
Synthesis of PS/PO-chimeric oligonucleotides using mixed oxathiaphospholane and phosphoramidite chemistry†
Ewa
Radzikowska
a and
Janina
Baraniak
*ab
aDepartment of Bioorganic Chemistry, Center of Molecular and Macromolecular Studies, Polish Academy of Sciences, Sienkiewicza 112, 90-363 Łódź, Poland
bInstitute of Chemistry, Environmental Protection and Biotechnology, Jan Długosz University, Armii Krajowej 13/15, 42-201 Częstochowa, Poland. E-mail: baraniak@cbmm.lodz.pl
First published on 14th October 2014
Abstract
Chimeric oligonucleotides containing phosphodiester and phosphorothioate linkages have been obtained using the solid phase synthesis. The oligonucleotide parts possessing natural internucleotide phosphate bonds were assembled using commercially available nucleoside 3′-O-(2-cyanoethyl-N,N-diisopropylamino)phosphoramidites 7 whereas the phosphorothioate segment was built using nucleoside 3′-O-(2-thio-1,3,2-oxathiaphospholanes) 3. The oxidation steps, crucial for the conversion of phosphite linkages into the phosphate moieties, were conducted using tert-butylperoxy-trimethylsilane, and this reagent was not harmful to the diester phosphorothioate linkages. When P-diastereopure nucleoside 3′-O-(2-thio-1,3,2-oxathiaphospholane) monomers were employed the resulting chimeric backbone retained the P-stereoregularity of the phosphorothioate units.
Introduction
The oligonucleotide based approach for downregulation of gene expression in cells is based on delivery of a synthetic oligonucleotide complementary to a target DNA or mRNA, with the intention to block either inhibition of transcription/translation (steric blockage) or enzymatic cleavage of the target mRNA (e.g. activation of RNase H).1 Both methodologies have gained remarkable attention and synthetic oligonucleotides have become an important class of potential therapeutic agents for the treatment of viral infections and genetic diseases.2 Unfortunately, both approaches have proven to be challenging in practical applications. A good oligonucleotide agent should be resistant to exo- and/or endonucleases, and should have suitable pharmacological and pharmacokinetic profiles, as well as high affinity for the target. However, susceptibility of unmodified phosphodiester oligonucleotides (PO-ODNs) to nucleolytic degradation by intracellular nucleases made them virtually useless for therapeutic purposes. To alleviate that problem, many chemical modifications have been introduced to the natural phosphodiester backbone to increase the stability of oligonucleotides in body fluids.3 Among them, phosphorothioate oligonucleotides (PS-ODNs), created by replacing one of the nonbridging oxygen atoms with a sulfur atom, are the major representatives of first generation DNA analogs.4PS-ODNs display several attractive features like nuclease resistance, activation of RNase H, and good pharmacokinetic properties.5 Their limitations include sequence-nonspecific binding to cellular proteins and concentration dependent competitive inhibition of many nucleases and polymerases.6 Because the uniform modification of oligonucleotides with phosphorothioate linkages is not required to confer enhanced stability,7 several efforts have been made to synthesize “chimeric” oligonucleotides possessing natural phosphate and phosphorothioate internucleotide linkages in the same molecule (PS/PO-ODNs).8 Two main types of such constructs have been reported. The first one consists of oligomers possessing the PS-linkages in alternate positions,9 while in the second the oligomer is composed of oligophosphate and oligophosphorothioate clusters.8a,b,c In the latter type it was assumed that the unmodified segments would preserve good hybridization properties of natural oligonucleotides, while the phosphorothioate segments would enhance the in vivo stability towards exo- and endonucleases compared to the unmodified PO-Oligos. Thus, the chimeric PS/PO-Oligos were expected to be advantageous over PS-Oligos8 with increased affinity towards the target oligonucleotides and reduced the non-specific interactions with proteins.
The actual biological activity of the oligonucleotide phosphorothioates (e.g., interactions with proteins or nucleic acids) may depend on stereochemical factors because the phosphorus atoms in the PS-linkages are stereogenic.10 One has to keep in mind that the phosphoramidite11 and H-phosphonate12 methodologies (commonly used to prepare PS-Oligos) are nonstereospecific and give a mixture of 2n diastereomers, where n is the number of internucleotide phosphorothioate functions. Thus, various methods have been elaborated to synthesize these P-chiral oligonucleotide analogs in a stereocontrolled manner,13 among them the oxathiaphospholane method developed by Stec et al.,14 the method utilizing nucleoside 3′-O-(3-N-acyl)oxazaphospholidine derivatives as monomer units,15 and the method based on a stereoselective synthesis of nucleoside 3′-O-oxazaphospholidine monomers16 are the most significant.
All these three methodologies can be used to synthesize PO/PS chimeric oligonucleotides although only the oxathiaphospholane method requires an additional set of 2-oxo-1,3,2-oxathiaphospholane monomers for the incorporation of PO-linkages.14b Taking into account the commercial availability of phosphoramidite nucleotides, it was tempting to combine the oxathiaphospholane method with the phosphoramidite one in order to obtain oligonucleotides having a stereoregular phosphate/phosphorothioate chimeric backbone. Unfortunately, the oxathiaphospholane method is incompatible with the phosphoramidite method of DNA synthesis, because the internucleotide phosphorothioate linkages (assembled using the oxathiaphospholane monomers) are diester moieties and they easily get oxidized during the treatment with an iodine–base–water solution,17 which is a typical way for the conversion of unstable phosphite (PIII) linkages into the stable phosphate (PV) moieties. To solve this problem one had to find an effective and strictly chemoselective reagent to oxidize the trivalent phosphorus atoms leaving untouched the labile phosphorothioate diesters present in the growing oligomer molecule. Here we present our results on making the oxathiaphospholane and phosphoramidite protocols compatible with the selection of tert-butylperoxy-trimethylsilane as an oxidizing reagent.
