
Pd-catalyzed oxidative C–H alkenylation for synthesizing arylvinyltriazole nucleosides†
Jingjie
Tang
a,
Mei
Cong
a,
Yi
Xia
ab,
Gilles
Quéléver
a,
Yuting
Fan
ac,
Fanqi
Qu
c and
Ling
Peng
*a
aAix-Marseille Université, CNRS, CINaM UMR 7325, 13288, Marseille, France. E-mail: ling.peng@univ-amu.fr; Fax: +(33) 4 91 82 93 01
bThe Vancouver Prostate Centre and Department of Urologic Sciences, University of British Columbia, 2660 Oak Street, Vancouver, BC V6H 3Z6, Canada
cCollege of Chemistry and Molecular Sciences, Wuhan University, 430072, Wuhan, P. R. China
First published on 30th September 2014
Abstract
Various arylvinyltriazole nucleoside analogues were synthesized using Pd-catalyzed oxidative Heck reaction. This method affords the corresponding and otherwise difficult to achieve arylvinyltriazole nucleosides with good yields and large functional group compatibility. These results further advocate the potential and practicality of this oxidative C–H alkenylation method for generating structurally challenging chemical entities in organic synthesis.
Introduction
An arylvinyl moiety is an important structural motif in many fine chemicals including pharmaceutics, agrochemicals and functional materials.1–3 Among various methods for incorporating an arylvinyl functionality, the Heck reaction is of particular interest and advantage by virtue of its high product yield and large functional group tolerance.4 This reaction consists in the coupling of an olefin to a halide with the aid of a Pd catalyst. However, the requirement of prehalogenated starting material, being neither atom economic nor environmentally friendly, largely limits the substrate scope. The recently fast-developing oxidative Heck reaction, also named the Fujiwara–Moritani reaction, aims to overcome this limitation by making use of C–H activation to directly couple a halogen-free arene with an alkene in the presence of a palladium(II) catalyst.5,6 This oxidative C–H alkenylation uses largely existing and low-cost halogen-free arenes as substrates, and is atom-economic, energy-saving and environmentally friendly. Nevertheless, electron-deficient heteroarenes remain notoriously difficult substrates for this coupling reaction. This is because the electron-deficient arenes often coordinate poorly with Pd to ensure an efficient catalytic process.7 Recently, You et al. disclosed Pd/Cu-catalyzed C–H alkenylation of biologically relevant heterocycles such as xanthines, purines, indolizines etc.,8 whereas Ong et al. further expanded the substrate scope with polycyclic aromatic and sterically hindered substrates.9 Here, we report the direct olefination of highly electron-deficient triazole heterocycles, via C–H activation, with styrene derivatives bearing manifold functional groups, for synthesizing arylvinyltriazole nucleoside analogues which are otherwise difficult to achieve.10 This is the first report on successful implementation of oxidative C–H alkenylation on triazole nucleosides, highlighting the aptness of the oxidative Heck reaction in establishing structurally challenging chemical entities.Results and discussion
Our motivation to develop Pd-catalyzed olefination on triazole nucleosides is impelled by our discovery of the appealing biological activities of aryl appended triazole nucleoside analogues,11 in particular several potent lead compounds bearing arylethynyltriazolyl as nucleobase entities. We are curious whether analogous paradigms with arylvinyltriazole nucleobases are also endowed with interesting biological activities. Therefore, synthesis of such arylvinyltriazole analogues is of primary importance, which can further help us to have an insightful understanding of the structure–activity relationship of aryl appended triazole nucleosides.To construct the arylvinyltriazole nucleosides, we first attempted the Heck reaction using bromotriazole nucleoside to couple with styrene, which was unable to afford the corresponding product (Scheme 1A and Table S1†). This finding was unsurprising since reported successful Heck reactions were primarily achieved with iodonucleoside derivatives and rarely with bromonucleoside analogues.12–15 It is also worth noting that triazole nucleosides are notably challenging substrates for metal-catalyzed reactions due to the low reactivity of the heterocyclic triazole ring, the labile glycosidic bond and the strongly coordinating N-atoms, which may attenuate the electrophilicity of the palladium, hence adversely affecting C–H activation.16
