
Synthesis of fully functionalized aglycone of lycoperdinoside A and B†
Balla
Chandrasekhar
,
Sudhakar
Athe
,
P. Purushotham
Reddy
and
Subhash
Ghosh
*
Organic and Biomolecular Chemistry Division, CSIR-Indian Institute of Chemical Technology, Hyderabad, India. E-mail: subhash@iict.res.in; Fax: +914027193275, +914027193108; Tel: +914027191604
First published on 1st October 2014
Abstract
This article reported the synthesis of fully functionalized aglycone of lycoperdinoside A and B. Pd-catalyzed Stille–Migita cross coupling between E-vinyl iodide 6 and E-vinyl stannane 23 established the highly substituted E,E-diene unit present in lycoperdinoside A and B. The other two Z-olefins present in the molecule were introduced by means of cis-selective Horner–Wadsworth–Emmons reaction with Still–Gennari phosphonate. Evans syn- and anti-aldol reactions were utilized to fix six of the seven stereo centres present in the aglycone.
Introduction
Myxomycetes are a unique group of organisms which produce a wide range of natural products having interesting structure and functions. In 2004 Řezanka and co-workers isolated lycoperdinoside A and B from the slime mold Enteridium lycoperdon.1 The detailed structure along with the absolute stereochemistry of lycoperdinoside A and B were established with the help of extensive NMR studies (Fig. 1). Although lycoperdinoside A and B were isolated almost ten years ago, surprisingly except for one synthetic study2 so far neither biological studies nor total synthesis have been reported. Because of their complex architecture and unknown biology, we became interested in developing a synthetic strategy for achieving the total synthesis of these molecules. Previously Chakraborty et al.2 reported the synthesis of C1–C9 and C10–C21 moieties of the aglycone of lycoperdinosides and proposed that their coupling can be achieved via the Suzuki coupling reaction. (S)-Roche ester was used as a chiral pool material to fix the two stereocenters (C4 and C8) out of the three present in the C1–C9 moiety, whereas the stereocenters present in the C10–C20 unit were introduced via the stereoselective aldol reaction (C17 and C18) and a Ti(III)-mediated opening of a trisubstituted epoxy alcohol (C15 and C16). In this article, we report the synthesis of the entire fully functionalized aglycone 30 of lycoperdinoside A and B, where a Pd-catalyzed Stille–Migita cross coupling reaction is used to install the highly substituted E,E-diene unit present in the molecule. The Z-olefins present in the molecule were introduced by means of Horner–Wadsworth–Emmons reaction using Still–Gennari phosphonate. C4 and C5 stereocenters present in the aglycone were introduced via the asymmetric Evans anti-aldol reaction, whereas the C15, C16, C17 and C18 stereocenters were fixed through the Evans asymmetric syn aldol reaction. The remaining C8 stereocenter in the aglycone was installed by means of Evans asymmetric alkylation.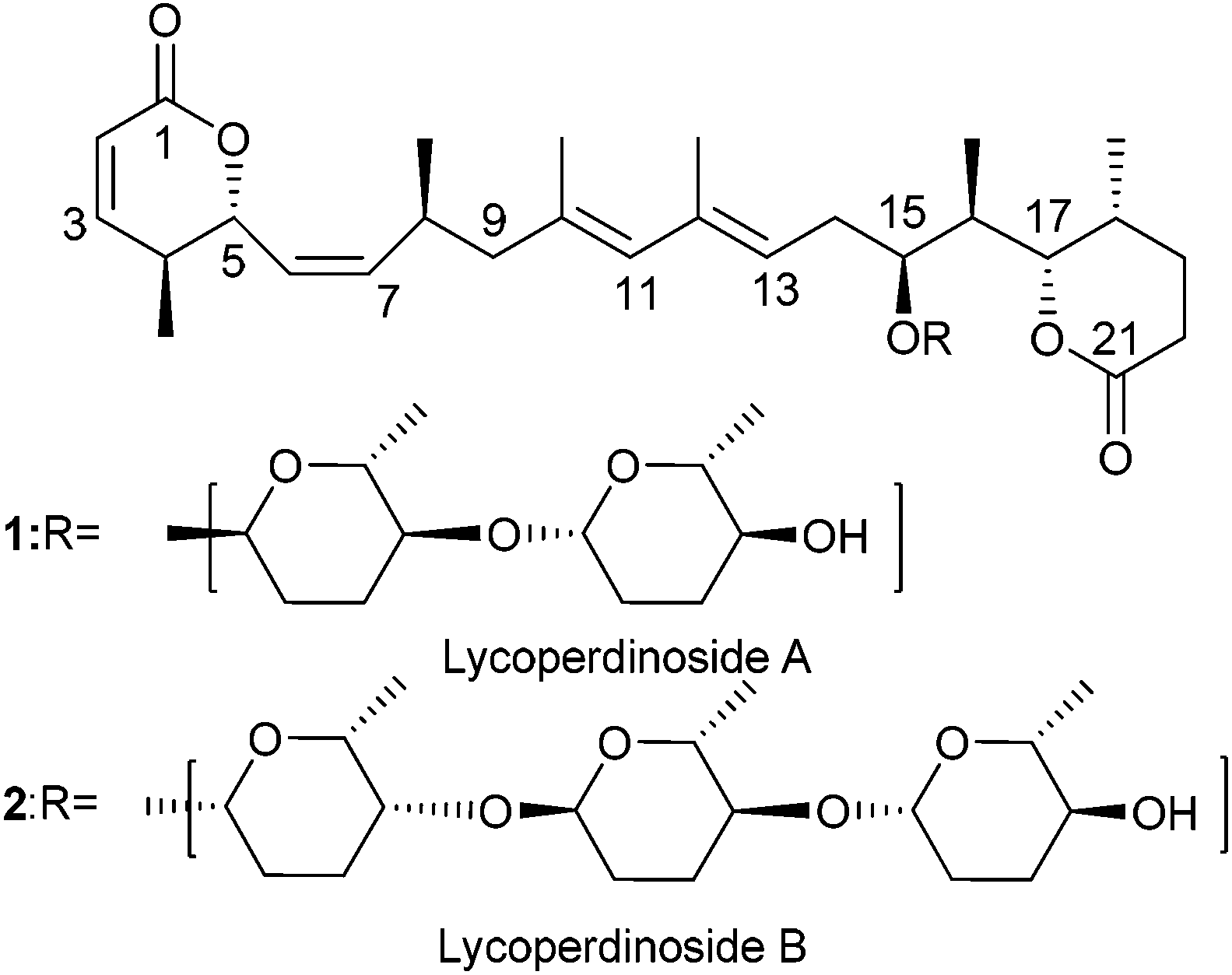 | ||
| Fig. 1 Structure of lycoperdinoside A and B. | ||
Results and discussion
Structural analysis of the aglycone of lycoperdinoside A and B revealed that it has a highly substituted 1,3-butadiene unit with E,E-configuration. We surmised that a Stille coupling3 between E-vinyl iodide 6 and E-vinyl stannane 5 could produce the diene unit with required stereochemistry. Vinyl stannane 5 could be synthesized from the alkyne compound 7, which in turn might be acquired from compound 8via the Evans aldol reaction.4 The vinyl iodide compound 6 could be obtained from compound 9, which in turn could be synthesized from compound 10 through the cis selective HWE reaction and magnesium chloride catalysed anti-aldol reaction developed by Evans et al.5 (Scheme 1).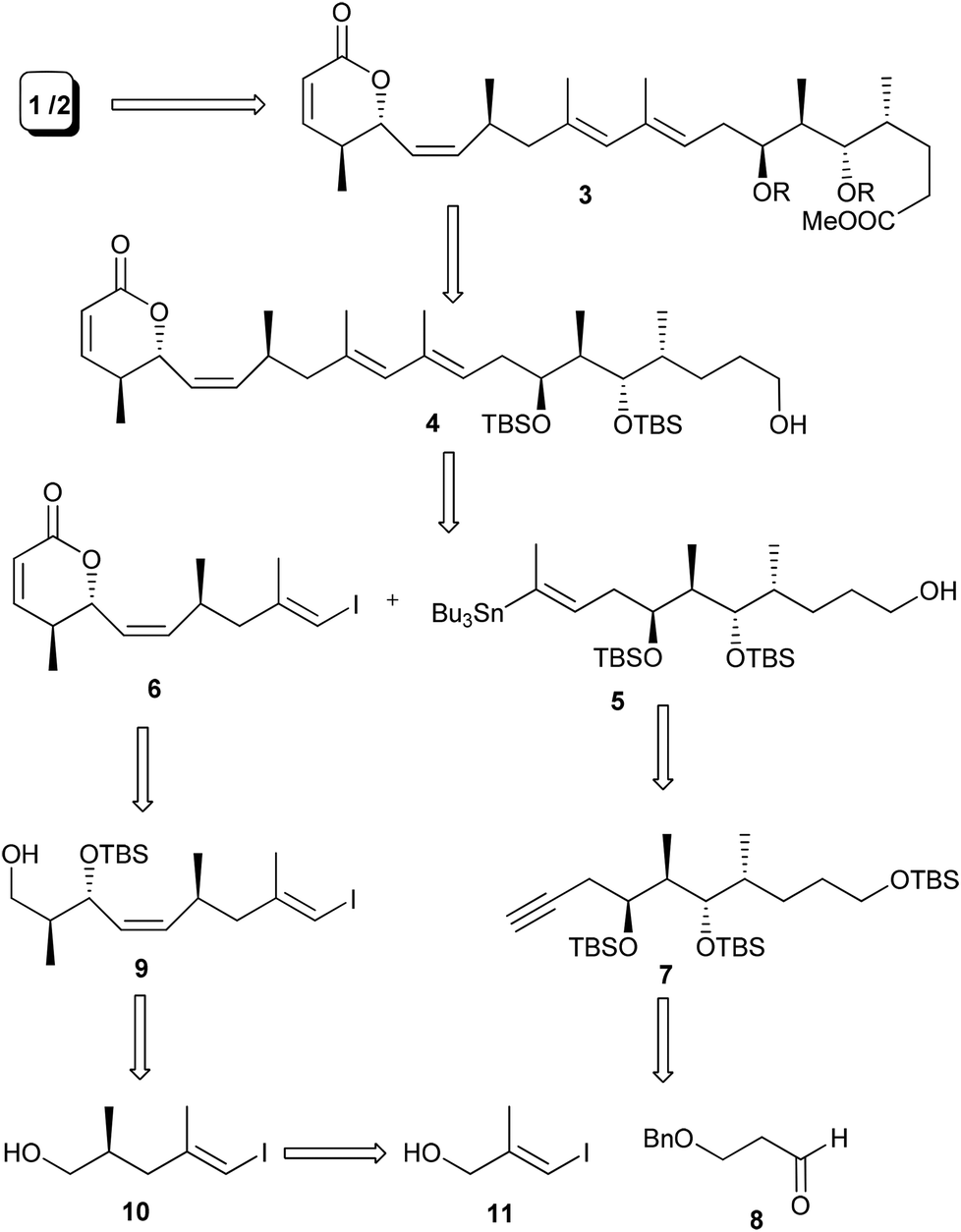 | ||
| Scheme 1 Retrosynthetic analysis of the aglycone of lycoperdinoside A and B. | ||
For the synthesis of fragment 5, compound 12 was prepared from compound 8 according to the reported procedure.6 Oxidation of the primary alcohol 12 under Swern conditions gave an aldehyde, which was subjected to Evans syn aldol reaction to provide compound 13 in 82% yield over two steps. Protection of the secondary alcohol with TBSOTf followed by removal of chiral auxiliary furnished alcohol 14 in 72% yield over two steps. Oxidation of the alcohol 14 followed by the Wittig reaction of the resulting aldehyde with two carbon stable ylide Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCOOEt afforded α,β-unsaturated ester 15 (E
CHCOOEt afforded α,β-unsaturated ester 15 (E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z = 98:2) in 80% yield over two steps. Reductions of the olefin as well as ester functional groups were carried out with LiBH4 to give primary alcohol 16 in 84% yield. TBS protection of the primary alcohol followed by debenzylation under hydrogenolysis conditions furnished primary alcohol 18 in 73% yield over two steps. Oxidation of the primary alcohol 18 with DMP gave an aldehyde which was subjected to Corey–Fuchs reaction7 to afford the alkyne compound 7 in 72% yield over three steps. Finally methylation of the alkyne compound 7 with BuLi, MeI and selective cleavage of the primary TBS ether with HF·Py provided the crucial intermediate 19 in 83% yield over two steps. Selectively primary TBS ether of 7 was cleaved at this stage with the thought that the stannane compound 5 will be polar, and will facilitate its separation from a highly non-polar unreacted stannane reactant. With the alkyne compound 19 in our hand, the stage was set for the crucial stannylation reaction. Accordingly compound 19 was subjected to stannylation under different conditions8 (Table 1) to provide the required stannane, however despite our best efforts we did not get the desired product 5 (Scheme 2).