Results and discussion
Searching for mild oxidizing systems we focused our attention to organic hydroperoxides. For initial studies we selected a solution of tert-butyl hydroperoxide (TBHP, 1) in decane. Its ability to oxidize PIII triesters to the corresponding phosphates was reported inter alia by Hayakawa et al.18 and Engels et al.19 To assess the chemical stability of the phosphorothioate diesters towards TBHP, the model phosphorothioate dinucleotide (Scheme 1, DMTTPSTOAc, 2; 31P NMR (CDCl3) two resonance signals with chemical shifts at ∼55 ppm) was synthesized on the reaction of thymidine-3′-O-(2-thio-4,4-dimethyl-1,3,2-oxathiaphospholane) (3d) with 3′-O-acetyl-thymidine (4), and its acetonitrile solution was treated with 10 equiv. of 1 at ambient temperature. After 30 minutes the 31P NMR analysis of the reaction mixture showed the presence of the oxidized DMTTPOTOAc (5, ca. 33%; 31P NMR (CD3CN) δ ∼ 0 ppm), but when 50 equiv. of TBHP was used complete disappearance of 2 was observed after 30 minutes (see ESI†). | ||
| Scheme 1 Synthesis of the model phosphorothioate dinucleotide 2 and its instability towards TBHP. | ||
In order to decrease the reactivity of TBHP, it was transformed into tert-butylperoxy-trimethylsilane (tert-BuOOSiMe3, 6). This reagent has been successfully applied by Salamończyk for selective oxidation of phosphite groups in dendrimers already possessing diester P = S and P = Se branching units within the same molecule.20 The trimethylsilane modification made 6 to be much less reactive than 1, thus 50 equiv. of the oxidant and 30 minutes time were needed for the quantitative oxidation of the model dithymidyl phosphite. It was demonstrated by 31P NMR spectroscopy that under these conditions no desulfurization of 2 occurred and that the phosphorothioate function in 2 was not temporarily protected (silylated) with the trimethylsilyl group delivered by 6 (see ESI†).
Having defined the conditions suitable for the reaction in solution, the analogous reaction was executed in solid-phase experiments on a 1 μmol scale. The reaction pathway leading to the desired PS/PO-ODNs is shown in Scheme 2. Commercially available nucleoside 3′-O-(2-cyanoethyl-N,N-diisopropylamino)phosphoramidites 7 were used for the introduction of natural phosphate nucleotide units. In turn, phosphorothioate nucleotide parts were introduced using nucleoside oxathiaphospholane monomers 3 (Table 1), which were obtained in the modified manner compared to the published procedure.14b In preliminary experiments the monomers 3 were used as mixtures of P-diastereomers.
 | ||
| Scheme 2 Synthesis of PS/PO-Oligos. Reagents: (i) 3, DBU, CH3CN; (ii) CHCl2COOH in CH2Cl2; (iii) 7, 1H-tetrazole, CH3CN; (iv) tert-BuOOSiMe3, (6); (v) m-fold repetition of steps ii–iv; (vi) piperidine, NH4OH. | ||
| Compound | Chromatographic mobility vs. configuration at P atom | 31P NMR (CDCl3) δ (ppm) | Yield (%) |
|---|---|---|---|
| 3a (Ade) | “Fast” (SP) | 103.57 | 42 |
| “Slow” (RP) | 103.65 | 33 | |
| 3b (Cyt) | “Fast” (SP) | 104.09 | 45 |
| “Slow” (RP) | 104.34 | 30 | |
| 3c (Gua) | “Fast” (SP) | 103.78 | 20 |
| “Slow” (RP) | 103.88 | 26 | |
| 3d (Thy) | “Fast” (RP) | 106.70 | 49 |
| “Slow” (SP) | 106.73 | 44 |
Using the procedure presented in Scheme 2, a series of protected chimeric PS/PO-oligonucleotides 13 was synthesized (Table 2). Starting from the 5′-end, the oligomers contained triester phosphate internucleotide linkages (1–5 units), followed by 1–3 diester phosphorothioate internucleotide linkages (Table 2). The starting nucleoside 8 was anchored to the controlled pore glass support using a DBU-resistant sarcosinyl-succinoyl linker.21 To build the phosphorothioate segments, the oxathiaphospholane derivatives 3 were condensed with the 5′-hydroxyl group of the growing oligomers. Then, the PO-segments were assembled via the phosphoramidite method using a 20-fold molar excess of the phosphoramidites 7 for each coupling, and at each of those cycles the phosphite moieties in oligonucleotide derivatives 11 were oxidized using a 50-fold molar excess (concentration: 0.33 μmol ml−1) of tert-BuOOSiMe3 (6) over 30 minutes to form the corresponding phosphates 12. Similarly, a 15-mer DMTTPSTPSTPS(TPO)8TPSTPSTPST was obtained containing an octameric phosphate segment at the middle of the chain and trimeric phosphorothioate segments at, both, 3′- and 5′-ends. Finally, the oligomers 12 were deprotected with concentrated NH4OH to form DMT-tagged chimeric PS/PO oligonucleotides 13, which without any further purification were subjected to MALDI-TOF MS analysis to assess the content of the oxidized products. For these compounds the molecular ions were accompanied by ions at m/z = M-304, attributed to the oligomers being partially detritylated due to the acidity of the matrices used.