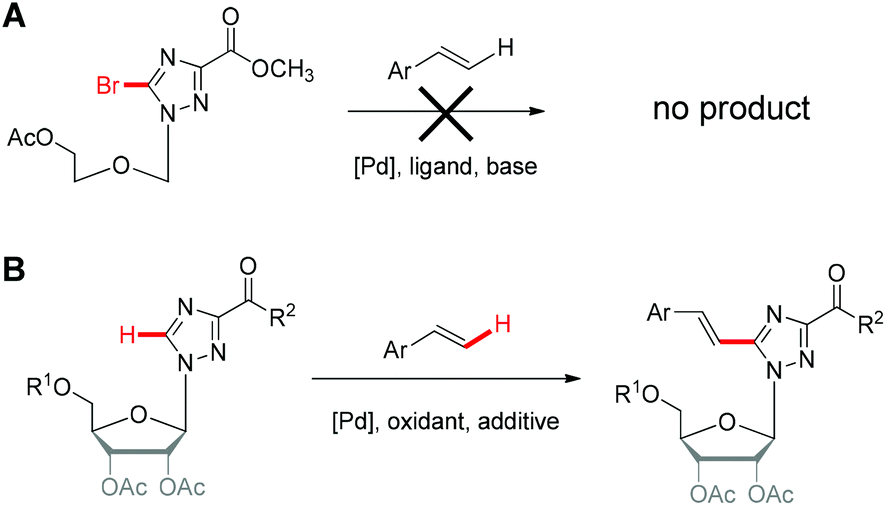 | ||
| Scheme 1 Synthesis of arylvinyltriazole nucleosides using (A) Heck reaction and (B) oxidative Heck reaction. | ||
We then turned our attention to the oxidative Heck reaction to couple, via C–H activation, the triazole nucleosides with styrene derivatives (Scheme 1B), firstly using triazole nucleoside 1 and styrene as model substrates (Table 1). The most frequently used and highly effective Pd source reported for C–H alkenylation is Pd(OAc)2, often with AcOH as a solvent and O2 as a co-oxidant.17–19 AcOH is also supposed to readily interact with the electron-rich nitrogen atoms of heterocycles, hence exempting the Pd catalyst from the unfavorable coordinating effect of nitrogen atoms.20 AgOAc is reported as a beneficial oxidant thanks to its favorable capacity to reoxidize palladium(0) to palladium(II) during the catalytic cycle.21,22 We therefore opted for Pd(OAc)2, AgOAc and AcOH as a catalyst, an oxidant and a solvent respectively to initiate our survey under the reaction conditions. Under a pure O2 atmosphere at 120 °C, while we did obtain the corresponding olefination product 1a with 23% yield (Table 1, entry 1), it was accompanied by a large quantity of unidentified by-products. Interestingly, performing the reaction in air promoted the reaction and considerably suppressed by-product formation (Table 1, entry 2), whereas reaction under Ar protection had no beneficial effect (Table 1, entry 3). We hence aimed towards further optimization in air, considering the associated ease, low cost and convenience of these conditions. Replacing AgOAc by other silver salts gave similar results (Table 1, entries 4 and 5), whereas oxidants such as Cu(OAc)2 and 1,4-benzoquinone (BQ) were either less effective or deleterious to the reaction (Table 1, entries 6 and 7). Increasing the amount of silver salts could also promote the reaction (Table 1, entries 2, 8 and 9), though we set AgOAc to 4.0 equiv. for subsequent optimizations as higher amounts gave no significant improvement. Switching AcOH to other solvents such as 1,4-dioxane, DME and DMF completely abrogated the reaction (Table 1, entries 10, 11 and 12), highlighting the critical role of AcOH as a solvent. This was further confirmed by replacing AcOH with other acids such as pivalic acid (PivOH) and propionic acid, which considerably reduced the product yields (Table 1, entries 13 and 14). Interestingly, the reaction yield could be enhanced when PivOH was used as an additive (Table 1, entries 8 and 15), while propionic acid and 2,4,6-trimethylbenzoic acid (MesCOOH) had no additive effect at all (Table 1, entries 8, 16 and 17). It has been reported that PivOH could favor the in situ formation of palladium(0)-PivO complexes, hence stabilizing the catalytic palladium(0) species and promoting the reaction.23,24 However, when we used Pd(OPiv)2 as the catalyst in our study, the product yield was rather reduced (Table 1, entries 15 and 18), implying that the role of PivOH might not simply be ascribed to the generation of palladium(0)-PivO complexes. Substituting Pd(OAc)2 with other Pd sources including both palladium(0) and palladium(II) could also furnish the desired product, nevertheless with somewhat lower yields (Table 1, entries 19, 20 and 21). Finally, the reaction yield could be further stepped up by increasing the temperature to a maximum of 130 °C (Table 1, entries 22, 23 and 24), whereas higher temperature led to considerable product decomposition (Table 1, entry 23). To summarize, the best conditions found were Pd(OAc)2 as a catalyst, AgOAc as an oxidant alongside PivOH as an additive in AcOH under air at 130 °C, giving the product 1a in the pure E-form with an isolated yield of 72%.
