
Synthesis of novel symmetrical 2-oxo-spiro[indole-3,4′-pyridines] by a reaction of oxindoles with 1,2-diaza-1,3-dienes†
Orazio
A. Attanasi
,
Linda
A. Campisi
,
Lucia
De Crescentini
,
Gianfranco
Favi
and
Fabio
Mantellini
*
Department of Biomolecular Sciences, Section of Organic Chemistry and Organic Natural Compounds, University of Urbino “Carlo Bo”, Via I Maggetti 24, 61029 Urbino (PU), Italy. E-mail: fabio.mantellini@uniurb.it
First published on 14th October 2014
Abstract
A simple reaction of some oxindole derivatives with 1,2-diaza-1,3-dienes to produce 2-oxo-spiro[indole-3,4′-pyridines] in good yields is described here. This transformation represents a practical two steps approach to new and biologically interesting 2-oxo-spiro[indole-3,4′-pyridine] scaffolds using a double Michael addition/cyclization sequence.
Introduction
Oxindoles constitutes a prominent class of heterocycles that represent important building blocks found in a wide variety of several pharmaceuticals and natural compounds, such as alkaloids.1 In particular, the synthesis of spiroxindoles is most targeted for many organic chemists, as several natural products that exhibit significant bioactivity possess this core in their structure.2,3 For example, the spirotryprostatins A and B, isolated from the fermentation broth of Aspergillus fumigatus, were found to completely inhibit the G2/M progression of cellular division in mammalian tsFT210 cells.3 Strychnofoline, belonging to a class of natural products isolated from the leaves of Strychnos usambarensis,4a displays antimitotic activity against cultures of mouse melanoma and Ehrlich tumor cells.4b Surugatoxin, isolated from the Japanese ivory shell, antagonizes the depolarizing effect of carbachol on isolated rat superior cervical ganglia, depresses the orthodromic transmission, and is a ganglionic blocker of nicotinic acetylcholine receptors.4c In the field of synthetic therapeutic agents, compounds containing the spiroxindole core, SOID-8, analogue of spirotryprostatin B, have been described as having anticancer activity against Melanoma cells,5 while NITD609 is a promising novel drug for the treatment of malaria6 (Fig. 1).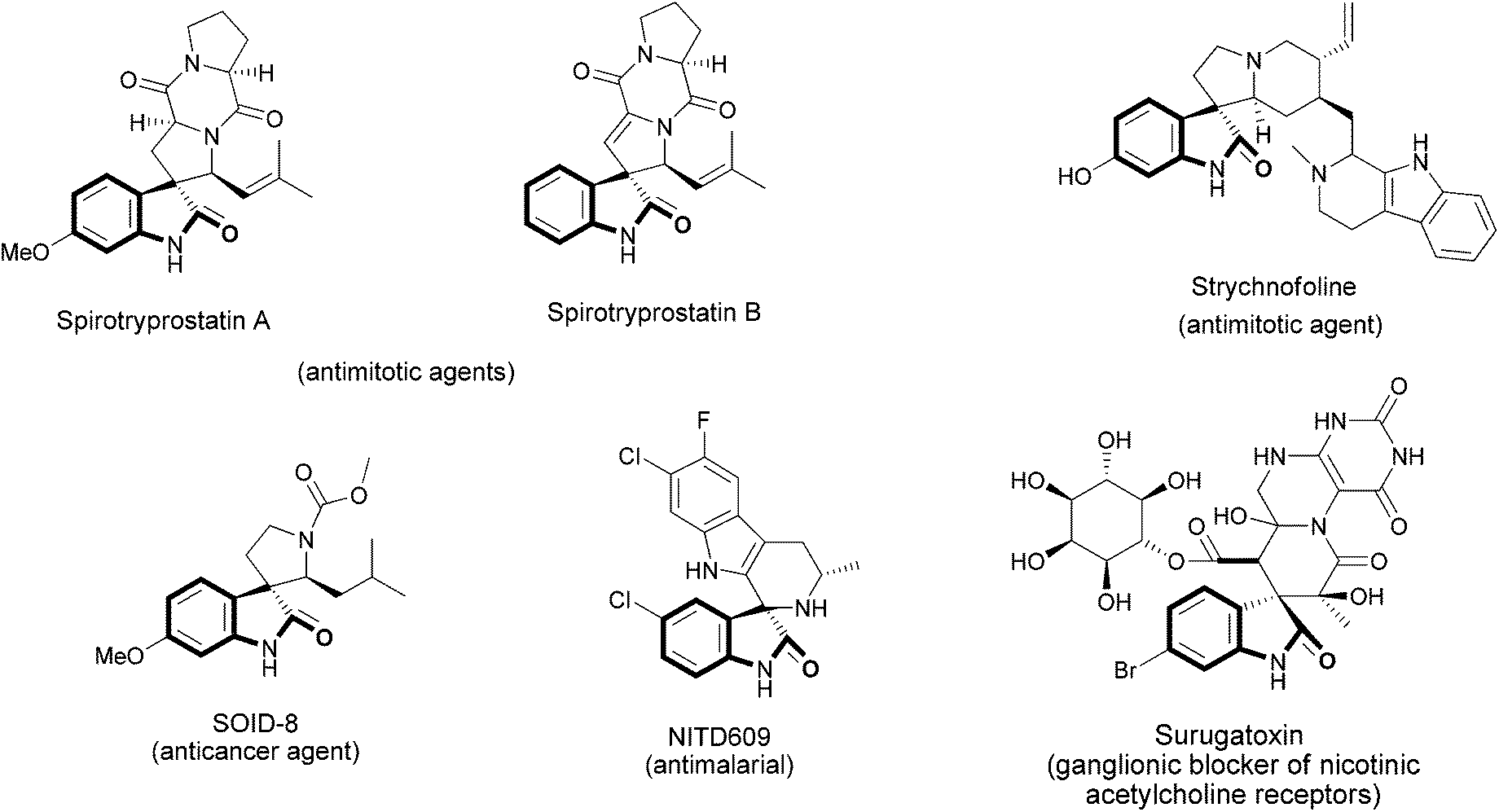 | ||
| Fig. 1 Examples of natural products and synthetic therapeutic agents containing a spiroxindole core. | ||
The common structural characteristic of these compounds is the spiro ring fusion at the 3-position of the oxindole nucleus with several five- or six-membered aza-heterocycles.