| Entry | Comp | Sequence | Theoretical mass (Da) | Observed mass MALDI-MS m/z (M − 1) | Peak (M − 16) contenta (%) |
|---|---|---|---|---|---|
| a The intensities of the (M − 1) peaks were taken as 100%. b HPA (3-hydroxypicolinic acid) matrix. c THA (2′,4′,6′-trihydroxyacetophenone) matrix. d Voyager-Elite MALDI-TOF mass spectrometer. e SHIMADZU Axima Performance MALDI-TOF mass spectrometer. f Below the detection limit. | |||||
| 1 | 13a | DMTTPOTPSTPST | 1488 | 1488.4 | 0b,d,f |
| 2 | 13b | DMTTPOTPOdCPST | 1457 | 1456.8 | 3.7c,d |
| 3 | 13c | DMTTPOTPOdGPST | 1497 | 1496.8 | 5.6c,d |
| 4 | 13d | DMTTPOTPOTPSTPST | 1792 | 1791.7 | 3.9b,d |
| 5 | 13e | DMTTPOTPOTPOAPSAPST | 2114 | 2113.1 | 4.8c,d |
| 6 | 13f | OHTPOTPOTPOAPSAPST | 1812 | 1811.3 | 7.0c,d |
| 7 | 13g | DMTTPOTPOTPOd(CPSCPS)T | 2066 | 2064.9 | 4.3b,d; 6.0c,d |
| 8 | 13h | OHTPOTPOTPOd(CPSCPS)T | 1764 | 1762.7 | 6.3b,d; 11.0c,d |
| 9 | 13i | DMTd(GPOGPOAPOAPOAPSA) | 2167 | 2167.0 | 10.6b,d |
| 10 | 13j | DMTd(GPOGPOAPOAPSAPSA) | 2183 | 2183.0 | 8.0c,d |
| 11 | 13k | DMTTPOTPOTPOd(APSAPSAPSA) | 2454 | 2454.1 | 1.3b,d |
| 12 | 13l | DMTTPOTPOTPOTPOd(APSAPSAPSA) | 3062 | 3061.5 | 0b,d,f |
| 13 | 13m | DMTTPOTPOTPOTPOTPOd(CPSCPSCPSC) | 2964 | 2963.5 | 25.9b,d |
| 14 | 13n | DMTTPSTPSTPS(TPO)8TPSTPSTPST | 4896 | 4895.1 | 0b,d,f |
| 15 | 13o | [all-Sp]-TPOTPOTPOTPOTPOd(APSAPSAPS)T | 2749 | 2751.1 | 20.0b,d; 16.0b,e |
| 16 | 13p | [all-Sp]-TPOTPOTPOTPOTPOd(CPSCPSCPS)T | 2677 | 2674.9 | 15.0b,d |
| 17 | 13r | [all-Rp]-TPOTPOTPOTPOTPOd(CPSCPSCPS)T | 2677 | 2674.3 | 5.6b,d; 3.0b,e |
| 18 | 13s | [all-Sp]-TPOTPOTPOTPOTPOd(GPSGPSGPS)T | 2797 | 2797.4 | 18.0b,d |
| 19 | 13t | [all-Rp]-TPOTPOTPOTPOTPOTPSTPSTPST | 2722 | 2721.5 | 6.0b,d |
Starting from pure P-diastereomers of 3, chimeric PS/PO-Oligos possessing stereodefined phosphorothioate linkages (of either RP or SP configuration)14b were synthesized (entries 15–19) and the MS data (Table 2) are given for the detritylated oligomers. During each synthesis of PS/PO-Oligo, the dimethoxytrityl cation absorbance was monitored and the crude product was characterized by means of RP HPLC (see ESI†). The repetitive yields calculated on the DMT+ cation absorbance decay were in the range of 94–96%.
One might consider that the yield of the synthesis of 13n should be lower than those for the other assembled oligomers. This is because, in this case, the attachment of the last PS segment by the oxathiaphospholane method inevitably leads to the elimination of the 2-cyanoethyl groups from the PO units and such generated phosphodiester linkages may be susceptible to side reactions. However, this can be avoided, because exactly the same situation had taken place during the synthesis of chimeric PS/PO-Oligos based only on the oxathiaphospholane method14b where the by-products related to transformations of the phosphodiester linkages were not observed.
An exemplary 31P NMR spectrum (recorded for crude 13r) indicates the presence of two groups of signals at the expected intensity ratio 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, corresponding to the phosphorothioate and phosphate moieties (δ 55.50 ppm and −0.97 ppm, respectively, Fig. 1).
:5, corresponding to the phosphorothioate and phosphate moieties (δ 55.50 ppm and −0.97 ppm, respectively, Fig. 1).
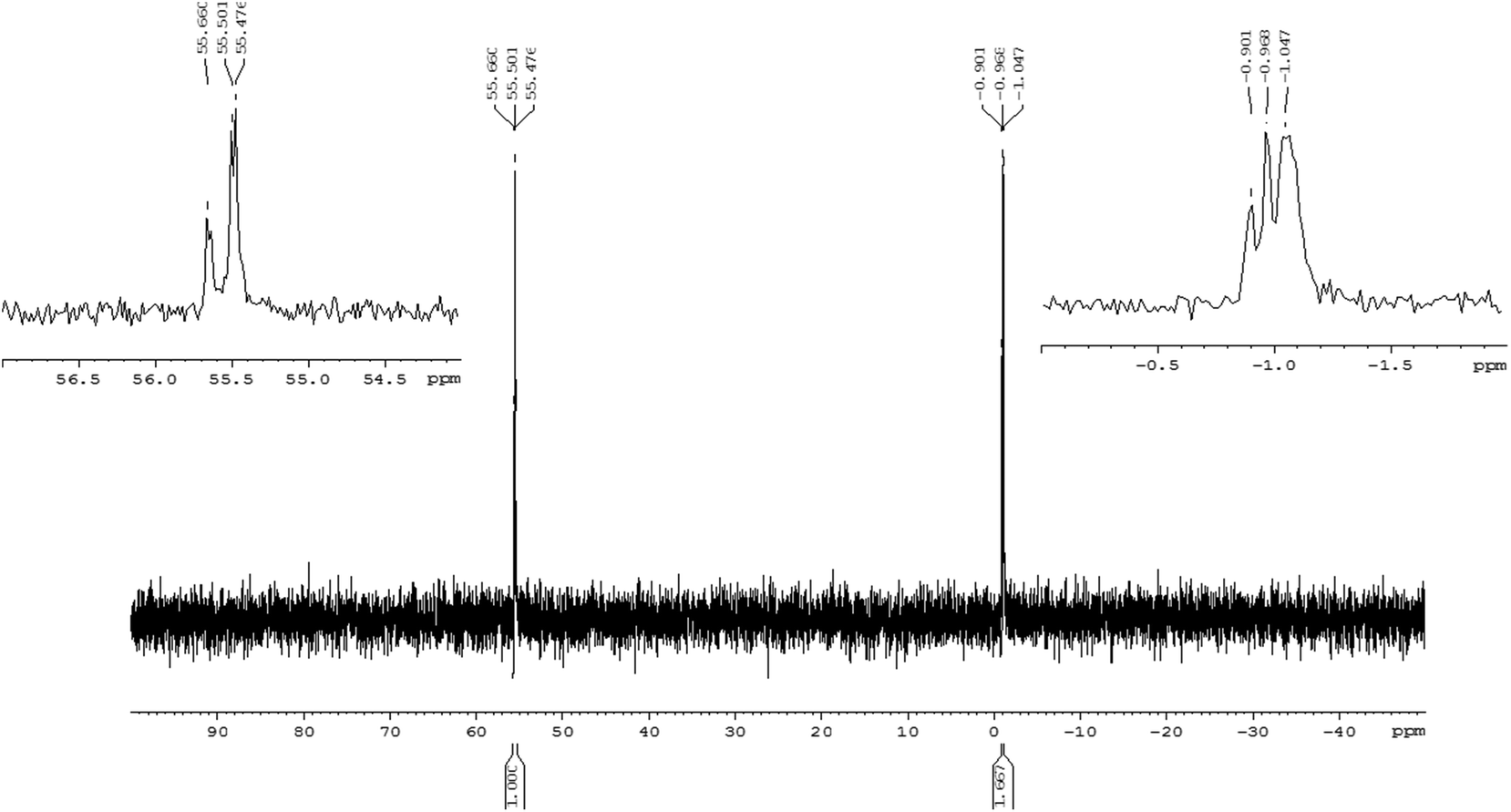 | ||
| Fig. 1 202 MHz 31P NMR spectrum of crude [all-Rp]-TPOTPOTPOTPOTPOd(CPSCPSCPS)T (13r). | ||
