
Evaluation of a focused virtual library of heterobifunctional ligands for Clostridium difficile toxins†
Carlos A.
Sanhueza
a,
Jonathan
Cartmell
a,
Amr
El-Hawiet
a,
Adam
Szpacenko
a,
Elena N.
Kitova
a,
Rambod
Daneshfar
a,
John S.
Klassen
a,
Dean E.
Lang
b,
Luiz
Eugenio
b,
Kenneth K.-S.
Ng
b,
Pavel I.
Kitov
a and
David R.
Bundle
*a
aDepartment of Chemistry, University of Alberta, Edmonton, Canada. E-mail: dave.bundle@ualberta.ca; Fax: (+1)-780-492-7705
bDepartment of Biological Sciences, University of Calgary, Calgary, Canada
First published on 21st October 2014
Abstract
A focused library of virtual heterobifunctional ligands was generated in silico and a set of ligands with recombined fragments was synthesized and evaluated for binding to Clostridium difficile toxins. The position of the trisaccharide fragment was used as a reference for filtering docked poses during virtual screening to match the trisaccharide ligand in a crystal structure. The peptoid, a diversity fragment probing the protein surface area adjacent to a known binding site, was generated by a multi-component Ugi reaction. Our approach combines modular fragment-based design with in silico screening of synthetically feasible compounds and lays the groundwork for future efforts in development of composite bifunctional ligands for large clostridial toxins.
Introduction
Clostridium difficile is a Gram-positive anaerobic bacterium that often colonizes the gut of mammals. In humans, this organism is the causative agent of Clostridium difficile infection (CDI).1,2 CDI imposes increasing burden on health care systems in industrialized countries often requiring patient isolation and, in extreme cases, resulting in hospital closures. The annual cost of hospital care for patients with CDI was estimated at $3.2 billion in the US.3 Although CDI is primarily a nosocomial infection other recent reports of livestock harboring pathogenic strains of C. difficile also suggest a food-borne link.4–6CDI manifests itself in symptoms ranging from mild diarrhea to life threatening pseudomembranous colitis that can lead to death in elderly and immuno-compromised patients. Growing resistance to antibiotics leaves few options for treatment other than the administration of either metronidazole or vancomycin.7 However, due to the ability of the bacterium to form endospores, the treatment with broad spectrum antibiotics is often inefficient since the suppression of regular intestinal microflora provides a competitive advantage to C. difficile, which often results in high recurrence rate (up to 20% of patients).2 Recently approved Fidaxomicin, a narrow scope antibiotic, causes less collateral damage to microflora and is indicated to high risk patients.8
Pathogenic strains of C. difficile express at least two UDP-glucosylating toxins A and B (TcdA and TcdB), and, in 6% of strains, also ADP-ribosylating binary toxin, which may increase severity but is unlikely to cause the disease in the absence of large clostridial toxins TcdA and TcdB.9 In contrast to TcdA and TcdB, the binary toxin does not have an internal cysteine protease domain and relies on trypsin for its activation. The cell surface receptor for TcdA and TcdB was identified in rodents as Gal(α1–3)Gal(β1–4)GlcNAc(β1-O) trisaccharide (αGalLacNAc).10 This glycan sequence, also known as “Galili antigen”11 for its role in xenotransplantation organ rejection, is found on the cell membrane of rabbit erythrocytes and other tissues in mammals but not in humans. Several oligosaccharide structures found in human milk12 or decorating the surface of human epithelial cells13,14 have been reported to bind clostridial toxins with weak affinities similar to that of the Galili antigen. The high potency of the toxins is attributed to multivalency-enhanced avidity due to simultaneous interaction of multiple carbohydrate-recognition domains located at the C-terminus with the host cell surface.15,16 Binding of the C-terminus initiates ligand-mediated endocytosis followed by auto-catalytic cleavage of the enzyme domain, which is translocated to the cytosol and glucosylates target proteins.17 Clostridial toxins covalently modify large number of proteins17 including Rho GTPases causing their functional inactivation and inducing disassembly of the actin cytoskeleton, which in turn leads to morphological changes and death of susceptible cells, and, ultimately, results in erosion and perforation of the intestinal epithelium.15,18
Considering the emergence of more virulent strains and resistance to antibiotics,19,20 for opportunistic pathogens such as Clostridia, nonlethal anti-infection therapies are required that target virulence factors rather than suppress bacterial growth. Passive immunotherapy21 and manipulation of the human microbiome by repopulating intestine with non-toxigenic strain of C. difficile22 are new approaches in managing CDI. Large clostridial toxins TcdA and TcdB also appear an attractive therapeutic target for development of both antibodies and chemical antidotes. A non-specific affinity agent, polystyrene-based sulfonate polymer Tolevamer that passively absorbs and neutralizes CD toxins in the gut is considered a proof-of-principle for such an approach. Although it has been shown to have lower efficacy than broad range antibiotics in clinical trials, treatment with Tolevamer led to dramatically reduced recurrence of CDI.23 Inhibitors of the glucosyltransferase24 and autocatalytic protease domain in TcdB have recently been reported.25
The crystal structure of TcdA-A2 fragment in a complex with Gal(α1–3)Gal(β1–4)GlcNAc(β1-O)26 (pdb code: 2G7C) provides a structural basis for the rational design of potential inhibitors. Inspection of the structure reveals a shallow groove located near the reducing terminus of the bound trisaccharide. The close proximity of this groove suggests an opportunity for design of heterobifunctional ligands, in which a variable fragment is linked to the trisaccharide via a short spacer (Fig. 1). Here we present a fragment-based strategy for developing ligands for clostridial toxins TcdA and TcdB, which interrogate a putative supplementary binding site on the protein surface adjacent to the known carbohydrate-binding site.
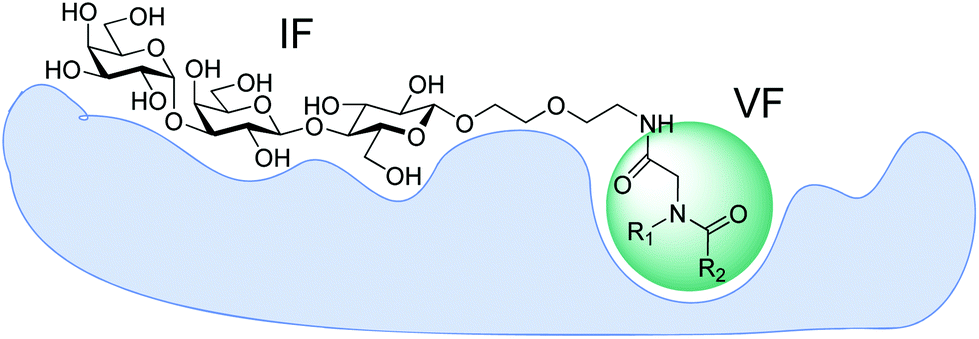 | ||
| Fig. 1 Design of heterobifunctional ligands for TcdA. Invariant fragment (IF) is presumed to occupy the same position as Gal(α1–3)Gal(β1–4)GlcNAc(β1-O(CH2)8COOMe) trisaccharide in the crystal structure while the variable fragment (VF) enhances binding through supplementary interactions with the putative binding site. | ||
Results
Design of ligands and virtual screening of a combinatorial library
Our approach combines fragment-based design and virtual screening of a combinatorial library. Trisaccharide Gal(α1–3)Gal(β1–4)Glc(β1-O) was chosen as an invariant fragment (IF) of the focused combinatorial library, while the structures of the variable fragment (VF) were suggested by computational screening of virtual combinatorial libraries of pseudo-dipeptides.To avoid uncertainty in stereo configuration of products we decided to vary only two components, the carboxylic acid and amine in construction of a virtual library of Ugi products and limit the third component to formaldehyde. Lists of fragments R1 and R2 were compiled from structures available from the Aldrich catalogue. Approximately 500 low molecular weight fragments of each type were chosen. Combinatorial library of 3D structures was generated with in-house scripts using Open Babel software.27 During preparation of the structures for docking appropriate positional constrains were used to prevent distortions in carbohydrate portion of the virtual ligands. Of the total library (∼250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 structures), approximately 20% representative randomly chosen structures were processed with Autodock Vina.28 Due to the large number of rotatable bonds multiple docking sessions were required for the majority of compounds to obtain poses with low RMSD with respect to the trisaccharide moiety found in the crystal structure. The resulting poses with RMSD less than 1.5 Å were sorted by Vina score and six hits containing different fragments were selected (Fig. 2). The fragments were recombined providing 36 structures for synthesis and activity evaluation.
000 structures), approximately 20% representative randomly chosen structures were processed with Autodock Vina.28 Due to the large number of rotatable bonds multiple docking sessions were required for the majority of compounds to obtain poses with low RMSD with respect to the trisaccharide moiety found in the crystal structure. The resulting poses with RMSD less than 1.5 Å were sorted by Vina score and six hits containing different fragments were selected (Fig. 2). The fragments were recombined providing 36 structures for synthesis and activity evaluation.
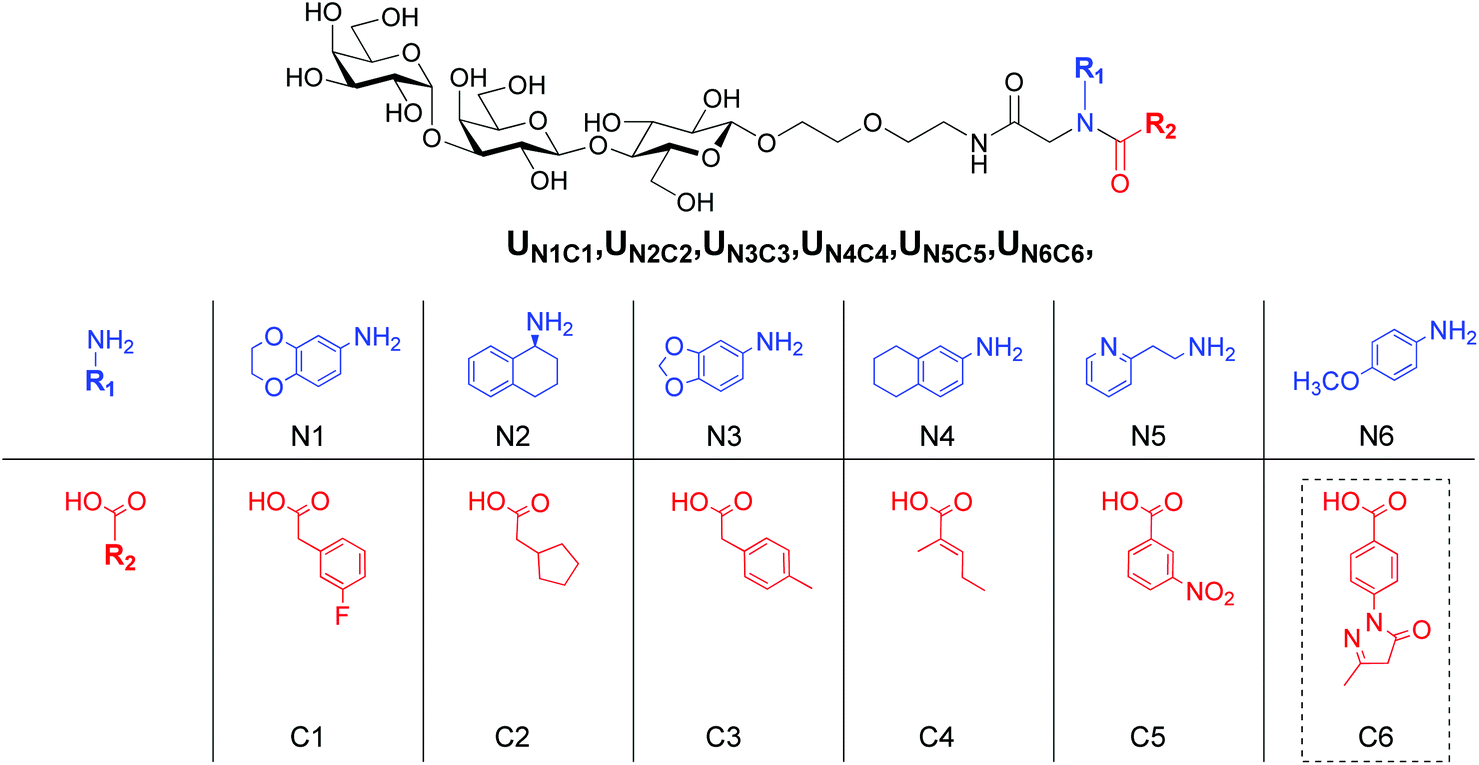 | ||
| Fig. 2 Structures of the original virtual hits and variable components, amines and carboxylic acids, chosen for Ugi condensation. Carboxylic acid C6 (shown in dashed frame) failed to form a product in the Ugi reaction. | ||
Synthesis of the ligands
For the preparation of the majority of the ligands Ugi condensation preceded the enzymatic step, while the remaining ligands were obtained via a route where the α-Gal moiety was introduced prior to formation of Ugi product. The former approach is more robust and convenient if an individual derivative is a target, while the latter is convergent and more suitable for preparation of a larger series of compounds provided the respective isocyanide derivative is readily available. The assembly of the Gal(α1–3)Gal(β1–4)Glc(β1-O) trisaccharide fragment was accomplished by enzymatic treatment of the corresponding lactose derivative with UDP-glucose in the presence of a fusion protein containing the enzymic activity (α1–3)-galactosyl transferase,29 and UDP-GalNAc(Gal)/GlcNAc(Glc) epimerase.30The synthesis started with the installation of the linker moiety into the oligosaccharide structure by boron trifluoride-catalyzed glycosylation of 2-(2-azidoethoxy)ethanol with lactose octaacetate 1 (Scheme 1). Deacetylation of the resulting azido-lactoside 2 and subsequent enzymatic processing followed by acetylation led to trisaccharide glycoside 3. Conversion of azido derivatives 2 and 3 to the corresponding isocyanides 6 and 7 was accomplished by a one-pot procedure including reduction, N-formylation and, finally, dehydration of the N-formate (Scheme 1). Although reduction of azide during the first step is coupled to in situ catalytic oxidation of formic acid31 we found that using a hydrogen atmosphere facilitates the conversion and improves the overall yield.
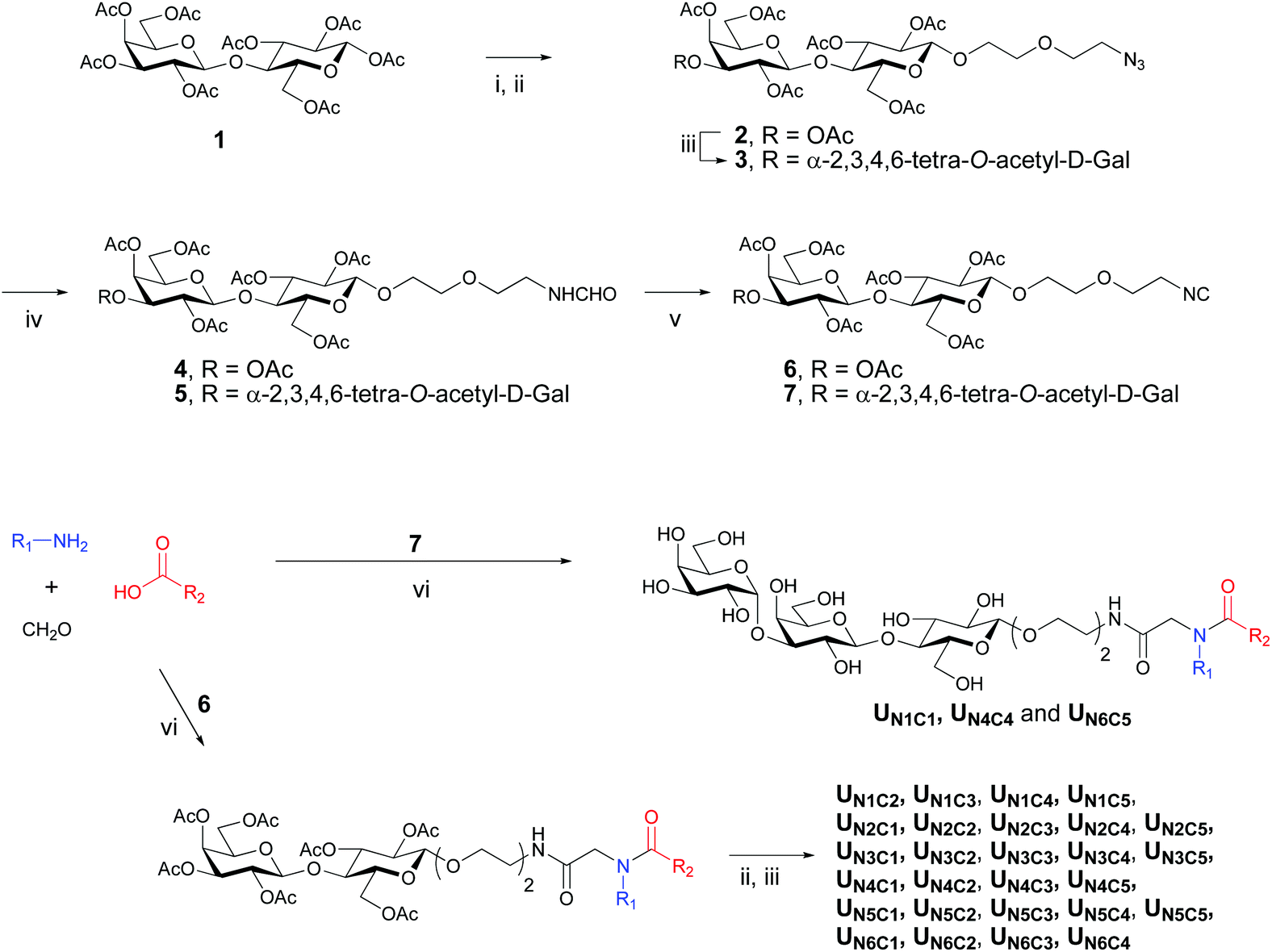 | ||
| Scheme 1 Synthesis of isocyanides 6 and 7 and Ugi products. Reagents and conditions: i. 2-(2-azidoethoxy)ethanol, BF3Et2O, DCM, 25 °C; ii. NaOMe, MeOH; iii. UDP-G1c, UDP-Glc/Gal epimerase, α-(1,4)Gal-transferase, 37 °C, 72 h. iv. Ac2O, Pyridine, 25 °C; (c) Pd/C, H2, ethyl formate, Et3N; v. POCl3, Et3N, DCM, 0–25 °C; vi. MeOH, 60 °C. | ||
Condensation of lactosyl isocyanide 6 with variable components, amines and carboxylic acids, in the presence of formaldehyde provided 27 Ugi products as shown in Scheme 1. One carboxylic acid, C6, failed to give the target products, presumably, due to the high reactivity of the pyrazolone ring, which represents a cyclic Schiff-type base and may also undergo condensations with isocyanides. However, it's amine counterpart N6 was utilized in the synthesis of the combinatorial products. The subsequent deacetylation and enzymatic treatment of lactosyl derivatives afforded the final products in up to 80% yield (Scheme 1). The remaining 3 derivatives of this series, UN1C1, UN4C4 and UN6C5, were obtained via the trisaccharide isocyanide 7 (Scheme 1).
