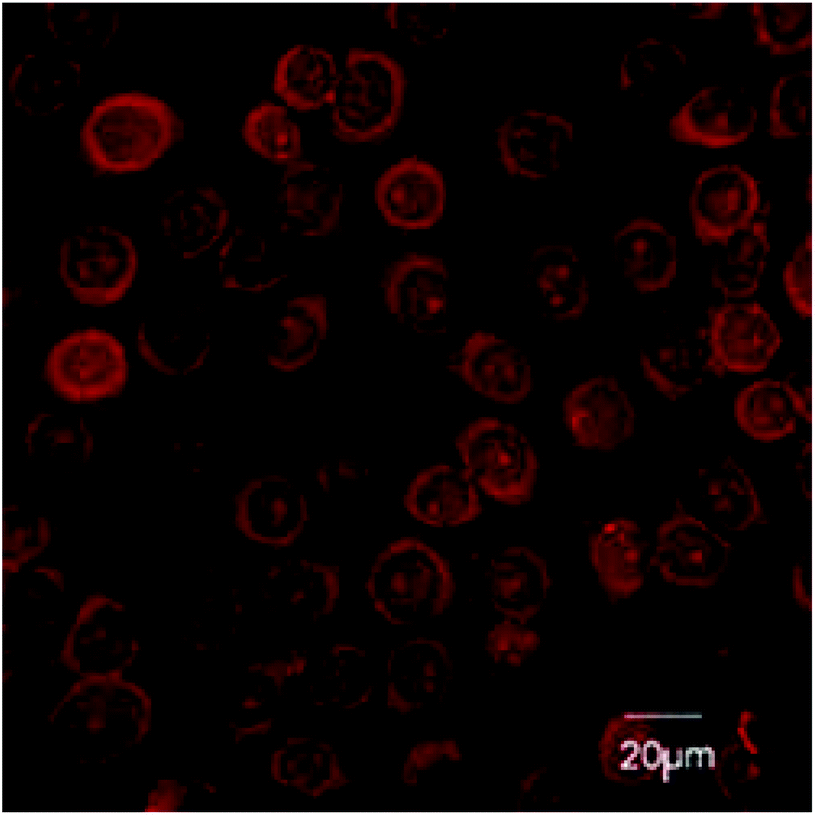 | ||
| Fig. 4 Confocal microscopy showing uptake of fluorescent 12a and 12b by oral epithelial cells. | ||
The surface receptor CD24 is highly expressed on diseased epithelium next to affected teeth.15,16 CD24-specific antibody attached to a peptide linker on the lysine side chain of 12a and 12b could localize the adducts to the tissue interface of disease sites. Studies are ongoing in our laboratories to further develop antibody CD24-mediated targeting.
Conclusions
A successful methodology for the synthesis of amino acid-linked porphyrin-nitroimidazole antibiotics targeting P. gingivalis has been developed. It involves a standard peptide-like coupling between the carboxylic acid of a free-base porphyrin DPIX 2 and an amino group of an antibiotic-appended amino acid or dipeptide and it affords the target compounds in good overall yields. DPIX-Lys(Boc)-Nim adducts 12a and 12b have been shown to have potential for selective action against P. gingivalis. The inactivity of adducts 17a and 17b suggests that certain modifications are not allowed with respect to HA2 recognition and highlights the need to refine the previously proposed HA2 binding site (Fig. 1) by indicating restriction in the space around the modified propionate. A new HA2 binding site model is proposed and is depicted in Fig. 5.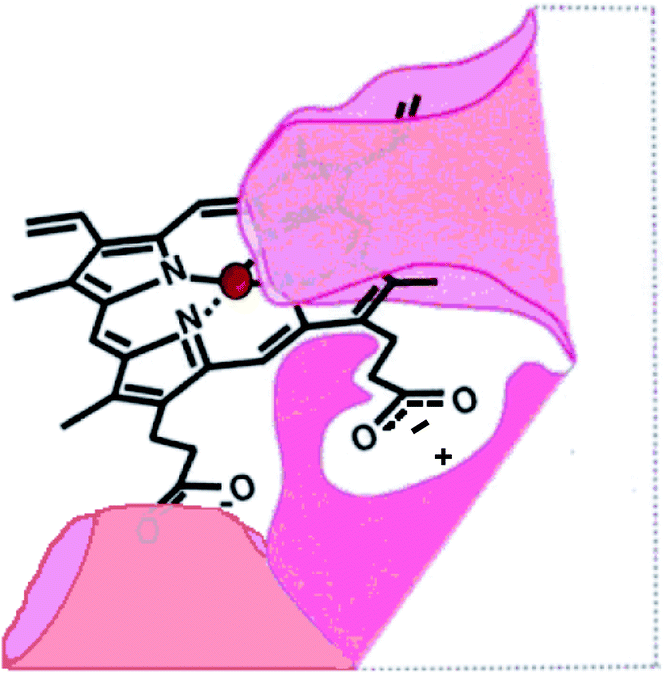 | ||
| Fig. 5 Newly proposed HA2 binding site showing space restrictions around the propionate region. | ||
It has been shown that some adducts, that bind rHA2 as well as haem 1, are ineffective in inhibiting the growth of P. gingivalis. This result highlights that not only is capture by HA2 necessary but for a drug to be active it needs to be successfully transported into the cell and/or utilised by the organism. The amino acid-linked porphyrin-nitroimidazole adducts with growth supporting or growth inhibitory activities are taken up by P. gingivalis. Findings from this study have also shown that Kgp is not critical for the activity of certain adducts as these adducts have the same albeit reduced growth inhibitory activities against both the Kgp− mutant and wild type of P. gingivalis.