:Z = 98:2) in 80% yield over two steps. Reductions of the olefin as well as ester functional groups were carried out with LiBH4 to give primary alcohol 16 in 84% yield. TBS protection of the primary alcohol followed by debenzylation under hydrogenolysis conditions furnished primary alcohol 18 in 73% yield over two steps. Oxidation of the primary alcohol 18 with DMP gave an aldehyde which was subjected to Corey–Fuchs reaction7 to afford the alkyne compound 7 in 72% yield over three steps. Finally methylation of the alkyne compound 7 with BuLi, MeI and selective cleavage of the primary TBS ether with HF·Py provided the crucial intermediate 19 in 83% yield over two steps. Selectively primary TBS ether of 7 was cleaved at this stage with the thought that the stannane compound 5 will be polar, and will facilitate its separation from a highly non-polar unreacted stannane reactant. With the alkyne compound 19 in our hand, the stage was set for the crucial stannylation reaction. Accordingly compound 19 was subjected to stannylation under different conditions8 (Table 1) to provide the required stannane, however despite our best efforts we did not get the desired product 5 (Scheme 2).
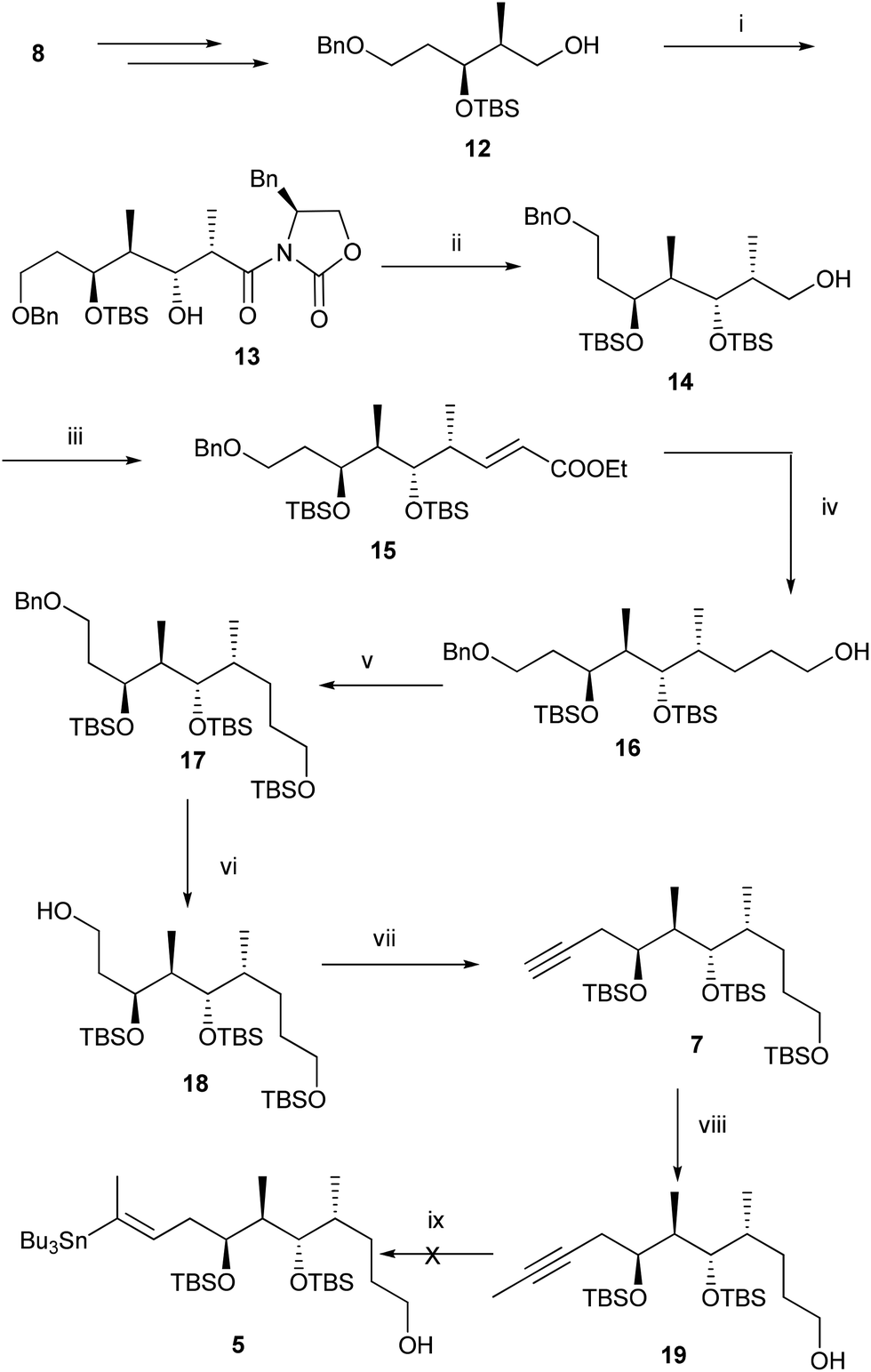 | ||
| Scheme 2
Reagents and conditions: (i) (a) (COCl)2, DMSO, Et3N, CH2Cl2, −78 °C to 0 °C, 1 h; (b) (S)-4-benzyl-3-propionyloxazolidin-2-one, Bu2BOTf, DIPEA, 0 °C, then aldehyde, −78 °C, 2.5 h, 82%; (ii) (a) TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C–rt, 24 h; (b) NaBH4, THF–H2O (5:1), 10 h, 72% over two steps; (iii) (a) DMP, CH2Cl2, 0 °C-rt, 1 h; (b) Ph3PCHCOOEt, C6H6, 90 °C, 1.5 h, 80%; (iv) LiBH4, THF, 0 °C–rt, 48 h, 84%; (v) TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C-rt, 30 min, 86%; (vi) H2/Pd–C, EtOAc, 5 h, 85%; (vii) (a) DMP, CH2Cl2, 0 °C–rt, 1 h; (b) PPh3, CBr4, Et3N, CH2Cl2, 0 °C, 2.5 h; (c) n-BuLi, THF, −78 °C to 0 °C, 1 h, 72% over three steps; (viii) (a) n-BuLi, MeI, THF, −78 °C to 0 °C, 1 h; (b) HF·Py, THF, 0 °C–rt, 6 h, 83% over two steps; (ix) see Table 1. | ||
| Entry | Conditions | Yield |
|---|---|---|
| 1 | PdCl2(PPh3)2, Bu3SnH, THF, rt | Traces |
| 2 | Bu3Sn(Bu)CuLi·LiCN, THF, MeOH, −78 °C to −10 °C | — |
| 3 | Bu3SnH, AIBN, Toluene, 80 °C | Traces |
This failure of stannylation forced us to develop an alternate strategy as shown in Scheme 3. In 2002, Marshall et al.9 reported that the electron withdrawing group like the –CH2OAc substituent at the terminal position facilitates the stannylation at the C-2 position. With this report, we thought that the stannylation reaction at the C2 position can be facilitated in a substrate like 22 and the CH2OAc can be converted to CH3 at the later stage of the synthesis. With this plan the alkyne 7 was reacted with HCHO in the presence of BuLi to furnish compound 20 which on acetylation afforded compound 21 in 72% yield over two steps. Selectively TBS ether of the primary alcohol was cleaved at this stage with HF·Py to give polar compound 22. Compound 22 on treatment with Bu3SnH in the presence of Pd(PPh3)2Cl2 in THF furnished chromatographically pure stannane compound 23 in 68% yield.
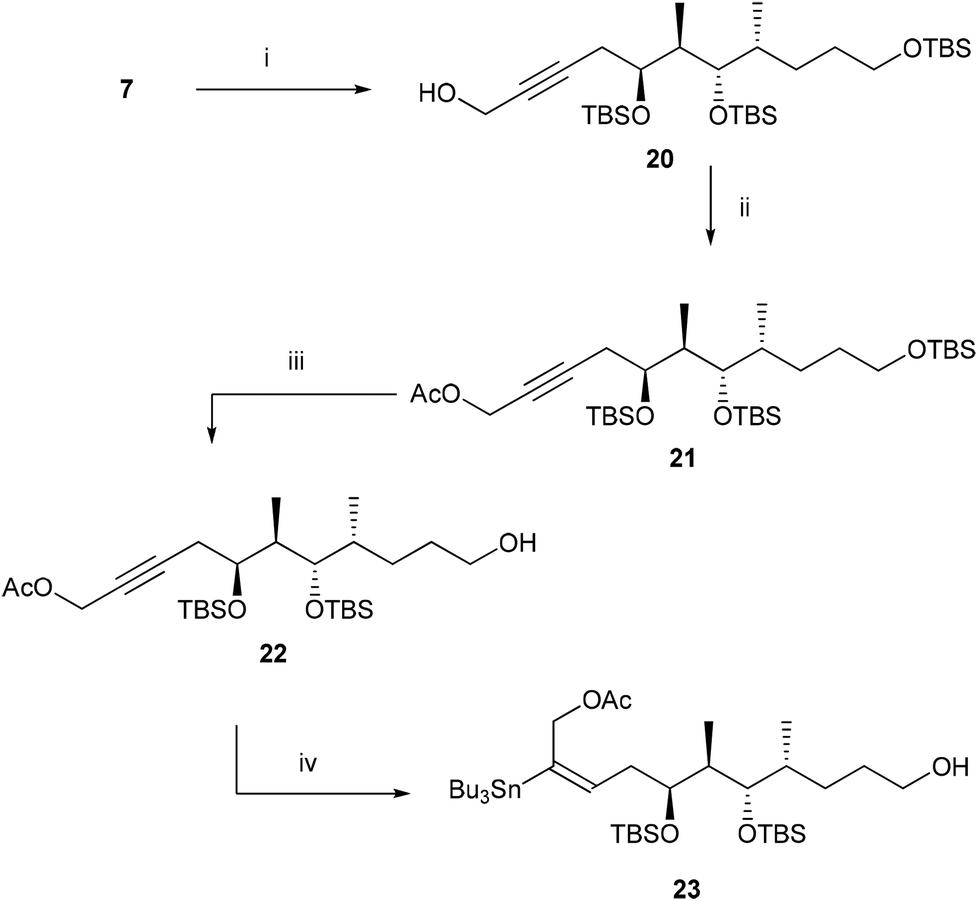 | ||
| Scheme 3 Reagents and conditions: (i) n-BuLi, (CH2O)n, THF, −78 °C–rt, overnight, 72%; (ii) Ac2O, Et3N, DMAP, CH2Cl2, 0 °C–rt, 30 min, quantitative yield; (iii) HF·Py, THF, 0 °C–rt, 6 h, 85%; (iv) Pd(PPh3)2Cl2, Bu3SnH, THF, rt, 30 min, 68%. | ||
Next we turn our attention for the synthesis of vinyl iodide 6. The Evans alkylation reaction10 with the known iodide 1111 furnished compound 24 (dr = 92:8) in 51% yield over three steps, which on reaction with NaBH4 in THF–H2O afforded known primary alcohol 1012 in 85%. Oxidation of alcohol 10 with DMP gave an aldehyde, which on cis selective Horner–Wadsworth–Emmons reaction with Still–Gennari phosphonate furnished compound 25 (Z:E = 95:5) in 75% over two steps. DIBAL-H reduction13 of 25 afforded allylic alcohol 26, which on oxidation followed by magnesium chloride catalysed anti aldol reaction under Evans protocol furnished compound 27 in 59% yield over three steps. Protection of the secondary alcohol with TBSOTf followed by the removal of chiral auxiliary from 27 gave primary alcohol 9, which on oxidation followed by the Z-selective HWE reaction afforded compound 28 (Z:E = 93:7) in 53% yield over four steps. Finally acid catalysed TBS group deprotection followed by in situ lactonization completed the synthesis of vinyl iodide fragment 6 (Scheme 4).