Conformational behaviour of glycopeptoids
Substitution at the amide nitrogen in the peptoid moiety of Ugi derivatives changed the preferred conformation and may affect recognition of ligands in biological systems. Due to additional steric interactions the restricted rotation about amide bond is less pronounced for peptoids resulting in a more complex potential energy landscape than for regular peptides.32 The amide bond readily undergoes transition at room temperature, therefore, the more thermodynamically stable configuration prevails. The energy barriers for transition between the two major conformations, cis and trans, are reduced in peptoids compared to peptides as was shown experimentally and by ab initio calculations for N-methyl and N,N-dimethyl formamides and acetamides.33 NMR spectra of all Ugi derivatives synthesized in this work, which contained aromatic substituents at the nitrogen atom (N1, N3, N4 and N6), have a single series of signals corresponding to the trans conformation of the terminal amide bond. In our set of compounds, most NMR spectra in D2O presented two series of signals corresponding to trans- and cis-configurations of the terminal peptidoid bond. The ratios varied depending on whether an aliphatic or aromatic substituent was present at the amide nitrogen atom. All compounds containing aromatic substituents at the nitrogen atom showed an approximate 20:1 ratio between major (trans) and minor (cis) conformers, whereas, the presence of an aliphatic substituent increased the relative abundance of the cis component up to 40%. Assignment of conformers was conducted for the representative compound UN1C2 by comparing inter-nuclear Rotating Frame Enhancement (ROE) cross-peak intensities (volume integrals) with estimated ROE effects based on Molecular Dynamics (MD) trajectories.
Our observations for N-aromatic amides are consistent with the previously reported trend in the conformational preferences for N-aryl substituted peptoids and ab initio calculations for substituted N-methylacetanilides.34 The observed NOE pattern for compound UN1C2 matches that obtained by MD simulation at 300 K for the trans conformer using the Amber force field (Table 1, Fig. 4). More bulky aliphatic substituents (N2 and N5) caused the corresponding Ugi derivatives to exist in both conformations. Up to 40% of the cis conformers were observed in the corresponding NMR spectra. This effect should be considered in virtual screening of peptoid structures and highly populated cis conformers should be tested for compounds containing amide bonds with aliphatic substituents at the amide nitrogen.
Evaluation of binding affinity
The affinities of the ligands were measured by direct electrospray ionization mass spectroscopy (ESI-MS) binding assay35 using relative abundance of ions corresponding to various bound states of the protein-receptor in the presence of a ligand as illustrated in Fig. 3. For example, Fig. 3a shows a representative ESI mass spectrum acquired for a solution of TcdA-A2 (50 μM), lysozyme (7 μM) and UN5C2 at 50 μM in aqueous ammonium acetate buffer (10 mM). Lysozyme served as Pref for the binding measurements performed on TcdA-A2. Inspection of the ESI mass spectrum reveals signals corresponding to protonated ions of free (unbound) A2 fragment as well as the A2 bound to one, two and three molecules of UN5C2, i.e. (TcdA-A2+qUN5C2)n+, where q = 0–3 and n = 9–11. Ions corresponding to unbound and bound Pref ions were also detected i.e. (Pref+qUN5C2)n+, where q = 0–2 and n = 8, 7 indicating that nonspecific binding of UN5C2 to TcdA-A2 occurred during the ESI process and contributed to the mass spectrum.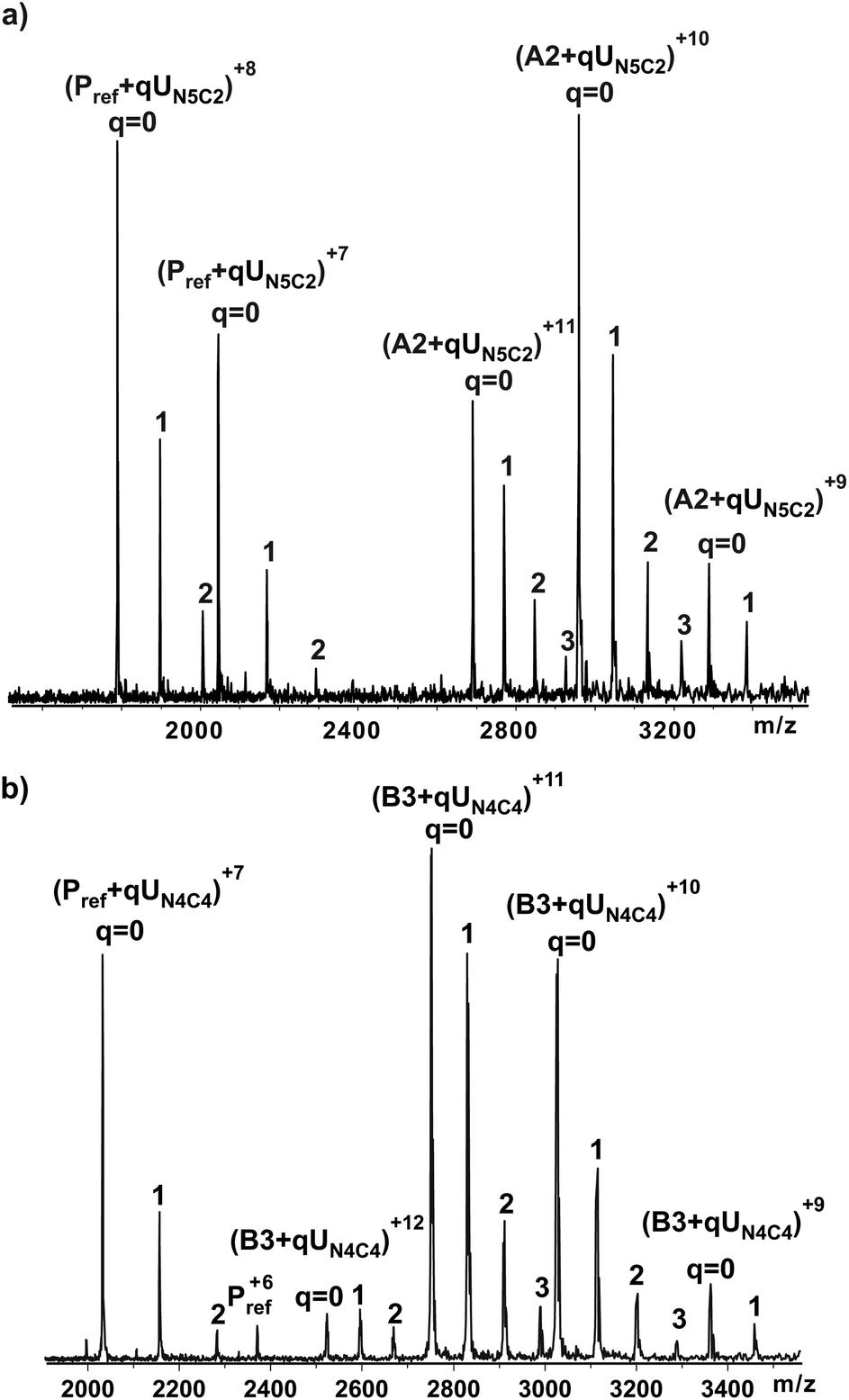 | ||
| Fig. 3 (a) ESI mass spectra of aqueous solutions containing 75 μM TcdA-A2, 100 μM UN5C2 at pH 7 and 25 °C. A Pref (10 μM) was added to quantify the extent of nonspecific protein-ligand binding during the ES process. (b) ESI mass spectra of aqueous solutions containing 15 μM B3, 50 μM UN4C4 at pH 7 and 25 °C. A Pref (10 μM) was added to quantify the extent of nonspecific protein-ligand binding during the ES process. | ||
Listed in Table 2 are the results of the ESI-MS binding measurements performed on the thirty compounds. Where binding was detected, the affinities were determined based on at least six measurements performed at multiple ligand concentrations. In all cases, the ESI mass spectra were corrected for nonspecific binding using the reference protein method.36 Low affinities have prevented an accurate measurement of the affinity for a number of ligands. For the purpose of data analysis, apparent binding constants for these ligands were assigned the value of 500 M−1 as was previously determined for the reference trisaccharide, Gal(α1–3)Gal(β1–4)GlcNAc(β1-O(CH2)8COOMe).11
| Compound | Apparent affinity to TcdA-A2 | Apparent affinity to TcdB-B3C |
|---|---|---|
| a No binding indicates low affinity and large error, not necessary complete absence of the complex in the spectrum. | ||
| Gal(α1–3)Gal(β1–4)GlcNAc(β1-O(CH2)8COOMe) | 50011 | |
| UN1C1 | NBa | 6500 ± 200 |
| UN1C2 | 3000 ± 600 | 9400 ± 1300 |
| UN1C3 | NB | 8300 ± 1700 |
| UN1C4 | 1800 ± 400 | 6300 ± 400 |
| UN1C5 | NB | 5700 ± 300 |
| UN2C1 | NB | 6000 ± 300 |
| UN2C2 | NB | 5800 ± 500 |
| UN2C3 | 900 ± 200 | 5000 ± 800 |
| UN2C4 | 3100 ± 400 | 6500 ± 200 |
| UN2C5 | NB | 6700 ± 1200 |
| UN3C1 | NB | 3900 ± 400 |
| UN3C2 | 900 ± 300 | 4900 ± 800 |
| UN3C3 | NB | 5600 ± 200 |
| UN3C4 | 1800 ± 200 | 8700 ± 300 |
| UN3C5 | NB | 6800 ± 600 |
| UN4C1 | NB | 7500 ± 800 |
| UN4C2 | NB | 4100 ± 200 |
| UN4C3 | NB | 5600 ± 700 |
| UN4C4 | 300 ± 200 | 5700 ± 600 |
| UN4C5 | NB | 4100 ± 700 |
| UN5C1 | 2300 ± 300 | 11300 ± 300 |
| UN5C2 | 2600 ± 200 | 7000 ± 400 |
| UN5C3 | 1800 ± 300 | 11000 ± 600 |
| UN5C4 | 2200 ± 300 | 11000 ± 500 |
| UN5C5 | 1300 ± 400 | 10100 ± 800 |
| UN6C1 | NB | 6000 ± 600 |
| UN6C2 | 2300 ± 600 | 7300 ± 800 |
| UN6C3 | NB | 7500 ± 100 |
| UN6C4 | 2300 ± 500 | 7000 ± 500 |
| UN6C5 | NB | 5700 ± 300 |
Crystal structure of TcdA bound to UN5C2
To further define the mode of binding predicted for the glycopeptoid ligands, a crystal structure was determined for a complex between a fragment of TcdA (TcdA-A2) containing two carbohydrate-binding sites and one of the ligands (UN5C2) with significant binding affinity to both TcdA and TcdB. The crystal structure was determined to 1.7 Å resolution and reveals a mode of binding for the trisaccharide that is essentially identical to that seen previously in the complex of the same fragment of TcdA2 bound to free trisaccharide (2G7C).26 Although the peptoid aglycone increases binding affinity slightly compared to the reference trisaccharide, most of the aglycone does not appear to adopt a single, well-ordered structure, as the electron density map in the region expected for the aglycone is quite weak and not readily interpretable (Fig. 5). Because two copies of the protein, each containing two carbohydrate-binding sites, are present in the asymmetric unit of this crystal form, four independent carbohydrate-binding sites are visible in the asymmetric unit of this crystal (same crystal form previously reported for 2G7C). | ||
| Fig. 4 Mapping of binding sites of large clostridial toxins. The residues contacting the carbohydrate ligand are highlighted in red. Peripheral groove area, the putative supplementary binding site, is highlighted in green. Lower: amino acid sequence alignment of TcdA binding sites in fragment (pdb code: 2G7C). Sequence consistency index (CI) was calculated for strain 630 and includes 7 homologous sites in TcdA and 4 sites in TcdB. Symbol (*) designates highest rank of 10. | ||
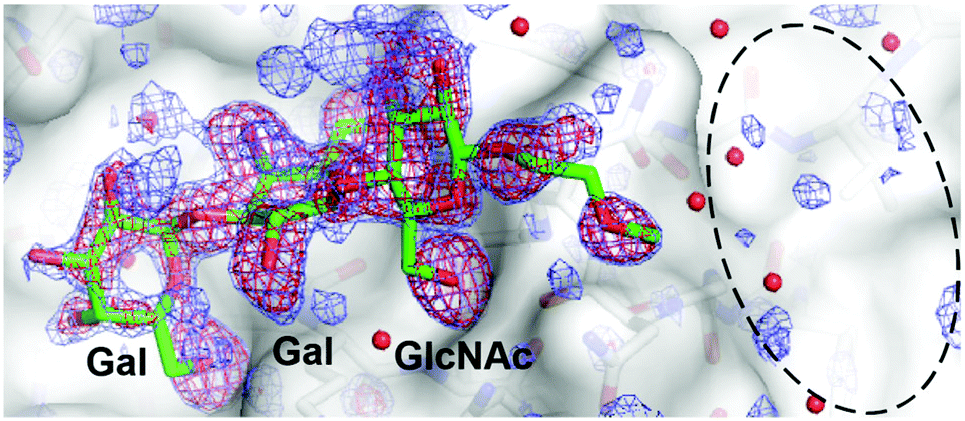 | ||
| Fig. 5 Crystal structure of TcdA-A2 bound to UN5C2. The trisaccharide and a short, partially ordered segment of the aglycone (carbon atoms colored green and oxygen atoms colored red) is modeled to fit the electron density at one of the four carbohydrate-binding sites in the asymmetric unit. This binding site seems most open and free from adjacent molecules in the crystal lattice, but very similar electron density for the trisaccharide and aglycone is seen at all four binding sites. Difference electron density maps are contoured at 2.8σ (red) and 2.0σ (blue), where σ = standard deviation above the mean. The refined structure of the trisaccharide is shown, but the coordinates were removed prior to the last round of refinement and map calculation to reduce model bias. The region where the aglycone is expected to interact with the protein is marked with a dashed black line. The solvent-accessible surface of the protein is drawn in a semi-transparent representation superimposed onto a stick representation. | ||
The electron density for only one of the four sites is shown in Fig. 5, but the electron density in the region expected for the aglycone in the other three sites is equally weak and uninterpretable, even though the electron density for the trisaccharide is quite strong in all sites. The lack of clear electron density for most of the aglycone suggests the presence of a significant amount of flexibility in the linker and the lack of a well-defined, single mode of binding. These observations are consistent with the results from NMR and molecular dynamics simulations described above, which confirm the flexibility of the linker and indicate that a fairly wide range of conformations can be adopted by the aglycone.
Discussion
The four-component Ugi reaction offers convenient access to highly diverse compound libraries. Synthetically, the condensation is robust and relatively independent of the nature of reactants and, thus, particularly suitable for the initial stage of a ligand discovery program, which relies on diversity-oriented virtual screening.The presence of multiple carbohydrate binding sites with homologous but not quite identical sequences of the repeats adds complexity to structure-based design of inhibitors for large clostridial toxins. Sequence alignment suggests the presence of 7 putative carbohydrate binding domains (CBDs) in TcdA and 4 CBDs in TcdB. Side chains of amino acids that form the binding sites differ significantly from site to site, and from strain to strain. This variation is even more significant between the binding sites found in TcdA versus TcdB. Polar side chains can be replaced with aromatic or hydrophobic ones, and charged side chains can be replaced with those carrying the opposite charge. The remarkable ability to maintain such diversity without loss of function may be beneficial for the bacteria for both evading the immune system and targeting a wide range of cell-surface receptors, but this significant degree of variation presents a formidable challenge for development of efficient antagonists for the receptor-binding domain of the large clostridial toxins.
The area on the protein surface that spans approximately 20 Å in diameter and can be accessed from the reducing terminal of the bound trisaccharide in the crystal structure 2G7C contains a groove composed of a relatively more conserved set of amino acids (Fig. 4, green highlighting). From a pharmacological prospective, targeting the relatively conserved region outside the native binding site appears to be an attractive strategy. Interaction with this supplementary site may be more uniform than with the highly variable principal binding site.