Mtz 3 killed all anaerobic bacteria tested. DPIX-Lys(Boc)-Nim adducts 12a and 12b (also 7a and 7b) kill P. gingivalis with a similar potency to that of Mtz 3. In contrast, all anaerobic bacteria tested grew in the presence of 12a and 12b in concentrations approximately ten fold higher than the lethal Mtz 3 concentration. Indeed, P. melaninogenica and T. forsythia grew in 50 μM 7a and 7b and 12a and 12b, respectively.
To facilitate comparisons between fastidious T. fosythia and other anaerobes, we elected to use nutrient rich eBHI media (which is more nutritious than CDC media). In eBHI, F. nucleatum was unaffected by 7a and 7b, whereas we previously demonstrated retarded growth in the presence of 7a and 7b in CDC media.6 Indeed, 12a and 12b were more effective for P. gingivalis in CDC than in eBHI media. We also note the rate of growth and final biomass was higher for all organisms tested in eBHI medium. We cannot discount increased fitness of the organisms in eBHI medium, although these results indicate competition for uptake of adducts with other porphyrin sources, via porphyrin uptake pathways. We have not explicitly proved this. Thus, the effectiveness of porphyrin adducts will be impacted by local concentration of nutrients (including porphyrin).
Data herein inform practical considerations for the selective control of bacteria using porphyrin-based antibiotics.
Experimental
General procedures
Melting points were recorded on a Gallenkamp melting point apparatus and are uncorrected. 1H Nuclear Magnetic Resonance (1H NMR) spectra were recorded on a Bruker Avance DPX 300 spectrometer at a frequency of 300.13 MHz at 300 K. J values are given in Hz. MALDI-TOF mass spectra were recorded on a Micromass Tof Spec 2E spectrometer. ElectroSpray Ionisation (ESI) mass spectra were recorded on a ThermoQuest Finnigan LCQ Deca ion trap mass spectrometer. Infrared absorption spectra were recorded on a Shimadzu Model 8400 FTIR spectrophotometer and electronic absorption spectra were carried out on a Cary 5E UV-Vis spectrophotometer. Analytical thin layer chromatography (TLC) was performed using Merck Kieselgel silica gel 60 F-254 precoated sheets (0.2 mm) and preparative column chromatography was carried out routinely on Merck Kieselgel 60 silica gel (SiO2, 0.040–0.063 mm). Analytical and preparative reverse phase HPLC was carried out on a Waters 600E solvent delivery system with a Rheodyne 7125 injector and a Waters 486 UV detector using the solvent system stated. Amino acids are all of the naturally occurring L-configuration and are referred to using their standard three-letter codes as monomers.Preparation of precursor protected L-amino acids and dipeptides
:1 Hexane–EtOAc ramping to neat EtOAc). The fractions that contained the first band were discarded. The fractions containing the second band were evaporated to dryness to yield Fmoc-Lys(Boc)-Nim 9 (953 mg, 80%) as an off-white solid. 1H NMR (300 MHz; CDCl3) δH 1.42 (9H, s, tBu-H), 2.48 (3H, s, N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–CH3), 3.08–3.10 (2H, overlapping t, –CH2), 3.57–3.60 (2H, overlapping t, –CH2), 4.07–4.23 (4H, overlapping t, –CH2), 4.19 (1H, t, J 3.1, –CH), 4.26 (1H, m, αH), 4.41–4.44 (6H, overlapping t, –CH2), 4.72 (1H, br t, NH), 5.47 (1H, br d, NH), 6.85 (1H, br t, NH), 7.27–7.78 (8H, m, Ar–H), 7.92 (1H, s, NO2CCH); m/z (ESI-MS) 621.1 [(M + H)+ requires 621.3]; m/z (FTICR-MS) 621.3034 [(M + H)+, calcd for [C32H40N6O7 + H+]: 621.3042].
:1, 20 cm3) and the mixture was stirred at room temperature under N2 for 3 h. The solvent was removed under reduced pressure and DMF (1 cm3) was added, followed by CH2Cl2 (9 cm3). To this was added Mtz-CO2H 5 (46 mg, 0.248 mmol), followed by HBTU (96 mg, 0.253 mmol) and DIPEA (0.18 cm3, 1.033 mmol) and the mixture was stirred at room temperature under N2 overnight. The solvents were removed and the residue was purified by silica gel chromatography eluting with CH3OH–CH2Cl2 (1:4) to yield Fmoc-Lys(Nim)-Nim 15 (120 mg, 72%) as an off-white solid. 1H NMR (300 MHz; CDCl3) δH 2.46/2.48 (6H, s, NC–CH3), 3.22–3.26 (2H, overlapping t, –CH2), 3.58–3.62 (2H, overlapping t, –CH2), 4.07–4.12 (2H, overlapping t, –CH2), 4.15–4.21 (2H, overlapping t, –CH2), 4.24 (1H, m, αH), 4.39–4.44 (6H, m, –CH2), 4.84 (2H, overlapping t, –CH2), 5.63 (1H, br s, NH), 6.77 (1H, br t, NH), 7.01 (1H, br t, NH), 7.24–7.76 (8H, m, Ar–H), 7.90/7.93 (2H, s, NO2CCH); m/z (ESI-MS) 688.5 [(M + H)+ requires 688.7].