Among them, 1,4-dihydropyridines are of particular interest, owing to their biological and pharmacological actions. First of all, they represent one of the most important groups of calcium-channel modulating agents and they are used in the treatment of cardiovascular diseases. In addition, they also show antibacterial, anticancer, antileishmanial, anticoagulant, anticonvulsant, antitubercular, antioxidant, antiulcer, CFTR, antimalarials and neuroprotection properties, as well as HIV-1 protease inhibitors, and antifertility activities.7
Thus, taking into account the potential biological properties of the 2-oxo-spiro[indole-3,4′-pyridine] system, the construction of complex molecular frameworks containing this core is a very challenging goal. The main reported methods for the synthesis of spiroxindole-pyridines require the employment of isatins as one of the starting materials, in multicomponent approaches (MCRs).4c
For example, Alizadeh and Mokhtari described a four component route to obtain spiro[indoline-3,4′-pyridine]-3′-carboxylates from isatin, 1-phenyl-2-(1,1,1-triphenyl-λ5-phosphanylidene)-1-ethanone, amines and dicarbonyl compounds.8a Yan et al. reported the synthesis of spiro[oxindole-dihydropyridines] from isatins, arylamines and cyclopentane-1,3-dione.8b The same group described a BF3·OEt2-catalyzed three component reaction of the same isatins, arylamines and acetylenedicarboxylate to obtain spiro[indoline-3,2′-pyridines] or spiro[indoline-3,4′-pyridines].8c Langer et al. prepared some spiro[indoline-3,4′-pyrrolo[2,3-b]pyridines] by a MCR starting with Meldrum's acid, isatins and 5-amino-3-cyanopyrroles, in acetic acid–ammonium acetate.8d Another recent example includes acid-catalyzed one-pot sequential reactions of isatins, arylamines and methyl propiolate to obtain spiro[indoline-3,4′-pyridines].8e
An alternative elegant route to spirocyclic dihydropyridines through an electrophile-induced dearomatizing spirocyclization of N-arylisonicotinamides was also reported by Clayden and co-workers.9
Results and discussion
Hence, by exploiting our experience in the field of 1,2-diaza-1,3-dienes (DDs) for the syntheses of five- and six-membered azaheterocycles,10 we focused on a strategy that aims at developing a simple methodology for the synthesis of the target 2-oxo-spiro[indole-3,4′-pyridine]. Our retrosynthetic analysis emphasizes three strategic disconnections of the pyridine ring. The first one is along the nitrogen–carbon N(1′)–C(2′) bond obtainable from two hydrazonic residues, using a N-heterocyclization (Scheme 1).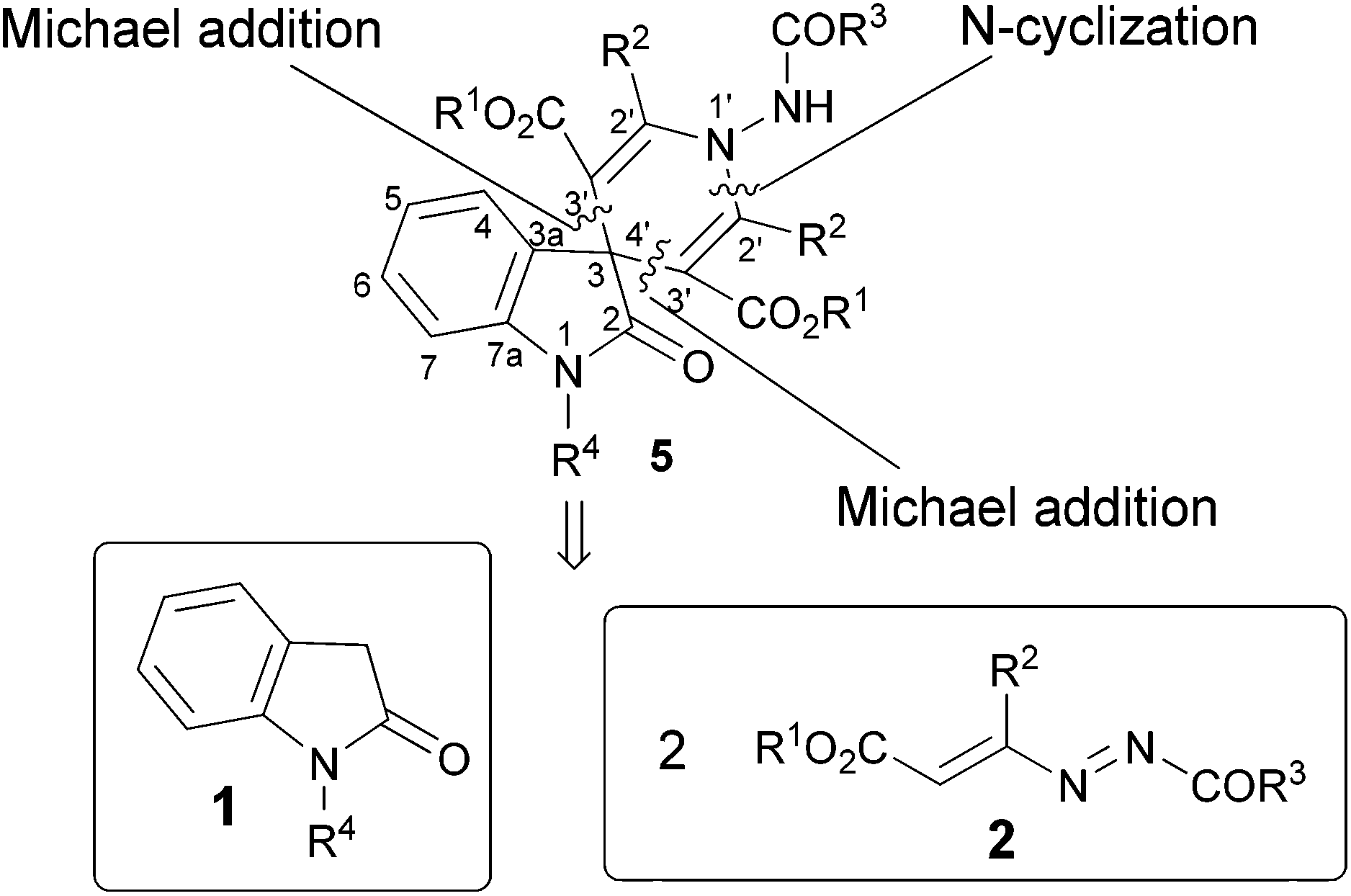 | ||
| Scheme 1 Retrosynthetic analysis to assemble 2-oxo-spiro[indole-3,4′-pyridines]. | ||
Two further disconnections can be envisaged between the spiro carbon C(3,4′) and the two C(3′) atoms. These can be connectable to the oxindoles 1 and two molecules of DDs 2, by a double Michael addition (Scheme 1).