Even though only a short linker is required to bridge the trisaccharide and the putative ligand in this groove, the resulting structures of heterobifunctional composite ligands are very flexible with approximately 20 rotatable bonds (∼30 if OH groups are included). Docking of such structures presents a considerable challenge; selection of the docked poses cannot be guided solely by a scoring function and additional information about conformational preferences of the structural elements has to be considered. For instance, the conformational behaviour of oligosaccharides is governed by the exo-anomeric effect37 as is apparent from thousands of available glycan crystal structures and NMR studies.38 In this work, we reduced the conformational search during the automated docking by restraining all internal degrees of freedom in the trisaccharide fragment. Additionally, the pose of the trisaccharide in crystal structure 2G7C was used as a reference for selection of plausible docked poses. Applying these constrains should permit evaluation of possible contributions by the auxiliary binding site assuming the carbohydrate fragment binds consistently.
Exhaustive computational evaluation of the entire combinatorial library may be hindered due to large computational expense while sampling of a fraction of the library may still permit identification of promising fragments. We tested this by recombining the fragments found in a small subset of original virtual hits and evaluating activities of these recombinant compounds. Out of five synthesized ligands that were originally identified by virtual screening against TcdA-A2, only one (UN5C5) has shown greater affinity than the reference trisaccharide while recombination of the Ugi reaction components gave significantly better results with more than one third of recombinant ligands showing higher activity than the reference trisaccharide (Table 1). Interestingly, all derivatives containing an amine fragment N5 were active when combined with any tested carboxylic acid fragment and against both TcdA and TcdB. Some fragments produce more active Ugi derivatives than the others, however, no synergy was observed and combination of “active” amines with “active” carboxylic acid did not produce a compound with outstanding activity.
Conclusions
To interrogate a supplementary binding site on the surface of large clostridial toxins a library of virtual molecules composed of a trisaccharide as an invariant fragment and a dipeptoid as a variable moiety was evaluated by docking. A set of 30 rearranged structures was synthesized and their binding activities with respect to two Clostridium difficile toxins, TcdA and TcdB, were measured by mass spectrometry. A number of ligands were found to bind better than the original trisaccharide to both C.difficile toxins thereby validating the underlying design approach, in which additional fragment targeting of a supplementary binding site enhances overall affinity.Experimental section
General methods
Commercially available reagents were used as supplied without further purification. Evaporation and concentration in vacuo was conducted under water–aspirator pressure. Reactions that required unhydrous conditions were carried out in argon atmosphere. Reactions were monitored by analytical thin-layer chromatography (TLC) with pre-coated silica gel 60 F254 glass plate (Merck). Plates were visualized under UV light or stained by treatment with cerium ammonium molybdate solution or 5% sulfuric acid in ethanol followed by heating at 180 °C. Amine containing compounds were visualized by treatment with 1% ninhydrin solution in ethanol followed by heating at 180 °C. Purification of products was conducted by column chromatography using silica gel SiliaFlashF60 (40–63 μm, 60 Å) from SiliCycle®Inc. Purification of deprotected final products was conducted by HPLC on reverse-phase C-18 column using water-acetonitrile gradient as eluent. NMR spectra were recorded at 500 or 600 MHz, at 27 °C in CDCl3 or D2O. Coupling constants are first order and chemical shifts are referenced to residual solvent (CDCl3) at 7.24 ppm for 1H and 77.0 ppm for 13C-spectra and relative to 0.1% external acetone at 2.225 ppm for 1H-spectra in D2O. IR data were recorded on Nic-Plan IR Microscope (solid film); only signals corresponding to indicative functional groups are reported. Electrospray ionization high resolution mass spectra (ESI-HRMS) were recorded on a Micromass Zabspec TOF-mass spectrometer.:1, 2 mL) and stirred for 5 h; then MeOH (1 mL) was added. The mixture was concentrated in vacuum and the residue was chromatographed on silica gel.
2-(2-Formamidoethoxy)ethyl 2,3,6-tri-O-acetyl-4-O-(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl)-β-D-glycopyranoside (4). Reduction and subsequent N-formylation of lactose-derived azide 2 (2.84 g, 3.79 mmol)41 was achieved according to the general procedure for formamide synthesis to give compound 4 (1.82 g, 2.42 mmol, 64%) as a colorless solid, Rf 0.38 (DCM–MeOH, 19
:1), [α]D −13.4 (c 1.3, CHCl3); IR (cm−1): 3389, 2941, 2877, 1753, 1237; 1H NMR (CDCl3, 600 MHz), δ (ppm): 8.22 (s, 1H, NHCHO), 6.25 (s, 1H, NHCHO), 5.35 (dd, 1H, J4,3 1.0, J4,5 3.6 Hz, H-4′), 5.22 (dd, 1H, J3,2 ≈ J3,4 9.0 Hz, H-3), 5.11 (dd, 1H, J2,1 7.8, J2,3 10.2 Hz, H-2′), 4.96 (dd, 1H, J3,4 3.6, J3,2 10.5 Hz, H-3′), 4.90 (dd, 1H, J2,1 7.8, J2,3 9.6 Hz, H-2), 4.55 (d, 1H, J1,2 7.8 Hz, H-1), 4.52 (dd, 1H, J6,5 2.1, J6,6 11.7 Hz, H-6a), 4.50 (d, 1H, J1,2 8.4 Hz, H-1′), 4.11 (m, 3H, H-6b, H-6a′, H-6b′), 3.92 (m, 1H), 3.89 (dd, 1H, J5,4 ≈ J5,6 7.8 Hz, H-5′), 3.80 (dd, 1H, J4,3 ≈ J4,5 9.6 Hz, H-4), 3.69 (m, 1H), 3.61 (m, 4H), 3.52 (m, 2H), 3.44 (m, 1H), 2.16 (s, 3H), 2.13 (s, 3H), 2.07 (s, 3H), 2.06 (s, 3H), 2.05 (s, 3H), 2.05 (s, 3H), 1.97 (s, 3H); 13C NMR (CDCl3, 125 MHz), δ (ppm): 170.3, 170.3, 170.1, 169.9, 169.8, 169.7, 169.0, 161.4 (NHCHO), 101.0 (C-1′), 100.7 (C-1), 76.1, 72.8, 72.5, 71.8, 70.9, 70.7, 69.8, 69.6, 69.1, 68.6, 66.5, 61.8, 60.8, 37.6, 20.8, 20.8, 20.7, 20.7, 20.7, 20.6, 20.4; ESI-HRMS: m/z: calcd for C31H45NNaO20: 774.2427; found: 774.2427.
2-(2-Isocyanoethoxy)ethyl 2,3,6-tri-O-acetyl-4-O-(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl)-β-D-glycopyranoside (4). Following the general procedure for isocyanide synthesis, dehydration of formamide 4 (1.22 g, 1.62 mmol) afforded isocyanide 6 (1.04 g, 1.41 mmol, 87%) as a white solid, Rf 0.48 (ethyl acetate–hexanes, 4
:1), [α]D −9.0 (c 1.1, CHCl3); IR (cm−1): 2945, 2883, 2153, 1752, 1223; 1H NMR (CDCl3, 600 MHz), δ (ppm): 5.35 (dd, 1H, J4,5 1.0, J4,3 3.6 Hz, H-4′), 5.21 (dd, 1H, J3,2 ≈ J3,4 9.6 Hz, H-3), 5.11 (dd, 1H, J2,1 7.8, J2,3 10.2 Hz, H-2′), 4.96 (dd, 1H, J3,4 4.6, J3,2 10.2 Hz, H-3′), 4.90 (dd, 1H, J2,1 7.8, J2,3 9.6 Hz, H-2), 4.55 (d, 1H, J1,2 7.8 Hz, H-1), 4.51 (dd, 1H, J6,5 1.8, J6,6 12.0 Hz, H-6a), 4.50 (d, 1H, J1,2 7.8 Hz, H-1′), 4.11 (m, 3H, H-6b, H-6a′, H-6b′), 3.93 (m, 1H), 3.88 (ddd, 1H, J5,6a 1.0, J5,6b ≈ J5,4 7.2 Hz, H-5′), 3.81 (dd, 1H, J4,3 ≈ J4,5 9.6 Hz, H-4), 3.72 (m, 1H), 3.67 (m, 4H), 3.62 (ddd, 1H, J5,6a 2.4, J5,6b 5.4, J5,4 10.2 Hz, H-5), 3.55 (m, 2H), 2.15 (s, 3H), 2.13 (s, 3H), 2.06 (s, 3H), 2.05 (s, 3H), 2.04 (s, 3H), 2.04 (s, 3H), 1.97 (s, 3H); 13C NMR (CDCl3, 125 MHz), δ (ppm): 170.2, 170.2, 170.0, 169.9, 169.6, 169.5, 168.9, 157.4 (-NC), 100.9 (C-1′), 100.5 (C-1), 76.1, 72.7, 72.6, 71.6, 70.9, 70.6, 70.4, 69.0, 68.9, 68.6, 66.6, 61.9, 60.8, 41.7, 20.8, 20.7, 20.7, 20.6, 20.6, 20.5, 20.4; ESI-HRMS: m/z: calcd for C31H43NNaO19: 756.2321; found: 756.2315.
2-(2-Azidoethoxy)ethyl 2,3,6-tri-O-acetyl-4-O-[3-O-(2,3,4,6-tetra-O-acetyl-α-D-galactopyranosyl)-2,4,6-tri-O-acetyl-β-D-galactopyranosyl]-β-D-glycopyranoside (3). Transesterification of lactose-derived azide 2 was achieved by addition of 1 M NaOMe (0.1 mL) to a solution of 2 (701.7 mg, 1.54 mmol) in dry MeOH (10 mL) followed by incubation for 16 h, quenching with DOWEX (H+) resin and concentration. Enzymatic α-galactosylation and O-acetylation of the residue afforded azide 3 (783.7 mg, 0.755 mmol, 49%) as a colorless solid, Rf 0.45 (ethyl acetate–hexanes, 4
:1), [α]D +41.0 (c 0.9, CHCl3); IR (cm−1, film): 3023, 2960, 1747, 1371, 1223; 1H NMR (CDCl3, 600 MHz), δ (ppm): 5.45 (dd, 1H, J4,5 1.8, J4,3 3.0 Hz, H-4′′), 5.33 (d, 1H, J4,5 1.2, J4,3 3.0 Hz, H-4′), 5.26 (dd, 1H, J2,1 3.6, J2,3 10.8 Hz, H-2′′), 5.25 (d, 1H, J1,2 3.6 Hz, H-1′′), 5.21 (dd, 1H, J3,2 ≈ J3,4 9.0 Hz, H-3), 5.16 (dd, 1H, J2,1 7.8, J2,3 10.2 Hz, H-2′), 5.09 (dd, 1H, J3,4 3.0, J3,2 10.8 Hz, H-3′′), 4.90 (dd, 1H, J2,1 7.8, J2,3 9.6 Hz, H-2), 4.57 (d, 1H, J1,2 7.8 Hz, H-1), 4.47 (dd, 1H, J6,5 2.4, J6,6 12.0 Hz, H-6a), 4.42 (d, 1H, J1,2 7.8 Hz, H-1′), 4.20 (dd, 1H, J5,4 1.8, J5,6 7.2 Hz, H-5′′), 4.10 (m, 5H, H-6b, H-6a′, H-6b′, H-6a′′, H-6b′′), 3.92 (m, 1H), 3.83 (dd, 1H, J3,4 3.0, J3,2 10.2 Hz, H-3′), 3.79 (m, 2H, H-4, 1H), 3.73 (m, 3H), 3.67 (m, 2H), 3.60 (m, 3H), 2.15 (s, 3H), 2.14 (s, 3H), 2.14 (s, 3H), 2.11 (s, 3H), 2.07 (s, 3H), 2.06 (s, 3H), 2.06 (s, 3H), 2.05 (s, 3H), 2.04 (s, 3H), 1.95 (s, 3H); 13C NMR (CDCl3, 125 MHz), δ (ppm): 170.4, 170.3, 170.2, 170.1, 169.9, 169.7, 169.7, 169.6, 169.6, 168.7, 101.1 (C-1′), 100.5 (C-1), 93.5 (C-1′′), 76.1, 72.9, 72.9, 72.7, 71.7, 71.4, 70.8, 70.3, 69.7, 69.1, 67.7, 67.2, 66.9, 66.5, 64.6, 62.0, 61.3, 61.0, 42.8, 20.8, 20.7, 20.7, 20.7, 20.6, 20.6, 20.6, 20.5, 20.5, 20.5; ESI-HRMS: m/z: calcd for C42H59N3NaO27: 1060.32282; found: 1060.32292.
2-(2-Formamidoethoxy)ethyl 2,3,6-tri-O-acetyl-4-O-[3-O-(2,3,4,6-tetra-O-acetyl-α-D-galactopyranosyl)-2,4,6-tri-O-acetyl-β-D-galactopyranosyl]-β-D-glycopyranoside (5). Following the general procedure for formamide synthesis, reduction and subsequent N-formylation of azide 3 (717.0 mg, 0.69 mmol) afforded compound 5 (420.7 mg, 0.40 mmol, 59%) of as colorless solid, Rf 0.40 (DCM–MeOH, 19
:1), [α]D +33.4 (c 0.13, CHCl3); IR (cm−1): 3392, 2943, 2878, 1747, 1229; 1H NMR (CDCl3, 600 MHz), δ (ppm): 8.22 (s, 1H, NHCHO), 6.26 (br s, 1H, NHCHO), 5.45 (dd, 1H, J4,5 1.2, J4,3 3.0 Hz, H-4′′), 5.33 (d, 1H, J4,3 2.4 Hz, H-4′), 5.26 (dd, 1H, J2,1 3.6, J2,3 10.8 Hz, H-2′′), 5.25 (d, 1H, J1,2 3.6 Hz, H-1′′), 5.21 (dd, 1H, J3,2 ≈ J3,4 9.0 Hz, H-3), 5.16 (dd, 1H, J2,1 7.8, J2,3 10.2 Hz, H-2′), 5.10 (dd, 1H, J3,4 3.0, J3,2 10.8 Hz, H-3′′), 4.89 (dd, 1H, J2,1 7.8, J2,3 9.0 Hz, H-2), 4.55 (d, 1H, J1,2 7.8 Hz, H-1), 4.48 (dd, 1H, J6,5 2.4, J6,6 12.0 Hz, H-6), 4.43 (d, 1H, J1,2 7.8 Hz, H-1′), 4.20 (dd, 1H, J5,4 ≈ J5,6 7.2 Hz, H-5′′), 4.10 (m, 5H), 3.93 (m, 1H), 3.83 (dd, 1H, J3,4 3.0, J3,2 10.2 Hz, H-3′), 3.80 (m, 2H, H-4, 1H), 3.68 (m, 1H), 3.60 (m, 2H), 3.58 (m, 2H), 3.52 (m, 2H), 3.44 (m, 1H), 2.15 (s, 3H), 2.14 (s, 3H), 2.13 (s, 3H), 2.11 (s, 3H), 2.07 (s, 3H), 2.06 (s, 3H), 2.06 (s, 3H), 2.05 (s, 3H), 2.04 (s, 3H), 1.95 (s, 3H); 13C NMR (CDCl3, 150 MHz), δ (ppm): 170.4, 170.3, 170.3, 170.3, 170.2, 170.1, 169.9, 169.8, 169.7, 169.6, 168.7, 101.1 (C-1′), 100.7 (C-1), 93.5 (C-1′′), 75.9, 72.9, 72.8, 72.6, 71.8, 70.8, 69.8, 69.7, 69.5, 68.9, 67.6, 67.2, 66.8, 66.5, 64.6, 61.9, 61.2, 60.9, 37.6, 20.8, 20.8, 20.8, 20.8, 20.7, 20.7, 20.6, 20.6, 20.5, 20.5; ESI-HRMS: m/z: calcd for C43H61NNaO28: 1062.3272; found: 1062.3263.
2-(2-Isocyanoethoxy)ethyl 2,3,6-tri-O-acetyl-4-O-[3-O-(2,3,4,6-tetra-O-acetyl-α-D-galactopyranosyl)-2,4,6-tri-O-acetyl-β-D-galactopyranosyl]-β-D-glycopyranoside (7). Dehydration of formamide 5 (420.7 mg, 0.40 mmol) afforded isocyanide 7 (328.3 mg, 0.32 mmol, 80%) as a white solid, Rf 0.44 (ethyl acetate–hexanes, 9
:1), [α]D +39.7 (c 1.3, CHCl3); IR (cm−1): 2961, 2152, 1743, 1220; 1H NMR (CDCl3, 600 MHz), δ (ppm): 5.45 (dd, 1H, J4,5 1.2, J4,3 3.0 Hz, H-4′′), 5.33 (d, 1H, J4,3 3.0 Hz, H-4′), 5.26 (dd, 1H, J2,1 3.6, J2,3 10.8 Hz, H-2′′), 5.24 (d, 1H, J1,2 3.6 Hz, H-1′′), 5.21 (dd, 1H, J3,2 ≈ J3,4 9.0 Hz, H-3), 5.16 (dd, 1H, J2,1 7.8, J2,3 10.2 Hz, H-2′), 5.10 (dd, 1H, J3,4 3.6, J3,2 10.8 Hz, H-3′′), 4.89 (dd, 1H, J2,1 7.8, J2,3 9.0 Hz, H-2), 4.55 (d, 1H, J1,2 7.8 Hz, H-1), 4.48 (dd, 1H, J6,5 2.4, J6,6 12.0 Hz, H-6), 4.43 (d, 1H, J1,2 8.4 Hz, H-1′), 4.21 (dd, 1H, J5,4 ≈ J5,6 6.4 Hz, H-5′′), 4.16 (m, 2H), 4.10 (m, 2H), 4.05 (m, 1H), 3.93 (m, 1H), 3.83 (dd, 1H, J3,4 3.6, J3,2 10.8 Hz, H-3′), 3.80 (m, 2H, H-4, 1H), 3.73 (m, 1H), 3.37 (m, 4H), 3.63 (ddd, 1H, J5,6a 2.4, J5,6b 5.4, J5,4 10.2 Hz, H-5), 3.55 (m, 2H), 2.15 (s, 3H), 2.14 (s, 3H), 2.13 (s, 3H), 2.11 (s, 3H), 2.06 (s, 3H), 2.06 (s, 3H), 2.06 (s, 3H), 2.05 (s, 3H), 2.04 (s, 3H), 1.95 (s, 3H); 13C NMR (CDCl3, 125 MHz), δ (ppm): 170.3, 170.2, 170.2, 170.1, 170.0, 169.8, 169.7, 169.6, 169.6, 168.7, 157.4 (–NC), 100.9 (C-1′), 100.5 (C-1), 93.4 (C-1′′), 75.9, 72.8, 72.7, 72.7, 71.7, 70.7, 70.4, 69.7, 68.9, 68.7, 67.6, 66.8, 66.4, 64.6, 61.9, 61.2, 60.9, 60.3, 41.8, 20.8, 20.8, 20.7, 20.7, 20.6, 20.6, 20.6, 20.5, 20.5, 20.4; ESI-HRMS: m/z: calcd for C43H59NNaO27: 1044.3167; found: 1044.3155.