:1 Hexane–EtOAc ramping to neat EtOAc). The fractions that contained the first band were discarded. The fractions containing the second band were evaporated to dryness to yield Fmoc-Pro-Nim 19 (620 mg, 85%) as an off-white solid. 1H NMR (300 MHz; CDCl3) δH 2.12–2.38 (4H, m, –CH2), 2.45 (3H, s, NC–CH3), 3.30–3.45 (2H, m, –CH2), 3.55–3.60 (2H, overlapping t, –CH2), 4.20 (1H, t, J 3.1, –CH), 4.23 (1H, m, αH), 4.37–4.41 (4H, overlapping t, –CH2), 6.55 (1H, br t, NH), 7.24–7.75 (8H, m, Ar–H), 7.88 (1H, s, NO2CCH); m/z (ESI-MS) 490.4 [(M + H)+ requires 490.2]; m/z (FTICR-MS) 490.2001 [(M + H)+, calcd for [C26H27N5O5 + H+]: 490.2012].
:1 Hexane–EtOAc ramping to neat EtOAc). The fractions which contained the first band were discarded. The fractions containing the second band were evaporated to dryness to yield Fmoc-Lys(Boc)-Pro-Nim 20 (117 mg, 81%) as an off-white solid. 1H NMR (300 MHz; CDCl3) δH 1.42 (9H, s, tBu-H), 2.11–2.36 (4H, m, –CH2), 2.47 (3H, s, NC–CH3), 3.08–3.10 (2H, overlapping t, –CH2), 3.30–3.46 (2H, m, –CH2), 3.55–3.61 (2H, overlapping t, –CH2), 4.08–4.22 (4H, overlapping t, –CH2), 4.20 (1H, t, J 3.1, –CH), 4.24 (1H, m, αH) 4.27 (1H, m, αH), 4.40–4.43 (6H, overlapping t, –CH2), 4.61 (1H, br t, NH), 5.45 (1H, br d, NH), 6.75 (1H, br t, NH), 7.25–7.77 (8H, m, Ar–H), 7.90 (1H, s, NO2CCH); m/z (ESI-MS) 718.2 [(M + H)+ requires 718.3]; m/z (FTICR-MS) 718.3461 [(M + H)+, calcd for [C37H47N7O8 + H+]: 718.3486].
C–CH3), 3.08–3.10 (2H, overlapping t, –CH2), 3.57–3.60 (2H, overlapping t, –CH2), 4.07–4.23 (4H, overlapping t, –CH2), 4.19 (1H, t, J 3.1, –CH), 4.26 (1H, m, αH), 4.41–4.44 (6H, overlapping t, –CH2), 4.72 (1H, br t, NH), 5.47 (1H, br d, NH), 6.85 (1H, br t, NH), 7.27–7.78 (8H, m, Ar–H), 7.92 (1H, s, NO2CCH); m/z (ESI-MS) 621.1 [(M + H)+ requires 621.3]; m/z (FTICR-MS) 621.3034 [(M + H)+, calcd for [C32H40N6O7 + H+]: 621.3042].
:1, 20 cm3) and the mixture was stirred at room temperature under N2 for 3 h. The solvent was removed under reduced pressure and DMF (1 cm3) was added, followed by CH2Cl2 (9 cm3). To this was added Mtz-CO2H 5 (46 mg, 0.248 mmol), followed by HBTU (96 mg, 0.253 mmol) and DIPEA (0.18 cm3, 1.033 mmol) and the mixture was stirred at room temperature under N2 overnight. The solvents were removed and the residue was purified by silica gel chromatography eluting with CH3OH–CH2Cl2 (1:4) to yield Fmoc-Lys(Nim)-Nim 15 (120 mg, 72%) as an off-white solid. 1H NMR (300 MHz; CDCl3) δH 2.46/2.48 (6H, s, NC–CH3), 3.22–3.26 (2H, overlapping t, –CH2), 3.58–3.62 (2H, overlapping t, –CH2), 4.07–4.12 (2H, overlapping t, –CH2), 4.15–4.21 (2H, overlapping t, –CH2), 4.24 (1H, m, αH), 4.39–4.44 (6H, m, –CH2), 4.84 (2H, overlapping t, –CH2), 5.63 (1H, br s, NH), 6.77 (1H, br t, NH), 7.01 (1H, br t, NH), 7.24–7.76 (8H, m, Ar–H), 7.90/7.93 (2H, s, NO2CCH); m/z (ESI-MS) 688.5 [(M + H)+ requires 688.7].