As reported above,8a–e the common building blocks for the preparation of the 2-oxo-spiro[indole-3,4′-pyridine] nucleus are represented by the isatins.4c In this case, the indole core participates with an electrophilic site in the construction of the spiro derivatives, as indicated in Scheme 2.
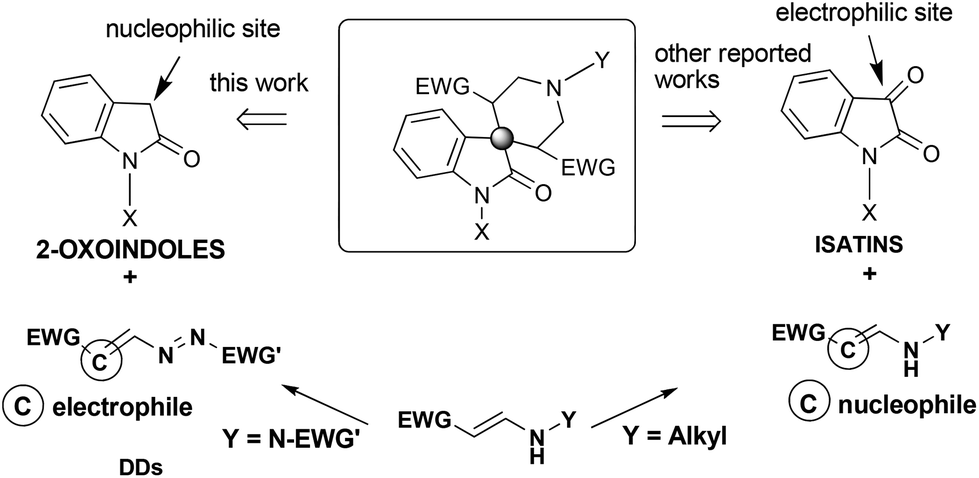 | ||
| Scheme 2 Different reactivity of isatins and oxindole derivatives, in the construction of 2-oxo-spiro[indole-3,4′-pyridines]. | ||
Using 2-oxindoles, the generated spiro carbon atom acts as a nucleophilic centre to react with two molecules of DDs (Scheme 2).
In fact, DDs 2 are good Michael acceptors because these substrates undergo nucleophilic additions at the terminal carbon atom of the azo-ene system.10 Therefore, the initial conjugate 1,4-addition of the carbon at 3-position of oxindoles 1 to two different molecules of DD 2 could permit the N(3′)–C(3,4′) junctions; the subsequent intramolecular cyclization could furnish the connection N(1′)–C(2′).
To verify our hypothesis for the synthesis of 2-oxo-spiro[indole-3,4′-pyridines] 5, we began our investigation by studying the reaction between oxindole 1a and DD 2b, which was chosen as a representative example (Table 1).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Catalyst (C) | Molar ratio 1a/2b/C | 3b yieldb (%) | 4b yieldb (%) | Time (h) |
| a The reactions were performed at 0.5 mmol scale of oxindole 1a in 3 mL of solvent. b Yields of isolated 3b and 4b, based on oxindole 1a. | ||||||
| 1 | THF | K2CO3 | 1/2/0.5 | 45 | 10 | 2 |
| 2 | CH3CN | K2CO3 | 1/2.2/1.1 | 13 | 32 | 2 |
| 3 | CH2Cl2 | K2CO3 | 1/2.2/1.1 | 16 | 31 | 1 |
| 4 | CH2Cl2 | DBU | 1/2/0.5 | 39 | 8 | 0.1 |
| 5 | CH3CN | DABCO | 1/2/0.5 | 33 | 11 | 0.1 |
| 6 | CH2Cl2 | DMAP | 1/2/0.5 | 26 | 13 | 0.1 |
| 7 | THF | CuCl2·2H2O | 1/2/0.5 | 38 | 5 | 0.1 |
| 8 | Toluene | DIPEA | 1/1/1 | 44 | 5 | 0.1 |
| 9 | THF | DIPEA | 1/2/0.5 | 29 | 18 | 0.1 |
| 10 | Toluene | DIPEA | 1/2.2/1.1 | 44 | 32 | 7 |
| 11 | Toluene | DIPEA | 1/3.3/2.2 | 18 | 55 | 5 |
| 12 | CH2Cl2 | DIPEA | 1/3.3/2.2 | 10 | 61 | 48 |
| 13 | CH2Cl2 | DIPEA | 1/4.4/2.2 | 4 | 75 | 18 |
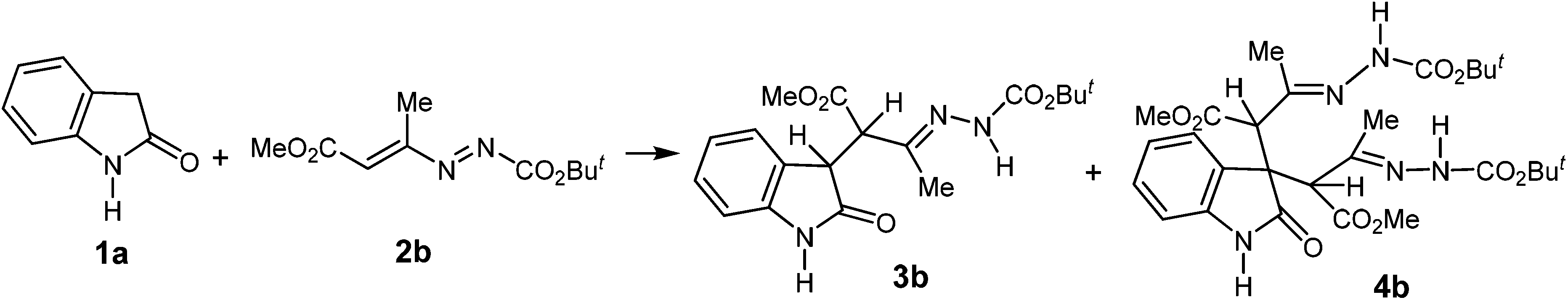
In the present screening, we tested several solvents, such as dichloromethane, tetrahydrofuran, toluene, and acetonitrile. Furthermore, a series of catalysts were used, such as K2CO3, DBU, DABCO, DMAP, CuCl2·2H2O and DIPEA and different molar ratios between oxindole/DD/catalyst were employed. In all these cases, two different products were achieved, which were isolated and characterized as the mono-hydrazonic adduct 3b, resulting from the nucleophilic attack (Michael-type) of the oxindole 1a to one equivalent of the DD 2b, and the bis-hydrazonic adduct 4b, resulting from a double Michael-type addition of the oxindole 1a to two equivalents of DD 2b.