UN1C1. Ugi coupling and subsequent deacetylation of trisaccharide isocyanide 7 (81.5 mg, 0.08 mmol) afforded UN1C1 (45.9 mg, 0.05 mmol, 63%) as a white solid, Rf 0.36 (DCM–MeOH–H2O, 7:3:0.5), [α]D +55.2 (c 0.5, MeOH); IR (cm−1, solid): 3376, 2929, 2883, 1653; 1H NMR (D2O, 600 MHz), δ (ppm): 7.25 (m, 1H), 6.97 (m, 1H), 6.89 (m, 1H), 6.82 (m, 1H), 6.79 (m, 1H), 6.72 (m, 2H), 5.13 (s, 1H, H-1′′), 4.49 (d, 1H, J1,2 7.2 Hz, H-1′), 4.43 (d, 1H, J1,2 7.2 Hz, H-1), 4.30 (m, 2H), 4.25 (m, 4H), 4.17 (m, 2H), 4.00 (m, 1H), 3.95 (m, 3H), 3.84 (m, 1H), 3.75 (m, 8 H), 3.65 (m, 3H), 3.61 (m, 2H), 3.57 (m, 5H), 3.37 (m, 2H), 3.30 (m, 1H); 13C NMR (D2O, 150 MHz), δ (ppm): 175.1, 171.1, 164.1–114.4 (aromatic Cs), 103.8 (C-1′), 103.0 (C-1), 96.3 (C-1′′), 79.6, 78.1, 75.9, 75.6, 75.3, 73.6, 71.7, 70.5, 70.4, 70.2, 70.0, 69.7, 69.7, 69.1, 65.7, 65.4, 65.3, 61.9, 61.8, 61.0, 53.8, 41.2, 39.9; ESI-HRMS: m/z: calcd for C40H55FN2NaO21: 941.3174; found: 941.3166.
The general procedure for Ugi coupling lactose-derived isocyanide 6 (126.3 mg, 0.17 mmol) followed by transesterification and enzymic α-galactosylation was exemplified to prepare compound UN1C2. Final product UN1C2 (40.0 mg, 0.045 mmol, 41%) was obtained as a white solid, Rf 0.36 (DCM–MeOH–H2O, 7:3:0.5), [α]D +53.4 (c 0.9, MeOH); IR (cm−1, solid): 3372, 2940, 2873, 2494, 1650; 1H NMR (D2O, 500 MHz), δ (ppm): 6.92 (d, 1H, J 8.5 Hz), 6.89 (s, 1H), 6.8 (d, 1H, J 8.5 Hz), 5.12 (d, 1H, J1,2 3.5 Hz, H-1′′), 4.49 (d, 1H, J1,2 8.0 Hz, H-1′), 4.44 (d, 1H, J1,2 8.0 Hz, H-1), 4.30 (s, 2H), 4.28 (s, 4H), 4.17 (m, 2H, H-4′, H-5′′), 3.96 (m, 4H, H-3′′, H-4′′, H-6a, H-6b), 3.84 (dd, 1H, J2,1 3.5, J2,3 10.5 Hz, H-2′′), 3.77 (m, 3H, H-3′, H-5, H-5′), 3.72 (m, 5H, H-6a′, H-6b′, H-6a′′, H-6b′′, 1H), 3.64 (m, 5H, H-2′, H-3, CH2, 1H), 3.57 (m, 3H, H-4, CH2), 3.38 (m, 2H), 3.31 (dd, 1H, J2,1 ≈ J2,3 8.0 Hz, H-2), 2.19 (d, 2H, J 7.5 Hz), 2.07 (m, 1H), 1.65 (m, 2H), 1.42 (m, 4H), 0.95 (m, 2H); 13C NMR (D2O, 125 MHz), δ (ppm): 177.7, 171.4, 144.4, 144.2, 136.5, 122.0, 118.9, 117.6, 103.9, (C-1′), 103.1 (C-1), 96.4 (C-1′′), 79.7, 78.2, 76.0, 75.7, 75.4, 73.7, 71.8, 70.6, 70.5, 70.3, 70.1, 69.8, 69.8, 69.2, 65.8, 65.5, 61.9, 61.9, 61.1, 53.7, 40.1, 39.9, 37.6, 32.8, 32.8, 25.3, 25.3; ESI-HRMS: m/z: calcd for C39H60N2NaO21: 915.3581; found: 915.3574.
UN1C3. Ugi coupling and subsequent deacetylation and enzymatic α-galactosylation of lactose-derived isocyanide 6 (173.6 mg, 0.24 mmol) afforded final product UN1C3 (83.7 mg, 0.092 mmol, 75%) as a white solid, Rf 0.36 (DCM–MeOH–H2O, 7:3:0.5), [α]D +51.9 (c 0.5, MeOH); IR (cm−1, solid): 3338, 2921, 2877, 1646; 1H NMR (D2O, 500 MHz), δ (ppm): 7.11 (d, 2H, J 8.0 Hz), 6.91 (m, 3H), 6.81 (m, 2H), 5.21 (d, 1H, J1,2 4.0 Hz, H-1′′), 4.57 (d, 1H, J1,2 8.0 Hz, H-1′), 4.50 (d, 1H, J1,2 8.0 Hz, H-1), 4.34 (s, 2H), 4.25 (m, 6H, H-4′, H-5′′, 2 × OCH2), 4.08 (d, 1H, J4,3 3.0 Hz, H-4′′), 4.01 (m, 3H, H-3′′, H-6a, H-6b), 3.93 (dd, 1H, J2,1 3.5, J2,3 10.0 Hz, H-2′′), 3.87 (m, 3H, H-3′, H-5, H-5′), 3.79 (m, 5H, H-6a′, H-6b′, H-6a′′, H-6b′′, 1H), 3.72 (m, 5H, H-2′, H-3, CH2, 1H), 3.62 (m, 3H, H-4, CH2), 3.54 (m, 2H), 3.44 (m, 2H), 3.39 (dd, 1H, J2,1 ≈ J2,3 9.0 Hz, H-2), 2.28 (s, 3H); 13C NMR (D2O, 125 MHz), δ (ppm): 175.6, 171.2, 144.4, 144.3, 137.7, 136.1, 132.6, 130.1, 130.1, 129.9, 129.9, 122.0, 118.8, 117.7, 103.9 (C-1′), 103.1 (C-1), 96.4 (C-1′′), 76.7, 78.2, 76.0, 75.7, 75.4, 73.7, 71.8, 70.6, 70.5, 70.3, 70.1, 69.8, 69.8, 69.2, 65.8, 65.5, 65.4, 61.9, 61.9, 61.1, 53.8, 41.0, 39.9, 21.1; ESI-HRMS: m/z: calcd for C41H58N2NaO21: 937.3424; found: 937.3418.
UN1C4. Ugi coupling and subsequent deacetylation and enzymatic α-galactosylation of lactose-derived isocyanide 6 (136.9 mg, 0.19 mmol) afforded final product UN1C2 (73.2 mg, 0.083 mmol, 75%) as a white solid, Rf 0.36 (DCM–MeOH–H2O, 7:3:0.5), [α]D +55.1 (c 0.3, MeOH); IR (cm−1, solid): 3399, 2932, 2878, 1663, 1068; 1H NMR (D2O, 600 MHz), δ (ppm): 6.90 (d, 1H, 3J 9.0 Hz), 6.84 (d, 1H, 4J 1.8 Hz), 6.80 (d, 1H, 3J 9.0, 4J 1.8 Hz), 5.68 (s, 1H), 5.14 (d, 1H, J1,2 3.6 Hz, H-1′′), 4.50 (d, 1H, J1,2 7.8 Hz, H-1′), 4.46 (d, 1H, J1,2 7.8 Hz, H-1), 4.42 (s, 2H), 4.29 (s, 4H), 4.19 (m, 2H, H-4′, H-5′′), 4.01 (d, 1H, J4,3 3.0 Hz, H-4′′), 3.95 (m, 3H, H-6a, H-6b, H-3′′), 3.85 (dd, 1H, J2,1 4.8, J2,3 10.2, H-2′′), 3.78 (m, 3H, H-3′, H-5, H-5′), 3.75 (m, 4H, H-6a′, H-6b′, H-6a′′, H-6b′′, 1H), 3.67 (m, 3H, H-2′, H-3, 1H), 3.64 (m, 2H), 3.58 (m, 3H, H-4, 2H), 3.04 (m, 2H), 3.32 (m, 1H, H-2), 1.90 (s, 2H), 1.59 (s, 3H), 0.72 (s, 3H); 13C NMR (D2O, 125 MHz), δ (ppm): 171.4, 171.4, 144.0, 143.5, 140.0, 130.1, 121.5, 118.5, 117.0, 103.9 (C-1′′), 103.1 (C-1), 96.4 (C-1′′), 79.6, 78.2, 76.0, 75.7, 75.4, 73.7, 71.8, 70.6, 70.5, 70.3, 70.1, 69.8, 69.8, 69.8, 69.2, 65.8, 65.5, 65.5, 65.5, 61.9, 61.9, 61.1, 39.9, 21.5, 14.0, 13.0; ESI-HRMS: m/z: calcd for C38H58N2NaO21: 901.3424; found: 901.3421.
UN1C5. Ugi coupling and subsequent deacetylation and enzymatic α-galactosylation of lactose-derived isocyanide 6 (156.2 mg, 0.21 mmol) afforded final product UN1C5 (69.0 mg, 0.074 mmol, 63%) as a white solid, Rf 0.34 (DCM–MeOH–H2O, 7:3:0.5), [α]D +50.5 (c 0.5, MeOH); IR (cm−1, solid): 3375, 2929, 2879, 1645; 1H NMR (D2O, 500 MHz), δ (ppm): 8.23 (s, 1H), 8.14 (d, 1H, J 8.0 Hz), 7.80 (d, 1H, J 8.0 Hz,), 7.51 (dd, 1H, J 8.0, J 8.0 Hz) 6.86 (s, 1H), 6.79 (d, 1H, J 9.0 Hz), 6.71 (d, 1H, J 8.5 Hz), 5.21 (d, 1H, J1,2 4.0 Hz, H-1′′), 4.66 (s, 2H, NHCH2), 4.58 (d, 1H, J1,2 7.5 Hz, H-1′), 4.54 (d, 1H, J1,2 8.0 Hz, H-1), 4.25 (m, 2H, H-4′, H-5′′), 4.17 (m, 4H), 4.09 (d, 1H, J4,3 3.0 Hz, H-4′′), 4.04 (m, 3H, H-3′′, H-6a, H-6b), 3.93 (dd, 1H, J2,1 4.0, J2,3 10.5 Hz, H-2′′), 3.86 (m, 3H, H-3′, H-5, H-5′), 3.81 (m, 4H, H-6a′, H-6b′, H-6a′′, H-6b′′), 3.78 (m, 6H, H-2′, H-3, 2 × CH2), 3.70 (m, 2H, H-4, 1H), 3.63 (m, 1H), 3.52 (m, 2H, CH2), 3.40 (dd, 1H, J2,1 ≈ J2,3 8.5 Hz, H-2); 13C NMR (D2O, 125 MHz), δ (ppm): 171.4, 171.9, 148.2, 144.2, 143.8, 136.9, 136.5, 135.5, 130.5, 125.9, 124.2, 122.1, 118.6, 117.5, 103.9 (C-1′), 103.2 (C-1), 96.4 (C-1′′), 79.7, 78.2, 76.0, 75.7, 75.4, 73.8, 71.8, 70.6, 70.5, 70.3, 70.1, 69.9, 69.8, 69.2, 65.8, 65.3, 61.9, 61.9, 61.9, 61.2, 54.3, 40.0; ESI-HRMS: m/z: calcd for C39H53N3NaO23: 954.2962; found: 954.2960.
UN2C1. Ugi coupling and subsequent deacetylation and enzymatic α-galactosylation of lactose-derived isocyanide 6 (164.1 mg, 0.22 mmol) afforded final product UN2C1 (86.0 mg, 0.094 mmol, 59%) as a white solid, Rf 0.36 (DCM–MeOH–H2O, 7:3:0.5), [α]D +34.3 (c 1.2, MeOH); IR (cm−1, solid): 3371, 2930, 2497, 1636; 1H NMR (D2O, 500 MHz), δ (ppm): 7.33 (m, 0.3H), 7.27 (m, 0.7H), 7.07 (m, 5H), 6.99 (m, 0.3H), 6.95 (m, 0.7H), 6.89 (d, 0.3H, J 8.0 Hz), 6.81 (d, 0.7H, J 8.0 Hz), 5.67 (dd, 0.3H, J 6.5, J 8.5 Hz), 5.16 (dd, 0.7H, J 6.5, J 8.5 Hz), 5.12 (d, 1H, J1,2 4.0 Hz, H-1′′), 4.48 (d, 1H, J1,2 7.5 Hz, H-1′), 4.43 (d, 0.7H, J1,2 8.0 Hz, H-1), 4.37 (d, 0.3H, J1,2 8.0 Hz, H-1), 4.17 (m, 2H, H-4′, H-5′′), 3.99 (d, 1H, J4,3 3.0 Hz, H-4′′), 3.92 (m, 5H, H-3′′, H-6a, H-6b, CH2), 3.84 (dd, 1H, J2,1 4.0, J2,3 10.5 Hz, H-2′′), 3.76 (m, 3H, H-3′, H-5, H-5′), 3.73 (s, 2H), 3.69 (m, 4H, H-6a′, H-6b′, H-6a′′, H-6b′′), 3.67 (m, 3H, H-2′, H-3, 1H), 3.56 (m, 6H, H-4, 2 × CH2, 1H), 3.33 (m, 2H), 3.25 (m, H-2), 2.66 (m, 2H), 1.98 (m, 1H), 1.84 (m, 1H), 1.61 (m, 2H); 13C NMR (D2O, 125 MHz), δ (ppm): 176.0, 175.2, 171.5, 171.5, 164.6–114.8 (aromatic Cs), 103.9 (C-1′), 103.1 (C-1), 96.4 (C-1′′), 79.7, 78.2, 76.0, 75.7, 75.4, 73.7, 71.8, 70.5, 70.5, 70.4, 70.3, 70.1, 69.8, 69.7, 69.2, 65.8, 61.9, 61.9, 61.1, 59.1, 55.4, 47.9, 41.0, 40.8, 39.9, 39.8, 29.7, 29.5, 29.4, 28.3, 22.2, 22.1; ESI-HRMS: m/z: calcd for C42H59FN2NaO19: 937.3588; found: 937.3585.
UN2C2. Ugi coupling and subsequent Zemplen deacetylation and enzymatic α-galactosylation of lactose-derived isocyanide 6 (181.1 mg, 0.25 mmol) afforded final product UN2C2 (67.0 mg, 0.075 mmol, 45%) as a white solid, Rf 0.40 (DCM–MeOH–H2O, 7:3:0.5), [α]D +42.5 (c 0.6, MeOH); IR (cm−1, solid): 3362, 2934, 2866, 1628; 1H NMR (D2O, 500 MHz), δ (ppm): 7.32 (m, 3H), 7.16 (d, 0.6H, J 7.3 Hz), 7.07 (d, 0.4H, J 7.3 Hz), 5.80 (dd, 0.4H, J 5.9, J 9.2 Hz), 5.39 (dd, 0.4H, J 5.9, J 9.2 Hz), 5.21 (d, 1H, J1,2 3.9 Hz, H-1′′), 4.57(d, 1H, J1,2 7.8 Hz, H-1′′), 4.55 (d, 0.6H, J1,2 8.0 Hz, H-1′), 4.53 (d, 0.4H, J1,2 8.0 Hz, H-1), 4.26 (m, 2H, H-4′, H-5′′), 4.08 (d, 1H, J4,3 3.0 Hz, H-4′′), 4.00 (m, 3H, H-3′′, H-6a, H-6b), 3.93 (dd, 1H, J2,1 3.9, J2,3 10.3 Hz, H-2′′), 3.85 (m, 3H, H-3′, H-5, H-5′), 3.76 (m, 4H, H-6a′, H-6b′, H-6a′′, H-6b′′), 3.71 (m, 6H, H-2′, H-3, 2 × CH2), 3.65 (m, 3H, H-4, CH2), 3.44 (m, 2H), 3.38 (m, H-2), 2.84 (m, 2H), 2.73 (m, 0.6H), 2.52 (m, 0.4H), 2.34 (m, 2H), 2.09 (m, 1H), 1.89 (m, 4H), 1.71 (m, 3H), 1.62 (m, 2H), 1.30 (m, 2H); 13C NMR (D2O, 125 MHz), δ (ppm): 178.9, 178.4, 172.0, 172.0, 140.5, 140.3, 135.2, 135.0, 130.6, 130.5, 128.7, 128.5, 128.1, 127.7, 127.3, 127.2, 103.8 (C-1′), 103.1 (C-1), 96.4 (C-1′′), 79.6, 78.2, 76.0, 75.7, 75.4, 75.4, 73.7, 71.8, 70.5, 70.5, 70.4, 70.3, 70.1, 69.8, 69.8, 69.7, 69.2, 65.8, 61.9, 61.9, 61.1, 58.9, 47.8, 40.4, 40.0, 39.8, 39.7, 37.7, 37.5, 33.2, 33.2, 33.0, 33.0, 29.7, 29.6, 29.5, 28.3, 25.5, 25.5, 25.4, 25.4, 22.3, 22.2; ESI-HRMS: m/z: calcd for C41H64N2NaO19: 911.3995; found: 911.3990.