|
|
||||
|---|---|---|---|---|
| Entry | Oxidant (equiv.) | Additive | Solvent | Yieldb (%) |
| a Unless otherwise noted, the reaction conditions were as follows: triazole 1 (0.10 mmol), styrene (0.50 mmol, 5.0 equiv.), Pd(OAc)2 (0.020 mmol, 20 mol %), additive (0.30 mmol, 3.0 equiv.), solvent (1.5 mL), 120 °C, 20 h, air. b Calculated by 1H NMR. c Under O2 atmosphere. d Under Ar atmosphere. e 101 °C. f 85 °C. g 20 mol% Pd(OPiv)2. h 20 mol% PdCl2. i 10 mol% Pd2dba3. j 20 mol% Pd(PhCN)2Cl2. k 110 °C. l 140 °C. m 130 °C. n Isolated yield. | ||||
| 1 | AgOAc (2.0) | — | AcOH | 23c |
| 2 | AgOAc (2.0) | — | AcOH | 36 |
| 3 | AgOAc (2.0) | — | AcOH | 35d |
| 4 | Ag2CO3 (1.0) | — | AcOH | 36 |
| 5 | Ag2O (1.0) | — | AcOH | 35 |
| 6 | Cu(OAc)2 (2.0) | — | AcOH | 19 |
| 7 | BQ (2.0) | — | AcOH | 0 |
| 8 | AgOAc (4.0) | — | AcOH | 54 |
| 9 | AgOAc (6.0) | — | AcOH | 55 |
| 10 | AgOAc (4.0) | — | Dioxane | 0e |
| 11 | AgOAc (4.0) | — | DME | 0f |
| 12 | AgOAc (4.0) | — | DMF | 0 |
| 13 | AgOAc (4.0) | — | PivOH | <1 |
| 14 | AgOAc (4.0) | — | EtCOOH | 37 |
| 15 | AgOAc (4.0) | PivOH | AcOH | 60 |
| 16 | AgOAc (4.0) | EtCOOH | AcOH | 54 |
| 17 | AgOAc (4.0) | MesCOOH | AcOH | 49 |
| 18 | AgOAc (4.0) | — | AcOH | 47g |
| 19 | AgOAc (4.0) | PivOH | AcOH | 42h |
| 20 | AgOAc (4.0) | PivOH | AcOH | 46i |
| 21 | AgOAc (4.0) | PivOH | AcOH | 52j |
| 22 | AgOAc (4.0) | PivOH | AcOH | 41k |
| 23 | AgOAc (4.0) | PivOH | AcOH | 47l |
| 24 | AgOAc (4.0) | PivOH | AcOH | 70m (72m,n) |
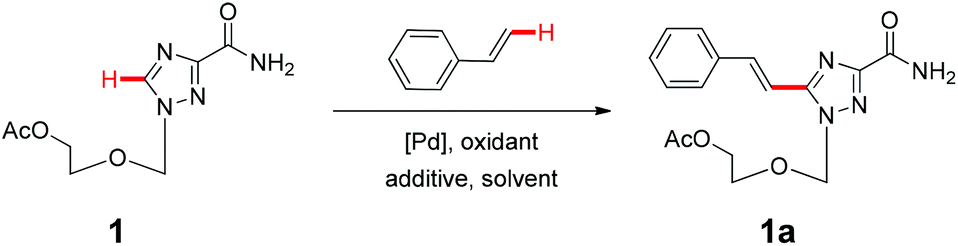
With the above optimized conditions in hand, we first explored the scope of olefin substrates and the representative results are summarized in Table 2. Diverse functional groups such as ester (Table 2, entries 2 and 3), amide (Table 2, entry 4), ether (Table 2, entry 5), nitro- (Table 2, entry 8) and chloro- (Table 2, entries 16–18) were tolerated under our conditions to afford the corresponding products in good yields. Likewise, polycyclic aromatic substrates such as 4-vinylbiphenyl and 2-vinylnaphthalene were also competent substrates (Table 2, entries 6 and 7). In addition, both electron-rich and electron-deficient styrene derivatives exhibited good reactivity for C–H alkenylation under our optimized conditions and smoothly delivered the desired products (Table 2). Even sterically hindered olefins with substituents at the ortho position readily reacted with 1 (Table 2, entries 12, 15 and 18). It is noted that all the alkenylation products could be easily purified using column chromatography, yielding the corresponding E isomers in the pure form, whereas no Z isomer could be isolated nor identified, as testified by NMR analysis. Altogether, these results indicate that under our optimized conditions, oxidative C–H alkenylation is effective and stereoselective with various styrene derivatives, showing good functional group compatibility. Even sterically unfavorable substrates bearing electron-donating or electron-withdrawing substituents could provide the corresponding products without any compromise in yields.