The structure of the mono-adduct 3b was unambiguously determined by 1H NMR spectrum, in which two doublets at 4.14 and 4.27 ppm, are the diagnostic peaks for the presence of CH–CH coupling.11
Though the reaction times with K2CO3, DBU, DABCO, DMAP, CuCl2·2H2O (Table 1, entries 1–7) are lower, the use of the DIPEA enhances the global yield of the reactions (Table 1, entries 8–13). In addition, by increasing the molar ratio of DD 2b from 1 to 4.4 equiv., the yield of the mono-adduct 3b significantly decreased, while the one of bis-adduct 4b drastically improved (Table 1, entries 8–13).
Thus, the best conditions we found to obtain the highest yield of 4b involve the use of 1 equiv. of the oxindole 1a, 4.4 equiv. of DD 2b and 2.2 equiv. of DIPEA as the promoter, in dichloromethane as the solvent (Table 1, entry 13).
Afterwards, we optimized the reaction conditions for the cyclization of compound 4b to tentatively obtain the corresponding 2-oxo-spiro[indole-3,4′-pyridines] 5b (Table 2).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Cat. | Amount of cat.b | Temp. | 5b yieldc (%) | Time (h) |
| a The reactions were performed at a 0.5 mmol scale of bis-hydrazone 4b in 3 mL of solvent. b Amount referred to 1 equiv. of 4b. c Yields of isolated 5b based on oxindole 4b. d TFA as trifluoroacetic acid. e Amb. as Amberlist 15H. | ||||||
| 1 | CH3CN | ZnCl2 | 0.2 equiv. | 25 | 55 | 24 |
| 2 | CH2Cl2 | ZnCl2 | 0.2 equiv. | Reflux | 40 | 10 |
| 3 | CH2Cl2 | TFAd | 0.1 equiv. | 25 | 85 | 5.0 |
| 4 | CH2Cl2 | TFA | 0.15 equiv. | 25 | 91 | 3.5 |
| 5 | CH2Cl2 | TFAd | 0.15 equiv | Reflux | 65 | 1.0 |
| 6 | CH2Cl2 | Amb.e | 0.5 equiv. | 25 | 35 | 8.0 |
| 7 | CH2Cl2 | Amb.e | 1.0 equiv. | 25 | 44 | 6.0 |
Acetonitrile and dichloromethane were tested as solvents and zinc(II) chloride,12 trifluoroacetic acid, and Amberlyst 15H as catalysts, in different molar ratios. The best conditions that were found, both in terms of lower reaction times and for the better yields of the product 5b (Table 2, entry 4), was the use of dichloromethane as the solvent and 0.15 equiv. of trifluoroacetic acid for one equiv. of 4b as the catalyst.
With these optimal conditions in hand, we explored the reactions of various oxindoles 1a–c and DDs 2a–i (Scheme 3, Table 3).