UN2C3. Ugi coupling and subsequent deacetylation and enzymatic α-galactosylation of lactose-derived isocyanide 6 (185.1 mg, 0.25 mmol) afforded final product UN2C3 (114.0 mg, 0.13 mmol, 52%) as a white solid, Rf 0.40 (DCM–MeOH–H2O, 7:3:0.5), [α]D +33.7 (c 0.8, MeOH); IR (cm−1, solid): 3339, 2928, 2502, 1653; 1H NMR (D2O, 600 MHz), δ (ppm): 7.13–6.70 (m, 8H), 5.65 (m, 0.4H), 5.13 (d, 1H, J1,2 4.2 Hz, H-1′′), 5.06 (m, 0.6H), 4.48 (d, 1H, J1,2 7.8 Hz, H-1′), 4.41 (d, 0.6H, J1,2 7.8 Hz, H-1), 4.35 (d, 0.4H, J1,2 7.8 Hz, H-1), 4.18 (m, 2H, H-5′′, H-4′), 3.99 (d, 1H, J4,3 3.0 Hz, H-4′′), 3.94 (m, 3H, H-3′′, H-6a, H-6b), 3.86 (dd, 1H, J2,1 3.6, J2,3 10.2 Hz, H-2′′), 3.80 (m, 5H, H-3′, H-5, H-5′, CH2), 3.71 (m, 5H, H-6a′, H-6b′, H-6a′′, H-6b′′, 1H), 3.68 (m, 6H, H-2′, H-3, 2 × CH2), 3.55 (m, 4H, H-4, CH2, 1H), 3.31 (m, 2H), 3.26 (m, H-2), 2.26 (m, 2H), 2.17 (s, 1.2H), 2.10 (s, 1.8H), 1.87 (m, 2H), 1.54 (m, 2H); 13C NMR (D2O, 125 MHz), δ (ppm): 176.2, 175.5, 171.4, 171.3, 140.3–126–9 (aromatic Cs), 103.9 (C-1′), 103.2 (C-1), 96.4 (C-1′′), 79.7, 78.3, 76.0, 75.7, 75.4, 73.7, 71.8, 70.6, 70.5, 70.4, 70.3, 70.1, 69.9, 69.7, 69.2, 65.8, 61.9, 61.9, 61.2, 59.1, 48.7, 47.8, 41.3, 41.0, 39.8, 29.7, 29.6, 29.4, 28.3, 22.4, 21.3, 21.3; ESI-HRMS: m/z: calcd for C43H62N2NaO19: 933.3839; found: 933.3834.
UN2C4. Ugi coupling and subsequent deacetylation and enzymatic α-galactosylation of lactose-derived isocyanide 6 (122.0 mg, 0.17 mmol) afforded final product UN2C4 (65.2 mg, 0.075 mmol, 64%) as a white solid, Rf 0.42 (DCM–MeOH–H2O, 7:3:0.5), [α]D +46.0 (c 0.5, MeOH); IR (cm−1, solid): 3370, 2929, 2874, 2492, 1662; 1H NMR (D2O, 500 MHz), δ (ppm): 7.24 (m, 2H), 7.19 (m, 1H), 7.11 (m, 1H), 5.81 (ddd, 0.8H, J 1.5, J 7.5, J 7.5 Hz), 5.71 (ddd, 0.2H, J 1.5, J 7.5, J 7.5 Hz), 5.59 (dd, 0.2H, J 6.0, 10.5 Hz), 5.24 (dd, 0.8H, J 6.0, 10.5 Hz), 5.13 (d, 1H, J1,2 4.0 Hz, H-1′′), 4.49 (d, 1H, J1,2 8.0 Hz, H-1′), 4.46 (d, 0.8H, J1,2 8.0 Hz, H-1), 4.44 (d, 0.2H, J1,2 8.0 Hz, H-1), 4.17 (m, 2H, H-4′, H-5′′), 4.00 (d, 1H, J4,3 3.5 Hz, H-4′′), 3.95 (m, 5H, H-3′′, H-6a, H-6b, CH2), 3.84 (dd, J2,1 3.5, J2,3 10.0 Hz, H-2′′), 3.77 (m, 3H, H-3′, H-5, H-5′), 3.68 (m, 4H, H-6a′, H-6b′, H-6a′′, H-6b′′), 3.61 (m, 6H, H-2′, H-3, 2 × CH2), 3.53 (m, 3H, H-4, CH2), 3.36 (m, 2H), 3.29 (m, H-2), 2.74 (m, 2H), 2.11 (m, 3H), 1.94 (m, 1H), 1.89 (s, 3H), 1.75 (m, 2H), 0.95 (dd, 3H, J 8.0, J 8.0); 13C NMR (D2O, 125 MHz), δ (ppm): 178.9, 178.8, 171.8, 171.5, 140.4, 140.3, 135.9, 135.1, 135.1, 135.0, 130.6, 130.5, 130.3, 128.7, 128.5, 127.9, 127.8, 127.4, 127.3, 103.9 (C-1′), 103.1 (C-1), 96.4 (C-1′′), 79.7, 78.2, 76.0, 75.7, 75.4, 73.7, 71.8, 70.6, 70.5, 70.3, 70.1, 69.8, 69.2, 65.8, 61.9, 61.9, 61.1, 60.3, 55.5, 50.4, 46.9, 39.8, 29.7, 29.6, 29.5, 28.2, 22.3, 22.2, 21.5, 21.5, 14.5, 13.5; ESI-HRMS: m/z: calcd for C40H62N2NaO19: 897.3839; found: 897.3836.
UN2C5. Ugi coupling and subsequent deacetylation and enzymatic α-galactosylation of lactose-derived isocyanide 6 (152.1 mg, 0.21 mmol) afforded final product UN2C5 (73.5 mg, 0.079 mmol, 52%) as a white solid, Rf 0.38 (DCM–MeOH–H2O, 7:3:0.5), [α]D +14.4 (c 0.5, MeOH); IR (cm−1, solid): 3378, 2933, 2883, 2504, 1628; 1H NMR (D2O, 500 MHz), δ (ppm): 8.30–6.85 (m, 8H), 5.67 (m, 0.2H), 5.13 (d, 1H, J1,2 4.0 Hz, H-1′′), 4.77 (m, 0.8H), 4.48 (d, 1H, J1,2 7.5 Hz, H-1′), 4.44 (d, 0.8H, J1,2 8.0 Hz, H-1′), 4.37 (d, 0.2H, J1,2 8.0 Hz, H-1′), 4.17 (m, 2H, H-5′′, H-4′), 4.00 (d, 1H, J4,3 3.0 Hz, H-4′′), 3.94 (m, 5H, H-3′′, H-6a, H-6b, CH2), 3.84 (dd, 1H, J2,1 4.0, J2,3 10.5 Hz, H-2′′), 3.77 (m, 3H, H-3′, H-5, H-5′), 3.69 (m, 4H, H-6a′, H-6b′, H-6a′′, H-6b′′), 3.62 (m, 6H, H-2′, H-3, 2 × CH2), 3.59 (m, 3H, H-4, CH2), 3.52 (m, 1H), 3.39 (m, 1H), 3.28 (dd, 0.8H, J2,1 8.0, J2,3 9.5 Hz, H-2), 3.26 (dd, 0.2H, J2,1 8.0, J2,3 9.5 Hz, H-2), 2.54 (m, 2H), 2.07 (m, 1H), 1.73 (m, 2H), 1.27 (m, 2H); 13C NMR (D2O, 125 MHz), δ (ppm): 173.6, 172.9, 170.8, 173.7, 148.8–122.1 (aromatic Cs), 103.8 (C-1′), 103.0 (C-1), 96.3 (C-1′′), 79.6, 78.1, 75.9, 75.6, 75.5, 75.2, 73.6, 71.7, 70.4, 70.3, 70.2, 70.1, 69.8, 69.6, 69.5, 69.4, 69.1, 65.7, 61.8, 61.8, 61.0, 60.5, 47.3, 39.8, 39.6, 29.6, 29.3, 22.0; ESI-HRMS: m/z: calcd for C41H57N3NaO21: 950.3377; found: 950.3371.
UN3C1. Ugi coupling and subsequent deacetylation and enzymatic α-galactosylation of lactose-derived isocyanide 6 (162.6 mg, 0.22 mmol) afforded final product UN3C1 (84.9 mg, 0.094 mmol, 76%) as a white solid, Rf 0.40 (DCM–MeOH–H2O, 7:3:0.5), [α]D +55.4 (c 0.5, MeOH); IR (cm−1, solid): 3389, 2892, 1655, 1075; 1H NMR (D2O, 600 MHz), δ (ppm): 7.27 (m, 1H), 6.98 (m, 1H), 6.87 (s, 1H), 6.85 (s, 1H), 6.79 (m, 2H), 6.76 (d, 1H, J 1.8 Hz), 5.99 (s, 2H), 5.13 (d, 1H, J1,2 4.2 Hz, H-1′′), 4.49 (d, 1H, J1,2 7.8 Hz, H-1′), 4.44 (d, 1H, J1,2 7.8 Hz, H-1), 4.32 (s, 2H), 4.19 (dd, 1H, J5,6a ≈ J5,6b 6.0 Hz, H-5′′), 4.17 (d, 1H, J4,3 3.0 Hz, H-4′), 4.01 (d, 1H, J4,3 3.6 Hz, H-4′′), 3.96 (m, 3H, H-3′′, H-6a, H-6b), 3.85 (d, 1H, J2,1 4.2, J2,3 10.2, H-2′′), 3.77 (m, 3H, H-3′, H-5, H-5′), 3.72 (m, 5H, H-6a′, H-6b′, H-6a′′, H-6b′′, 1H), 3.66 (m, 3H, H-2′, H-3, 1H), 3.59 (m, 6H, H-4, 2 × CH2, 1H), 3.54 (m, 1H), 3.39 (m, 2H), 3.30 (m, 1H, H-2); 13C NMR (D2O, 150 MHz), δ (ppm): 175.2, 171.2, 163.4–109 (aromatic Cs), 103.8 (C-1′), 103.0, 102.9 (C-1), 96.3 (C-1′′), 79.6, 78.1, 75.9, 75.6, 75.3, 73.6, 71.7, 70.5, 70.4, 70.2, 70.0, 69.7, 69.7, 69.1, 61.9, 61.8, 61.0, 53.8, 41.1, 39.9; ESI-HRMS: m/z: calcd for C39H53FN2NaO21: 927.3017; found: 927.3011.
UN3C2. Ugi coupling and subsequent deacetylation and enzymatic α-galactosylation of lactose-derived isocyanide 6 (122.6 mg, 0.17 mmol) afforded final product UN3C2 (54.4 mg, 0.062 mmol, 81%) as a white solid, Rf 0.36 (DCM–MeOH–H2O, 7:3:0.5), [α]D +56.9 (c 0.6, MeOH); IR (cm−1, solid): 3388, 2944, 1658, 1074; 1H NMR (D2O, 600 MHz), δ (ppm): 6.90 (d, 1H, J 8.4 Hz), 6.89 (d, 1H, J 1.8 Hz), 6.85 (dd, 1H, J 1.8, J 8.4 Hz), 6.02 (s, 2H), 5.14 (d, 1H, J1,2 4.2 Hz, H-1′′), 4.50 (d, 1H, J1,2 8.4 Hz, H-1′), 4.46 (d, 1H, J1,2 7.8 Hz, H-1), 4.32 (s, 2H), 4.18 (m, 2H, H-5′′, H4′), 4.00 (m, 2H, H-4′′, H-6a), 3.95 (m, 2H, H-6b, H-3′′), 3.85 (dd, 1H, J2,1 3.9, J2,3 10.2, H-2′′), 3.78 (m, 3H, H-5, H-5′, H-3′), 3.73 (m, 4H, H-6a′, H-6b′, H-6a′′, H-6b′′), 3.68 (m, 2H, H-2′, H-3), 3.62 (m, 4H), 3.60 (dd, 1H, J4,5 ≈ J4,3 8.4 Hz, H-4), 3.57 (m, 2H), 3.41 (m, 2H), 3.31 (m, 1H), 2.23 (d, 2H, J 7.2 Hz), 2.09 (m, 1H), 1.67 (m, 2H), 1.44 (m, 4H), 0.99 (m, 2H); 13C NMR (D2O, 150 MHz), δ (ppm): 177.8, 171.4, 148.8, 148.1, 136.7, 122.4, 109.5, 109.4, 103.7 (C-1′), 103.1, 102.9 (C-1), 96.3 (C-1′′), 79.6, 78.1, 75.9, 75.6, 75.3, 73.7, 71.7, 70.5, 70.4, 70.2, 70.0, 69.7, 69.7, 69.1, 65.7, 61.9, 61.8, 61.1, 53.7, 40.1, 39.8, 37.5, 32.7, 32.7, 25.2, 25.2; ESI-HRMS: m/z: calcd for C38H58N2NaO21: 901.3424; found: 901.3420.
UN3C3. Ugi coupling and subsequent deacetylation and enzymatic α-galactosylation of lactose-derived isocyanide 6 (182.7 mg, 0.25 mmol) afforded final product UN3C3 (78.0 mg, 0.087 mmol, 54%) as a white solid, Rf 0.38 (DCM–MeOH–H2O, 7:3:0.5), [α]D +55.8 (c 0.7, MeOH); IR (cm−1, solid): 3369, 2890, 1650; 1H NMR (D2O, 500 MHz), δ (ppm): 7.12 (d, J 8.0 Hz, 2H), 6.93 (d, J 8.0 Hz, 2H), 6.86 (d, J 8.0 Hz, 1H), 6.78 (dd, J 2.0, J 8.0 Hz, 1H), 6.74 (d, J 2.0 Hz, 1H), 5.99 (s, 2H), 5.12 (d, J1,2 4.0 Hz, H-1′′), 4.48 (d, J1,2 7.5 Hz, H-1′), 4.43 (d, J1,2 7.5 Hz, H-1), 4.13 (s, NHCH2), 4.17 (m, H-4′, H-5′′), 4.00 (d, J4,3 3.0 Hz, H-4′′), 3.94 (m, H-3′′, H-6a, H-6b), 3.84 (dd, J2,1 3.5, J2,3 10.0 Hz, H-2′′), 3.75 (m, H-3′, H-5, H-5′), 3.70 (m, H-6a′, H-6b′, H-6a′′, H-6b′′), 3.64 (m, H-2′, H-3, 2 × CH2), 3.58 (m, H-4, 2 × CH2), 3.39 (m, CH2), 3.29 (dd, J2,1 8.0, J2,3 9.5 Hz, H-2), 2.27 (s, 3H); 13C NMR (D2O, 125 MHz), δ (ppm): 175.8, 171.3, 148.8, 148.2, 137.9, 136.5, 132.6, 130.1, 130.1, 129.9, 129.9, 122.6, 109.5, 109.5, 103.9 (C-1′), 103.1 (C-1), 102.9, 96.4 (C-1′′), 79.7, 78.2, 76.0, 75.7, 75.4, 73.7, 71.8, 70.5, 70.5, 70.3, 70.1, 69.8, 69.8, 69.2, 65.8, 61.9, 61.9, 61.1, 53.9, 40.9, 39.9, 21.1; ESI-HRMS: m/z: calcd for C40H56N2NaO21: 923.3268; found: 923.3260.