Analysis of the MALDI-TOF MS data revealed that in the majority of spectra the signal corresponding to the molecular ion (taken as 100%) was accompanied by a signal at m/z (M-16) of low intensity (typically <10%), which we attributed to the molecules with one of the sulfur atoms being randomly replaced with an oxygen atom. However, if the PS→PO exchange was caused by tert-BuOOSiMe3, the phosphorothioate diester linkages should be exposed to a cumulative treatment over up to eight synthetic cycles. Thus, one should observe a correlation between the number of phosphoramidite condensation/oxidation steps and the extent of the PS→PO exchange. But none of the recorded spectra contained signals attributable to the oxidation of two (m/z = M-32) or three (m/z = M-48) P-S linkages in a single molecule. Moreover, the MS analysis of 13k (entry 11) showed the presence of the M-16 signal, whereas in the MS spectra recorded for 13l (which contains one more thymidine phosphate residue) and for 13n (eight consecutive oxidation steps) the relevant M-16 signals were not detected at all. On the other hand, it has been noticed that the extent of desulfurization for a given oligomer varied depending on the spectrometer and on the matrix used for MS analysis (entry 15 or 17). When THA was employed, a more intense signal (M-16) was observed compared to that detected for the HPA matrix (entry 7 or 8).
It is worth noting that when oligo 13o-mix was synthesized using only commercially available deoxyribonucleoside phosphoramidites 7 (B1 = Thy, AdeBz) and either 3H-1,2-benzodithiol-3-one1,1-dioxide or iodine–water to oxidize the trivalent phosphorus atoms, MALDI-TOF MS analysis of deprotected oligo revealed desulfurization of phosphorothioate diesters to the extent of 22%.
Therefore, these studies have shown that to a certain extent the desulfurization occurred under the MALDI-TOF MS operating conditions. To the best of our knowledge it is the first example demonstrating that unexpected desulfurization can take place even under common negative MALDI-TOF MS operating conditions. On the other hand desulfurization of phosphorothioate oligonucleotides occurring during negative electrospray ionization (ESI-MS) has been described in the literature.22 Thus, a comparative analysis of the desulfurization of oligo 13k (entry 11) based on spectra recorded by means of ESI and MALDI techniques (see ESI†) revealed the greater amount of its desulfurization product in the ESI-MS spectrum (ca. 8% vs. 1.3%).
To identify the other factors which might be responsible for desulfurization we focused our attention on the paper published by Beaucage et al. concerning the desulfurization of PS-Oligos synthesized by the phosphoramidite method.23 Their studies based on 31P NMR revealed that under solid-phase synthesis conditions the loss of the 2-cyanoethyl protecting group from the triester thiophosphate linkage (occurring to the extent of ∼5–13%) depended on the capping reagents being used.24 Generated in this way phosphorothioate diesters underwent desulfurization by phosgene detected in, both, fresh and aged solutions of the detritylating reagent (3% TCA in CH2Cl2). Since the internucleotide phosphorothioate linkages assembled by the oxathiaphospholane method were unprotected we have taken into consideration the influence of the detritylating reagent on the desulfurization of the PS/PO-Oligos. Hence, oligos 13-e and 13-g were synthesized on the solid support (entries 5 and 7) and MS analysis showed that the observed desulfurization increased for oligos which were released from the support after removal of the DMT group (entries 6 and 8).
These examples have shown that the process of detritylation plays a role in the desulfurization process. This phenomenon should not be neglected, because the DMT group is repeatedly removed during PS/PO-oligonucleotide chain assembly. As a remedy to avoid the negative effect of phosgene on the quality of the PS/PO-Oligos a freshly prepared 3% TCA in toluene may be used.