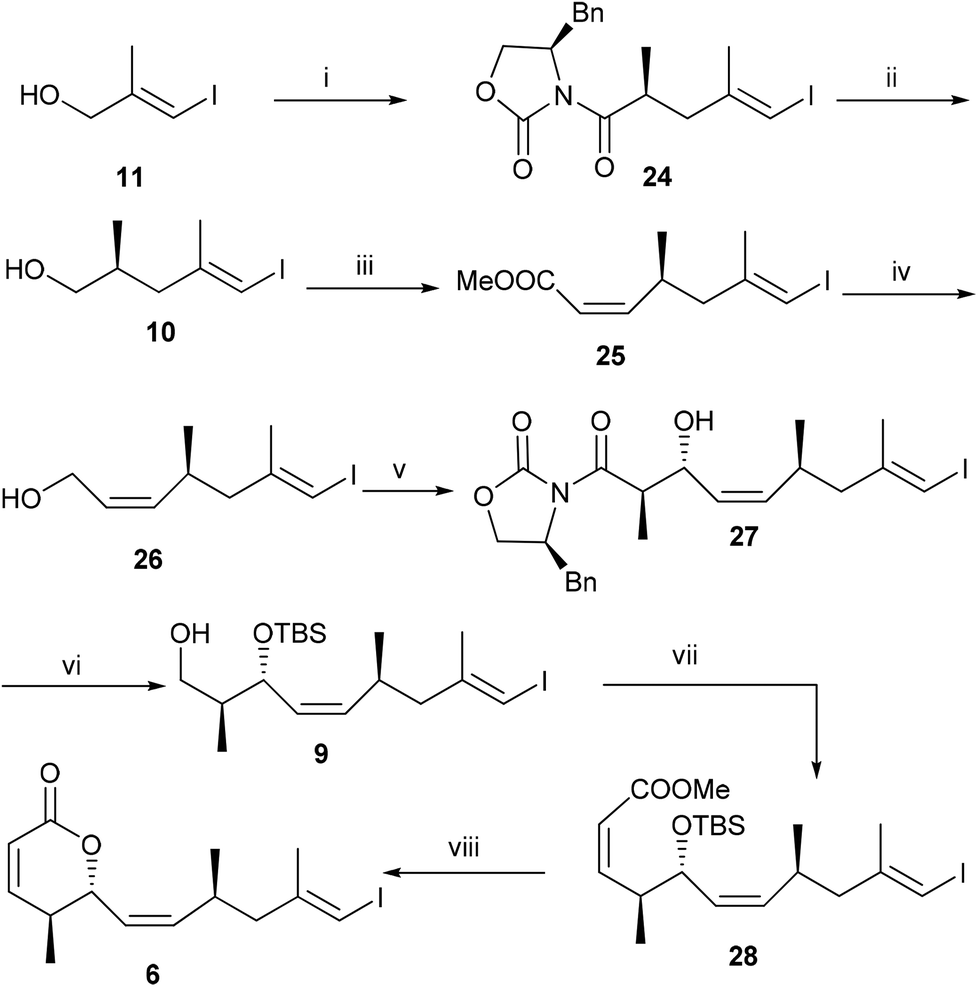 | ||
| Scheme 4
Reagents and conditions: (i) (a) MsCl, Et3N, DMAP, CH2Cl2, 0 °C–rt, 6 h; (b) NaI, THF, rt, 3 h; (c) (R)-4-benzyl-3-propionyloxazolidin-2-one, NaHMDS, −78 °C to 0 °C, overnight, 51% over three steps; (ii) NaBH4, THF–H2O (5:1), 10 h, 85%; (iii) (a) DMP, CH2Cl2, 0 °C–rt; (b) CH3O2CCH2P(O)(OCH2CF3)2, NaH, −78 °C, 6 h, 75%; (iv) 1 M DIBAL-H, CH2Cl2, −78 °C, 1 h, 84%; (v) (a) DMP, CH2Cl2, 0 °C–rt; (b) (S)-4-benzyl-3-propionyloxazolidin-2-one, MgCl2, Et3N, TMSCl, rt, 24 h, 70%; (vi) (a) TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C-rt, 1 h; (b) NaBH4, THF–H2O (5:1), 0 °C–rt, overnight, 74% over two steps. (vii) (a) DMP, CH2Cl2, 0 °C–rt, (b) CH3O2CCH2P(O)(OCH2CF3)2, NaH, −78 °C, 6 h, 72%; (viii) CSA, MeOH, 0 °C–rt, 6 h, 86%. | ||
Having both the fragments in hand, the crucial Pd-catalyzed Stille coupling14 between 23 and 6 was tried under different reaction conditions (Table 2). The best result was obtained when the coupling reaction was carried out under Stille–Migita cross coupling conditions15 using the combination of catalytic amounts of Pd(PPh3)4, copper thiophene-2-carboxylate (CuTC)16 and [Ph2PO2][NBu4]17 in a THF and DMF mixture to give compound 29 in 45% yield (Scheme 5). The geometry of the newly formed diene unit in 29 was confirmed by NOE studies.18 Finally, two-step oxidation of the primary alcohol followed by esterification of the resulting acid completed the synthesis of C1–C21 aglycone 30, of lycoperdinoside A and B.
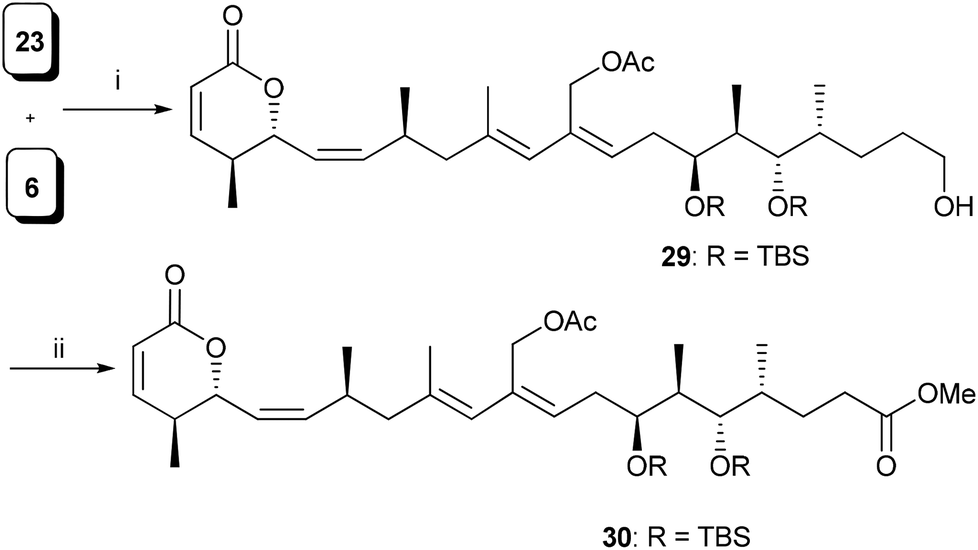 | ||
| Scheme 5
Reagents and conditions: (i) (a) see Table 2, (ii) (a) DMP, CH2Cl2, 0 °C–rt, (b) NaClO2, NaH2PO4, t-BuOH:H2O, 2-methyl-2-butene, 0 °C–rt, 1 h; (c) CH2N2, Et2O, 0 °C, 30 min, 70% over three steps. | ||
| Entry | Catalyst (10 mol%) | Conditions | Yield |
|---|---|---|---|
| 1 | Pd(CH3CN)2Cl2 | LiCl, Hunig's base, DMF, rt, 10 h | 10% |
| 2 | Pd(PPh3)4 | CuTC, DMF, THF, rt, 10 h | 20% |
| 3 | Pd(PPh3)4 | CuTC, [Ph2PO2][NBu4], DMF, THF, rt, 4 h | 45% |
Conclusions
In conclusion, we have developed a highly convergent synthetic strategy for the synthesis of aglycone of lycoperdinoside A and B. Key features of our strategy include Pd-catalysed Stille–Migita cross coupling, the cis selective HWE reaction and the aldol reaction. We are currently working with this strategy to achieve the total synthesis of lycoperdinoside A, which will be reported in due course.Experimental section
(S)-4-Benzyl-3-((2S,3R,4S,5S)-7-(benzyloxy)-5-(tert-butyldimethylsilyloxy)-3-hydroxy-2,4-dimethylheptanoyl)oxazolidin-2-one (13)
To a stirred solution of (COCl)2 (1.93 mL, 22.15 mmol) in CH2Cl2 (40 mL) at −78 °C was added DMSO (3.36 mL, 47.26 mmol) with stirring under a nitrogen atmosphere. After 20 min, a solution of compound 12 (5 g, 14.77 mmol) in CH2Cl2 (20 mL) was added to the reaction mixture via a cannula and stirred for 30 minutes. Then Et3N (10.3 mL, 73.85 mmol) was added to the reaction mixture and stirred for another 30 min at −78 °C and 30 min at 0 °C. The reaction mixture was quenched with saturated aqueous NH4Cl (20 mL) and extracted with EtOAc (2 × 100 mL). The combined organic extracts were washed with water (50 mL), saturated aqueous NaCl (50 mL) and dried over Na2SO4. Evaporation of the solvent under reduced pressure provided an aldehyde which was passed through a silica gel column and used for the next reaction.Di-n-butylborontriflate (1 M in CH2Cl2, 17.7 mL, 17.72 mmol) was added to a solution of (S)-4-benzyl-3-propionyloxazolidin-2-one (3.79 g, 16.24 mmol) in CH2Cl2 (40 mL) at 0 °C under a nitrogen atmosphere, followed by the addition of DIPEA (3.34 mL, 19.2 mmol). After stirring at 0 °C for 1 h, a solution of the above aldehyde in CH2Cl2 (10 mL, 2×) was added at −78 °C. The resulting pale yellow solution was stirred at −78 °C for 1.5 h and at 0 °C for 30 minutes. The reaction mixture was quenched at 0 °C with phosphate buffer (pH = 7, 21 mL) followed by MeOH (75 mL), resulting in a homogeneous solution. After 5 min, 21 mL of 30% aqueous H2O2 in MeOH (30 mL) was added over a period of 30 min. After stirring at 0 °C for 1 h, the solvent was removed by rotary evaporation and the resulting residual oil was extracted with EtOAc (2 × 100 mL). The combined organic extracts were washed with saturated aqueous NaHCO3 (50 mL), water (50 mL), saturated aqueous NaCl (50 mL) and dried over Na2SO4. Evaporation of the solvent under reduced pressure furnished the crude product which on purification via column chromatography (silica gel, 15% EtOAc in petroleum ether as an eluent) provided the aldol product 13 (6.89 g, 82%) as a colourless liquid.