:1 Hexane–EtOAc ramping to neat EtOAc). The fractions that contained the first band were discarded. The fractions containing the second band were evaporated to dryness to yield Fmoc-Pro-Nim 19 (620 mg, 85%) as an off-white solid. 1H NMR (300 MHz; CDCl3) δH 2.12–2.38 (4H, m, –CH2), 2.45 (3H, s, NC–CH3), 3.30–3.45 (2H, m, –CH2), 3.55–3.60 (2H, overlapping t, –CH2), 4.20 (1H, t, J 3.1, –CH), 4.23 (1H, m, αH), 4.37–4.41 (4H, overlapping t, –CH2), 6.55 (1H, br t, NH), 7.24–7.75 (8H, m, Ar–H), 7.88 (1H, s, NO2CCH); m/z (ESI-MS) 490.4 [(M + H)+ requires 490.2]; m/z (FTICR-MS) 490.2001 [(M + H)+, calcd for [C26H27N5O5 + H+]: 490.2012].
:1 Hexane–EtOAc ramping to neat EtOAc). The fractions which contained the first band were discarded. The fractions containing the second band were evaporated to dryness to yield Fmoc-Lys(Boc)-Pro-Nim 20 (117 mg, 81%) as an off-white solid. 1H NMR (300 MHz; CDCl3) δH 1.42 (9H, s, tBu-H), 2.11–2.36 (4H, m, –CH2), 2.47 (3H, s, NC–CH3), 3.08–3.10 (2H, overlapping t, –CH2), 3.30–3.46 (2H, m, –CH2), 3.55–3.61 (2H, overlapping t, –CH2), 4.08–4.22 (4H, overlapping t, –CH2), 4.20 (1H, t, J 3.1, –CH), 4.24 (1H, m, αH) 4.27 (1H, m, αH), 4.40–4.43 (6H, overlapping t, –CH2), 4.61 (1H, br t, NH), 5.45 (1H, br d, NH), 6.75 (1H, br t, NH), 7.25–7.77 (8H, m, Ar–H), 7.90 (1H, s, NO2CCH); m/z (ESI-MS) 718.2 [(M + H)+ requires 718.3]; m/z (FTICR-MS) 718.3461 [(M + H)+, calcd for [C37H47N7O8 + H+]: 718.3486].
Synthesis of deuteroporphyrin-metronidazole adducts
:97 CH3OH–CH2Cl2) to yield DPIX-Lys(Boc)-Nim methyl esters 11a and 11b (47 mg, 90%) as a reddish-black solid. m/z (ESI-MS) 905.5 [(M + H)+ requires 905.5]; m/z (FTICR-MS) 905.4671 [(M + H)+, calcd for [C48H60N10O8 + H+]: 905.4669]. Hydrolysis of 11a and 11b (100 mg, 0.110 mmol) occurred with LiOH (222 mg, 9.30 mmol) in CH3OH–THF–H2O (3:4:1, 20 cm3) with stirring overnight under N2 at room temperature. The solvent was removed to give DPIX-Lys(Boc)-Nim adducts 12a and 12b (99 mg, quant.) as a black solid. λmax (CH3OH)/nm 399 (logε dm3 mol−1 cm−1 5.18), 497 (4.04), 530 (3.87), 565 (3.78), 619 (3.44); νmax (KBr)/cm−1 3453w (NH), 3040m (OH), 2990m (aromatic CH), 2860m (aliphatic CH), 1711s (CO), 1651/1463m (CC), 1513/1367w (NO), 1166m (C–O); 1H NMR (300 MHz; DMSO-D6) δH −4.00 (2H, s, inner NH), 1.36 (9H, s, tBu-H), 1.85/1.99 (3H, s, NC–CH3), 2.52–2.55 (4H, overlapping t, –CH2), 3.05–3.17 (4H, overlapping t, –CH2), 3.25–3.30 (2H, overlapping t, –CH2), 3.67 (3H, s, –CH3), 3.71 (3H, s, –CH3), 3.76/3.78 (3H, s, –CH3), 3.76–3.81 (2H, overlapping t, –CH2), 3.80/3.81 (3H, s, –CH3), 4.15–4.20 (2H, overlapping t, –CH2), 4.22 (1H, m, αH), 4.40–4.45 (6H, overlapping t, –CH2), 6.40 (1H, br s, OC–NH), 7.89/7.92 (1H, s, NO2CCH), 7.99 (1H, br s, –NH), 9.34/9.37 (2H, s, β-pyrrolic H), 10.30–10.38 (4H, overlapping s, meso H), 12.45 (1H, br s, OC–OH); m/z (ESI-MS) 891.4 [(M + H)+ requires 891.5]; m/z (FTICR-MS) 891.4532 [(M + H)+, calcd for [C47H58N10O8 + H+]: 891.4512].