Conclusions
PS/PO-Oligos having a phosphate/stereoregular phosphorothioate chimeric backbone have been synthesized using mixed oxathiaphospholane and phosphoramidite chemistry. The oligonucleotide segments possessing natural internucleotide phosphate bonds were assembled using commercially available nucleoside 3′-O-(2-cyanoethyl-N,N-diisopropylamino)phosphoramidites 7 whereas phosphorothioate parts were built based on nucleoside-2-thio-1,3,2-oxathiaphospholanes 3. The oxidation step, crucial for the formation of PO-linkages, was conducted by means of tert-butylperoxy-trimethylsilane and phosphorothioate linkages were stable towards this reagent. Nonetheless, low intensity signals at m/z (M-16) (typically <10%) attributed to the molecules with one of sulfur atoms along the chain randomly replaced with oxygen were present in the majority of MALDI-TOF MS spectra. But, in our opinion, the occurrence of desulfurization during the recording of the spectrum seems to be the most likely explanation of this observation because the extent of desulfurization varied depending on the spectrometer and on the matrix used. No data on this subject have been found in the literature. Additionally, phosgene present in the reagent (3% TCA in CH2Cl2) used for the 5′-deblocking step in solid phase oligonucleotide synthesis seems to be in part responsible for the desulfurizing of the PS-linkages. Thus the use of toluene instead of CH2Cl2 may be beneficial in the preparation of PS/PO-Oligos.Experimental
General experimental
The nuclear magnetic resonance spectra were recorded on a Brüker AC-200 instrument (200 MHz, TMS internal standard for 1H and 85% H3PO4 as the external standard for 31P) unless stated otherwise. The FAB-MS spectra (13 keV, Cs+) were recorded on a Finnigan MAT 95 spectrometer, negative ion MALDI mass spectra were recorded on a Voyager-Elite instrument (PerSeptive Biosystems Inc., Framingham, USA) and on a Shimadzu Axima Performance MALDI-TOF mass spectrometer, while electrospray mass spectrometry analyses were done using a Brüker amaZon speed ETD instrument. A mixture of 0.05 M solution of 2,4,6-trihydroxyacetophenone (THA) or 3-hydroxypicolinic acid (HPA) in 50% acetonitrile and 0.2 M solution of diammonium hydrogen citrate in water (8:1, v/v) was used as a matrix. The mass spectrum was accumulated from at least 50 laser shots and processed using the Data Explorer ver. 4 program (Applied Biosystems, Foster City, CA).
Deoxyribonucleosides were purchased from Pharma Waldhof (Germany). Tetrazole solution, 1,4-diazabicyclo[5.4.0]undec-7-ene (DBU), 2-cyanoethanol, succinic anhydride, bis(trimethylsilyl)acetamide, acetic anhydride, dimethyl-aminopyridine and piperidine were purchased from Aldrich (USA). Fmoc-sarcosine was purchased from Bachem (Switzerland). Long chain alkylamine controlled pore glass (500 Å, mesh 80–120) was supplied by Sigma (USA). Phosphorus trichloride, ethyl acetate, butyl acetate, chloroform, dichloromethane, benzene and triethylamine were purchased from POCH (Poland). Elemental sulfur was dried under high vacuum for 12 h. Acetonitrile was used as a solvent for DBU and oxathiaphospholane monomers was dried over P2O5 (5 g L−1) and distilled through a 20 cm Vigreux column under an atmosphere of dry argon and stored over 4 Å molecular sieves. At least one-third of the initial volume must be left in the flask. Acetonitrile dried in this way must be transferred using gastight syringes under an atmosphere of dry argon, or by vacuum line. Phosphoramidites of 5′-O-DMT-nucleosides were purchased from Glen Research and tert-butyl hydroperoxide [a solution in decane (ca. 5.5 M over molecular sieves 4 Å)] was obtained from Fluka. 2-Chloro-1,3,2-oxathiaphospholane (14)26 and 2-chloro-4,4-dimethyl-1,3,2-oxathiaphospholane (15)14b were prepared according to the procedures described previously.
Phosphitylation of the protected nucleosides with 2-chloro-1,3,2-oxathiaphospholane (14) and 2-chloro-4,4-dimethyl-1,3,2-oxathiaphospholane (15) – a general procedure
To a mixture of appropriately protected 2′-deoxyribonucleoside (1 mmol) (ABz, GiBu, T, or CBz), elemental sulfur (2 mmol) and dry pyridine (4 ml) 14 (1.2 mmol; in the case of CBz, ABz, GiBu) or 15 (1.2 mmol, for T) were added dropwise. The reaction mixture was stirred at room temperature for 3 h (monitored by TLC). Then the solvent was removed in vacuo and into it the residual acetonitrile (5 mL) was added. Excess sulfur was filtered off and the filtrate was condensed in vacuo. The product 3 was isolated by silica gel column chromatography (230–400 mesh, 0→3% methanol in chloroform with 0.1% addition of pyridine) as a mixture of diastereomers.5′-O-(4,4′-Dimethoxytrityl)-N6-benzoyl-2′-deoxyadenosine-3′-O-(2-thio-1,3,2-oxathiaphospholane) (3a)
Yield 75%; 31P NMR (CDCl3) δ: 103.62, 103.71; 1H NMR (200 MHz, CD3CN, for diastereomeric mixture) δ: 8.56 (s, 1H), 8.24 (s, 1H), 7.98 (br.d, 2H), 7.63–7.16 (m, 12H), 6.80 (dd, 4H), 6.44 (t, 1H), 5.61–5.44 (m, 1H), 4.62–4.28 (m, 3H), 4.13–3.98 (m, 1H), 3.73 (s, 6H), 3.39–3.14 (m, 3H), 2.80–2.65 (m, 1H), 2.29–2.21 (m, 1H) FAB-MS m/z: (M − 1) 794.3; HR-MS (FAB-MS) m/z for C40H37N5O7PS2 calculated m/z: 794.1882, found m/z: 794.1872.5′-O-(4,4′-Dimethoxytrityl)-N4-benzoyl-2′-deoxycytidine-3′-O-(2-thio-1,3,2-oxathiaphospholane) (3b)