R f = 0.5 (SiO2, 30% EtOAc in petroleum ether); [α]24D = +14.52 (c 1.37, CHCl3); IR νmax 3456, 2929, 2856, 2363, 2333, 1778, 1703, 1458, 1384, 1206, 1103, 836, 770, 741, 700 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.29–7.18 (m, 10H), 4.62 (m, 1H), 4.49 (d, J = 12.0 Hz, 1H), 4.41 (d, J = 12.0 Hz, 1H), 4.18–4.09 (m, 3H), 3.97–3.88 (m, 2H), 3.80 (qd, J = 6.8, 1.5 Hz, 1H), 3.48 (dd, J = 5.4, 6.6 Hz, 2H), 3.35 (dd, J = 13.2, 2.6 Hz, 1H), 2.70 (dd, J = 13.0, 10.0 Hz, 1H), 1.92–1.72 (m, 3H), 1.18 (d, J = 6.7 Hz, 3H), 0.88 (s, 9H), 0.85 (d, J = 7.1 Hz, 3H), 0.11 (s, 3H), 0.06 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 175.8, 153.1, 138.3, 135.4, 129.4(2), 128.9(2), 128.3(2), 127.6(2), 127.5, 127.2, 73.4, 73.0, 72.8, 66.9, 66.1, 55.8, 40.6, 39.8, 37.7, 32.0, 25.8, 17.9, 12.3, 8.3, −4.5, −5.0; MS (ESI) m/z 570 [M + H]+, 592 [M + Na]+; HRMS (ESI, QSTAR-TOF) calcd for C32H47O6NSiNa [M + Na]+ 592.3064, found 592.3062.
(2R,3S,4R,5S)-7-(Benzyloxy)-3,5-bis(tert-butyldimethylsilyloxy)-2,4-dimethylheptan-1-ol (14)
To a solution of compound 13 (6.6 g, 11.58 mmol) in CH2Cl2 (35 mL), 2,6-lutidine (4.05 mL, 34.74 mmol) and TBSOTf (2.95 mL, 12.74 mmol) were added sequentially at 0 °C, under a nitrogen atmosphere. The reaction mixture was allowed to warm to room temperature and stirred for 24 h. Then the reaction mixture was quenched by adding saturated aqueous NaHCO3 (20 mL). The resulting mixture was extracted with EtOAc (2 × 100 mL) and the combined organic extracts were washed with saturated aqueous CuSO4 solution (2 × 20 mL), water (50 mL), brine (50 mL), dried over Na2SO4 and concentrated in vacuo to afford the TBS protected compound as a yellowish liquid which was used as such for the next reaction.To a solution of the above TBS protected compound in a mixture of THF and water (5:1) (60 mL) was added NaBH4 (2.19 g, 57.90 mmol) at 0 °C. The reaction mixture was allowed to attain room temperature and stirred overnight. Then the reaction mixture was quenched by the addition of saturated aqueous NH4Cl (20 mL) at 0 °C. The solvent was removed by rotary evaporation and the resulting residual oil was extracted with EtOAc (2 × 100 mL). The combined organic layers were washed with water (50 mL), saturated aqueous NaCl (50 mL), dried (Na2SO4), filtered and concentrated under vacuum. Purification by column chromatography (silica gel, 5–8% EtOAc in the petroleum ether eluent) afforded pure compound 14 (4.26 g, 72% over two steps) as light yellow oil.
R f = 0.45 (SiO2, 10% EtOAc in petroleum ether); [α]24D = −5.33 (c 1.12, CHCl3); IR νmax 3419, 2952, 2929, 2856, 2363, 1463, 1363, 1252, 1079, 1039, 833, 771, 672 cm−1; 1H NMR (300 MHz, CDCl3): δ 7.34–7.25 (m, 5H), 4.48 (ABq, J = 12.8 Hz, 2H), 3.91 (dd, J = 6.0, 1.5 Hz, 1H), 3.84 (q, J = 5.2 Hz, 1H), 3.53–3.38 (m, 4H), 1.96–1.65 (m, 6H), 0.97–0.81 (m, 24H), 0.08–0.04 (m, 12H); 13C NMR (75 MHz, CDCl3): δ 138.4, 128.3, 127.7, 127.5, 73.0, 72.4, 71.3, 66.9, 66.8, 44.0, 38.0, 35.5, 26.0, 25.9, 18.3, 18.2, 11.2, 11.1, −3.6, −3.7, −3.9, −4.5; MS (ESI) m/z 511 [M + H]+, 533 [M + Na]+; HRMS (ESI, QSTAR-TOF) calcd for C28H54O4Si2Na [M + Na]+ 533.3458, found 533.3462.
(4R,5S,6R,7S,E)-Ethyl-9-(benzyloxy)-5,7-bis(tert-butyldimethylsilyloxy)-4,6-dimethylnon-2-enoate (15)
To a solution of compound 14 (3.5 g, 6.85 mmol) in CH2Cl2 (20 mL) at 0 °C, NaHCO3 (1.15 g, 13.7 mmol) was added followed by Dess–Martin periodinane (4.36 g, 10.27 mmol) stirring under a nitrogen atmosphere. The reaction mixture was allowed to attain room temperature and stirred for 1 h. Saturated aqueous NaHCO3 (20 mL) and saturated aqueous Na2S2O3 (20 mL) were added. The resulting biphasic mixture was stirred for 15 min and then extracted with EtOAc (2 × 75 mL). The combined organic phases were washed with water (30 mL), brine (30 mL), dried (Na2SO4) and concentrated in vacuo. The crude aldehyde (Rf = 0.5, 5% EtOAc in petroleum ether) obtained was directly used for the next step without any further characterization.To the above aldehyde in benzene (25 mL) was added Wittig ylide Ph3PCHCOOEt (4.8 g, 13.7 mmol) at room temperature and heated to reflux at 90 °C. After 1.5 h, the solvent was evaporated in vacuo and the residue was purified by column chromatography (silica gel, 2% EtOAc in the petroleum ether eluent) to afford the pure compound 15 (3.17 g, 80%) as yellow oil.
R f = 0.52 (SiO2, 5% EtOAc in petroleum ether); [α]28D = +10.61 (c 3.16, CHCl3); IR νmax 2953, 2932, 2857, 2363, 1720, 1648, 1464, 1365, 1256, 1180, 1075, 1031, 835, 773 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.34–7.26 (m, 5H), 6.98 (dd, J = 16.0, 7.0 Hz, 1H), 5.78 (d, J = 16.0 Hz, 1H), 4.50 (d, J = 12.0 Hz, 1H), 4.47 (d, J = 12.0 Hz, 1H), 4.21–4.17 (m, 2H), 3.93 (q, J = 5.0 Hz, 1H), 3.71 (dd, J = 6.0, 3.0 Hz, 1H), 3.48 (t, J = 7.0 Hz, 2H), 2.51 (m, 1H), 1.88–1.84 (m, 2H), 1.67 (m, 1H), 1.30–1.27 (m, 3H), 1.04 (d, J = 7.0 Hz, 3H), 0.88 (s, 18H), 0.87 (d, J = 7.0 Hz, 3H), 0.06 (s, 6H), 0.04 (s, 3H), 0.02 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 166.6, 153.6, 138.3, 128.3, 127.6, 127.5, 120.3, 76.5, 73.0, 70.5, 66.9, 60.1, 43.7, 39.6, 35.5, 26.0, 25.9, 18.3, 18.2, 14.2, 13.2, 10.6, −3.6, −3.7, −3.9, −4.1; MS (ESI) m/z 601 [M + Na]+; HRMS (ESI, QSTAR-TOF) calcd for C32H58O5Si2Na [M + Na]+ 601.3715, found 601.3718.
(4R,5S,6R,7S)-9-(Benzyloxy)-5,7-bis(tert-butyldimethylsilyloxy)-4,6-dimethylnonan-1-ol (16)
To a solution of ester 15 (2.0 g, 3.45 mmol) in THF (15 mL), LiBH4 (226 mg, 10.36 mmol) was added portion wise under a nitrogen atmosphere at 0 °C with gentle stirring. After completion of addition, the reaction mixture was warmed to room temperature and stirred for 48 h. The reaction mixture was cooled to 0 °C and carefully quenched with saturated aqueous solution of NH4Cl (15 mL). The resulting mixture was extracted with EtOAc (50 mL), washed with water (10 mL), brine (10 mL), dried (Na2SO4), filtered and concentrated in vacuo. Column chromatography of the residue (SiO2, 10% EtOAc in the petroleum ether eluent) furnished pure alcohol 16 (1.56 g, 84%) as a colourless liquid.R f = 0.5 (SiO2, 20% EtOAc in petroleum ether); [α]24D = +3.89 (c 0.72, CHCl3); IR νmax 3564, 2931, 2857, 2363, 1515, 1463, 1253, 1056, 835, 772, 739, 697 cm−1; 1H NMR (400 MHz, CDCl3): δ 7.29–7.23 (m, 5H), 4.48 (d, J = 11.7 Hz, 1H), 4.45 (d, J = 11.7 Hz, 1H), 3.91 (q, J = 5.4 Hz, 1H), 3.62–3.55 (m, 3H), 3.45 (t, J = 6.5 Hz, 2H), 1.84–1.78 (m, 2H), 1.65–1.33 (m, 6H), 1.24 (m, 1H), 0.88–0.82 (m, 24H), 0.06–0.02 (m, 12H); 13C NMR (75 MHz, CDCl3): δ 138.4, 128.3, 127.6, 127.5, 76.4, 73.0, 70.8, 67.0, 63.2, 43.7, 35.7, 35.5, 31.3, 30.9, 26.1, 25.9, 18.5, 18.2, 14.0, 11.0, −3.5, −3.6, −3.8, −3.9; MS (ESI) m/z 561 [M + Na]+; HRMS (ESI, QSTAR-TOF) calcd for C30H58O4Si2Na [M + Na]+ 561.3771, found 561.3778.
(5S,6R,7S,8R)-5-(2-(Benzyloxy)ethyl)-7-(tert-butyldimethylsilyloxy)-2,2,3,3,6,8,13,13,14,14-decamethyl-4,12-dioxa-3,13-disilapentadecane (17)
Alcohol 16 (1.4 g, 2.59 mmol) was reacted with TBSOTf (0.7 mL, 2.85 mmol) under similar experimental conditions as stated earlier to give TBS protected compound 17 (1.45 g, 86%) as a yellowish liquid.R f = 0.5 (SiO2, 2% EtOAc in petroleum ether); [α]24D = +2.73 (c 1.13, CHCl3); IR νmax 2931, 2953, 2858, 1465, 1384, 1253, 1101, 836, 774, 670 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.33–7.25 (m, 5H), 4.50 (d, J = 11.9 Hz, 1H), 4.47 (d, J = 11.9 Hz, 1H), 3.92 (q, J = 4.9 Hz, 1H), 3.62–3.57 (m, 3H), 3.48 (t, J = 6.9 Hz, 2H), 1.85 (q, J = 6.9 Hz, 2H), 1.64–1.32 (m, 5H), 1.25 (m, 1H), 0.89–0.82 (m, 33H), 0.06, 0.04 and 0.038 (three s, 18H); 13C NMR (125 MHz, CDCl3): δ 138.4, 128.2, 127.6, 127.4, 76.6, 73.0, 70.8, 67.0, 63.5, 43.5, 35.7, 35.6, 31.6, 31.1, 26.1, 25.9(2), 18.5, 18.3, 18.2, 13.8, 10.9, −3.5, −3.6, −3.9(2), −5.3(2); MS (ESI) m/z 653 [M + H]+, 675 [M + Na]+; HRMS (ESI, QSTAR-TOF) calcd for C36H72O4Si3Na [M + Na]+ 675.4636, found 675.4641.