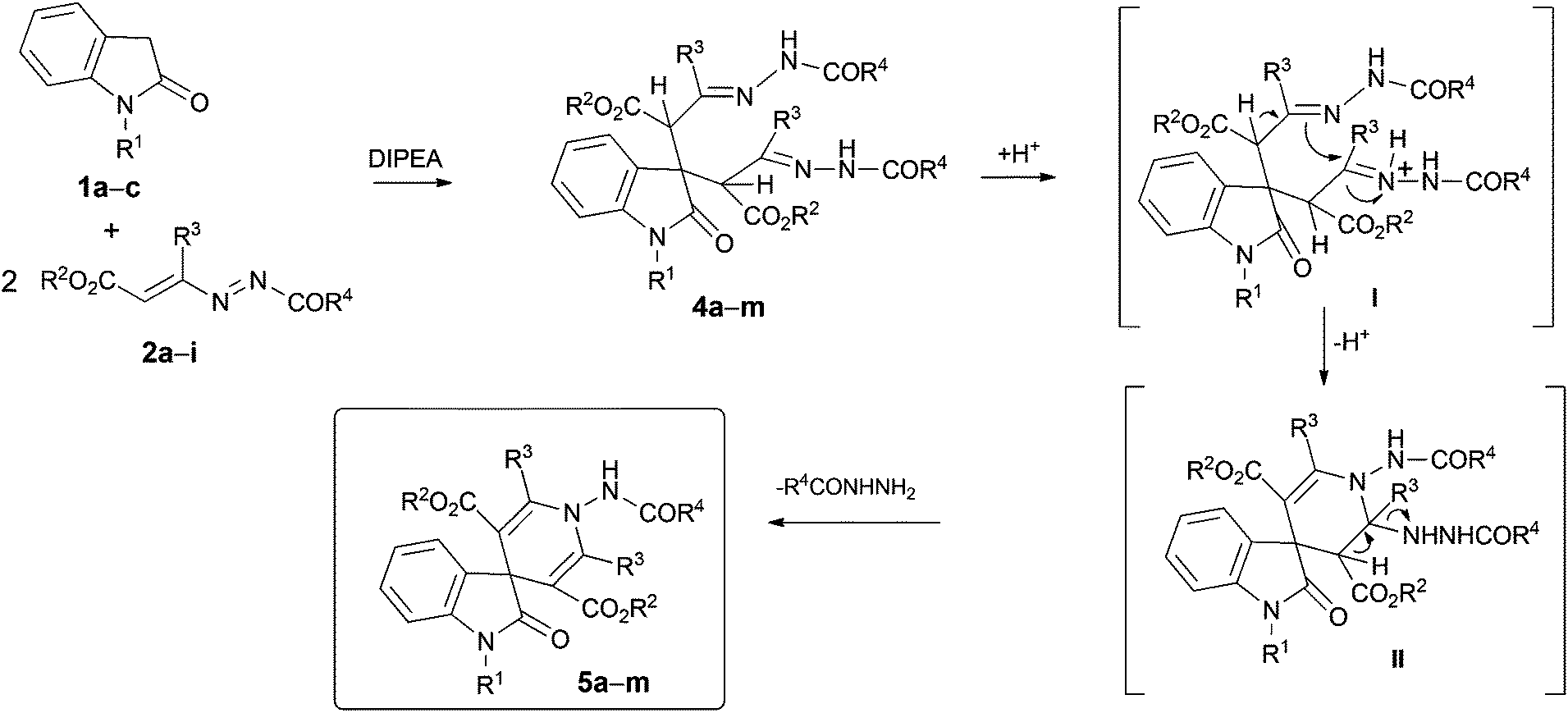 | ||
| Scheme 3 Synthesis of bis-hydrazones 4a–m, and of 2-oxo-spiro[indole-3,4′-pyridines] 5a–m. | ||
| Entry | 1 | R1 | 2 | R2 | R3 | R4 | 4 | Yieldb (%) | Time (h) | 5 | Yieldc (%) | Time (h) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Reagents and conditions: 1a–c (1.0 mmol), 2a–i (4.4 mmol), DIPEA (2.2 mmol). b Yield of pure isolated bis-hydrazones 4 referred to 1a–c. c Yield of pure isolated 2-oxo-spiro[indole-3,4′-pyridines] 5 referred to 4a–m. | ||||||||||||
| 1 | 1a | H | 2a | Me | Me | OEt | 4a | 72 | 14.0 | 5a | 89 | 4.0 |
| 2 | 1a | H | 2b | Me | Me | OBut | 4b | 75 | 18.0 | 5b | 91 | 3.5 |
| 3 | 1a | H | 2c | Et | Me | OEt | 4c | 65 | 14.5 | 5c | 88 | 5.0 |
| 4 | 1a | H | 2d | Et | Me | OBut | 4d | 71 | 16.0 | 5d | 90 | 3.5 |
| 5 | 1a | H | 2e | Et | Me | NHPh | 4e | 70 | 17.0 | 5e | 52 | 3.0 |
| 6 | 1a | H | 2f | i-Pr | Me | OBut | 4f | 69 | 18.0 | 5f | 88 | 3.0 |
| 7 | 1b | Me | 2b | Me | Me | OBut | 4g | 74 | 14.5 | 5g | 75 | 4.0 |
| 8 | 1b | Me | 2g | Me | Me | NHPh | 4h | 72 | 15.0 | 5h | 92 | 4.5 |
| 9 | 1c | Ph | 2b | Me | Me | OBut | 4i | 71 | 15.0 | 5i | 87 | 4.5 |
| 10 | 1c | Ph | 2d | Et | Me | OBut | 4j | 75 | 16.0 | 5j | 73 | 5.0 |
| 11 | 1c | Ph | 2f | i-Pr | Me | OBut | 4k | 69 | 17.5 | 5k | 80 | 3.0 |
| 12 | 1c | Ph | 2h | Me | Et | OBut | 4l | 68 | 16.0 | 5l | 64 | 3.0 |
| 13 | 1c | Ph | 2i | Allyl | Me | OBut | 4m | 73 | 17.5 | 5m | 88 | 4.0 |
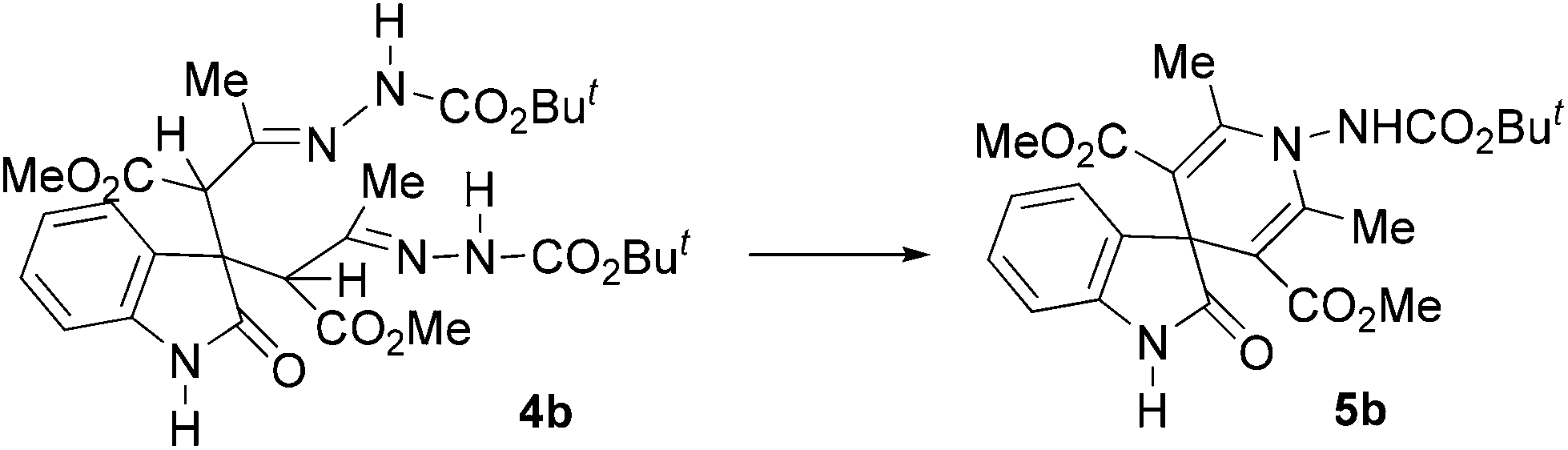
Bis-hydrazones 4a–m11 were obtained in good yields (65%–75%) and the reactions were completed in 14.0–18.0 h. The final acidic treatment of compounds 4a–m, under the conditions above described furnished the desired 2-oxo-spiro[indole-3,4′-pyridines] 5a–m, in 3.0–5.0 h in good to excellent yields (52%–92%).
The plausible mechanism of this reaction involves the preliminary double nucleophilic attack (Michael-type) of the carbon atom in the 3 position of the oxindole 1 to the terminal carbon atom of the azo-ene system of two molecules of the DD 2, with the formation of the bis-hydrazonic intermediate 4. The intramolecular ring closure occur using nucleophilic attack of the sp2 hydrazonic nitrogen to the other hydrazone moiety, activated by the acidic treatment, with the formation of the non-isolable intermediate II (Scheme 3). This process is promoted by the loss of the proton at the α position to the hydrazone acting as nucleophile. The final loss of the hydrazines furnishes the desired spiro[indole-3,4′-pyridines] 5a–m.11
Experimental
Materials and methods