Yield 74%; 31P NMR (CDCl3) δ: 104.20; 104.31; 1H NMR (200 MHz CD3CN, for diastereomeric mixture) δ: 8.11 (d, 1H), 7.93 (d, 2H), 7.64–7.23 (m, 13H), 6.89 (d, 4H), 6.15 (t, 1H), 5.29–5.17 (m, 1H), 4.58–4.28 (m, 3H), 3.76 (s, 6H), 3.56–3.44 (m, 3H), 3.39 (dd, 1H), 2.80–2.64 (m, 1H), 2.50–2.34 (m, 1H) FAB-MS m/z: (M − 1) 770. HR-MS (FAB-MS) m/z for C39H37N3O8PS2 calculated m/z: 770.1773, found m/z: 770.1760.5′-O-(4,4′-Dimethoxytrityl)-N2-isobutyryl-2′-deoxyguanosine-3′-O-(2-thio-1,3,2-oxathiaphospholane) (3c)
Yield 51%; 31P NMR (CDCl3) δ: 103.54, 103.71; 1H NMR (200. MHz, CDCl3, for diastereomeric mixture) δ: 7.82 (s, 1H), 7.41–7.14 (m, 9H), 6.78 (dd, 4H), 6.22 (t, 1H), 5.42–5.27 (m, 1H), 4.55–4.20 (m, 2H), 3.72 (s, 6H), 3.58–3.27 (m, 3H), 3.17–2.99 (m, 1H), 2.69–2.46 (m, 4H), 1.15 (dd, 6H), FAB-MS m/z: (M – 1) 776.4. HR-MS (FAB-MS) m/z for C37H39N5O8PS2 calculated m/z: 776.1973, found m/z: 776.1978.5′-O-Dimethoxytritylthymidine-3′-O-(2-thio-4,4-dimethyl-1,3,2-oxathiaphospholane) (3d)
Yield 92%; 31P NMR (CDCl3) δ: 106.71, 106.75; 1H NMR (200 MHz, CDCl3, for diastereomeric mixture) δ: 7.61 (s, 1H), 7.45–7.19 (m, 9H), 6.85 (d, 4H), 6.44 (t, 1H), 5.64–5.50 (m, 1H), 4.30–4.23 (m, 1H), 4.18–4.11 (m, 1H), 4.08–4.03 (m, 1H), 3.78 (s, 6H), 3.59–3.33 (m, 2H), 2.68–2.33 (m, 2H), 1.62 (s, 1H), 1.59 (s, 3H), 1.39 (s, 3H), FAB-MS m/z: (M − 1) 709. HR-MS (FAB-MS) m/z for C35H38N2O8PS2 calculated m/z 709.1820, found 709.1807.Separation of the diastereomers of 3a–d
A solution of 100–200 mg of monomer 3 in 1.0 mL of an appropriate eluent was applied onto a column (75 × 2 cm) containing 200 g of silica gel (Merck, particle size 200–300 mesh). The column was eluted with 300 mL of ethyl acetate–butyl acetate (1:1 v/v for the dA and dC derivatives – 3a and 3b), ethyl acetate (for the dG monomer – 3c) or butyl acetate (for the T monomer – 3d) and fractions of 10–12 mL were collected. The control of the eluate was performed on TLC plates. Typically for the T monomer one passage gave 93% of separated P-epimers of 100% diastereomeric purity while for dA and dC separation was obtained in 75% yield. For the dG derivative isomers “fast” and “slow” were obtained in 46% summary yield (both of 100% diastereomeric purity). Yields and 31P NMR chemical shifts of the isolated P-epimers of the compounds 3a–d are given in Table 1.
“In Solution” synthesis of phosphorothioate dinucleotide 2
5′-O-DMT-thymidine-3′-O-(2-thio-4,4-dimethyl-1,3,2-oxathiaphospholane) (3d, 1 mmol) was dissolved in dry acetonitrile (5 mL), and then a solution of 3′-O-acetylthymidine (4, 1.1 mmol) and DBU (1.25 mmol) in dry acetonitrile (3 mL) was added. After 3 hours, the reaction mixture was concentrated under reduced pressure. The product 2 was isolated by column chromatography (0→12% methanol in chloroform with 0.1% addition of pyridine) as a mixture of diastereomers. Yield 85% (769 mg); 31P NMR (CD3CN) δ: 55.26, 55.16 ppm; MALDI-MS m/z: (M − 1) 905.Trimethyl(tert-butylperoxy)silane (6)
To bis-(trimethylsilyl)acetamide (1.5 mmol) tert-butyl hydroxy-peroxide (1 mmol) was added dropwise at 4 °C. The reaction mixture was stirred at room temperature for 24 hours. The crude product was purified via distillation under reduced pressure, and a fraction boiling at 40–41 °C/25 mmHg was collected: 1H NMR (CDCl3); δ: 1.25 (s, 9H), 0.20 (s, 9H); yield 79%; CI-MS m/z: (M + 1) 163.Solid phase synthesis of PS/PO-Oligos 13
All oligonucleotides syntheses were carried out manually. For this purpose gastight syringes were used while all reagents and solvents were stored under an atmosphere of dry argon in vials capped with rubber septa. A column containing a solid support was equipped with two Teflon adapters: one for the insertion of a syringe to deliver reagents and the second with a needle to allow for safe collection of wastes. After each wash step the excess of solvent was removed from the column by suction while dry argon was delivered from the opposite side.In the column 5′-O-DMT-nucleoside 8 (1 μmol) anchored to the LCA CPG support using a DBU-resistant sarcosinyl-succinoyl linker21 was detritylated with 3% solution of TCA (trichloroacetic acid) in methylene chloride and washed with 4 mL of dry acetonitrile and then with 4 mL of dry methylene chloride and dried under high vacuum.
For the coupling step, in the case of introduction of phosphorothioate internucleotide linkage (PS), a solution of an appropriate oxathiaphospholane monomer 3 (20 μmol) and DBU (50 μmol) in dry acetonitrile (150 μL) was prepared and instantly introduced into the column. After 15 minutes of intensive swirling the column was washed with dry acetonitrile (4 mL) and dry methylene chloride (4 mL) and dried under argon. Unreacted 5′-hydroxyl groups were capped using the standard DMAP–Ac2O–lutidine solution in THF.