:4:1, 20 cm3) with stirring overnight under N2 at room temperature. The solvent was removed to give DPIX-Lys(Boc)-Nim adducts 12a and 12b as a black solid. To this was added CH2Cl2 (10 cm3) and CF3CO2H (2 cm3) and the mixture was stirred for 3 h at room temperature under N2. The solvent was removed and sodium hydrogen carbonate was added until the pH of the solution was approximately 6–7. The solvent was removed and the residue was passed through a reverse phase Zorbax Rx-SIL column eluting with CH3OH–H2O–CH3CO2H (80:20:0.5) over a gradient to CH3OH–CH3CO2H (100:0.5) to yield DPIX-Lys(H)-Nim adducts 14a and 14b (17 mg, 69%) as a black solid. λmax (CHCl3)/nm 395 (logε dm3 mol−1 cm−1 4.40), 495 (3.69), 528 (3.45), 565 (3.35), 618 (3.14); νmax (KBr)/cm−1 3492w (NH), 3065m (OH), 2980m (aromatic CH), 2864m (aliphatic CH), 1710s (CO), 1680/1466m (CC), 1573/1365w (NO), 1190m (C–O); 1H NMR (300 MHz; DMSO-D6) δH −4.01 (2H, s, inner NH), 1.78/1.91 (3H, s, NC–CH3), 2.39–2.45 (4H, overlapping t, –CH2), 3.00–3.13 (4H, overlapping t, –CH2), 3.21–3.26 (2H, overlapping t, –CH2), 3.63 (3H, s, –CH3), 3.67 (3H, s, –CH3), 3.72/3.73 (3H, overlapping s, –CH3), 3.72–3.77 (2H, overlapping t, –CH2), 3.75/3.77 (3H, s, –CH3), 4.05–4.11 (2H, overlapping t, –CH2), 4.15 (1H, m, αH), 4.35–4.40 (6H, overlapping t, –CH2), 6.32 (1H, br s, OC–NH), 7.32–7.44 (2H, br s, –NH), 7.86/7.87 (1H, s, NO2CCH), 9.32/9.34 (2H, s, β-pyrrolic H), 10.26–10.32 (4H, overlapping s, meso H), 12.45 (1H, br s, OC–OH); m/z (ESI-MS) 791.5 [(M + H)+ requires 791.9]; m/z (FTICR-MS) 791.3988 [(M + H)+, calcd for [C42H50N10O6 + H+]: 791.4004]. Retention time: 9.2 min; ZorbaxRx-SIL975.9; 1 mL min−1.
Method 2. DPIX-Lys(Boc)-Nim methyl esters 11a and 11b (30 mg, 0.033 mmol) was dissolved in CH2Cl2 (10 cm3). To this was added CF3CO2H (2 cm3) and the mixture was stirred for 3 h at room temperature under N2. The solvent was removed to yield DPIX-Lys(H)-Nim methyl esters 13a and 13b as a black solid m/z (ESI-MS) 805.47, [(M + H)+ requires 805.42]. To the residue was added LiOH (222 mg, 9.30 mmol) in CH3OH–THF–H2O (3:4:1, 20 cm3) and the mixture was stirred at room temperature under N2 overnight. The solvent was removed to yield a black solid. The residue was passed through a reverse phase Zorbax Rx-SIL column eluting with CH3OH–H2O–CH3CO2H (80:20:0.5) over a gradient to CH3OH–CH3CO2H (100:0.5) to yield DPIX-Lys(H)-Nim adducts 14a and 14b (17 mg, 65%) as a black solid with identical characterisation to the product from Method 1.
:95 CH3OH–CH2Cl2) to yield DPIX-Lys(Nim)-Nim methyl esters 16a and 16b (50 mg, 90%) as a black solid. m/z (ESI-MS) 972.9 [(M + H)+ requires 972.4]; m/z (FTICR-MS) 972.4454 [(M + H)+, calcd for [C49H57N13O9 + H+]: 972.4475]. Hydrolysis of 16a and 16b (40 mg, 0.042 mmol) occurred with LiOH (222 mg, 9.30 mmol) in CH3OH–THF–H2O (3:4:1, 10 cm3) with stirring overnight at room temperature. The solvent was removed to give adducts 17a and 17b (39 mg, quant.) as a black residue. The residue was passed through a reverse phase Zorbax Rx-SIL column eluting with CH3OH–H2O–CH3CO2H (80:20:0.5) over a gradient to CH3OH–CH3CO2H (100:0.5) to yield DPIX-Lys(Nim)-Nim adducts 17a and 17b as a black solid. λmax (CH3OH)/nm 398 (logε dm3 mol−1 cm−1 5.19), 497 (4.03), 528 (3.89), 564 (3.79), 618 (3.45); νmax (KBr)/cm−1 3455w (NH), 3048m (OH), 2992m (aromatic CH), 2863m (aliphatic CH), 1715s (CO), 1650/1462m (CC), 1512/1369w (NO), 1164m (C–O); 1H NMR (300 MHz; DMSO-D6) δH −4.04 (2H, s, inner NH), 2.27/2.30 (6H, s, NC–CH3), 2.84–2.90 (2H, overlapping t, –CH2), 3.06–3.13 (4H, overlapping t, –CH2), 3.15–3.18 (2H, overlapping t, –CH2), 3.30–3.55 (4H, overlapping t, –CH2), 3.60 (3H, s, –CH3), 3.64 (3H, s, –CH3), 3.72 (3H, s, –CH3), 3.75 (3H, s, –CH3), 3.80–3.89 (2H, overlapping t, –CH2), 3.97–4.01 (2H, overlapping t, –CH2), 4.15–4.19 (1H, m, αH), 4.30–4.39 (6H, overlapping t, –CH2), 7.86/7.88 (2H, s, NO2CCH), 8.32–8.36 (2H, m, –NH), 9.12–9.18 (1H, br s, –NH), 9.30/9.32 (2H, s, β-pyrrolic H), 10.24–10.28 (4H, overlapping s, meso H), 10.68 (1H, broad s, OC–OH); m/z (ESI-MS) 958.5 [(M + H)+ requires 958.4]; m/z (FTICR-MS) 958.4500 [(M + H)+, calcd for [C48H55N13O9 + H+]: 958.4530].