|
|
||
|---|---|---|
| Entry | Ar- | Yieldb (%) |
| a Unless otherwise noted, the reaction conditions were as follows: triazole 1 (0.20 mmol), olefin (1.2 mmol, 6.0 equiv.), Pd(OAc)2 (0.040 mmol, 20 mol %), AgOAc (0.80 mmol, 4.0 equiv.), PivOH (0.60 mmol, 3.0 equiv.), AcOH (3.0 mL), 130 °C, 20 h, air. b Isolated yield. c 5.0 equiv. (1.0 mmol) of olefin were used. d 4.0 equiv. (0.80 mmol) of olefin were used. e 8.0 equiv. (1.6 mmol) of olefin were used.25 | ||
| 1 |
|
72c |
| 2 |
|
70 |
| 3 |
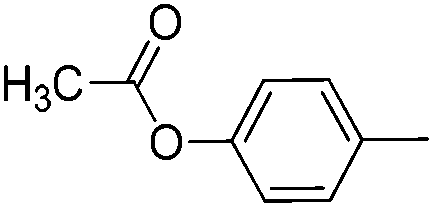
|
60 |
| 4 |
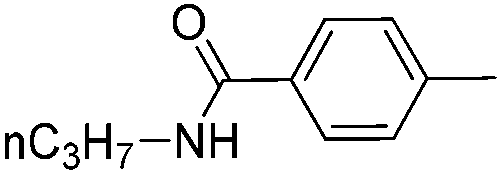
|
63d |
| 5 |
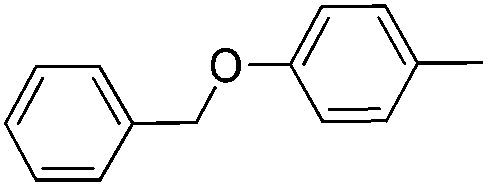
|
52c |
| 6 |
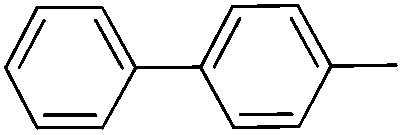
|
62d |
| 7 |
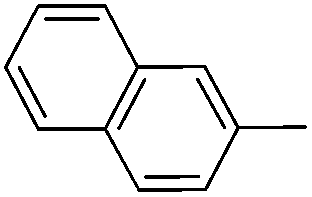
|
62c |
| 8 |
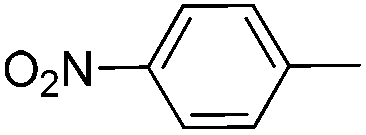
|
61 |
| 9 |
|
57e |
| 10 |
|
68d |
| 11 |
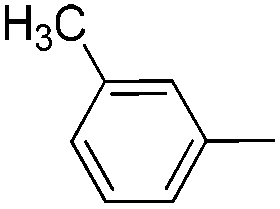
|
67d |
| 12 |
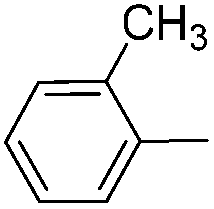
|
67 |
| 13 |
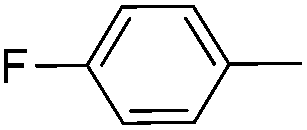
|
69 |
| 14 |
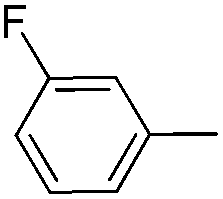
|
66 |
| 15 |
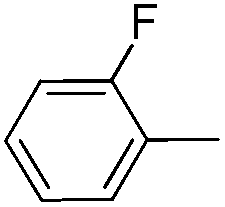
|
62 |
| 16 |
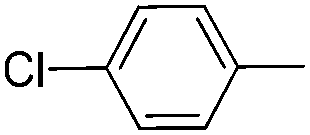
|
65 |
| 17 |
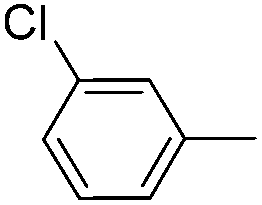
|
63 |