UN3C4. Ugi coupling and subsequent deacetylation and enzymatic α-galactosylation of lactose-derived isocyanide 6 (141.1 mg, 0.19 mmol) afforded final product UN3C4 (81.5 mg, 0.094 mmol, 70%) as a white solid, Rf 0.34 (DCM–MeOH–H2O, 7:3:0.5), [α]D +56.9 (c 0.7, MeOH); IR (cm−1, solid): 3390, 2928, 2891, 1662, 1066; 1H NMR (D2O, 600 MHz), δ (ppm): 6.87 (d, 1H, J 8.4 Hz), 6.84 (d, 1H, J 1.8 Hz), 6.78 (dd, 1H, J 1.8, J 8.4 Hz), 5.99 (s, 2H), 5.71 (s, 1H), 5.14 (d, 1H, J1,2 3.6 Hz, H-1′′), 4.50 (d, 1H, J1,2 7.8 Hz, H-1′), 4.46 (d, 1H, J1,2 8.4 Hz, H-1), 4.42 (s, 2H), 4.18 (m, 2H, H-5′′, H4′), 4.01 (d, 1H, J4,3 3.6 Hz, H-4′′), 3.95 (m, 3H, H-3′′, H-6a, H-6b), 3.85 (dd, 1H, J2,1 4.2, J2,3 10.8, H-2′′), 3.77 (m, 3H, H-3′, H-5, H-5′), 3.72 (m, 5H, H-6a′, H-6b′, H-6a′′, H-6b′′, 1H), 3.67 (m, 3H, H-2′, H-3, 1H), 3.63 (m, 2H), 3.59 (dd, 1H, J4,5 ≈ J4,3 8.4 Hz, H-4), 3.56 (m, 2H), 3.41 (m, 2H), 3.32 (dd, 1H, J1,2 7.8, J2,3 5.4 Hz, H-2), 1.92 (s, 2H), 1.59 (s, 3H), 0.74 (s, 3H); 13C NMR (D2O, 125 MHz), δ (ppm): 171.3, 171.3, 148.4, 147.4, 140.0, 130.1, 121.8, 109.2, 109.1, 109.0, 103.8 (C-1′), 103.0, 102.7 (C-1), 96.3 (C-1′′), 79.5, 78.1, 75.9, 75.6, 75.3, 73.6, 71.7, 70.4, 70.4, 70.2, 70.0, 70.0, 69.7, 69.7, 69.1, 65.7, 61.8, 61.8, 61.0, 39.8, 21.3, 13.9, 12.9; ESI-HRMS: m/z: calcd for C37H56N2NaO21: 887.3268; found: 887.3263.
UN3C5. Ugi coupling and subsequent deacetylation and enzymatic α-galactosylation of lactose-derived isocyanide 6 (108.5 mg, 0.15 mmol) afforded final product UN3C5 (24.0 mg, 0.026 mmol, 18%) as a white solid, Rf 0.38 (DCM–MeOH–H2O, 7:3:0.5), [α]D +54.9 (c 0.5, MeOH); IR (cm−1, solid): 3382, 2889, 1646; 1H NMR (D2O, 600 MHz), δ (ppm): 8.21 (s, 1H), 8.14 (d, 1H, J 8.4 Hz), 7.74 (d, 1H, J 7.8 Hz), 7.48 (dd, 1H, J 7.8, J 7.8 Hz), 6.81 (s, 1H), 6.72 (dd, 1H, J 1.8, J 7.8 Hz), 6.66 (d, 1H, J 8.4 Hz), 5.87 (s, 2H), 5.13 (d, 1H, J 3.6 Hz, H-1′′), 4.49 (d, 1H, J 7.8 Hz, H-1′), 4.47 (d, 1H, J 7.8 Hz, H-1), 4.18 (m, 2H, H-4′, H-5′′), 4.00 (d, 1H, J 3.0 Hz, H-4′′), 3.99 (dd, 1H, J3,4 4.2, J3,2 8.4, Hz, H-3′′), 3.96 (m, 2H, H-6a, H-6b), 3.85 (dd, 1H, J2,1 3.6, J2,3 10.2, Hz, H-2′′), 3.78 (m, 5H, H-3′, H-5, H-5′, CH2), 3.73 (m, 4H, H-6a′, H-6b′, H-6a′′, H-6b), 3.67 (m, 6H, H-2′, H-3, 2 × CH2), 3.62 (m, 2H, H-4, 1H), 3.57 (m, 1H), 3.46 (m, 2H), 3.31 (dd, 1H, J2,1 ≈ J2,3 9.0, Hz, H-2); 13C NMR (D2O, 125 MHz), δ (ppm): 171.7, 171.0, 148.7, 148.3, 147.8, 136.9, 136.8, 135.4, 130.6, 125.9, 124.2, 122.8, 109.4, 109.3, 103.9 (C-1′), 103.1 (C-1), 102.9, 96.4 (C-1′′), 79.7, 78.2, 76.0, 75.7, 75.4, 73.7, 71.8, 70.5, 70.5, 70.3, 70.1, 69.8, 69.8, 69.2, 65.8, 61.9, 61.9, 61.1, 54.4, 40.0; ESI-HRMS: m/z: calcd for C38H51N3NaO23: 940.2806; found: 940.2799.
UN4C1. Ugi coupling and subsequent deacetylation and enzymatic α-galactosylation of lactose-derived isocyanide 6 (148.9 mg, 0.20 mmol) afforded final product UN4C1 (57.4 mg, 0.063 mmol, 58%) as a white solid, Rf 0.40 (DCM–MeOH–H2O, 7:3:0.5), [α]D +54.8 (c 0.8, MeOH); IR (cm−1, solid): 3367, 2929, 2508, 1653; 1H NMR (D2O, 500 MHz), δ (ppm): 7.03 (m, 1H), 6.91 (d, 1H, J 7.0 Hz), 6.89 (d, 1H, J 7.0 Hz), 6.69 (m, 3H), 6.35 (d, 1H, J 9.5 Hz), 5.12 (d, 1H, J1,2 3.5 Hz, H-1′′), 4.48 (d, 1H, J1,2 7.5 Hz, H-1′), 4.40 (d, 1H, J1,2 8.0 Hz, H-1), 4.17 (m, 4H), 3.99 (d, 1H, J4,3 3.0 Hz, H-4′′), 3.93 (m, 3H, H-3′′, H-6a, H-6b), 3.84 (dd, 1H, J2,1 3.5, J2,3 10.5 Hz, H-2′′), 3.78 (m, 3H, H-3′, H-5, H-5′), 3.72 (m, 6H, H-6a′, H-6b′, H-6a′′, H-6b′′, CH2), 3.61 (m, 4H, H-2′, H-3, CH2), 3.50 (m, 3H, H-4, CH2), 3.31 (m, 5H), 2.39 (m, 4H), 1.42 (m, 4H); 13C NMR (D2O, 125 MHz), δ (ppm): 173.6, 170.4, 164–114.1 (aromatic Cs), 103.9 (C-1′), 103.2 (C-1), 96.4 (C-1′′), 79.7, 78.3, 76.0, 75.7, 75.4, 73.8, 71.8, 70.6, 70.5, 70.3, 70.1, 69.9, 69.7, 69.2, 65.8, 61.9, 61.9, 61.2, 53.7, 41.2, 39.9, 29.7, 29.5, 23.6, 23.5; ESI-HRMS: m/z: calcd for C42H59FN2NaO10: 937.3588; found: 937.3583.
UN4C2. Ugi coupling and subsequent deacetylation and enzymatic α-galactosylation of lactose-derived isocyanide 6 (158.8 mg, 0.22 mmol) afforded final product UN4C2 (78.0 mg, 0.088 mmol, 79%) as a white solid, Rf 0.40 (DCM–MeOH–H2O, 7:3:0.5), [α]D +58.0 (c 0.5, MeOH); IR (cm−1, solid): 3352, 2932, 2866, 1653; 1H NMR (D2O, 500 MHz), δ (ppm): 7.09 (m, 2H), 7.00 (d, 1H, J 8.0 Hz), 5.21 (d, 1H, J1,2 4.0 Hz, H-1′′), 4.58 (d, 1H, J1,2 7.5 Hz, H-1′), 4.52 (d, 1H, J1,2 8.0 Hz, H-1), 4.35 (s, 2H), 4.26 (m, 2H, H-4′, H-5′′), 4.08 (d, 1H, J4,3 3.0 Hz, H-4′′), 4.03 (m, 3H, H-3′′, H-6a, H-6b), 3.93 (dd, 1H, J2,1 3.5, J2,3 10.0 Hz, H-2′′), 3.86 (m, 3H, H-3′, H-5, H-5′), 3.81 (m, 5H, H-6a′, H-6b′, H-6a′′, H-6b′′, 1H), 3.72 (m, 5H, H-2′, H-3, CH2, 1H), 3.63 (m, 3H, H-4, CH2), 3.45 (m, 2H), 3.41 (dd, 1H, J2,1 ≈ J2,3 8.5 Hz, H-2), 2.66 (m, 4H), 2.17 (m, 3H), 1.64 (m, 6H), 1.38 (m, 4H), 0.88 (m, 2H); 13C NMR (D2O, 125 MHz), δ (ppm): 175.9, 170.7, 141.2, 139.3, 137.8, 131.1, 129.1, 125.9, 103.9 (C-1′), 103.2 (C-1), 96.4 (C-1′′), 79.7, 78.3, 76.0, 75.8, 75.4, 73.8, 71.8, 70.6, 70.6, 70.3, 70.1, 69.9, 69.7, 69.2, 65.8, 61.9, 61.9, 61.2, 53.7, 40.2, 39.9, 37.6, 33.0, 33.0, 29.9, 29.7, 25.5, 25.5, 23.8, 23.7; ESI-HRMS: m/z: calcd for C41H64N2NaO19: 911.3995; found: 911.3992.
UN4C3. Ugi coupling and subsequent deacetylation and enzymatic α-galactosylation of lactose-derived isocyanide 6 (167.3 mg, 0.23 mmol) afforded final product UN4C3 (56.5 mg, 0.062 mmol, 56%) as a white solid, Rf 0.40 (DCM–MeOH–H2O, 7:3:0.5), [α]D +49.3 (c 0.5, MeOH); IR (cm−1, solid): 3353, 2926, 1652; 1H NMR (D2O, 500 MHz), δ (ppm): 7.02 (d, 1H, J 7.0 Hz), 6.85 (m, 4H), 6.76 (d, 2H, J 7.5 Hz), 5.21 (d, 1H, J1,2 3.5 Hz, H-1′′), 4.57 (d, 1H, J1,2 8.0 Hz, H-1′), 4.48 (d, 1H, J1,2 8.0 Hz, H-1), 4.26 (m, 4H, H-4′, H-5′′, CH2), 4.08 (d, 1H, J4,3 3.0 Hz, H-4′′), 4.02 (m, 3H, H-3′′, H-6a, H-6b), 3.93 (dd, 1H, J2,1 3.5, J2,3 10.5 Hz, H-2′′), 3.86 (m, 3H, H-3′, H-5, H-5′), 3.78 (m, 6H, H-6a′, H-6b′, H-6a′′, H-6b′′, CH2), 3.70 (m, 4H, H-2′, H-3, CH2), 3.58 (m, 3H, H-4, CH2), 3.40 (m, 4H), 3.34 (m, 1H, H-2), 2.42 (m, 4H), 2.08 (s, 3H), 1.45 (m, 4H); 13C NMR (D2O, 125 MHz), δ (ppm): 174.2, 170.4, 140.8, 139.0, 137.8, 136.8, 132.9, 130.9, 129.8, 129.8, 129.8, 129.3, 125.9, 125.9, 103.9 (C-1′), 103.2 (C-1), 96.4 (C-1′′), 79.8, 78.4, 78.3, 76.0, 75.7, 75.4, 73.8, 71.8, 70.6, 70.3, 70.1, 69.9, 69.7, 69.2, 65.8, 61.9, 61.9, 61.2, 53.8, 40.9, 39.9, 29.7, 29.7, 23.7, 23.5, 21.4; ESI-HRMS: m/z: calcd for C43H62N2NaO19: 933.3839; found: 933.3832.
UN4C4. Ugi coupling and subsequent Zemplen deacetylation of trisaccharide isocyanide 7 (70.1 mg, 0.069 mmol) afforded UN4C4 (40.2 mg, 0.046 mmol, 67%) as a white solid, Rf 0.38 (DCM–MeOH–H2O, 7:3:0.5), [α]D +54.6 (c 0.2, MeOH); IR (cm−1, solid): 3390, 2928, 2891, 1662, 1066; 1H NMR (D2O, 600 MHz), δ (ppm): 7.13 (d, J 9.0 Hz, 1H), 7.00 (m, 2H), 5.70 (s, 1H), 5.14 (d, J1,2 1.8 Hz, H-1′′), 4.51 (d, J1,2 7.8 Hz, H-1′), 4.45 (m, H-1, NHCH2), 4.19 (m, H-5′′, H-4′), 4.02 (m, H-4′′), 3.96 (m, H-3′′, H-6a, H-6b), 3.86 (dd, J2,1 1.8, J2,3 10.2 Hz, H-2′′), 3.79 (m, H-3′, H-5, H-5′′), 3.74 (m, H-6a′, H-6b′, H-6a′′, H-6b′′, 1H), 3.65 (m, H-2′, H-3, –CH2, 1H), 3.58 (m, H-4, CH2), 3.40 (m, CH2), 3.32 (dd, J2,1 ≈ J2,3 7.8 Hz, H-2), 2.73 (m, 4H), 1.90 (m, 2H), 1.75 (m, 4H), 1.59 (s, 3H), 0.71 (s, 3H); 13C NMR (D2O, 125 MHz), δ (ppm): 171.4, 171.4, 141.6, 139.5, 138.0, 130.7, 130.3, 128.4, 124.8, 103.9 (C-1′), 103.1 (C-1), 96.4 (C-1′′), 79.6, 78.2, 76.0, 75.7, 75.4, 73.7, 71.8, 70.6, 70.5, 70.3, 70.1, 69.8, 69.8, 69.8, 69.2, 65.8, 61.9, 61.9, 61.1, 53.8, 39.9, 29.8, 29.5, 23.6, 23.5, 21.5, 14.1, 12.9; ESI-HRMS: m/z: calcd for C40H62N2NaO19: 897.3839; found: 897.3828.
UN4C5. Ugi coupling and subsequent deacetylation and enzymatic α-galactosylation of lactose-derived isocyanide 6 (143.2 mg, 0.20 mmol) afforded final product UN4C5 (85.0 mg, 0.092 mmol, 69%) as a white solid, Rf 0.38 (DCM–MeOH–H2O, 7:3:0.5), [α]D +56.2 (c 0.8, MeOH); IR (cm−1, solid): 3363, 2930, 1646; 1H NMR (D2O, 500 MHz), δ (ppm): 8.04 (s, 1H), 7.77 (m, 2H), 7.2 (s, 1H), 6.90 (m, 2H), 6.62 (m, 1H), 5.21 (d, 1H, J1,2 4.0 Hz, H-1′′), 4.57 (m, 2H, H-1′, 1H), 4.51 (d, 1H, J1,2 8.0 Hz, H-1), 4.26 (m, 2H, H-4′, H-5′′), 4.08 (d, 1H, J4,3 3.5 Hz, H-4′′), 4.01 (m, 3H, H-3′′, H-6a, H-6b), 3.93 (dd, 1H, J2,1 3.5, J2,3 10.5 Hz, H-2′′), 3.88 (m, 3H, H-3, H-5, H-5′), 3.81 (m, 5H, H-6a′, H-6b′, H-6a′′, H-6b′′, 1H), 3.71 (m, 6H, H-2′, H-3, 2 × CH2), 3.62 (m, 3H, H-4, CH2), 3.47 (m, 2H), 3.41 (dd, 1H, J2,1 ≈ J2,3 8.0 Hz, H-2), 2.40 (m, 4H), 1.41 (m, 4H); 13C NMR (D2O, 125 MHz), δ (ppm): 170.5, 170.2, 147.8, 140.9, 139.3, 137.5, 130.8, 130.4, 128.8, 125.7, 125.6, 125.3, 125.3, 125.1, 103.9 (C-1′), 103.2 (C-1), 96.4 (C-1′′), 79.8, 78.3, 76.0, 75.7, 75.4, 73.7, 71.8, 70.6, 70.6, 70.3, 70.1, 69.9, 69.7, 69.2, 65.8, 61.9, 61.9, 61.2, 53.7, 39.9, 29.7, 29.4, 23.5, 23.2; ESI-HRMS: m/z: calcd for C41H57N3NaO21: 950.3377; found: 950.3368.
UN5C1. Ugi coupling and subsequent deacetylation and enzymatic α-galactosylation of lactose-derived isocyanide 6 (129.7 mg, 0.18 mmol) afforded final product UN5C1 (88.5 mg, 0.10 mmol, 79%) as a white solid, Rf 0.28 (DCM–MeOH–H2O, 7:3:0.5), [α]D +57.3 (c 0.7, MeOH); IR (cm−1, solid): 3384, 2930, 2882, 1645, 1075; 1H NMR (D2O, 600 MHz), δ (ppm): 8.46 (d, 0.6H, J 4.8 Hz), 8.36 (d, 0.4H, J 4.8 Hz), 7.81 (m, 0.6H), 7.72 (m, 0.4H), 7.31 (m, 3H), 7.05 (m, 0.4H), 7.01 (m, 0.6H), 6.99 (d, 0.6H, J 7.2 Hz), 6.85 (d, 0.4H, J 7.2 Hz), 6.80 (d, 0.6H, J 7.2 Hz), 6.75 (d, 0.4H, J 7.2 Hz), 5.13 (d, 1H, J1,2 4.2 Hz, H-1′′), 4.50 (d, 0.6H, J1,2 7.8 Hz, H-1′), 4.48 (d, 0.4H, J 7.8 Hz), 4.46 (d, 0.6H, J 7.8 Hz, H-1), 4.42 (d, 0.4H, J 7.8 Hz), 4.18 (m, 2H, H-4′, H-5′′), 4.08 (s, 0.8H), 4.05 (s, 1.20H), 3.98 (m, 4H, H-3′′, H-4′′, H-6a, H-6b), 3.86 (m, 2H, H-2′′, 1H), 3.76 (m, 4H, H-3′, H-5, H-5′, 1H), 3.73 (m, 4H, H-6a′, H-6b′, H-6a′′, H-6b′′), 3.69 (m, 6H, H-2′, H-3, 2 × CH2), 3.61 (m, 3H, H-4, CH2), 3.55 (m, 1H), 3.52 (m, 3H), 3.34 (dd, 0.6H, J2,1 7.8, J4,3 9.0 Hz, H-2), 3.29 (dd, 0.4H, J2,1 7.8, J4,3 9.0 Hz, H-2), 3.04 (dd, 1.2H, J 6.6, J 6.6 Hz), 2.98 (dd, 0.8H, J 6.6, J 6.6 Hz); 13C NMR (D2O, 125 MHz), δ (ppm): 175.1, 171.7, 171.2, 164.4–114.6 (aromatic Cs), 103.7 (C-1′), 102.9 (C-1), 96.3 (C-1′′), 79.5, 78.1, 75.9, 75.6, 75.3, 73.6, 71.7, 70.4, 70.4, 70.3, 70.2, 69.9, 69.7, 69.6, 69.1, 65.7, 61.8, 61.8, 61.0, 51.9, 50.7, 50.4, 48.5, 40.3, 39.8, 39.4, 36.4, 35.4; ESI-HRMS: m/z: calcd for C39H56FN3NaO19: 912.3384; found: 912.3379.