During the introduction of phosphate linkages for the coupling step a solution of appropriate phosphoramidite monomer 7 (20 μmol) and 1-H-tetrazole (20 μmol) in dry acetonitrile (150 μL) was prepared and instantly introduced into the column. After 15 minutes of intensive swirling the column was washed with dry acetonitrile (4 mL) and dry methylene chloride (4 mL) and dried under argon. Unreacted 5′-hydroxyl groups were capped using the standard DMAP–Ac2O–lutidine solution in THF. For oxidation of newly formed phosphite to phosphate groups t-BuOOSiMe3 (50 μmol) was used as solution in acetonitrile (150 μL).
When the synthesis was complete, the oligomer was cleaved from the support under standard conditions (25% NH4OH, 2 h) and the protecting groups from nucleobases were removed at 55 °C over 12 h. The sample was concentrated under reduced pressure in a Speed-Vac concentrator.
A single cycle of chain elongation was as follows:
1) detritylation, 3% TCA in methylene chloride (2 × 5 ml),
2) wash, acetonitrile (4 ml), methylene chloride (4 ml), drying under high vacuum,
3) coupling (monomers 3 or 7),
4) wash, acetonitrile (4 ml), methylene chloride (4 ml),
5) capping, acetic anhydride/DMAP/2,6-lutidine/THF (0.15 ml; 2 min),
6)* oxidation, t-BuOOSiMe3 (6) (50 μmol) in acetonitrile (150 μL) (30 min),
7) wash, acetonitrile (4 ml), methylene chloride (4 ml),
8) removing of β-cyanoethyl groups, 10% piperidine in acetonitrile (v/v) (1 hour),
9) wash, acetonitrile (4 ml), methylene chloride (4 ml), drying under high vacuum.
* Only when phosphoramidite monomers 7 were used.
Acknowledgements
These studies were supported financially by the Statutory Funds of CMMS PAS.Notes and references
- (a) N. Dias and C. A. Stein, Mol. Cancer Ther., 2002, 1, 347 CrossRef CAS; (b) R. Kole, A. R. Krainer and S. Altman, Nat. Rev. Drug Discovery, 2012, 11, 125 CAS.
- (a) S. Agrawal, Tibitech, 1996, 14, 376 CrossRef CAS; (b) D. E. Szymkowski, Drug Discovery Today, 1996, 1, 415 CrossRef CAS.
- P. D. Cook, Anti-Cancer Drug Des., 1991, 6, 585 CAS.
- (a) F. Eckstein, Annu. Rev. Biochem., 1985, 54, 367 CrossRef CAS PubMed; (b) F. Eckstein, Antisense Nucleic Acid Drug Dev., 2000, 10, 117 CrossRef CAS.
- (a) J. M. Campbell, T. A. Bacon and E. Wickstrom, J. Biochem. Biophys. Methods, 1990, 20, 259 CrossRef CAS; (b) M. I. Phillips and Y. C. Zhang, Methods Enzymol., 2000, 313, 46 CAS; (c) S. T. Crooke, Methods Enzymol., 2000, 313, 3 CAS.
- (a) S. T. Crooke, Therapeutic Application of Oligonucleotides, R. G. Landes Co., Austin, TX, 1995, pp. 63–79 Search PubMed; (b) M. A. Guvakova, L. A. Yakubov, I. Vlodavsky, J. L. Tonkinson and C. A. Stein, J. Biol. Chem., 1995, 270, 2620 CrossRef CAS PubMed; (c) W. M. Galbraith, W. C. Hobson, D. C. Giclas, P. J. Schechter and S. Agrawal, Antisense Res. Dev., 1994, 4, 201 CAS; (d) D. A. Brown, S.-H. Kang, S. M. Gryaznov, L. DeDionisio, O. Heidenreich, S. Sullivan, X. Xu and M. I. Neerenberg, J. Biol. Chem., 1994, 43, 26801 Search PubMed.
- (a) J. P. Shaw, K. Kent, J. Bird, J. Fishback and B. Froehler, Nucleic Acids Res., 1991, 19, 747 CrossRef CAS PubMed; (b) T. M. Woolf, C. G. B. Jennings, M. Rebagliati and D. A. Melton, Nucleic Acids Res., 1990, 18, 1763 CrossRef CAS PubMed; (c) G. D. Hoke, K. Draper, S. M. Freier, C. Gonzalez, V. B. Driver, M. C. Zounes and D. J. Ecker, Nucleic Acids Res., 1991, 19, 5743 CrossRef CAS PubMed.
- (a) C. A. Stein, Ch. Subasinghe, K. Shinozuka and J. S. Cohen, Nucleic Acids Res., 1988, 16, 3209 CrossRef CAS PubMed; (b) M. K. Ghosh, K. Ghosh and J. S. Cohen, Anti-Cancer Drug Des., 1993, 8, 15 CAS; (c) H. Soreq, D. Patinkin, E. Lev-Lehman, M. Grifman, D. Ginzberg, F. Eckstein and H. Zakut, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 7907 CrossRef CAS; (d) S. Agrawal, Z. Jiang, Q. Zhao, D. Shaw, Q. Cai, A. Roskey, L. Channavajjala, C. Saxinger and R. Zhang, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2620 CrossRef CAS; (e) M. C. Uzagare, K. J. Padiya, M. M. Salunkhe and Y. S. Sanghvi, Bioorg. Med. Chem. Lett., 2003, 13, 3537 CrossRef CAS; (f) M. A. Maier, A. P. Guzaev and M. Manoharan, Org. Lett., 2000, 2, 1819 CrossRef CAS PubMed; (g) N. U. Mohe, K. J. Padiya and M. M. Salunkhe, Bioorg. Med. Chem., 2003, 11, 1419 CrossRef CAS.