(3S,4R,5S,6R)-3,5,9-Tris(tert-butyldimethylsilyloxy)-4,6-dimethylnonan-1-ol (18)
To a solution of compound 17 (1.1 g, 1.68 mmol) in EtOAc (10 mL), 10% Pd–C (110 mg) was added and the mixture was subjected to hydrogenolysis using H2-filled balloons for 5 h. It was then filtered through a short pad of Celite and the filter cake was washed with EtOAc (10 mL). The filtrate and washings were combined and concentrated in vacuo. Purification of the crude product by column chromatography (SiO2, 5% EtOAc in the petroleum ether eluent) eluted compound 18 (805 mg, 85%) as a colourless oil.R f = 0.5 (SiO2, 10% EtOAc in petroleum ether); [α]24D = +2.15 (c 0.32, CHCl3); IR νmax 3619, 2931, 2857, 2363, 2332, 1516, 1464, 1252, 1101, 835, 773 cm−1; 1H NMR (400 MHz, CDCl3): δ 3.91 (q, J = 5.4 Hz, 1H), 3.80 (m, 1H), 3.67 (m, 1H), 3.58 (t, J = 6.2 Hz, 3H), 1.89–1.69 (m, 4H), 1.55–1.32 (m, 4H), 1.24 (m, 1H), 0.90 and 0.89 (two s, 30H), 0.86 (d, J = 6.2 Hz, 3H), 0.11 (s, 3H), 0.08 (s, 6H), 0.06 (s, 3H), 0.04 (s, 6H); 13C NMR (75 MHz, CDCl3): δ 76.5, 72.4, 63.4, 59.9, 43.1, 37.2, 35.6, 31.7, 31.1, 26.1(2), 26.0(3), 14.2, 11.9(2), −3.5, −3.7, −4.0, −4.1, −5.2(2); MS (ESI) m/z 563 [M + H]+, 585 [M + Na]+; HRMS (ESI, QSTAR-TOF) calcd for C29H66O4Si3Na [M + Na]+ 585.4166, found 585.4165.
(5S,6R,7S,8R)-7-(tert-Butyldimethylsilyloxy)-2,2,3,3,6,8,13,13,14,14-decamethyl-5-(prop-2-ynyl)-4,12-dioxa-3,13-disilapentadecane (7)
Alcohol 18 (350 mg, 0.62 mmol) was oxidised with Dess–Martin periodinane following the same experimental procedure as stated earlier to give an aldehyde (Rf = 0.54, 5% EtOAc in petroleum ether), which was used directly for the next reaction.To a solution of CBr4 (412 mg, 1.24 mmol) in dry CH2Cl2 (10 mL) was added PPh3 (651 mg, 2.48 mmol) at 0 °C and this mixture was stirred for 30 min. To the resulting orange-red solution, Et3N (0.86 mL, 6.21 mmol) was added and stirred for 10 min at the same temperature. Aldehyde in CH2Cl2 (3 mL, ×2) was cannulated to the above reaction mixture and stirred for 2.5 h at 0 °C. Then the reaction mixture was poured into the petroleum ether (30 mL). The solvent was decanted from the sticky precipitate and the residue was dissolved again in CH2Cl2 (5 mL). The solution was again poured into petroleum ether (20 mL) and the solvent was again decanted from the sticky precipitate. This procedure was repeated twice. The precipitate was then discarded. The solvent fractions were combined and concentrated in vacuo. The resulting crude product (Rf = 0.80, 5% EtOAc in petroleum ether) was subjected to silica gel column chromatography (silica was basified with 1% Et3N in petroleum ether) (SiO2, 2% EtOAc in the petroleum ether eluent) to provide a pure dibromo compound, as a syrupy liquid. This dibromo compound was used for next step without further characterization.
To a solution of dibromide in dry THF (4 mL) at −78 °C, n-BuLi (1.6 M in hexane, 0.78 mL, 1.25 mmol) was added slowly with stirring under a nitrogen atmosphere. The mixture was slowly warmed to 0 °C and held at this temperature for 1 h. The reaction mixture was then quenched with saturated aqueous NH4Cl (5 mL) and extracted with EtOAc (15 mL). The combined organic extracts were washed with water (5 mL), brine (5 mL), dried (Na2SO4), filtered and concentrated in vacuo. Column chromatography (SiO2, 1% EtOAc in the petroleum ether eluent) gave pure compound 7 (250 mg, 72% over three steps) as a yellow liquid.
R f = 0.5 (SiO2, 2% EtOAc in petroleum ether); [α]26D = +6.47 (c 0.86, CHCl3); IR νmax 2932, 2858, 2362, 1465, 1392, 1253, 1097, 1033, 834, 773 cm−1; 1H NMR (500 MHz, CDCl3): δ 3.92 (q, J = 5.0 Hz, 1H), 3.60–3.58 (m, 3H), 2.39 (dd, J = 5.9, 2.0 Hz, 2H), 2.00–1.95 (m, 2H), 1.58–1.51 (m, 2H), 1.47 (m, 1H), 1.37 (m, 1H), 1.25 (m, 1H), 0.90 and 0.89 (two s, 27H), 0.87 (d, J = 6.9 Hz, 3H), 0.86 (d, J = 6.0 Hz, 3H), 0.09 (s, 6H), 0.08 (s, 3H), 0.06 (s, 3H), 0.05 (s, 6H); 13C NMR (75 MHz, CDCl3): δ 81.4, 77.1, 71.6, 70.3, 63.5, 42.7, 35.9, 31.6, 31.1, 26.2(2), 26.0, 25.9, 18.5, 18.4, 18.2, 14.0, 11.0, −3.5, −3.7, −3.8, −4.2, −5.3(2); MS (ESI) m/z 579 [M + Na]+; HRMS (ESI, QSTAR-TOF) calcd for C30H64O3Si3Na [M + Na]+ 579.4055, found 579.4044.
(4R,5S,6R,7S)-5,7-Bis(tert-butyldimethylsilyloxy)-4,6-dimethylundec-9-yn-1-ol (19)
To a stirred solution of compound 7 (100 mg, 0.18 mmol) in anhydrous THF (2 mL) was added n-BuLi (0.17 mL, 0.27 mmol) at −78 °C and stirred for 5 min. Then MeI (0.06 mL, 0.89 mmol) was added to the reaction mixture and stirred at 0 °C for 1 h. The reaction mixture was then quenched with saturated aqueous NH4Cl (5 mL) and extracted with EtOAc (15 mL). The combined organic extracts were washed with water (5 mL), brine (5 mL), dried (Na2SO4), filtered and concentrated in vacuo and used as such for the next reaction.The above methylated compound dissolved in dry THF (3 mL) was transferred to a plastic container fitted with a magnetic stirrer bar and treated with HF·Py (23 µL) at 0 °C. After stirring for 6 h at room temperature, saturated aqueous NaHCO3 (2 mL) was added to the reaction mixture and extracted with EtOAc (2 × 10 mL). The combined organic extracts were washed with water (10 mL), brine (10 mL), dried (Na2SO4), filtered and concentrated in vacuo. Purification of the residue by column chromatography (SiO2, 10% EtOAc in the petroleum ether eluent) afforded the pure product 19 (68 mg, 83% over two steps) as colourless oil.
R f = 0.5 (SiO2, 15% EtOAc in petroleum ether); [α]24D = +2.0 (c 1.4, CHCl3); IR νmax 3309, 2930, 2857, 1465, 1384, 1252, 1058, 834, 773, 672 cm−1; 1H NMR (500 MHz, CDCl3): δ 3.85 (td, J = 6.1, 3.9 Hz, 1H), 3.63 (t, J = 6.7 Hz, 2H), 3.59 (dd, J = 6.4, 1.7 Hz, 1H), 2.31–2.29 (m, 2H), 1.97 (m, 1H), 1.75 (t, J = 2.4 Hz, 3H), 1.62–1.52 (m, 4H), 1.37 (m, 1H), 1.28 (m, 1H), 0.90 and 0.87 (two s, 18H), 0.87 (d, J = 6.0 Hz, 3H), 0.85 (d, J = 7.1 Hz, 3H), 0.09 (s, 3H), 0.08 (s, 3H), 0.07 (s, 3H), 0.06 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 77.5, 77.0, 76.1, 72.2, 63.4, 42.9, 35.7, 31.6, 31.0, 26.3, 26.1, 25.9, 18.5, 18.2, 14.1, 10.5, 3.5, −3.6, −3.6, −3.8, −4.2; MS (ESI) m/z 457 [M + H]+; HRMS (ESI, QSTAR-TOF) calcd for C25H53O3Si2 [M + H]+ 457.35277, found 457.35467.
(5S,6R,7S,8R)-5,7,11-Tris(tert-butyldimethylsilyloxy)-6,8-dimethylundec-2-yn-1-ol (20)
To a solution of alkyne 7 (150 mg, 0.269 mmol) in THF (5 mL), n-BuLi (1.6 M in hexane, 0.2 mL, 0.32 mmol) was added at −78 °C under a nitrogen atmosphere. The reaction mixture was stirred for 5 min at −78 °C and 30 minutes at 0 °C. Paraformaldehyde (24 mg, 0.81 mmol) was added and stirred at room temperature for 12 h. The reaction was quenched with saturated aqueous NH4Cl (2 mL) and extracted with EtOAc (2 × 10 mL). The combined organic extracts were washed with water (10 mL), brine (10 mL), dried (Na2SO4), filtered and concentrated in vacuo. Column chromatography (SiO2, 10% EtOAc in the petroleum ether eluent) gave pure compound 20 (113 mg, 72%) as a syrupy liquid.R f = 0.5 (SiO2, 20% EtOAc in petroleum ether); [α]24D = +12.11 (c 1.09, CHCl3); IR νmax 3620, 2931, 2858, 2362, 2333, 1465, 1253, 1098, 1030, 834, 774, 671 cm−1; 1H NMR (500 MHz, CDCl3): δ 4.22 (s, 2H), 3.89 (dd, J = 9.9 Hz, 5.9 Hz, 1H), 3.61–3.57 (m, 3H), 2.43–2.42 (m, 2H), 1.94 (m, 1H), 1.59–1.41 (m, 3H), 1.38 (m, 1H), 1.23 (m, 1H), 0.91, 0.90 and 0.89 (three s, 27H), 0.87 (d, J = 7.0 Hz, 3H), 0.86 (d, J = 7.0 Hz, 3H), 0.10, 0.09, 0.08, 0.07 (four s, 12H), 0.05 (s, 6H); 13C NMR (75 MHz, CDCl3): δ 83.4, 80.4, 77.3, 71.7, 63.5, 51.3, 42.8, 36.0, 31.5, 31.1, 26.4, 26.1, 26.0, 25.9, 18.5, 18.3, 18.2, 14.1, 11.2, −3.5, −3.7, −3.8, −4.2, −5.3(2); MS (ESI) m/z 604 [M + NH4]+; HRMS (ESI, QSTAR-TOF) calcd for C31H70O4Si3N [M + NH4]+ 604.4613, found 604.4615.