| 18 |
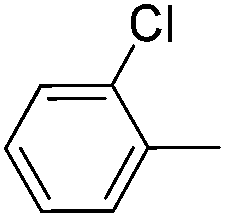
|
52e |
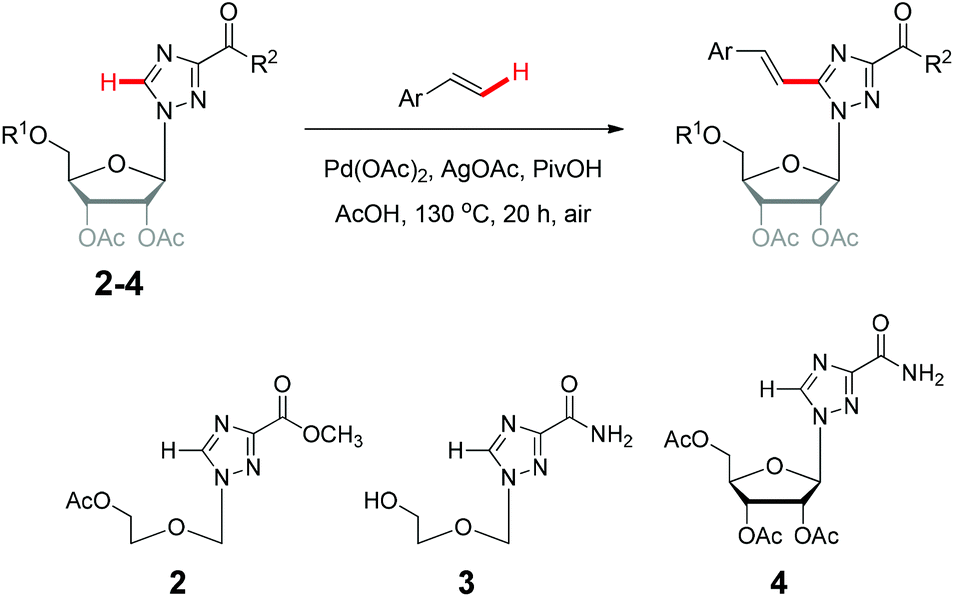
We next examined the direct olefination of various triazole nucleosides 2–4 including sterically challenging ribonucleoside 4 as substrates (Table 3). All triazole nucleosides 2–4 were found to readily undergo the C–H alkenylation with different styrene derivatives to furnish the corresponding products in good yields (Table 3). Interestingly, triazole nucleoside 3 underwent acylation of the free hydroxyl group of the sugar moiety in parallel to the oxidative Heck reaction on the triazole ring, giving the same nucleoside products as those obtained with 1 (Scheme 2). It is noteworthy that the sterically challenging triazole ribonucleoside 4, a derivative of the broad-spectrum antiviral drug ribavirin,26 could also deliver the desired C–H alkenylation products without considerably compromising the yields. This could constitute a useful means to construct novel arylvinyl substituted ribavirin analogues. Collectively, the successful application of oxidative C–H alkenylation to the functionalization of the highly electron deficient and low reactive triazole structure, speaks well for the potentiality of further applications.