All of the commercially available reagents and solvents were used without further purification. 1,2-Diaza-1,3-dienes 2a–i were synthesized as a mixture of E/Z isomers as previously reported.13,14 Oxindole (1a), 1-methyloxindole (1b), and 1-phenyloxindole (1c) are commercial materials and were used without further purification. The chromatographic purification of compounds was carried out on silica gel (60–200 μm). TLC analysis was performed on pre-loaded (0.25 mm) glass supported silica gel plates (Kieselgel 60); compounds were visualized by exposure to UV light and by dipping the plates in 1% Ce(SO4)·4H2O, 2.5% (NH4)6Mo7O24·4H2O in 10% sulphuric acid followed by heating on a hot plate. All 1H NMR and 13C NMR spectra were recorded at 400 and 100.56 MHz, respectively. Proton and carbon spectra were internally referenced to solvent signals, using values of δ = 2.50 ppm for proton (middle peak) and δ = 39.50 ppm for carbon (middle peak) in DMSO-d6 and δ = 7.27 ppm for proton and δ = 77.00 ppm for carbon (middle peak) in CDCl3. The following abbreviations are used to describe peak patterns: s = singlet, d = doublet, t = triplet q = quartet, sex = sextet, m = multiplet and br = broad signal. All coupling constants (J) are given in Hz. FTIR spectra were obtained as Nujol mulls. Mass spectra were recorded in the EI mode (70 eV). Melting points were determined in open capillary tubes and are uncorrected.Representative general procedure for the synthesis of compound 4b
A mixture of oxindole 1a (1 mmol), 1,2-diaza-1,3-diene 2b (4.4 mmol), and DIPEA (2.2 mmol) was stirred at room temperature in CH2Cl2 (6 mL) for 18 h, until the disappearance of reagent 1 (TLC monitoring). Then, the crude mixture was purified by column chromatography on silica gel to afford the products 4b, which was crystallized from diethyl ether. Moreover, a little amount (4%) of mono-adduct 3b was obtained.Spectral data of compound 4b
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80) in 75% yield. Pale yellow solid; mp: 129–131 °C; 1H NMR (400 MHz, DMSO-d6, 25 °C): δ = 1.30–1.36 (m, 24H), 3.50 (s, 6H), 4.10 (s, 1H), 4.26 and 4.57 (2 br, 1H), 6.71 (d, J = 7.6 Hz, 1H), 6.84–6.95 (m, 1H), 7.11–7.19 (m, 1H), 7.77–7.89 (m, 1H), 9.37 and 9.45 (2 br, 1H), 9.56 (s, 1H), 10.44 and 10.52 (2 br, 1H); 13C NMR (100 MHz, DMSO-d6, 25 °C): δ = 15.0 (q), 15.2 (q), 28.0 (q), 51.9 (q), 54.3 (s), 57.6 (d), 79.2 (s), 109.0 (d), 120.8 (d), 126.3 (d), 128.6 (d), 128.9 (s), 142.9 (s), 146.3 (s), 152.6 (s), 169.4 (s), 176.7 (s), 177.1 (s); IR (nujol): νmax = 3514, 3460, 3294, 1751, 1726, 1685 cm−1; MS m/z (%): 589 (M+) (9), 544 (6), 499 (6), 319 (45), 283 (41), 255 (29), 200 (32), 185 (38), 161 (64), 133 (100); anal. calcd for C28H39N5O9 (589.63): C 57.03, H 6.67, N 11.88; found: C 57.01, H 6.71, N 11.85.