(5S,6R,7S,8R)-5,7,11-Tris(tert-butyldimethylsilyloxy)-6,8-dimethylundec-2-ynyl acetate (21)
To a solution of compound 20 (85 mg, 0.15 mmol) in CH2Cl2 (5 mL), Et3N (0.06 mL, 0.44 mmol), Ac2O (0.02 mL, 0.22 mmol) and DMAP (2 mg, 0.01 mmol) were added sequentially at 0 °C, under a nitrogen atmosphere. After stirring for 30 min at the same temperature, the reaction mixture was quenched using saturated aqueous NaHCO3 (2 mL) and extracted with EtOAc (2 × 15 mL). The organic extracts were washed with water (10 mL), brine (10 mL), dried (Na2SO4), filtered and concentrated in vacuo. Purification by column chromatography (SiO2, 5–10% EtOAc in the petroleum ether eluent) furnished 21 (90 mg, quantitative yield) as a colourless liquid.R f = 0.7 (SiO2, 15% EtOAc in petroleum ether); [α]24D = +2.09 (c 0.96, CHCl3); IR νmax 2928, 2857, 2406, 2363, 1741, 1707, 1647, 1516, 1464, 1252, 1096, 1030, 835, 774 cm−1; 1H NMR (500 MHz, CDCl3): δ 4.64 (s, 2H), 3.91 (q, J = 5.2 Hz, 1H), 3.60–3.56 (m, 3H), 2.41 (br, 2H), 2.07 (s, 3H), 1.91 (m, 1H), 1.49–1.33 (m, 3H), 0.90–0.85 (m, 35H); 0.08–0.04 (m, 18H); 13C NMR (75 MHz, CDCl3): δ 170.3, 84.7, 77.1, 75.8, 71.6, 63.5, 52.8, 43.0, 35.8, 31.6, 31.1, 26.4, 26.1, 26.0, 25.8, 20.8, 18.5, 18.3, 18.1, 13.9, 10.8, −3.6, −3.7, −3.8, −4.2, −5.3(2); MS (ESI) m/z 651 [M + Na]+; HRMS (ESI, QSTAR-TOF) calcd for C33H68O5Si3Na [M + Na]+ 651.4266, found 651.4271.
(5S,6R,7S,8R)-5,7-Bis(tert-butyldimethylsilyloxy)-11-hydroxy-6,8-dimethylundec-2-ynyl acetate (22)
To compound 21 (60 mg, 0.09 mmol) in dry THF (3 mL) in a plastic container was added HF·Py (12 µL) at 0 °C and stirred for 6 h at room temperature. Saturated aqueous NaHCO3 (2 mL) was added to the reaction mixture and extracted with EtOAc (2 × 10 mL). The combined organic extracts were washed with water (10 mL), brine (10 mL), dried (Na2SO4), filtered and concentrated in vacuo. Purification of the crude product by column chromatography (SiO2, 10% EtOAc in the petroleum ether eluent) afforded the pure product 22 (41 mg, 85%) as a colourless oil.R f = 0.5 (SiO2, 20% EtOAc in petroleum ether); [α]24D = −6.07 (c 0.89, CHCl3); IR νmax 3565, 2931, 2858, 2363, 2332, 1741, 1693, 1516, 1463, 1030, 835, 773, 670 cm−1; 1H NMR (500 MHz, CDCl3): δ 4.64 (s, 2H), 3.90 (q, J = 5.3 Hz, 1H), 3.63 (t, J = 6.5 Hz, 2H), 3.57 (dd, J = 6.0, 2.0 Hz, 1H), 2.43–2.42 (m, 2H), 2.06 (s, 3H), 1.93 (m, 1H), 1.63–1.48 (m, 4H), 1.41 (m, 1H), 0.93–0.85 (m, 25H), 0.09 (s, 6H), 0.08 (s, 3H), 0.07 (s, 3H), 0.06 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 170.4, 84.6, 77.1, 75.9, 71.6, 63.3, 52.8, 42.8, 36.0, 31.2, 31.0, 26.5, 26.1, 25.8, 20.8, 18.5, 18.2, 14.2, 11.2, −3.6, −3.7, −3.9, −4.2; MS (ESI) m/z 537 [M + Na]+; HRMS (ESI, QSTAR-TOF) calcd for C27H54O5Si2Na [M + Na]+ 537.3402, found 537.3391.
(5S,6R,7S,8R,E)-5,7-Bis(tert-butyldimethylsilyloxy)-11-hydroxy-6,8-dimethyl-2-(tributylstannyl)undec-2-enyl acetate (23)
To a stirred solution of propargylic acetate 22 (30 mg, 0.06 mmol) in 3 mL of degassed THF, (PPh3)2PdCl2 (catalytic) was added at room temperature under an argon atmosphere. The reaction mixture was stirred for 5 min and Bu3SnH (0.03 mL, 0.09 mmol) was added. The solution turned black and H2 was evolved during the addition of Bu3SnH. The reaction mixture was stirred for 30 min and concentrated under reduced pressure. Column chromatography (silica was basified with 1% Et3N in PE prior purification) (SiO2, 5% EtOAc in the petroleum ether eluent) afforded vinyl stannane 23 (32 mg, 68%) as a colourless liquid.R f = 0.7 (SiO2, 20% EtOAc in petroleum ether); [α]24D = +5.11 (c 0.90, CHCl3); IR νmax 3564, 2925, 2856, 2362, 2333, 1741, 1531, 1462, 1250, 1053, 835, 773, 671 cm−1; 1H NMR (300 MHz, CDCl3): δ 5.74 (t, J = 6.5 Hz, 1H), 4.80 (q, J = 13.8 Hz, 2H), 3.86 (q, J = 5.1 Hz, 1H), 3.62 (t, J = 6.8 Hz, 2H), 3.55 (d, J = 6.6 Hz, 1H), 2.34–2.30 (m, 2H), 2.07 (s, 3H), 1.71–1.29 (m, 6H), 0.91–0.84 (m, 52H), 0.07 (s, 3H), 0.06 (s, 3H), 0.04 (s, 3H), 0.02 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 170.9, 140.6, 137.5, 76.0, 72.6, 67.0, 63.2, 43.0, 35.6, 31.9, 31.6, 29.1, 27.4, 26.2, 25.9, 22.7, 21.0, 18.5, 18.2, 14.1, 13.7, 11.2, 10.0, −3.4(2), −3.9, −4.0; MS (ESI) m/z 807 [M + H]+, 829 [M + Na]+; HRMS (ESI, QSTAR-TOF) calcd for C39H82O5Si2SnNa [M + Na]+ 829.4615, found 829.4613.
(R)-4-Benzyl-3-((S,E)-5-iodo-2,4-dimethylpent-4-enoyl)oxazolidin-2-one (24)
To a stirred solution of compound 11 (3 g, 15.15 mmol) in dry CH2Cl2 (30 mL) were added Et3N (4.2 mL, 30.3 mmol) and methanesulfonyl chloride (1.76 mL, 22.72 mmol) followed by DMAP (185 mg, 1.51 mmol) at 0 °C under a nitrogen atmosphere. Then the reaction mixture was slowly warmed to room temperature and stirred for 6 h. The reaction mixture was filtered through a short pad of silica and the solvent was evaporated under vacuum to afford the mesylated compound in quantitative yield. This compound (Rf = 0.8, 20% EtOAc in petroleum ether) was used directly for the next step without any further purification.To the above mesylated compound dissolved in dry THF (40 mL) was added NaI (3.41 g, 22.72 mmol) at room temperature and stirred for 3 h. Then the reaction mixture was filtered through a short pad of silica and concentrated in vacuo. The iodo compound (Rf = 0.45, 10% EtOAc in petroleum ether) was used directly for next step without any further purification.
To a stirred solution of (R)-4-benzyl-3-propionyloxazolidin-2-one (7.07 g, 30.3 mmol) in THF (60 mL) was added NaHMDS (1 M in THF, 30.3 mL, 30.3 mmol) at −78 °C. The resulting dark yellow coloured solution was stirred for 1 h at −78 °C. Then the iodo compound dissolved in THF (10 mL, ×2) was cannulated to the above reaction mixture and stirred for 2 h at −78 °C and 12 h at −40 °C. It was then quenched by adding saturated NH4Cl (20 mL) and extracted with EtOAc (2 × 100 mL). The organic extracts were washed with water (20 mL), brine (20 mL), dried (Na2SO4), filtered and concentrated in vacuo. Purification of the crude product by column chromatography (SiO2, 10% EtOAc in the petroleum ether eluent) furnished 24 (3.19 g, 51% over three steps) as a colourless liquid.
R f = 0.4 (SiO2, 20% EtOAc in petroleum ether); [α]24D = −25.66 (c 0.6, CHCl3); IR νmax 2922, 2852, 2322, 1775, 1694, 1453, 1383, 1273, 1237, 1208, 1102, 1014, 970, 760, 745, 700 cm−1; 1H NMR (300 MHz, CDCl3): δ 7.30–7.29 (m, 2H), 7.28–7.27 (m, 1H), 7.20–7.19 (m, 2H), 6.0 (d, J = 0.6 Hz, 1H), 4.64 (m, 1H), 4.22–4.15 (m, 2H), 4.05 (m, 1H), 3.25 (dd, J = 13.6, 3.0 Hz, 1H), 2.76–2.70 (m, 2H), 2.26 (dd, J = 12.8, 7.6 Hz, 1H), 1.91 (s, 3H), 1.15 (d, J = 6.8 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 176.2, 153.1, 145.2, 135.2, 129.4, 129.0, 127.3, 77.4, 66.0, 55.2, 43.3, 38.0, 35.7, 23.7, 16.6; MS (ESI) m/z 436 [M + Na]+; HRMS (ESI, QSTAR-TOF) calcd for C17H20INO3Na [M + Na]+ 436.0380, found 436.0389.
(S,2Z,6E)-Methyl 7-iodo-4,6-dimethylhepta-2,6-dienoate (25)
To a solution of compound 24 (2.8 g, 6.77 mmol) in a mixture of THF and water (5:1) (20 mL) was added NaBH4 (1.28 g, 33.88 mmol) portionwise at 0 °C. The resulting reaction mixture was allowed to attain room temperature and stirred overnight. Then the reaction mixture was quenched by the addition of saturated aqueous NH4Cl (15 mL) at 0 °C. The solvent was removed by rotary evaporation and the resulting residual oil was extracted with EtOAc (2 × 75 mL). The combined organic extract was washed with water (15 mL), saturated aqueous NaCl (15 mL), dried (Na2SO4), filtered and concentrated under vacuum. Purification by column chromatography (silica gel, 10–15% EtOAc in the petroleum ether eluent) afforded the known alcohol 10 (1.38 g, 85%) as light yellow oil.
Alcohol 10 (1.2 g, 5.0 mmol) was subjected to DMP mediated oxidation under similar experimental conditions as stated earlier to give an aldehyde (Rf = 0.7, 10% EtOAc in petroleum ether), which was used for the next step after passing through a short pad of column.
To a solution of phosphonate, CH3O2CCH2P(O)(OCH2CF3)2 (1.17 mL, 5.5 mmol) in THF (15 mL) at 0 °C, NaH (60% dispersion in oil, 179 mg, 4.5 mmol) was added and stirred for 40 min. The reaction mixture was then cooled to −78 °C and the above aldehyde, dissolved in THF (5 mL, ×2) was cannulated, stirred at this temperature for 6 h and slowly warmed to 0 °C. It was then quenched with saturated aqueous NH4Cl (10 mL) and extracted with EtOAc (2 × 75 mL). The combined organic extracts were washed with water (15 mL), brine (15 mL), dried (Na2SO4), filtered and concentrated in vacuo. Purification of the crude product by column chromatography (SiO2, 5% EtOAc in the petroleum ether eluent) afforded the pure compound 25 (1.175 g, 80%) as a colourless liquid.