 | ||
| Scheme 2 The direct C–H alkenylation of triazole nucleoside (3) is accompanied by the acylation of the free hydroxyl group of the sugar component. | ||
|
|
|||
|---|---|---|---|
| Ar- | Product yieldsb (%) | ||
| 2 | 3 | 4 | |
| a Unless otherwise noted, the reaction conditions were as follows: triazoles 2–4 (0.20 mmol), olefin (1.2 mmol, 6.0 equiv.), Pd(OAc)2 (0.040 mmol, 20 mol %), AgOAc (0.80 mmol, 4.0 equiv.), PivOH (0.60 mmol, 3.0 equiv.), AcOH (3.0 mL), 130 °C, 20 h, air. b Isolated yield. c 5.0 equiv. (1.0 mmol) of olefin were used. d 4.0 equiv. (0.80 mmol) of olefin were used. e 10 equiv. (2.0 mmol) of olefin were used. | |||
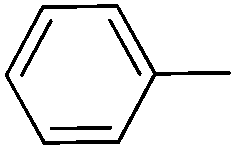
|
70c | 70c | 56 |
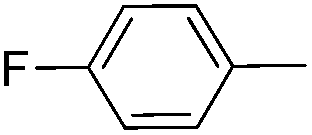
|
61 | 65 | 57e |
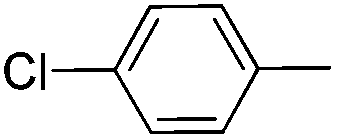
|
64 | 62 | 56e |
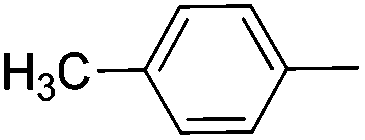
|
65c | 60d | 55 |
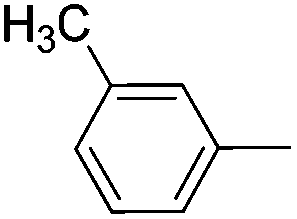
|
66d | 68d | 60c |
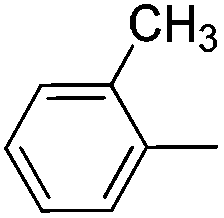
|
54 | 60 | 53e |
Conclusions
In summary, we have reported here a convenient and useful method for direct oxidative C–H alkenylation of the highly electron-deficient triazole nucleosides. A variety of styrene derivatives bearing different functional groups including ester, amide, ether, nitro- and chloro-, as well as polycyclic aromatic substrates were tolerated to supply the desired products favorably with good yields and unique stereoselectivity. Even relatively sterically unfavorable styrene reagents could be successfully coupled onto the triazole heterocyclic nucleobases, offering the corresponding arylvinyltriazole nucleosides. Moreover, triazole nucleosides with either a cyclic ribose or acyclic sugar component underwent smooth oxidative olefination. Even the unprotected triazole nucleoside substrate could furnish the desired coupling products with high yields. To our knowledge, this is the first report on successful implementation of oxidative Heck reaction to construct arylvinyltriazole nucleoside derivatives, which are otherwise difficult to achieve using the conventional Heck reaction. Therefore, this oxidative C–H alkenylation method may constitute a helpful means to creating novel arylvinyl substituted nucleoside analogues. Our results presented here further empower and advocate the potential and practicality of the oxidative Heck reaction via C–H activation for generating structurally challenging chemical entities, which are nevertheless in high demand in medicinal and agrochemical research as well as in material sciences.Experimental section
General
All the reactions were carried out using Schlenk tubes. All the chemicals were purchased from Sigma Aldrich or Alfa Aesar and used directly without any purification. Triazole nucleosides 1–4,27,11b methyl 4-vinylbenzoate,28N-propyl-4-vinyl-benzamide,29 1-(benzyloxy)-4-vinylbenzene30 and 4-nitrostyrene31 were synthesized following the reported procedures. All the solvents used in the reactions were dried according to the described methods and distilled before use except DMF. All the products were purified by flash chromatography on silica gel (Merck 200–300 mesh). 1H NMR spectra were recorded at 250, 300 or 400 MHz and 13C NMR spectra were recorded at 62.5 or 100 MHz on Bruker Avance II 250, Bruker Avance III 300, Bruker Avance III 400 or JEOL ECS 400 spectrometers. Chemical shifts (δ) are expressed in parts per million (ppm) with the residual peak of CHCl3 at 7.26 ppm or TMS at 0.00 ppm as an internal reference. The high resolution mass spectra (HRMS) were obtained with an electrospray ionization (ESI) using the mass spectrometer QStar Elite (Applied Biosystems SCIEX). The exact mass measurement was done in triplicate with a double internal calibration. Analytical thin layer chromatographies (TLC) were performed using silica gel 60 F254 plates 0.2 mm thick with UV light (254 and 364 nm) as a revelator.General procedure of synthesis
Triazoles 1–4 (0.20 mmol), alkene (0.80–2.0 mmol), Pd(OAc)2 (0.040 mmol), AgOAc (0.80 mmol), PivOH (0.60 mmol) and AcOH (3.0 mL) were refluxed at 130 °C for 20 hours under an air atmosphere. The solvent was then removed under reduced pressure and the crude residue was purified by flash chromatography on silica gel (eluent: cyclohexane–EtOAc or CH2Cl2–CH3OH) affording the desired products.Acknowledgements
Financial support was provided by INCa, Cancéropôle PACA, CNRS and Wuhan University. J. T., M. C. and Y. F. are supported by overseas PhD fellowship from China Scholarship Council. We thank Pascal Raynal at Aix-Marseille University for his help in the NMR experiments.Notes and references
- A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945 CrossRef CAS PubMed.