:97 CH3OH–CH2Cl2) to yield DPIX-Lys(Boc)-Pro-Nim mono-methyl esters 21a and 21b (43 mg, 75%) as a reddish-black solid. m/z (ESI-MS) 1002.4 [(M + H)+ requires 1002.5]; m/z (FTICR-MS) 1002.5120 [(M + H)+, calcd for [C53H67N11O9 + H+]: 1002.5123]. Hydrolysis of methyl esters 21a and 21b (20 mg, 0.0200 mmol) occurred with LiOH (100 mg, 4.19 mmol) in CH3OH–THF–H2O (3:4:1, 20 cm3) with stirring overnight under N2 at room temperature. The solvent was removed to give DPIX-Lys(Boc)-Pro-Nim 22a and 22b (19 mg, quant.) as a black solid. λmax (CH3OH)/nm 398 (logε dm3 mol−1 cm−1 5.15), 495 (4.05), 530 (3.83), 563 (3.75), 617 (3.43); νmax (KBr)/cm−1 3454w (NH), 3042m (OH), 2992m (aromatic CH), 2863m (aliphatic CH), 1714s (CO), 1650/1465m (CC), 1513/1365w (NO), 1170m (C–O); 1H NMR (300 MHz; DMSO-D6) δH −3.98 (2H, s, inner NH), 1.34 (9H, s, tBu-H), 2.10–2.33 (4H, m, –CH2), 2.46 (3H, s, NC–CH3), 2.72–2.77 (2H, overlapping t, –CH2), 3.09–3.18 (4H, overlapping t, –CH2), 3.21–3.28 (2H, overlapping t, –CH2), 3.33–3.49 (2H, m, –CH2), 3.55–3.62 (2H, overlapping t, –CH2), 3.66 (3H, s, –CH3), 3.69 (3H, s, –CH3), 3.72/3.75 (3H, s, –CH3), 3.78/3.81 (3H, s, –CH3), 4.09–4.21 (4H, overlapping t, –CH2), 4.22 (1H, m, αH) 4.26 (1H, m, αH), 4.41–4.45 (6H, overlapping t, –CH2), 4.64 (1H, br t, NH), 5.43 (1H, br d, NH), 6.60 (1H, br t, NH), 7.92 (1H, s, NO2CCH), 9.22/9.26 (2H, s, β-pyrrolic H), 10.03–10.10 (4H, overlapping s, meso H), 12.32 (1H, br s, OC–OH); m/z (ESI-MS) 988.7 [(M + H)+ requires 988.5]; m/z (FTICR-MS) 988.4966 [(M + H)+, calcd for [C52H65N11O9 + H+]: 988.4967].
:97 CH3OH–CH2Cl2) to yield DPIX-Pro-Nim methyl esters 23a and 23b (37 mg, 84%) as a reddish-black solid. m/z (ESI-MS) 774.5 [(M + H)+ requires 774.4]; m/z (FTICR-MS) 774.3647 [(M + H)+, calcd for [C42H47N9O6 + H+]: 774.3649]. Hydrolysis of methyl esters 23a and 23b (20 mg, 0.026 mmol) occurred with LiOH (100 mg, 4.19 mmol) in CH3OH–THF–H2O (3:4:1, 20 cm3) with stirring overnight under N2 at room temperature. The solvent was removed to give DPIX-Pro-Nim adducts 24a and 24b (19 mg, quant.) as a black solid. λmax (CH3OH)/nm 398 (logε dm3 mol−1 cm−1 5.15), 495 (4.02), 531 (3.88), 565 (3.76), 620 (3.43); νmax (KBr)/cm−1 3456w (NH), 3041m (OH), 2994m (aromatic CH), 2862m (aliphatic CH), 1712s (CO), 1648/1460m (CC), 1512/1364w (NO), 1164m (C–O); 1H NMR (300 MHz; DMSO-D6) δH −3.98 (2H, s, inner NH), 2.11–2.36 (4H, m, –CH2), 2.43 (3H, s, NC–CH3), 2.58–2.61 (2H, overlapping t, –CH2), 3.01–3.09 (2H, overlapping t, –CH2), 3.21–3.26 (2H, overlapping t, –CH2), 3.30–3.44 (2H, m, –CH2), 3.53–3.55 (2H, m, –CH2), 3.62 (3H, s, –CH3), 3.65 (3H, s, –CH3), 3.70/3.72 (3H, s, –CH3), 3.78/3.81 (3H, s, –CH3), 4.11–4.15 (2H, overlapping t, –CH2), 4.22 (1H, m, αH), 4.35–4.40 (2H, overlapping t, –CH2), 6.60 (1H, br t, NH), 7.75 (1H, s, NO2CCH), 9.22/9.26 (2H, s, β-pyrrolic H), 10.02–10.10 (4H, overlapping s, meso H), 12.28 (1H, br s, OC–OH); m/z (ESI-MS) 760.5 [(M + H)+ requires 760.3]; m/z (FTICR-MS) 760.3487 [(M + H)+, calcd for [C41H45N9O6 + H+]: 760.3493].