R f = 0.6 (SiO2, 10% EtOAc in petroleum ether); [α]28D = −69.65 (c 0.72, CHCl3); IR νmax 2922, 2853, 1723, 1646, 1516, 1462, 1202, 1176, 822, 670 cm−1; 1H NMR (300 MHz, CDCl3): δ 5.97–5.87 (m, 2H), 5.72 (d, J = 12.0 Hz, 1H), 3.77 (m, 1H), 3.70 (s, 3H), 2.22–2.19 (m, 2H), 1.83 (d, J = 1.5 Hz, 3H), 0.97 (d, J = 6.8 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 166.5, 154.7, 146.0, 118.4, 76.0, 51.1, 46.7, 30.8, 23.7, 19.7; Anal. Calcd for C10H15IO2: C, 40.83; H, 5.14. Found: C, 41.19; H, 5.54.
(S,2Z,6E)-7-Iodo-4,6-dimethylhepta-2,6-dien-1-ol (26)
To a solution of compound 25 (900 mg, 3.06 mmol) in dry CH2Cl2 (10 mL) at −78 °C, DIBAL-H (1 M solution in toluene, 7.96 mL, 7.96 mmol) was added slowly with stirring under a nitrogen atmosphere. After stirring for 1 h at the same temperature, the reaction mixture was quenched with H2O (2 mL). MTB ether (30 mL) was added to the reaction mixture and stirred for 15 min. 4 N NaOH (5 mL) and water (2 mL) were added and stirred vigorously for 30 min at room temperature. Combined organic layers were separated and dried over Na2SO4. Filtration followed by evaporation of the solvent afforded the crude product. Purification of the crude product by column chromatography (SiO2, 10–15% EtOAc in the petroleum ether eluent) provided compound 26 (667 mg, 82%) as pale yellow liquid.R f = 0.4 (SiO2, 20% EtOAc in petroleum ether); [α]24D = −31.9 (c 0.50, CHCl3); IR νmax 3565, 2922, 2854, 2362, 2333, 1741, 1706, 1516, 1036, 669 cm−1; 1H NMR (500 MHz, CDCl3): δ 5.85 (s, 1H), 5.56 (m, 1H), 5.26 (t, J = 10.1 Hz, 1H), 4.14 (d, J = 6.7 Hz, 2H), 2.66 (m, 1H), 2.22–2.11 (m, 2H), 1.82 (s, 3H), 1.20 (br s, 1H), 0.95 (d, J = 6.8 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 146.1, 137.7, 127.7, 76.1, 58.7, 47.2, 30.6, 24.0, 20.8; Anal. Calcd for C9H15IO: C, 40.62; H, 5.68. Found: C, 41.33; H, 5.94.
(S)-4-Benzyl-3-((2R,3R,4Z,6S,8E)-3-hydroxy-9-iodo-2,6,8-trimethylnona-4,8-dienoyl)oxazolidin-2-one (27)
Alcohol 26 (430 mg, 1.62 mmol) was oxidized to an aldehyde with Dess–Martin periodinane (1.028 g, 2.43 mmol) as stated earlier and used as such for the next reaction.To a stirred solution of (S)-4-benzyl-3-propionyloxazolidin-2-one (565 mg, 2.42 mmol) in dry EtOAc (5 mL) was added MgCl2 (46 mg, 0.48 mmol) followed by Et3N (4.5 mL, 32.32 mmol) at room temperature under a nitrogen atmosphere. Then the aldehyde dissolved in EtOAc (3 mL, ×2) was added via a cannula, followed by TMS-Cl (3.08 mL, 24.24 mmol). After stirring for 24 h at room temperature the reaction mixture was filtered through a short pad of silica and the silica bed was washed with EtOAc (20 mL). Combined filtrate and washings were concentrated under reduced pressure. The residual oil was dissolved in MeOH (10 mL) and treated with TFA (1 drop) and stirred for 30 min. The solvent was removed under reduced pressure. Purification of the crude product by column chromatography (SiO2, 10–15% EtOAc in the petroleum ether eluent) provided aldol compound 27 (578 mg, 72%) as yellow liquid.
R f = 0.4 (SiO2, 20% EtOAc in petroleum ether); [α]26D = +6.30 (c 1.00, CHCl3); IR νmax 3565, 2963, 2924, 2362, 2333, 1778, 1705, 1647, 1485, 1388, 1211, 1008, 754, 700 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.34–7.24 (m, 5H), 5.88 (m, 1H), 5.41–5.33 (m, 2H), 4.71 (m, 1H), 4.53 (t, J = 8.4 Hz, 1H), 4.22–4.16 (m, 2H), 3.91 (m, 1H), 3.33 (dd, J = 13.6, 3.3 Hz, 1H), 2.82 (dd, J = 13.6, 9.3 Hz, 1H), 2.78 (m, 1H), 2.21 (d, J = 7.5 Hz, 2H), 1.85 (d, J = 0.9 Hz, 3H), 1.13 (d, J = 6.8 Hz, 3H), 0.95 (d, J = 6.7 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 176.3, 153.5, 146.2, 139.2, 135.2, 129.5, 129.0, 128.9, 127.3, 76.2, 70.8, 66.0, 55.5, 47.3, 43.6, 37.7, 30.8, 24.0, 20.5, 14.5; MS (ESI) m/z 520 [M + Na]+; HRMS (ESI, QSTAR-TOF) calcd for C22H28IO4NNa [M + Na]+ 520.0955, found 520.0949.
(2S,3R,4Z,6S,8E)-3-(tert-Butyldimethylsilyloxy)-9-iodo-2,6,8-trimethylnona-4,8-dien-1-ol (9)
Alcohol 27 (510 mg, 1.02 mmol) was protected as TBS ether with TBSOTf via a similar experimental procedure as described earlier and used as such for the next reaction.To a solution of the above TBS protected compound in a mixture of THF and water (5:1) (5 mL) was added NaBH4 (194 mg, 5.13 mmol) at 0 °C. The reaction mixture was allowed to attain room temperature and stirred overnight. Then the reaction mixture was quenched by the addition of saturated aqueous NH4Cl (5 mL) at 0 °C. The solvent was removed by rotary evaporation and the resulting residual oil was extracted with EtOAc (2 × 10 mL). The combined organic layers were washed with water (5 mL), saturated aqueous NaCl (5 mL), dried (Na2SO4), filtered and concentrated under vacuum. Purification by column chromatography (silica gel, 10% EtOAc in the petroleum ether eluent) afforded pure compound 9 (332 mg, 74% over two steps) as light yellow oil.
R f = 0.65 (SiO2, 20% EtOAc in petroleum ether); [α]26D = −2.99 (c 0.30, CHCl3); IR νmax 3565, 2955, 2926, 2362, 2333, 1694, 1647, 1516, 1463, 1253, 1071, 1029, 838, 778, 673 cm−1; 1H NMR (500 MHz, CDCl3): δ 5.91 (s, 1H), 5.41 (dd, J = 11.3, 8.3 Hz, 1H), 5.19 (t, J = 10.6 Hz, 1H), 4.41 (dd, J = 8.5, 5.6 Hz, 1H), 3.81 (m, 1H), 3.57 (m, 1H), 2.76 (m, 1H), 2.57 (m, 1H), 2.25 (dd, J = 13.4, 5.4 Hz, 1H), 2.10 (dd, J = 13.6, 9.0 Hz, 1H), 1.83 (s, 3H), 0.98 (d, J = 6.8 Hz, 3H), 0.88 (brs, 12H), 0.09 (s, 3H), 0.05 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 145.7, 135.2, 131.4, 76.5, 74.1, 65.7, 47.0, 41.7, 30.6, 25.8, 23.9, 20.1, 18.0, 14.3, −3.9, −4.9; MS (ESI) m/z 461 [M + Na]+; HRMS (ESI, QSTAR-TOF) calcd for C18H35IO2SiNa [M + Na]+ 461.1343, found 461.1349.
(2Z,4S,5R,6Z,8S,10E)-Methyl-5-(tert-butyldimethylsilyloxy)-11-iodo-4,8,10-trimethylun deca-2,6,10-trienoate (28)
Oxidation of alcohol 9 (150 mg, 0.34 mmol) with Dess–Marin periodinane followed by cis-selective HWE olefination of the resulting aldehyde with CH3O2CCH2P(O)(OCH2CF3)2 (0.08 mL, 0.376 mmol) was performed following the same experimental procedure as stated earlier to give olefin 28 (121 mg, 72%) as a colourless liquid.R f = 0.5 (SiO2, 2% EtOAc in petroleum ether); [α]28D = +4.13 (c 0.63, CHCl3); IR νmax 2927, 2856, 2363, 2332, 1690, 1627, 1516, 1462, 1180, 1029, 866, 775 cm−1; 1H NMR (500 MHz, CDCl3): δ 6.23 (t, J = 10.5 Hz, 1H), 5.89 (s, 1H), 5.78 (d, J = 11.9 Hz, 1H), 5.25 (dd, J = 10.9, 8.9 Hz, 1H), 5.12 (m, 1H), 4.35 (dd, J = 8.9, 4.0 Hz, 1H), 3.68 (s, 3H), 3.57 (m, 1H), 2.58 (m, 1H), 2.22 (dd, J = 13.0, 6.0 Hz, 1H), 2.10 (dd, J = 13.0, 8.0 Hz, 1H), 1.83 (s, 3H), 1.05 (d, J = 7.0 Hz, 3H), 0.88 (brs, 12H), 0.07 (s, 3H), 0.03 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 166.6, 152.0, 146.0, 134.8, 131.2, 119.1, 76.3, 71.9, 51.0, 47.1, 40.2, 30.7, 25.8, 23.9, 20.2, 18.1, 16.6, −4.0, −4.8; MS (ESI) m/z 515 [M + Na]+; HRMS (ESI, QSTAR-TOF) calcd for C21H37IO3SiNa [M + Na]+ 515.1448, found 515.1453.
(5S,6R)-6-((S,1Z,5E)-6-Iodo-3,5-dimethylhexa-1,5-dienyl)-5-methyl-5,6-dihydro-2H-pyran-2-one (6)
To a stirred solution of compound 28 (80 mg, 0.162 mmol) in dry MeOH (2 mL) was added CSA (38 mg, 0.162 mmol) at 0 °C under a nitrogen atmosphere. Then the reaction mixture was slowly warmed to room temperature and stirred for 6 h. Saturated aqueous NaHCO3 (2 mL) was added to the reaction mixture and extracted with EtOAc (2 × 10 mL). The combined organic extracts were washed with water (5 mL), brine (5 mL), dried (Na2SO4), filtered and concentrated in vacuo. Purification of the crude product by column chromatography (SiO2, 20–25% EtOAc in the petroleum ether eluent) afforded the pure lactone product 6 (48 mg, 86%) as yellowish oil.R f = 0.5 (SiO2, 40% EtOAc in petroleum ether); [α]28D = +21.05 (c 0.47, CHCl3); IR νmax 2921, 2853, 2363, 2332, 1685, 1516, 1461, 1229, 1009, 812 cm−1; 1H NMR (500 MHz, CDCl3): δ 6.68 (dd, J = 10.1, 2.0 Hz, 1H), 5.99 (dd, J = 10.1, 2.0 Hz, 1H), 5.92 (s, 1H), 5.52 (dd, J = 11.2, 10.2 Hz, 1H), 5.43 (dd, J = 11.2, 9.1 Hz, 1H), 4.77 (dd, J = 10.1, 9.1 Hz, 1H), 2.69 (m, 1H), 2.50 (m, 1H), 2.28 (dd, J = 13.2, 6.1 Hz, 1H), 2.16 (dd, J = 13.2, 8.1 Hz, 1H), 1.83 (s, 3H), 1.09 (d, J = 7.1 Hz, 3H), 0.93 (d, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 163.8, 150.9, 145.7, 141.8, 124.6, 120.5, 79.2, 76.5, 46.9, 33.8, 30.7, 23.8, 20.0, 15.9; MS (ESI) m/z 347 [M + H]+, 369 [M + Na]+; HRMS (ESI, QSTAR-TOF) calcd for C14H19IO2Na [M + Na]+ 369.0321, found 369.0324.