- K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442 CrossRef CAS PubMed.
- J. G. de Vries, Can. J. Chem., 2001, 79, 1086 CrossRef CAS.
- For reviews about the Heck reaction, see: (a) I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009 CrossRef CAS PubMed; (b) D. Mc Cartney and P. J. Guiry, Chem. Soc. Rev., 2011, 40, 5122 RSC; (c) R. F. Heck, Acc. Chem. Res., 1979, 12, 146 CrossRef CAS; (d) C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062 CrossRef CAS PubMed; (e) T. M. Shaikh and F.-E. Hong, Beilstein J. Org. Chem., 2013, 9, 1578 CrossRef PubMed.
- For reviews about the oxidative Heck reaction, see: (a) J. Le Bras and J. Muzart, Chem. Rev., 2011, 111, 1170 CrossRef CAS PubMed; (b) B. Karimi, H. Behzadnia, D. Elhamifar, P. F. Akhavan, F. K. Esfahani and A. Zamani, Synthesis, 2010, 1399 CrossRef CAS PubMed; (c) X. Chen, K. M. Engle, D. Wang and J. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed; (d) V. Ritleng, C. Sirlin and M. Pfeffer, Chem. Rev., 2002, 102, 1731 CrossRef CAS PubMed; (e) I. Moritani and Y. Fujiwara, Synthesis, 1973, 524 CrossRef CAS.
- For selected examples about the oxidative Heck reaction, see: (a) F. Klotter and A. Studer, Angew. Chem., Int. Ed., 2014, 53, 2473 CrossRef CAS PubMed; (b) L. Meng, C. Liu, W. Zhang, C. Zhou and A. Lei, Chem. Commun., 2014, 50, 1110 RSC; (c) L. Yang, G. Zhang and H. Huang, Adv. Synth. Catal., 2014, 356, 1509 CrossRef CAS; (d) C.-H. Ying, S.-B. Yan and W.-L. Duan, Org. Lett., 2014, 16, 500 CrossRef CAS PubMed; (e) C. Pi, Y. Li, X. Cui, H. Zhang, Y. Han and Y. Wu, Chem. Sci., 2013, 4, 2675 RSC; (f) G. G. Pawar, G. Singh, V. K. Tiwari and M. Kapur, Adv. Synth. Catal., 2013, 355, 2185 CrossRef CAS; (g) X. Cong, H. Tang, C. Wu and X. Zeng, Organometallics, 2013, 32, 6565 CrossRef CAS; (h) L. Meng, K. Wu, C. Liu and A. Lei, Chem. Commun., 2013, 49, 5853 RSC; (i) M. Min, Y. Kim and S. Hong, Chem. Commun., 2013, 49, 196 RSC.
- X. Zhang, S. Fan, C. He, X. Wan, Q. Min, J. Yang and Z. Jiang, J. Am. Chem. Soc., 2010, 132, 4506 CrossRef CAS PubMed.
- Y. Huang, F. Song, Z. Wang, P. Xi, N. Wu, Z. Wang, J. Lan and J. You, Chem. Commun., 2012, 48, 2864 RSC.
- W.-C. Lee, T.-H. Wang and T.-G. Ong, Chem. Commun., 2014, 50, 3671 RSC.
- We also tried the conditions reported in ref. 8 and 9. Unfortunately, no corresponding triazole nucleoside product could be obtained.