:99 CH3OH–CH2Cl2) and the solvent was removed to yield Fmoc-Lys(Boc)-DMA 27 (1.12 g, 77%) as an off-white solid. m/z (ESI-MS) 559.1 [(M + Na)+ requires 559.3]. Fmoc-Lys(Boc)-DMA 27 (160 mg, 0.298 mmol) was dissolved in CH2Cl2 (20 cm3). To this was added DBU (0.50 cm3, 0.322 mmol) and the mixture was stirred at room temperature under N2 for 1 h. HOBt (45 mg, 0.335 mmol) was added followed by DPIX mono–methyl esters 10 (120 mg, 0.229 mmol), HBTU (173 mg, 0.458 mmol) and DIPEA (0.157 cm3, 0.901 mmol). The mixture was stirred at room temperature under N2 overnight. Solvent was removed and the residue was purified by silica gel chromatography using CH3OH–CH2Cl2 (2:98). The solvent was removed to give the desired compounds 28a and 28b (154 mg, 82%) as a black solid. m/z (ESI-MS) 821.3 [(M + H)+ requires 821.0]; m/z (FTICR-MS) 821.4596 [(M + H)+, calcd for [C47H60N6O7 + H+]: 821.4602]. Hydrolysis of 28a and 28b (50 mg, 0.061 mmol) occurred with LiOH (222 mg, 9.30 mmol) in CH3OH–THF–H2O (3:4:1, 20 cm3) with stirring overnight at room temperature. The solvent was removed to give DPIX-Lys(Boc)OH regioisomers 29a and 29b (49 mg, quant.) as a black residue. The residue was passed through a reverse phase Zorbax Rx-SIL column eluting with CH3OH–H2O–CH3CO2H (80:20:0.5) over a gradient to CH3OH–CH3CO2H (100:0.5) to yield 29a and 29b as a black solid. λmax (CH3OH)/nm 399 (logε dm3 mol−1 cm−1 5.17), 496 (4.04), 529 (3.86), 564 (3.78), 618 (3.44); νmax (KBr)/cm−1 3455w (NH), 3041m (OH), 2988m (aromatic CH), 2858m (aliphatic CH), 1712s (CO), 1649/1460m (CC), 1510/1364w (NO), 1165m (C–O); 1H NMR (300 MHz; DMSO-D6) δH −4.19 (2H, s, inner NH), 1.27 (9H, s, tBu-H), 3.08–3.22 (4H, overlapping t, –CH2), 3.29–3.33 (2H, overlapping t, –CH2), 3.49/3.50 (3H, s, –CH3), 3.54/3.58 (3H, s, –CH3), 3.54–3.58 (2H, overlapping t, –CH2), 3.70 (3H, s, –CH3), 3.71 (3H, s, –CH3), 3.92–3.96 (1H, m, αH), 4.23–4.30 (2H, overlapping, –CH2), 4.32–4.38 (4H, overlapping t, –CH2), 4.42–4.50 (2H, overlapping t, –CH2), 5.11–5.22 (1H, overlapping d, –NH), 6.21–6.23 (1H, br t, –NH), 9.03 (2H, s, β-pyrrolic H), 9.90–10.02 (4H, overlapping s, meso H); m/z (ESI-MS) 738.9 [(M + H)+ requires 738.9]; m/z (FTICR-MS) 738.9243 [(M + H)+, calcd for [C41H50N6O7 + H+]: 738.9250].
General procedures for biological testing
Recombinant HA2 (rHA2) preparation, purification and ELISA were performed as described previously.1,2,6For growth curves, P. gingivalis was cultured in CDC broth supplemented with menadione and haemin (at 37 °C in an atmosphere containing CO2 (5%), H2 (10%) and N2 (85%)).2 A kgp− mutant of P. gingivalis (KDP 129) was cultured in BHI broth containing brain heart infusion (37 g L−1, Oxoid), yeast extract (5 g L−1, Oxoid), L-cysteine (0.5 mg L−1, Fluka) supplemented with menadione (1 mg L−1) and haemin (5 mg L−1) 1. Fusobacterium nucleatum ATCC 25586, Tannerella forsythia ATCC 43037, Prevotella melaninogenica ATCC 25845 and Bacteroides fragilis ATCC 25285 were grown in ‘enriched BHI’ (eBHI), which was BHI with additional fetal bovine serum (5% v/v) and N-acetylmuramic acid (10 mg L−1, Sigma) supplementation.