:80) in 75% yield. Pale yellow solid; mp: 129–131 °C; 1H NMR (400 MHz, DMSO-d6, 25 °C): δ = 1.30–1.36 (m, 24H), 3.50 (s, 6H), 4.10 (s, 1H), 4.26 and 4.57 (2 br, 1H), 6.71 (d, J = 7.6 Hz, 1H), 6.84–6.95 (m, 1H), 7.11–7.19 (m, 1H), 7.77–7.89 (m, 1H), 9.37 and 9.45 (2 br, 1H), 9.56 (s, 1H), 10.44 and 10.52 (2 br, 1H); 13C NMR (100 MHz, DMSO-d6, 25 °C): δ = 15.0 (q), 15.2 (q), 28.0 (q), 51.9 (q), 54.3 (s), 57.6 (d), 79.2 (s), 109.0 (d), 120.8 (d), 126.3 (d), 128.6 (d), 128.9 (s), 142.9 (s), 146.3 (s), 152.6 (s), 169.4 (s), 176.7 (s), 177.1 (s); IR (nujol): νmax = 3514, 3460, 3294, 1751, 1726, 1685 cm−1; MS m/z (%): 589 (M+) (9), 544 (6), 499 (6), 319 (45), 283 (41), 255 (29), 200 (32), 185 (38), 161 (64), 133 (100); anal. calcd for C28H39N5O9 (589.63): C 57.03, H 6.67, N 11.88; found: C 57.01, H 6.71, N 11.85.
Representative general procedure for the synthesis of compound 5b
To a magnetically stirred solution of bis-hydrazone 4b (1 mmol) in CH2Cl2 (6 mL), 1.5 equiv. of TFA were added. The mixture was allowed to stand in these conditions for 3.5 h, until the disappearance of the 4 (TLC monitoring). The crude mixture was purified by column chromatography on silica gel to afford the product 5b, which was crystallized from diethyl ether–light petroleum (bp 40–60 °C).Spectral data of compound 5b
Dimethyl 1′-[(tert-butoxycarbonyl)amino]-2′,6′-dimethyl-2-oxo-1,2-dihydro-1′H-spiro[indole-3,4′-pyridine]-3′,5′-dicarboxylate (5b): 5b was isolated by column chromatography (acetate–cyclohexane 20:80) in 91% yield. White solid; mp: 205–207 °C; 1H NMR (400 MHz, DMSO-d6, 25 °C): δ = 1.46 (s, 9H), 2.00 (s, 6H), 3.28 (s, 6H), 6.51 (d, J = 8.0 Hz, 1H), 6.82 (t, J = 7.6 Hz, 1H), 7.06 (t, J = 7.2 Hz, 1H), 7.38 (d, J = 7.2 Hz, 1H), 9.93 (s, 1H), 10.08 (s, 1H); 13C NMR (100 MHz, DMSO-d6, 25 °C): δ = 15.6 (q), 27.9 (q), 50.7 (q), 50.8 (s), 80.8 (s), 104.5 (s), 108.3 (d), 121.1 (d), 123.8 (d), 127.9 (d), 136.6 (s), 141.8 (s), 146.5 (s), 155.0 (s), 166.3 (s), 180.4 (s); IR (nujol): νmax = 3208, 1735, 1720, 1700, 1685 cm−1; MS m/z (%): 457 (M+) (100), 396 (16), 382 (23), 356 (15), 322 (23), 296 (14), 281 (14); anal. calcd for C23H27N3O7 (457.47): C 60.38, H 5.95, N 9.19; found: C 60.40, H 5.98, N 9.21.
Conclusions
In conclusion, we report a practical and very facile two-step methodology, which leads to the formation of a set of 2-oxo-spiro[indole-3,4′-pyridines], starting with simple 2-oxindole derivatives and DDs. Further investigations are in progress to expand the utility of this procedure to other oxindole spiro-heterocycles, including non-symmetrical 2-oxo-spiro[indole-3,4′-pyridines].Acknowledgements
This work was supported by financial assistance from the Ministero dell'Università, dell'Istruzione e della Ricerca (MIUR)–Roma, and the Università degli Studi di Urbino “Carlo Bo”.Notes and references
- (a) A. U. Rahman and A. Basha, Indole Alkaloids, Harwood Academic publishers, Amsterdam, 1997 Search PubMed; (b) C. Marti and E. M. Carreire, Eur. J. Org. Chem., 2003, 2209–2219 CrossRef CAS.
- (a) C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748–8758 CrossRef CAS PubMed; (b) D. Cheng, Y. Ishihara, B. Tan and C. F. III Barbas, ACS Catal., 2014, 4, 743–762 CrossRef CAS.
- K. Ding, Y. Lu, Z. Nikolovska-Coleska, S. Qui, Y. Ding, W. Gao, J. Stuckey, K. Krajewski, P. P. Roller, Y. Tomita, D. A. Parrish, J. R. Deschams and S. Wang, J. Am. Chem. Soc., 2005, 127, 10130–10131 CrossRef CAS PubMed.
- (a) L. Angenot, Plant Med. Phytother., 1978, 12(2), 123–129 CAS; (b) R. Bassleer, M. C. Depauw-Gillet, B. Massart, J.-M. Marnette, P. Wiliquet, M. Caprasse and L. Angenot, Planta Med., 1982, 45, 123–126 CrossRef CAS PubMed; (c) G. S. Singh and Z. Y. Desta, Chem. Rev., 2012, 112, 6104–6155 CrossRef CAS PubMed , and references cited therein.
- Y. Tian, S. Nam, L. Liu, F. Yakushijin, K. Yakushijin, R. Buettner, W. Liang, F. Yang, Y. Ma, D. Horne and R. Jove, PLoS One, 2012, 7, 1–8 CrossRef.