- (a) B. T. Monia, J. F. Johnston, H. Sasmor and L. L. Cummins, J. Biol. Chem., 1996, 271, 14533 CrossRef CAS PubMed; (b) S. V. Patil, R. B. Mane and M. M. Salunkhe, Bioorg. Med. Chem. Lett., 1994, 4, 2663 CrossRef CAS.
- W. J. Stec and A. Wilk, Angew. Chem., 1994, 106, 747 ( Angew. Chem., Int. Ed. Engl. , 1994 , 33 , 709 ) CrossRef CAS.
- (a) L. J. McBride and M. H. Caruthers, Tetrahedron Lett., 1983, 24, 245 CrossRef CAS; (b) S. Beaucage and R. P. Iyer, Tetrahedron, 1992, 48, 2223 CrossRef CAS.
- (a) B. C. Froehler, Tetrahedron Lett., 1986, 27, 5575 CrossRef CAS; (b) B. C. Froehler, in Methods in Molecular Biology, Vol. 20: Protocols for Oligonucleotides and Analogues, ed., S. Agrawal, Humana Press Inc., Totowa, NJ, 1993, pp. 63–80 Search PubMed.
- (a) T. Wada, N. Kobayashi, T. Mori and M. Sekine, Nucleosides Nucleotides, 1998, 17, 351 CrossRef CAS; (b) K. Seio, K. Kumura, J.-C. Bologna and M. Sekine, J. Org. Chem., 2003, 68, 3849 CrossRef CAS PubMed; (c) H. Almer, J. Stawinski and R. Strömberg, J. Chem. Soc., Chem. Commun., 1994, 1459 RSC; (d) Y. Hayakawa, Y. Hirabayashi, M. Hyodo, S. Yamashita, T. Matsunami, D.-M. Cui, R. Kawai and H. Kodama, Eur. J. Org. Chem., 2006, 3834 CrossRef CAS; (e) R. P. Iyer, M.-J. Guo, D. Yu and S. Agrawal, Tetrahedron Lett., 1998, 39, 2491 CrossRef CAS; (f) H. Almer, T. Szabo and J. Stawinski, Chem. Commun., 2004, 290 RSC; (g) V. A. Efimov, N. S. Molchanova and O. G. Chakhmakhcheva, Nucleosides, Nucleotides Nucleic Acids, 2007, 26, 1087 CrossRef CAS PubMed.
- (a) W. J. Stec, A. Grajkowski, M. Koziołkiewicz and B. Uznański, Nucleic Acids Res., 1991, 19, 5883 CrossRef CAS PubMed; (b) W. J. Stec, B. Karwowski, M. Boczkowska, P. Guga, M. Koziołkiewicz, M. Sochacki, M. W. Wieczorek and J. Błaszczyk, J. Am. Chem. Soc., 1998, 120, 7156 CrossRef CAS; (c) P. Guga, A. Okruszek and W. J. Stec, Top. Curr. Chem., 2002, 220, 169 CrossRef CAS.
- A. Wilk, A. Grajkowski, L. R. Phillips and S. L. Beaucage, J. Am. Chem. Soc., 2000, 122, 2149 CrossRef CAS.
- (a) N. Oka, T. Wada and K. Saigo, J. Am. Chem. Soc., 2002, 124, 4962 CrossRef CAS PubMed; (b) N. Oka, T. Wada and K. Saigo, J. Am. Chem. Soc., 2003, 125, 8307 CrossRef CAS PubMed; (c) N. Oka, M. Yamamoto, T. Sato and T. Wada, J. Am. Chem. Soc., 2008, 130, 16031 CrossRef CAS PubMed; (d) N. Oka, M. Yamamoto, T. Sato and T. Wada, Nucleic Acids Symp. Ser., 2008, 52, 335 CrossRef CAS PubMed; (e) N. Oka and T. Wada, Chem. Soc. Rev., 2011, 40, 5829 RSC.
- (a) P. M. J. Burgers and F. Eckstein, Biochemistry, 1979, 18, 450 CrossRef CAS; (b) F. Eckstein and B. V. L. Potter, J. Biol. Chem., 1984, 259, 14243 Search PubMed; (c) F. Eckstein, B. A. Connolly and A. Pingoud, J. Biol. Chem., 1984, 259, 10760 Search PubMed.
- Y. Hayakawa, M. Uchiyama and R. Noyori, Tetrahedron Lett., 1986, 27, 4191 CrossRef CAS.
- (a) J. Engels and A. Jäger, Angew. Chem., Int. Ed. Engl., 1982, 21, 912 CrossRef; (b) J. Engels and A. Jäger, Tetrahedron Lett., 1984, 25, 1437 CrossRef.
- G. M. Salamończyk, M. Kuźnikowski and E. Poniatowska, Tetrahedron Lett., 2002, 43, 1747 CrossRef.
- T. Brown, C. E. Pritchard, G. Turner and S. A. Salisbury, J. Chem. Soc., Chem. Commun., 1989, 891 RSC.
- L. Wu, D. E. White, C. Ye, F. G. Vogt, G. J. Terfloth and H. Matsuhashi, J. Mass. Spectrom., 2012, 47, 836 CrossRef CAS PubMed.
- J. Cieślak, C. Austin, M. K. Chmielewski, J. S. Kauffman, J. Snyder, A. Del-Grosso and S. L. Beaucage, J. Org. Chem., 2005, 70, 3303 CrossRef PubMed.
- The 2-cyanoethyl thiophosphate protecting group was lost to the level of ∼5% when 1-methylimidazole was present in the capping solution, and to the extent of ∼13% when 4-(dimethylamino)pyridine was used.25.
- 4-(Dimethylamino)pyridine was used during the solid phase synthesis of PO/PS chimeric oligos listed in Table 2.
- I. V. Martynov, Y. L. Kruglyak, G. A. Leibovskaya, Z. I. Khromova and O. G. Strukov, Zh. Obshch. Khim., 1969, 39, 996 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ob01837k |
| This journal is © The Royal Society of Chemistry 2015 |