- (a) R. Zhu, M. Wang, Y. Xia, F. Qu, J. Neyts and L. Peng, Bioorg. Med. Chem. Lett., 2008, 18, 3321 CrossRef CAS PubMed; (b) J. Wan, Y. Xia, Y. Liu, M. Wang, P. Rocchi, J. Yao, F. Qu, J. Neyts, J. L. Iovanna and L. Peng, J. Med. Chem., 2009, 52, 1144 CrossRef CAS PubMed; (c) Y. Xia, Y. Liu, J. Wan, M. Wang, P. Rocchi, F. Qu, J. L. Iovanna and L. Peng, J. Med. Chem., 2009, 52, 6083 CrossRef CAS PubMed; (d) Y. Xia, F. Qu and L. Peng, Mini-Rev. Med. Chem., 2010, 10, 806 CrossRef CAS; (e) Y. Fan, Y. Xia, J. Tang, P. Rocchi, F. Qu, J. L. Iovanna and L. Peng, Org. Lett., 2010, 12, 5712 CrossRef CAS PubMed; (f) M. Wang, R. Zhu, Z. Fan, Y. Fu, L. Feng, J. Yao, A. Maggiani, Y. Xia, F. Qu and L. Peng, Bioorg. Med. Chem. Lett., 2011, 21, 354 CrossRef CAS PubMed; (g) Y. Xia, M. Wang, O. Demaria, J. Tang, P. Rocchi, F. Qu, J. L. Iovanna, L. Alexopoulou and L. Peng, J. Med. Chem., 2012, 55, 5642 CrossRef CAS PubMed; (h) Y. Xia, Y. Liu, P. Rocchi, M. Wang, Y. Fan, F. Qu, J. L. Iovanna and L. Peng, Cancer Lett., 2012, 318, 145 CrossRef CAS PubMed.
- L. A. Agrofoglio, I. Gillaizeau and Y. Saito, Chem. Rev., 2003, 103, 1875 CrossRef CAS PubMed.
- J. H. Cho and K. H. Shaughnessy, Synlett, 2011, 2963 CAS.
- J. Dadová, P. Vidláková, R. Pohl, L. Havran, M. Fojta and M. Hocek, J. Org. Chem., 2013, 78, 9627 CrossRef PubMed.
- We were unable to get iodotriazole nucleosides in our hands, mainly because of the notoriously unreactive features of the triazole heterocycles.
- G. Wu, J. Zhou, M. Zhang, P. Hu and W. Su, Chem. Commun., 2012, 48, 8964 RSC.
- I. Moritanl and Y. Fujiwara, Tetrahedron Lett., 1967, 8, 1119 CrossRef.
- Y. Fujiwara, I. Noritani, S. Danno, R. Asano and S. Teranishi, J. Am. Chem. Soc., 1969, 91, 7166 CrossRef CAS.
- C. Jia, T. Kitamura and Y. Fujiwara, Acc. Chem. Res., 2001, 34, 633 CrossRef CAS PubMed.
- G. Meng, H. Niu, G. Qu, J. S. Fossey, J. Li and H. Guo, Chem. Commun., 2012, 48, 9601 RSC.
- B. Liu, Y. Huang, J. Lan, F. Song and J. You, Chem. Sci., 2013, 4, 2163 RSC.
- M. Miyasaka, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2010, 75, 5421 CrossRef CAS PubMed.
- L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed.
- M. Lafrance and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 16496 CrossRef CAS PubMed.
- The amount of olefins also affected the product yields, depending on the electronic and steric properties of the styrene derivatives.
- (a) R. W. Sidwell, J. H. Huffman, G. P. Khare, L. B. Allen, J. T. Witkowski and R. K. Robins, Science, 1972, 177, 705 CAS; (b) J. D. Graci and C. E. Cameron, Rev. Med. Virol., 2006, 16, 37 CrossRef CAS PubMed.
- R. Zhu, F. Qu, G. Quéléver and L. Peng, Tetrahedron Lett., 2007, 48, 2389 CrossRef CAS PubMed.
- M. Schedler, D.-S. Wang and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 2585 CrossRef CAS PubMed.
- G. Quéléver, P. Kachidian, C. Melon, C. Garino, Y. Laras, N. Pietrancosta, M. Sheha and J. L. Kraus, Org. Biomol. Chem., 2005, 3, 2450 Search PubMed.
- Z.-H. Shan, J. Liu, L.-M. Xu, Y.-F. Tang, J.-H. Chen and Z. Yang, Org. Lett., 2012, 14, 3712 CrossRef CAS PubMed.
- B. Schmidt and R. Berger, Adv. Synth. Catal., 2013, 355, 463 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, analytical data and spectra of all synthesized compounds. See DOI: 10.1039/c4ob01836b |
| This journal is © The Royal Society of Chemistry 2015 |