(5S,6R,7S,8R,Z)-5,7-Bis(tert-butyldimethylsilyloxy)-2-((S,1E,5Z)-2,4-dimethyl-6-((2R,3S)-3-methyl-6-oxo-3,6-dihydro-2H-pyran-2-yl)hexa-1,5-dienyl)-11-hydroxy-6,8-dimethylundec-2-enyl acetate (29)
To a stirred solution of iodo compound 6 (10 mg, 0.027 mmol) and stannane 23 (20 mg, 0.024 mmol) in degassed DMF (2 mL) under argon was added a solution of Pd(PPh3)4 (1.5 mg, 0.001 mmol) in degassed THF (1 mL) followed by CuTC (7.1 mg, 0.037 mmol) and [Ph2PO2][NBu4] (13.7 mg, 0.029 mmol) at room temperature. After stirring for 4 h at room temperature, the reaction mixture was quenched with saturated aqueous NH4Cl (1 mL) and extracted with EtOAc (2 × 5 mL). The organic extract was washed with water (2 mL), brine (2 mL), dried (Na2SO4), filtered and concentrated in vacuo. Column chromatography (SiO2, 20–25% EtOAc in the petroleum ether eluent) gave pure compound 29 (7.7 mg, 45%) as a syrupy liquid.R f = 0.3 (SiO2, 40% EtOAc in petroleum ether); [α]26D = +37.90 (c 0.29, CHCl3); IR νmax 3619, 2926, 2856, 2362, 2333, 1737, 1546, 1463, 1250, 1055, 1021, 835, 773, 671 cm−1; 1H NMR (600 MHz, CDCl3): δ 6.69 (dd, J = 9.8, 2.2 Hz, 1H), 5.99 (dd, J = 9.8, 2.4 Hz, 1H), 5.61 (d, J = 1.3 Hz, 1H), 5.54 (t, J = 10.7 Hz, 1H), 5.53 (t, J = 6.7 Hz, 1H), 5.38 (dd, J = 10.7, 10.0 Hz, 1H), 4.80 (t, J = 10 Hz, 1H), 4.65 (d, J = 12.5 Hz, 1H), 4.56 (d, J = 12.5 Hz, 1H), 3.86 (m, 1H), 3.62 (m, 2H), 3.56 (dd, J = 6.8, 1.8 Hz, 1H), 2.67 (ddqd, J = 10.7, 8.6, 7.0, 6.0 Hz, 1H), 2.50 (dqt, J = 10.0, 7.0, 2.5 Hz, 1H), 2.40 (m, 2H), 2.15 (dd, J = 13.5, 6.0 Hz, 1H), 2.03 (s, 3H), 1.95 (dd, J = 13.5, 8.6 Hz, 1H), 1.72 (d, J = 1.3 Hz, 3H), 1.68–1.51 (m, 4H), 1.38 (m, 1H), 1.24 (m, 1H), 1.09 (d, J = 7.3 Hz, 3H), 0.90 (d, J = 6.5 Hz, 3H), 0.88–0.86 (m, 18H), 0.85 (d, J = 6.9 Hz, 3H), 0.84 (d, J = 6.7 Hz, 3H), 0.07–0.06 (s, 6H), 0.03–0.02 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 171.1, 163.9, 150.9, 142.8, 135.9, 132.8, 130.5, 127.1, 123.9, 120.5, 79.3, 76.3, 72.7, 63.2, 62.6, 47.8, 42.9, 35.7, 34.3, 33.8, 31.4, 31.0, 30.7, 26.1, 25.9, 21.0, 19.8, 18.5, 18.2, 17.9, 15.9, 14.1, 10.9, −3.4, −3.5, −4.0, −4.1; MS (ESI) m/z 735 [M + H]+, 757 [M + Na]+; HRMS (ESI, QSTAR-TOF) calcd for C41H74O7Si2Na [M + Na]+ 757.4865, found 757.4859.
(4R,5S,6R,7S,9Z,11E,14S,15Z)-Methyl-10-(acetoxymethyl)-5,7-bis(tert-butyldimethyl silyloxy)-4,6,12,14-tetramethyl-16-((2R,3S)-3-methyl-6-oxo-3,6-dihydro-2H-pyran-2-yl)hexadeca-9,11,15-trienoate (30)
Alcohol 29 (5 mg, 0.006 mmol) was oxidized with Dess–Martin periodinane under the same experimental procedure as stated earlier to give an aldehyde which was dissolved in a mixture of t-BuOH (1 mL) and 2-methyl-2-butene (0.1 mL) and cooled to 0 °C. Aqueous solution of NaClO2 (1.3 mg, 0.014 mmol) and NaH2PO4·2H2O (6.4 mg, 0.04 mmol) was added to the reaction mixture and stirred at the same temperature for 0.5 h. Then the solvent was removed under reduced pressure and saturated NH4Cl (1 mL) was added to the residue. It was extracted with EtOAc (3 × 2 mL) and the combined organic extracts were washed with water (1 mL), brine (1 mL), dried (Na2SO4), filtered and concentrated in vacuo. The crude acid thus obtained was dissolved in ether (1 mL) and treated with an ethereal solution of CH2N2 at 0 °C, till a yellow colour persisted. After 15 min, the solvent was evaporated and the residue was purified by silica gel column chromatography (SiO2, 15% EtOAc in the petroleum ether eluent) to afford compound 30 (3.6 mg, 70% over three steps) as a colourless oil.R f = 0.8 (SiO2, 40% EtOAc in petroleum ether); [α]26D = +12.75 (c 0.4, CHCl3); IR νmax 3743, 2921, 2851, 1740, 1550, 1464, 1219, 772 cm−1; 1H NMR (500 MHz, CDCl3): δ 6.68 (dd, J = 9.7, 2.3 Hz, 1H), 5.99 (dd, J = 9.7, 2.4 Hz, 1H), 5.61 (s, 1H), 5.54 (t, J = 10.7 Hz, 1H), 5.49 (t, J = 7.3 Hz, 1H), 5.39 (dd, J = 10.3, 9.9 Hz, 1H), 4.80 (m, 1H), 4.66 (d, J = 12.0 Hz, 1H), 4.56 (d, J = 12.0 Hz, 1H), 3.92 (m, 1H), 3.66 (s, 3H), 3.57 (dd, J = 7.1, 0.6 Hz, 1H), 2.67 (m, 1H), 2.50 (dqt, J = 10.0, 7.0, 2.5 Hz, 1H), 2.41 (t, J = 6.8 Hz, 2H), 2.38–2.23 (m, 4H), 2.15 (dd, J = 13.0, 5.8 Hz, 1H), 2.03 (s, 3H), 1.95 (dd, J = 13.0, 8.5 Hz, 1H), 1.72 (d, J = 1.0 Hz, 3H), 1.10 (d, J = 7.3 Hz, 3H), 0.91 (d, J = 5.0 Hz, 3H), 0.90–0.86 (m, 20H), 0.85–0.80 (m, 6H), 0.08–0.07 (m, 6H), 0.06 (s, 3H), 0.05 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 174.2, 170.9, 163.8, 150.8, 142.7, 135.9, 133.0, 130.3, 127.2, 124.0, 120.5, 79.3, 72.4, 62.5, 51.5, 47.8, 42.5, 35.3, 34.3, 33.8, 32.3, 31.9, 30.7, 30.5, 26.2, 25.9, 21.0, 19.7, 18.5, 18.2, 17.9, 15.9, 13.2, 10.3, −3.3, −3.4, −3.9, −4.0; MS (ESI) m/z 785 [M + Na]+; HRMS (ESI, QSTAR-TOF) calcd for C42H74O8Si2Na [M + Na]+ 785.4814, found 785.4812.
Acknowledgements
The authors wish to thank UGC (B. C.), CSIR (S. A. and P. P. R.), New Delhi for research fellowships. S. G. thanks CSIR, New Delhi for financial support as part of the XII Five Year plan programme under the title ORIGIN (CSC-0108). We are also thankful to Dr A. C. Kunwar for useful discussions.Notes and references
- T. Řezanka, R. Dvořáková, L. O. Hanuš and V. M. Dembitsky, Eur. J. Org. Chem., 2004, 5, 995 CrossRef.
- T. K. Chakraborty, R. K. Goswami and M. Sreekanth, Tetrahedron Lett., 2007, 48, 4075 CrossRef CAS PubMed.
- J. K. Stille, Angew. Chem., Int. Ed. Engl., 1986, 25, 508 CrossRef.
- D. A. Evans, J. Bartroli and T. L. Shih, J. Am. Chem. Soc., 1981, 103, 2127 CrossRef CAS.
- D. A. Evans, J. S. Tedrow, J. T. Shaw and C. W. Downey, J. Am. Chem. Soc., 2002, 124, 392 CrossRef CAS PubMed.
- (a) C. Bauder, Eur. J. Org. Chem., 2010, 32, 6207 CrossRef; (b) V. K. Nyavanandi, S. Nanduri, R. V. Dev, A. Naidu and J. Iqbal, Tetrahedron Lett., 2006, 47, 6667 CrossRef CAS PubMed.
- E. J. Corey and P. L. Fuchs, Tetrahedron Lett., 1972, 13, 3769 CrossRef.
- J. F. Betzer, F. Delaloge, B. Muller, A. Pancrazi and J. Prunet, J. Org. Chem., 1997, 62, 7768 CrossRef CAS.
- J. A. Marshall and M. P. Bourbeau, Org. Lett., 2002, 4, 3931 CrossRef CAS.
- (a) D. A. Evans, M. D. Ennis and D. J. Mathre, J. Am. Chem. Soc., 1982, 104, 173 Search PubMed; (b) A. Schmauder, S. Muller and M. E. Maier, Tetrahedron, 2008, 64, 6263 CrossRef CAS PubMed.
- (a) X. Li and X. Zeng, Tetrahedron Lett., 2006, 47, 6839 CrossRef CAS PubMed; (b) Y. Xiao and P. Liu, Angew. Chem., Int. Ed., 2008, 47, 9722 CrossRef CAS PubMed.
- (a) F. Kleinbeck, G. J. Fettes, L. D. Fader and E. M. Carreira, Chem. – Eur. J., 2012, 18, 3598 CrossRef CAS PubMed; (b) F. Kleinbeck and E. M. Carreira, Angew. Chem., Int. Ed., 2009, 48, 578 CrossRef CAS PubMed.
- U. Bhatt, M. Christmann, M. Quitschalle, E. Claus and M. Kalesse, J. Org. Chem., 2001, 66, 1885 CrossRef CAS PubMed.
- T. Kajikawa, K. Aoki, R. S. Singh, T. Iwashita, T. Kusumoto, H. A. Frank, H. Hashimoto and S. Katsumura, Org. Biomol. Chem., 2009, 7, 3723 CAS.
- A. Fürstner, J. A. Funel, M. Tremblay, L. C. Bouchez, C. Nevado, M. Waser, J. Ackerstaff and C. C. Stimson, Chem. Commun., 2008, 25, 2873 RSC.
- G. D. Allred and L. S. Liebeskind, J. Am. Chem. Soc., 1996, 118, 2748 CrossRef CAS.
- J. Srogl, G. D. Allred and L. S. Liebeskind, J. Am. Chem. Soc., 1997, 119, 12376 CrossRef CAS.
- See the ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available: The copies of 1H and 13C NMR spectra for compounds 13–19, 7, 20–28, 9, 6, 29 and 30. See DOI: 10.1039/c4ob01716a |
| This journal is © The Royal Society of Chemistry 2015 |