UN5C2. Ugi coupling and subsequent deacetylation and enzymatic α-galactosylation of lactose-derived isocyanide 6 (116.9 mg, 0.16 mmol) afforded final product UN5C2 (69.0 mg, 0.08 mmol, 78%) as a white solid, Rf 0.28 (DCM–MeOH–H2O, 7:3:0.5), [α]D +57.8 (c 0.5, MeOH); IR (cm−1, solid): 3399, 2939, 2872, 1640, 1075; 1H NMR (D2O, 600 MHz), δ (ppm): 8.47 (d, 0.6H, J 4.8 Hz), 8.41 (d, 0.4H, J 4.8 Hz), 7.80 (m, 1H), 7.33 (m, 2H), 5.13 (d, 1H, J1,2 3.6 Hz, H-1′′), 4.50 (d, 1H, J1,2 7.8 Hz, H-1′), 4.49 (d, 0.6H, J1,2 8.4 Hz, H-1), 4.47 (d, 0.4H, J1,2 8.4 Hz, H-1), 4.18 (m, 2H, H-4′, H-5′′), 3.96 (m, 6H, H-3′′, H-4′′, H-6a, H-6b, CH2), 3.85 (dd, 1H, J2,1 3.6, J2,3 10.0 Hz, H-2′′), 3.82 (m, 3H, H-3′, H-5, H-5′), 3.76 (m, 4H, H-6a′, H-6b′, H-6a′′, H-6b′′), 3.72 (m, 6H, H-2′, H-3, 2 × CH2), 3.62 (m, 5H, H-4, 2 × CH2), 3.57 (m, 1H), 3.41(m, 2H), 3.33 (m, 1H, H-2), 3.06 (dd, 1.2H J 6.6, J 6.6 Hz), 2.99 (dd, 0.8H J 6.6, J 6.6 Hz), 2.26 (d, 0.8H, J 7.8 Hz), 1.97 (d, 1.2H, J 7.8 Hz), 1.96 (m, 0.4H), 1.89 (m, 0.6H), 1.61 (m, 2H), 1.45 (m, 4H), 1.02 (m, 0.8H), 0.87 (m, 1.2H); 13C NMR (D2O, 125 MHz), δ (ppm): 177.9, 177.8, 172.1, 171.8, 159.0, 158.6, 149.6, 149.2, 139.4, 139.1, 125.8, 125.3, 123.7, 123.4, 103.9 (C-1′), 103.1 (C-1), 96.4 (C-1′′), 79.6, 78.2, 76.0, 75.7, 75.4, 73.8, 71.8, 70.6, 70.5, 70.4, 71.3, 70.1, 69.8, 69.8, 69.2, 65.8, 61.9, 61.9, 61.1, 51.9, 50.7, 50.4, 39.9, 39.6, 38.9, 37.2, 36.7, 36.6, 35.7, 32.9, 32.9, 32.9, 25.4, 25.4, 25.3; ESI-HRMS: m/z: calcd for C38H61N3NaO19: 886.3791; found: 886.3788.
UN5C3. Ugi coupling and subsequent deacetylation and enzymatic α-galactosylation of lactose-derived isocyanide 6 (121.5 mg, 0.17 mmol) afforded final product UN5C3 (77.1 mg, 0.087 mmol, 73%) as a white solid, Rf 0.32 (DCM–MeOH–H2O, 7:3:0.5), [α]D +54.2 (c 0.9, MeOH); IR (cm−1, solid): 3396, 2926, 2886, 1639, 1075; 1H NMR (D2O, 600 MHz), δ (ppm): 8.45 (d, 0.6H, J 4.8 Hz), 8.36 (d, 0.4H, J 4.8 Hz), 7.80 (m, 0.6H), 7.71 (m, 0.4H), 7.36 (m, 0.6H), 7.28 (m, 1H), 7.23 (d, 0.4H, J 7.8 Hz), 7.18 (d, 0.8H, J 7.8 Hz), 7.15 (d, 1.2H, J 7.8 Hz), 6.98 (d, 0.8H, J 7.8 Hz), 6.91 (d, 1.2H, J 7.8 Hz), 5.13 (d, 1H, J1,2 4.2 Hz, H-1′′), 4.49 (d, 0.6H, J1,2 7.8 Hz, H-1′), 4.48 (d, 0.4H, J1,2 7.8 Hz, H-1′), 4.45 (d, 0.6H, J1,2 8.4 Hz, H-1), 4.40 (d, 0.4H, J1,2 8.4 Hz, H-1), 4.17 (m, 2H, H-4′, H-5′′), 4.03 (s, 2H), 3.94 (m, 4H, H-3′′, H-4′′, H-6a, H-6b), 3.85 (dd, 1H, J2,1 4.2, J2,3 10.8 Hz, H-2′′), 3.78 (m, 3H, H-3′, H-5, H-5′), 3.74 (m, 6H, H-6a′, H-6b′, H-6a′′, H-6b′′, CH2), 3.68 (m, 6H, H-2′, H-3, 2 × CH2), 3.55 (m, 4H), 3.51 (m, 1H), 3.39 (m, 3H, H-2, CH2), 3.01 (dd, 1.2H, J 6.6, J 6.6 Hz), 2.97 (dd, 0.8H, J 6.6, J 6.6 Hz), 2.31 (s, 1.2H), 2.28 (s, 1.8H); 13C NMR (D2O, 125 MHz), δ (ppm): 177.5, 177.5, 173.5, 172.9, 160.4, 160.0, 151.2, 150.8, 140.9, 140.6, 139.8, 139.7, 133.8, 133.5, 131.9, 131.9, 131.8, 131.7, 131.6, 131.5, 127.2, 126.9, 125.3, 124.9, 105.4 (C-1′), 104.6 (C-1), 97.9 (C-1′′), 81.2, 79.8, 77.6, 77.3, 76.9, 75.3, 75.3, 73.4, 72.1, 72.1, 71.9, 71.9, 71.7, 71.4, 71.2, 70.8, 67.4, 63.5, 63.5, 62.7, 53.7, 52.4, 52.2, 50.2, 42.2, 41.5, 41.1, 38.1, 37.1, 22.7; ESI-HRMS: m/z: calcd for C40H59N3NaO19: 908.3635; found: 908.3630.
UN5C4. Ugi coupling and subsequent deacetylation and enzymatic α-galactosylation of lactose-derived isocyanide 6 (150.8 mg, 0.21 mmol) afforded final product UN5C4 (99.2 mg, 0.11 mmol, 57%) as a white solid, Rf 0.30 (DCM–MeOH–H2O, 7:3:0.5), [α]D +57.8 (c 0.8, MeOH); IR (cm−1, solid): 3376, 2930, 2877, 1663, 1075; 1H NMR (D2O, 600 MHz), δ (ppm): 8.44 (m, 1H), 7.79 (m, 1H), 7.32 (m, 2H), 5.37 (dd, 0.5H, J 6.6, J 6.6 Hz), 5.13 (d, 1H, J1,2 4.2 Hz, H-1′′), 5.09 (dd, 0.5H, J 6.6, J 6.6 Hz), 4.49 (m, 2H, H-1′, H-1), 4.19 (m, 2H, H-4′, H-5′′), 4.09 (m, 1H), 4.04 (s, 2H), 4.01 (d, 1H, J4,3 3.6 Hz, H-4′′), 3.95 (m, 2H, H-6a, H-6b), 3.85 (m, 2H, H-3′′, H-2′′), 3.79 (m, 3H, H-3′, H-5, H-5′), 3.74 (m, 4H, H-6a′, H-6b′, H-6a′′, H-6b′′), 3.71 (m, 6H, H-2′, H-3, 2 × CH2), 3.64 (m, 5H, H-4, 2 × CH2), 3.57 (m, 1H), 3.41 (m, 2H), 3.34 (m, 1H, H-2), 2.00 (m, 2H), 1.63 (s, 1.5H), 1.51 (s, 1.5H), 0.90 (m, 3H); 13C NMR (D2O, 125 MHz), δ (ppm): 179.8, 179.5, 173.5, 173.2, 160.5, 160.1, 151.2, 150.7, 140.8, 140.7, 137.4, 136.7, 131.6, 131.1, 127.1, 127.0, 125.2, 125.0, 105.4 (C-1′), 104.7 (C-1), 97.9 (C-1′′), 81.2, 79.8, 77.6, 77.3, 76.9, 75.3, 73.4, 72.1, 72.0, 71.9, 71.7, 71.4, 71.4, 70.8, 67.4, 63.5, 63.5, 62.7, 54.5, 53.3, 50.9, 49.1, 41.5, 41.5, 38.1, 36.9, 22.9, 15.5, 15.1, 14.9; ESI-HRMS: m/z: calcd for C37H59N3NaO19: 872.3635; found: 872.3631.
UN5C5. Ugi coupling and subsequent deacetylation and enzymatic α-galactosylation of lactose-derived isocyanide 6 (125.0 mg, 0.17 mmol) afforded final product UN5C5 (87.1 mg, 0.097 mmol, 57%) as a white solid, Rf 0.32 (DCM–MeOH–H2O, 7:3:0.5), [α]D +54.4 (c 0.5, MeOH); IR (cm−1, state): 3394, 2924, 2886, 1633, 1075; 1H NMR (D2O, 600 MHz), δ (ppm): 8.49 (d, 0.3H, J 4.8 Hz), 8.33 (dd, 0.3H, J 1.2, J 8.4 Hz), 8.27 (dd, 0.7H, J 1.2, J 8.4 Hz), 8.17 (d, 0.7H, J 4.8 Hz), 8.08 (m, 0.3H), 7.84 (m, 0.3H), 7.72 (m, 0.7H), 7.69 (dd, 0.3H, J 8.4, J 8.4 Hz), 7.66 (m, 0.7), 7.63 (m, 0.7H), 7.60 (dd, 0.7H, J 8.4, J 8.4 Hz), 7.47 (m, 0.3H), 7.42 (d, 0.3H, J 7.8 Hz), 7.35 (m, 0.3H), 7.26 (m, 0.7H), 7.17 (d, 0.7H, J 7.8 Hz), 5.13 (d, 1H, J1,2 4.2 Hz, H-1′′), 4.50 (d, 0.3H, J1,2 7.8 Hz, H-1′), 4.49 (d, 0.7H, J1,2 7.8 Hz, H-1′), 4.48 (d, 0.7H, J1,2 8.4 Hz, H-1), 4.44 (d, 0.3H, J1,2 8.4 Hz, H-1), 4.33 (s, 2H), 4.18 (m, 2H, H-4′, H-5′′), 4.02 (d, 1H, J4,3 3.0 Hz, H-4′′), 3.94 (m, 3H, H-3′′, H-6a, H-6b), 3.86 (m, 2H, H-2, 1H), 3.80 (m, 3H, H-3′, H-5. H-5′), 3.76 (m, 6H, H-6a′, H-6b′, H-6a′′, H-6b′′, CH2), 3.69 (m, 4H, H-2′, H-3, CH2), 3.62 (m, 3H, H-4, CH2), 3.54 (m, 3H), 3.31 (m, 1H, H-2), 3.15 (dd, 0.6H, J 6.6, J 6.6 Hz), 2.94 (dd, 1.4H, J 6.6, J 6.6 Hz); 13C NMR (D2O, 125 MHz), δ (ppm): 172.9, 172.8, 171.2, 170.9, 158.9, 157.8, 149.4, 149.3, 148.6, 148.5, 139.1, 139.1, 136.8, 135.9, 133.7, 133.6, 131.4, 131.0, 125.9, 125.8, 125.6, 125.4, 123.6, 123.5, 123.5, 122.6, 122.2, 103.8 (C-1′), 103.0 (C-1), 96.3 (C-1′′), 79.6, 79.5, 78.1, 75.9, 75.6, 75.3, 73.7, 71.7, 70.5, 70.4, 70.3, 70.2, 70.0, 69.8, 69.7, 69.5, 69.1, 65.7, 61.9, 69.8, 61.0, 53.1, 52.2, 50.0, 48.3, 39.9, 39.8, 36.2, 35.3; ESI-HRMS: m/z: calcd for C38H54N4NaO21: 925.3173; found: 925.3165.
UN6C1. Ugi coupling and subsequent deacetylation and enzymatic α-galactosylation of lactose-derived isocyanide 6 (123.4 mg, 0.17 mmol) afforded final product UN6C1 (40.9 mg, 0.046 mmol, 60%) as a white solid, Rf 0.36 (DCM–MeOH–H2O, 7:3:0.5), [α]D +52.5 (c 0.6, MeOH); IR (cm−1, solid): 3370, 2930, 2532, 1647; 1H NMR (D2O, 500 MHz), δ (ppm): 7.25 (m, 1H), 7.20 (d, J 9.0 Hz, 2H), 7.08 (m, 1H), 6.96 (d, J 9.0 Hz, 2H), 6.82 (m, 1H), 6.75 (m, 1H), 5.12 (d, J1,2 4.0 Hz, H-1′′), 4.48 (d, J1,2 8.0 Hz, H-1′′), 4.42 (d, J1,2 8.0 Hz, H-1), 4.33 (s, NHCH2), 4.16 (m, H-5′′, H-4′), 4.00 (d, J4,3 3.5 Hz, H-4′′), 3.93 (m, H-3′′, H-6a, H-6b), 3.84 (dd, J2,1 3.5, J2,3 10.0 Hz, H-2′′), 3.79 (s, 3H), 3.76 (m, H-3′, H-5, H-5′), 3.69 (m, H-6a′, H-6b′, H-6a′′, H-6b′′, 1H), 3.65 (m, H-2′, H-3, CH2, 1H), 3.55 (m, H-4, 2 × CH2), 3.35 (m, CH2), 3.29 (dd, J2,1 8.0, J2,3 9.5 Hz, H-2); 13C NMR (D2O, 125 MHz), δ (ppm): 175.3, 171.3, 164–114 (aromatics Cs), 103.8 (C-1′), 103.1 (C-1), 96.4 (C-1′′), 79.6, 78.2, 76.0, 75.7, 75.4, 73.7, 71.8, 70.6, 70.5, 70.3, 70.1, 69.8, 69.8, 69.2, 65.8, 61.9, 61.9, 61.1, 56.6, 53.9, 41.2, 39.9; ESI-HRMS: m/z: calcd for C39H55FN2NaO20: 913.3224; found: 913.3218.
UN6C2. Ugi coupling and subsequent deacetylation and enzymatic α-galactosylation of lactose-derived isocyanide 6 (120.7 mg, 0.17 mmol) afforded final product UN6C2 (41.5 mg, 0.048 mmol, 57%) as a white solid, Rf 0.40 (DCM–MeOH–H2O, 7:3:0.5), [α]D +20.4 (c 1.2, MeOH); IR (cm−1, solid): 3365, 2937, 2869, 2491, 1652; 1H NMR (D2O, 500 MHz), δ (ppm): 7.25 (d, 2H, J 8.9 Hz), 6.99 (d, 2H, J 8.9 Hz), 5.11 (d, 1H, J1,2 3.9 Hz, H-1′′), 4.47 (d, 1H, J1,2 7.9 Hz, H-1′), 4.42 (d, 1H, J1,2 8.0 Hz, H-1), 4.33 (d, 1H, 2J 16.1 Hz), 4.30 (d, 1H, 2J 16.1 Hz), 4.15 (m, 2H, H-4′, H-5′′), 3.98 (d, 1H, J4,3 3.7 Hz, H-4′′), 3.94 (m, 3H, H-3′′, H-6a, H-6b), 3.81 (dd, 1H, J2,1 3.9, J2,3 10.3 Hz, H-2′′), 3.81 (s, 3H), 3.74 (m, 3H, H-3′, H-5, H-5′), 3.67 (m, 4H, H-6a′, H-6b′, H-6a′′, H-6b′′), 3.60 (m, 6H, H-2′, H-3, 2 × CH2), 3.54 (m, 3H, H-4, CH2), 3.36 (m, 2H), 3.29 (dd, 1H, J2,1 8.0, J2,3 9.3 Hz, H-2), 2.17 (d, 2H, J 7.5 Hz), 2.05 (m, 1H), 1.64 (m, 2H), 1.42 (m, 4H), 0.96 (m, 2H); 13C NMR (D2O, 125 MHz), δ (ppm): 177.9, 171.5, 159.7, 136.1, 130.0, 130.0, 115.9, 115.9, 103.9 (C-1′), 103.1 (C-1), 96.4 (C-1′′), 79.6, 78.2, 76.0, 75.7, 75.4, 73.7, 71.8, 70.6, 70.5, 70.3, 70.1, 69.8, 69.8, 69.2, 65.8, 61.9, 61.9, 61.1, 56.6, 53.7, 40.2, 39.9, 37.6, 32.8, 32.8, 25.2, 25.2; ESI-HRMS: m/z: calcd for C38H60N2NaO20: 887.3632; found: 887.3626.