All rate of kill experiments were performed on solid eBHI with sheep blood (4% v/v) and agar (15 g L−1). For experimental evaluation log phase cultures were passaged as a 1% (v/v) inoculum (at 5 × 105 cfu mL−1) into media supplemented with menadione only. Under this condition the bacteria had sufficient haem 1 reserves to grow through a complete cycle as reported previously.2,6
Bacterial growth was monitored at 600 nm (with correction against reagent blanks). Data show mean values and standard error of the mean. Porphyrins, adducts and metronidazole derivatives were prepared as 5–10 mM stock solutions in DMSO (Sigma) for dilution into growth media. To control the possible inhibition of bacterial growth by DMSO, all cultures were adjusted to contain levels of this solvent equivalent to the highest concentration used in the experiment (1% v/v). All biological testing results were representative of at least two experiments.
Kgp (1 μL of a 1 μM solution) was incubated with porphyrin adducts (200 μM in 100μl) in the presence of cysteine (1 μM) for 1 h at 37 °C and the products of the reaction were monitored by HPLC-MS using a solvent system of CH3CN–H2O–CF3CO2H (10:90:0.5) over a gradient to CH3CN–H2O–CF3CO2H (60:40:0.5). In a control experiment, the Kgp was first inactivated with TLCK (4 mM) for 30 min before incubation with porphyrin adducts (200 μM) in the presence of cysteine (1 μM) for 1 h at 37 °C (see ESI†).
For confocal studies, monolayer cultures of H413 oral epithelial cells were incubated for 15 min with 20 μM adduct, washed and examined by confocal microscopy.
Extensive details of H413 cell culture and confocal microscopy were recently described by us.15,16
Acknowledgements
This work was supported by a Linkage Project Grant (LP0562660) from the Australian Research Council and a grant from The Australian Dental Research Foundation. We thank Professor K. Nakayama (Osaka University), for the kgp deletion mutant of P. gingivalis strain 33277.Notes and references
- M. Paramaesvaran, K.-A. Nguyen, E. Caldon, J. A. McDonald, S. Najdi, G. Gonzaga, D. B. Langley, A. DeCarlo, M. J. Crossley and N. Hunter, J. Bacteriol., 2003, 185, 2528–2537 CrossRef CAS PubMed.
- A. A. DeCarlo, M. Paramaesvaran, P. L. Yun, C. Collyer and N. Hunter, J. Bacteriol., 1999, 181, 3784–3791 CAS.
- N. Hunter, K.-A. Nguyen, J. A. McDonald, M. J. Quinn, D. B. Langley, M. J. Crossley and C. A. Collyer, J. Porphyrins Phthalocyanines, 2002, 6, 774–782 CrossRef CAS.
- H. E. Jakobsson, C. Jernberg, A. F. Andersson, M. Sjölund-Karlsson, J. K. Jansson and L. Engstrand, PLoS One, 2010, 5, e9836 Search PubMed.
- C. Jernberg, S. Löfmark, C. Edlund and J. K. Jansson, Microbiology, 2010, 156, 3216–3223 CrossRef CAS PubMed.
- B. C.-M. Yap, G. L. Simpkins, C. A. Collyer, N. Hunter and M. J. Crossley, Org. Biomol. Chem., 2009, 7, 2855–2863 CAS.
- D. B. Langley, C. A. Collyer and N. Hunter, WO Pat, 2002061091, 2002 Search PubMed.
- M. J. Crossley, B. C.-M. Yap, P. Thordarson, N. Hunter and C. A. Collyer, WO Pat, 2006005137, 2006 Search PubMed.
- P. Arnoux, R. Haser, N. Izadi, A. Lecroisey, M. Delepierre, C. Wandersman and M. Czjzek, Nat. Struct. Biol., 1999, 6, 516–520 CrossRef CAS PubMed.
- M. P. Hay, W. R. Wilson, J. W. Moselen, B. D. Palmer and W. A. Denny, J. Med. Chem., 1994, 37, 381–391 CrossRef CAS PubMed.
- S. M. Sondhi, A. Magan and J. W. Lown, Bull. Chem. Soc. Jpn., 1992, 65, 579–585 CrossRef CAS.
- M. Sedighi, S. Çalimsiz and M. A. Lipton, J. Org. Chem., 2006, 71, 9517–9518 CrossRef CAS PubMed.
- B. B. Willard and M. Kinter, J. Am. Soc. Mass Spectrom., 2001, 12, 1262–1271 CrossRef CAS PubMed.
- Y. Shi, D. B. Ratnayake, K. Okamoto, N. Abe, K. Yamamoto and K. Nakayama, J. Biol. Chem., 1999, 274, 17955–17960 CrossRef CAS PubMed.
- P. Ye, M. Simonian, M. Nadkarni, A. Decarlo, C. Chapple and N. Hunter, Clin. Exp. Immunol., 2005, 139, 328–337 CrossRef CAS PubMed.
- P. Ye, M. A. Nadkarni, M. Simonian and N. Hunter, BMC Cell Biol., 2009, 10, 2 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S6: Growth inhibition of P. gingivalis by the substrates 12a/12b, 14a/14b, 17a/17b, 22a/22b, 24a/24b and 29a/29b, respectively, Fig. S7: Enzyme activity studies using lysine-specific gingipain protease, NMR spectra and HRMS of new compounds. See DOI: 10.1039/c4ob01841a |
| ‡ Authors’ contribution was equal. |
| This journal is © The Royal Society of Chemistry 2015 |