- (a) M. Rottmann, C. McNamara, B. K. S. Yeung, M. C. S. Lee, B. Zou, B. Russell, P. Seitz, D. M. Plouffe, N. V. Dharia, J. Tan, S. B. Cohen, K. R. Spencer, G. E. González-Páez, S. B. Lakshminarayana, A. Goh, R. Sueanarusk, T. Jegla, E. K. Schmitt, H.-P. Beck, R. Brun, F. Nosten, L. Renia, V. Dartois, T. H. Keller, D. A. Fidock, E. A. Winzeler and T. T. Diagana, Science, 2010, 329, 1175–1180 CrossRef CAS PubMed; (b) B. Zou, P. Yap, L.-S. Sonntag, S. Y. Leong, B. K. S. Yeung and T. H. Keller, Molecules, 2012, 17, 10131–10141 CrossRef CAS PubMed.
- (a) For some examples in the field of pharmacologically properties of 1,4-dihydropyridazines, see:; (b) G. M. Reddy, M. Shiradkar and A. K. Chakravarthy, Curr. Org. Chem., 2007, 11, 847–852 Search PubMed; (c) E. Carosati, P. Ioan, M. Micucci, F. Broccatelli, G. Cruciani, B. S. Zhoror, A. Chiarini and R. Budriesi, Curr. Med. Chem., 2012, 19, 4306–4323 CrossRef CAS; (d) S. A. Khedkar and P. B. Auti, Mini-Rev. Med. Chem., 2014, 14, 282–290 CrossRef CAS , and references cited therein.
- (a) A. Alizadeh and J. Mokhtari, Tetrahedron, 2011, 67, 3519–3523 CrossRef CAS PubMed; (b) Y. Sun, J. Sun and C.-G. Yan, Beilstein J. Org. Chem., 2013, 9, 8–14 CrossRef CAS PubMed; (c) H. Gao, J. Sun and C.-G. Yan, J. Org. Chem., 2014, 79, 4131–4136 CrossRef CAS PubMed; (d) M. Vilches-Herrera, A. Spannenberg and P. Langer, Tetrahedron, 2013, 69, 5955–5967 CrossRef CAS PubMed; (e) H. Gao, J. Sun and C.-G. Yan, Mol. Diversity, 2014, 18, 511–519 CrossRef CAS PubMed.
- G. Arnott, H. Brice, J. Clayden and E. Blaney, Org. Lett., 2008, 10, 3089–3092 CrossRef CAS PubMed.
- For a review on the chemistry of DDs, see: O. A. Attanasi, L. De Crescentini, G. Favi, P. Filippone, F. Mantellini, F. R. Perrulli and S. Santeusanio, Eur. J. Org. Chem., 2009, 3109–3127 CrossRef CAS . For some recent examples, see: (a) O. A. Attanasi, G. Favi, A. Geronikaki, F. Mantellini, G. Moscatelli and A. Paparisva, Org. Lett., 2013, 15, 2624–2627 CrossRef CAS PubMed; (b) O. A. Attanasi, L. Bianchi, L. A. Campisi, L. De Crescentini, G. Favi and F. Mantellini, Org. Lett., 2013, 15, 3646–3649 CrossRef CAS PubMed; (c) J.-R. Chen, W.-R. Dong, M. Candy, F.-F. Pan, M. Jörres and C. Bolm, J. Am. Chem. Soc., 2012, 134, 6924–6927 CrossRef CAS PubMed; (d) O. A. Attanasi, G. Favi, F. Mantellini, G. Moscatelli and S. Santeusanio, J. Org. Chem., 2011, 76, 2860–2866 CrossRef CAS PubMed; (e) O. A. Attanasi, G. Favi, F. Mantellini, G. Moscatelli and S. Santeusanio, Adv. Synth. Catal., 2011, 353, 595–605 CrossRef CAS; (f) O. A. Attanasi, S. Berretta, L. De Crescentini, G. Favi, G. Giorgi, F. Mantellini and S. Nicolini, Adv. Synth. Catal., 2011, 353, 1519–1524 CrossRef CAS; (g) J. M. Hatcher and D. M. Coltart, J. Am. Chem. Soc., 2010, 132, 4546–4547 CrossRef CAS PubMed; (h) O. A. Attanasi, G. Favi, P. Filippone, F. Mantellini, G. Moscatelli and F. R. Perrulli, Org. Lett., 2010, 12, 468–471 CrossRef CAS PubMed; (i) O. A. Attanasi, L. De Crescentini, G. Favi, P. Filippone, G. Giorgi, F. Mantellini, G. Moscatelli and M. S. Behalo, Org. Lett., 2009, 11, 2265–2268 CrossRef CAS PubMed; (j) O. A. Attanasi, S. Berretta, L. De Crescentini, G. Favi, G. Giorgi and F. Mantellini, Adv. Synth. Catal., 2009, 351, 715–719 CrossRef CAS.
- For the detailed spectral data, see ESI.†.
- (a) G. Deng, Z. Li, S.-Y. Peng, L. Fang and Y.-C. Li, Tetrahedron, 2007, 63, 4630–4635 CrossRef CAS PubMed; (b) G. Deng, Z. Huang, X. Zhao, Z. Li, Y. Li and B. Jiang, Chin. J. Chem., 2013, 31, 15–17 CrossRef CAS.
- S. Sommer, Tetrahedron Lett., 1977, 18, 117–120 CrossRef.
- (a) O. A. Attanasi, P. Filippone, A. Mei and S. Santeusanio, Synthesis, 1984, 671–672 CrossRef CAS; (b) O. A. Attanasi, P. Filippone, A. Mei and S. Santeusanio, Synthesis, 1984, 873–874 CrossRef CAS; (c) L. Preti, O. A. Attanasi, E. Caselli, G. Favi, C. Ori, P. Davoli, F. Felluga and F. Prati, Eur. J. Org. Chem., 2010, 4312–4320 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ob01959h |
| This journal is © The Royal Society of Chemistry 2015 |