UN6C3. Ugi coupling and subsequent deacetylation and enzymatic α-galactosylation of lactose-derived isocyanide 6 (130.6 mg, 0.18 mmol) afforded final product UN6C3 (84.2 mg, 0.095 mmol, 53%) as a white solid, Rf 0.40 (DCM–MeOH–H2O, 7:3:0.5), [α]D +57.1 (c 0.4, MeOH); IR (cm−1, solid): 3347, 2921, 1644; 1H NMR (D2O, 500 MHz), δ (ppm): 7.23 (d, 2H, J 9.0 Hz), 7.12 (d, 2H, J 8.0 Hz), 6.97 (d, 2H, J 9.0 Hz), 6.93 (d, 2H, J 8.0 Hz), 5.21 (d, 1H, J1,2 4.0 Hz, H-1′′), 4.57 (d, 1H, J1,2 8.0 Hz, H-1′), 4.49 (d, 1H, J1,2 7.5 Hz, H-1), 4.38 (s, 2H), 4.26 (m, 2H, H-4′, H-5′′), 4.08 (d, 1H, J4,3 3.0 Hz, H-4′′), 4.02 (m, 3H, H-3′′, H-6a, H-6b), 3.93 (dd, 1H, J2,1 3.5, J2,3 10.0 Hz, H-2′′), 3.87 (m, 3H, H-3′, H-5, H-5′), 3.83 (s, 3H), 3.79 (m, 4H, H-6a′, H-6b′, H-6a′′, H-6b′′), 3.72 (m, 6H, H-2′, H-3, 2 × CH2), 3.62 (m, 3H, H-4, CH2), 3.52 (s, 2H), 3.44 (m, 2H), 3.38 (dd, 1H, J2,1 ≈ J2,3 8.5 Hz, H-2), 2.29 (s, 3H); 13C NMR (D2O, 125 MHz), δ (ppm): 175.8, 171.3, 159.8, 137.8, 135.8, 132.6, 130.1, 130.1, 130.0, 130.0, 129.9, 129.9, 115.8, 115.8, 103.9 (C-1′), 103.1 (C-1), 96.4 (C-1′′), 79.7, 78.2, 76.0, 75.7, 75.4, 73.7, 71.8, 70.6, 70.5, 70.3, 70.1, 69.8, 69.2, 65.8, 61.9, 61.9, 61.1, 56.5, 56.5, 53.9, 40.9, 39.9, 21.1; ESI-HRMS: m/z: calcd for C40H58N2NaO20: 909.3475; found: 909.3569.
UN6C4. Ugi coupling and subsequent deacetylation and enzymatic α-galactosylation of lactose-derived isocyanide 6 (124.0 mg, 0.17 mmol) afforded final product UN6C4 (80.0 mg, 0.094 mmol, 55%) as a white solid, Rf 0.34 (DCM–MeOH–H2O, 7:3:0.5), [α]D +61.7 (c 0.5, MeOH); IR (cm−1, solid): 3352, 2930, 1662; 1H NMR (D2O, 500 MHz), δ (ppm): 7.28 (d, 2H, J 9.0 Hz), 7.04 (d, 2H, J 9.0 Hz), 5.75 (m, 1H), 5.21 (d, 1H, J1,2 4.0 Hz, H-1′′), 4.58 (d, 1H, J1,2 7.5 Hz, H-1′), 4.53 (d, 1H, J1,2 8.0 Hz, H-1′), 4.51 (s, 2H), 4.26 (m, 2H, H-4′, H-5′′), 4.08 (d, 1H, J4,3 3.5 Hz, H-4′′), 4.03 (m, 3H, H-3′′, H-6a, H-6b), 3.93 (d, 1H, J2,1 3.5, J2,3 10.0 Hz, H-2′′), 3.88 (s, 3H), 3.84 (m, 3H, H-3′, H-5, H-5′), 3.80 (m, 4H, H-6a′, H-6b′, H-6a′′, H-6b′′), 3.71 (m, 6H, H-2′, H-3, 2 × CH2), 3.63 (m, 3H, H-4, CH2), 3.45 (m, 2H), 3.39 (d, 1H, J2,1 ≈ J2,3 8.5 Hz, H-2′′), 1.95 (m, 2H), 1.62 (s, 3H), 0.76 (s, 3H); 13C NMR (D2O, 125 MHz), δ (ppm): 171.4, 171.4, 159.0, 130.3, 129.4, 129.4, 115.5, 103.9 (C-1′), 103.1 (C-1), 96.4 (C-1′′), 79.6, 78.2, 76.0, 75.7, 75.4, 73.7, 71.8, 70.6, 70.5, 70.3, 70.1, 69.8, 69.8, 69.8, 69.2, 65.8, 61.9, 61.9, 61.1, 56.5, 56.5, 39.9, 21.4, 14.1, 13.0; ESI-HRMS: m/z: calcd for C37H58N2NaO20: 873.3475; found: 873.3469.
UN6C5. Ugi coupling and subsequent Zemplen deacetylation of trisaccharide isocyanide 7 (69.3 mg, 0.068 mmol) afforded UN6C5 (46.7 mg, 0.052 mmol, 76%) as a white solid, Rf 0.36 (DCM–MeOH–H2O, 7:3:0.5), [α]D +54.0 (c 0.4, MeOH); IR (cm−1, solid): 3349, 2931, 1636; 1H NMR (D2O, 600 MHz), δ (ppm): 8.18 (s, 1H), 8.13 (d, 1H, J 8.4 Hz), 7.71 (d, 1H, J 7.8 Hz), 7.47 (dd, 1H, J 7.8, 7.8 Hz), 7.18 (d, 2H, J 8.4 Hz), 6.82 (d, 2H, J 8.4 Hz), 5.13 (d, 1H, J 3.6 Hz, H-1′′), 4.64 (s, 2H, NHCH2), 4.50 (d, 1H, J 7.8 Hz, H-1′), 4.45 (d, 1H, J 7.8 Hz, H-1), 4.18 (m, 2H, H-5′′, H-4′), 3.96 (m, 4H, H-4′′, H-3′′, H-6a, H-6b), 3.85 (dd, J2,1 3.6, J2,3 10.2 Hz, H-2′′), 3.79 (m, 3H, H-3′, H-5, H-5′), 3.72 (m, 5H, H-6a′, H-6b′, H-6a′′, H-6b′′, 1H), 3.69 (s, 3H), 3.67 (m, 3H, H-2′, H-3, 1H), 3.62 (m, 4H, H-4, CH2, 1H), 3.55 (m, 1H), 3.44 (m, 2H), 3.31 (dd, J2,1 ≈ J2,3 7.8 Hz, H-2); 13C NMR (D2O, 125 MHz), δ (ppm): 171.9, 171.1, 159.1, 148.2, 136.9, 136.1, 135.4, 130.7, 130.0, 130.0, 125.9, 124.3, 115.7, 115.7, 103.9 (C-1′′), 103.1 (C-1), 96.4 (C-1′′), 79.7, 78.2, 76.0, 75.2, 75.4, 73.7, 71.8, 70.6, 70.5, 70.3, 70.1, 69.8, 69.8, 69.2, 65.8, 61.9, 61.9, 61.1, 56.5, 54.2, 40.0; ESI-HRMS: m/z: calcd for C38H53N3NaO22: 926.3013; found: 926.3008.
Evaluation of binding activity by mass spectrometry
For the ESI-MS assay, complete details of the experimental methodology and data analysis are described elsewhere.42 The TcdA-A2 fragment (A2, MW 29580 Da) and TcdB-B3C fragment (B3C, MW 30240 Da) were expressed in E.coli and purified as previously described.43,44 Apparent association constants (Ka,app) for the fragments TcdA-A2 and TcdB-B3C binding to the library of thirty compounds were evaluated using the direct ESI-MS assay. Prior to analysis, the TcdA-A2 and TcdB-B3C solutions were diluted with 50 mM ammonium acetate (pH 7.2) and concentrated using Amicon Ultra-4 centrifugal filters with a molecular weight cut-off of 10000 Da (Millipore). The concentrations of the TcdA-A2 and TcdB-B3C solutions were measured by UV absorption. Each ESI solution was prepared from stock solutions of protein (TcdA-A2 and TcdB-B3C) and one of carbohydrate ligands. Lysozyme and α-lactalbumin were used as reference proteins (Pref) to distinguish specific from nonspecific ligand binding with TcdA-A2 and TcdB-B3C, respectively, during the ESI-MS measurements. Electrospray ionization mass spectrometry (ESI-MS) measurements were conducted using an Apex-Qe 9.4 T Fourier transform ion cyclotron resonance mass spectrometer (FT-ICR MS) (Bruker, Billerica, MA) equipped with an external Nano-ESI source. NanoESI-MS was carried out with NanoMate 100 (Advion Bio-Sciences, Ithaca, NY). Data acquisition and analysis were performed using ApexControl, version 4.0 (Bruker Daltonics). A minimum of 25 transients was used for each acquisition.
Conformational analysis
We conducted MD simulations using Amber 11 with the General Amber Force Field (gaff) and partial charges assigned by the sqm program using the AM1-BCC charge method. Molecules were placed in a rectangular box to allow at least 12 Å distance from any atom to the box border and the box was saturated with water. After minimization (first, 1000 steps of steepest descent then 1000 steps with the conjugate gradients with restrained solute (500 kcal mol−1 Å−2) the system was heated to 300 K with weak restrains to solute (10 kcal mol−1 Å−2) and equilibrated for 30 ps with controlled pressure and temperature. Production runs were performed at 1 atm, 300 K using the Langevin heat bath with 1 ps−1 collision frequency. Non-bonded interactions were evaluated with a 8 Å cut off and the SHAKE constraint system for bonds to hydrogen was enabled. Long-range electrostatics were treated with particle-mesh Ewald (PME) periodic boundary conditions. Trajectories were saved every 2 ps. For all trajectories, distances of interest were extracted, expected NOE effects were summed up as reciprocal six power of the distance (r−6) and normalized assuming the reference distance H-1(Glc)–H-5(Glc) to be 100%.Construction of combinatorial library of virtual compounds
Library enumeration and generation of 3D structures was accomplished using in-house software (see description in ESI,† also available from authors upon request) based on Open Babel and Pybel modules in Python. Each compound was generated in the form of two alternative isomers differing in the configuration of the terminal amide bond, designated as “cis” and “trans” according to the conventional definition for peptide bond conformations, that is, ignoring substituent at the nitrogen atom in a peptoid moiety. All structures were subjected to energy minimization using the MMFF94s force field and steepest gradient (500 steps) followed by conjugate gradient (until convergence or 5000 steps). In order to avoid folding and ring distortions during minimization, positional constrains were applied to the trisaccharide atoms, linker and peptoid scaffold. The protein structure was prepared from the crystal structure (pdb entry: 2G7C) by removing all low molecular weight species and the protein chain B. Protons were added to all hetero atoms of the remaining protein chain A using MGLTools followed by generating a pdbqt file of the receptor. Automatic molecular docking was performed on multi-CPU cluster computers using Autodock Vina28 with an exhaustiveness set at 64. The grid box dimensions were 20 × 20 × 20 Å and it was centered on the coordinates (x, y, z) = (25.45, 15.1, 5.9) Å. Each ligand was resubmitted several times until at least one pose was obtained, in which the position of the trisaccharide fragment matched the position of the trisaccharide in the crystal structure with a RMSD less than 1.5 Å.In silico ranking of ligands
Preparation of molecular structures was conducted as described for preparation of the virtual library except the torsional constrains for peptoid amide bond were removed in order to increase the docking success rate. Ten automated docking sessions performed with Autodock Vina (different random seeds, exhaustiveness set at 64) yielded from 2 to 15 poses per compound, in which the position of the trisaccharide matched that found in the crystal structure with RMSD lower than 0.5 Å.Crystal structure determination of the TcdA-A2 complex with UN5C2
TcdA-A2 was expressed, purified and crystallized as previously described.26,43 UN5C2 (5 μL × 50 mM) was added to 10 μL of a 1.5× strength precipitant solution to generate 15 μL of precipitant solution containing 16.7 mM UN5C2 (Solution A). Crystals (∼0.4 × 0.01 × 0.01 mm) were soaked in Solution A for 60 minutes before flash freezing under nitrogen gas (100 K) and storage in liquid nitrogen. The frozen crystal was mounted at the Canadian Macromolecular Crystallography Facility Beamline 08B1-1, where diffraction data (400 images × 0.5° rotation) were measured using a Marmosaic MX300 CCD X-ray detector. Data were processed and scaled using XDS and XSCALE (40–1.7 Å, Rsym = 0.057, I/σ = 13.9).45 Crystals were isomorphous to the TcdA-A2 trisaccharide complex (Space group P21, a = 38.89, b = 65.69, c = 132.85, β = 100.75°),26 and the structure was refined using REFMAC (Rwork = 0.184, Rfree = 0.215).46 Electron density maps were calculated using FFT and COOT, and model building was performed using COOT.47Fig. 4 and 5 were prepared using PyMOL (Schrödinger LLC).Acknowledgements
We thank the Alberta Glycomics Centre for financial support, Compute Canada for providing access to WestGrid clusters, and Dr W. W. Wakarchuk of the National Research Council of Canada for providing cell-lines expressing α-(1–3)-galactosyl transferase and UDP-glucose 4-epimerase used for the synthesis of the trisaccharide. X-ray diffraction data were measured at beamline 08B1-1 of the Canadian Light Source (CLS), which is supported by the Natural Sciences and Engineering Research Council of Canada, the National Research Council Canada, the Canadian Institutes of Health Research, the Province of Saskatchewan, Western Economic Diversification Canada, and the University of Saskatchewan.References
- J. G. Bartlett, Clin. Infect. Dis., 1994, 18, S265–S272 CrossRef PubMed.
- M. Kachrimanidou and N. Malisiovas, Crit. Rev. Microbiol., 2011, 37, 178–187 CrossRef CAS PubMed.
- E. R. Dubberke and M. A. Olsen, Clin. Infect. Dis., 2012, 55(Suppl 2), S88–S92 CrossRef PubMed.
- M. C. Hammitt, D. A. Bueschel, A. K. Kee, R. D. Glock, P. Cuneo, D. W. DeYoung, C. Reggiardo, H. T. Trinh and J. G. Songer, Vet. Microbiol., 2008, 127, 343–352 CrossRef CAS PubMed.
- A. Goorhuis, D. Bakker, J. Corver, S. B. Debast, C. Harmanus, D. W. Notermans, A. A. Bergwerff, F. W. Dekker and E. J. Kuijper, Clin. Infect. Dis., 2008, 47, 1162–1170 CrossRef CAS PubMed.
- M. A. Jhung, A. D. Thompson, G. E. Killgore, W. E. Zukowski, G. Songer, M. Warny, S. Johnson, D. N. Gerding, L. C. McDonald and B. M. Limbago, Emerging Infect. Dis., 2008, 14, 1039–1045 CrossRef CAS PubMed.
- E. J. Kuijper, J. T. van Dissel and M. H. Wilcox, Curr. Opin. Infect. Dis., 2007, 20, 376–383 Search PubMed.
- K. Mullane, Ther. Adv. Chronic Dis., 2014, 5, 69–84 CrossRef CAS PubMed.
- B. Geric, R. J. Carman, M. Rupnik, C. W. Genheimer, S. P. Sambol, D. M. Lyerly, D. N. Gerding and S. Johnson, J. Infect. Dis., 2006, 193, 1143–1150 CrossRef CAS PubMed.
- H. C. Krivan, G. F. Clark, D. F. Smith and T. D. Wilkins, Infect. Immun., 1986, 53, 573–581 CAS.
- X. Y. Wu and R. R. Schmidt, J. Org. Chem., 2004, 69, 1853–1857 CrossRef CAS PubMed.
- (a) A. El-Hawiet, E. N. Kitova, P. I. Kitov, L. Eugenio, K. K. Ng, G. L. Mulvey, T. C. Dingle, A. Szpacenko, G. D. Armstrong and J. S. Klassen, Glycobiology, 2011, 21, 1217–1227 CrossRef CAS PubMed; (b) T. Dingle, S. Wee, G. L. Mulvey, A. Greco, E. N. Kitova, J. Sun and S. Lin, et al. , Glycobiology, 2008, 18, 698–706 CrossRef CAS PubMed.
- S. Teneberg, I. Lonnroth, J. F. T. Lopez, U. Galili, M. O. Halvarsson, J. Angstrom and K. A. Karlsson, Glycobiology, 1996, 6, 599–609 CrossRef CAS PubMed.
- K. D. Tucker and T. D. Wilkins, Infect. Immun., 1991, 59, 73–78 CAS.
- I. Just, F. Hofmann, H. Genth and R. Gerhard, Int. J. Med. Microbiol., 2001, 291, 243–250 CrossRef CAS PubMed.
- R. N. Pruitt, M. G. Chambers, K. K. S. Ng, M. D. Ohi and D. B. Lacy, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13467–13472 CrossRef CAS PubMed.
- (a) T. Jank, T. Giesemann and K. Aktories, Glycobiology, 2007, 17, 15R–22R CrossRef CAS PubMed; (b) J. Zeiser, R. Gerhad, I. Just and A. Pich, J. Proteome Res., 2013, 12, 1604–1618 CrossRef CAS PubMed.
- I. Just and R. Gerhard, Rev. Physiol., Biochem., Pharmacol., 2004, 152, 23–47 CAS.
- M. Rupnik, FEMS Microbiol. Rev., 2008, 32, 541–555 CrossRef CAS PubMed.
- P. Spigaglia, F. Barbanti and P. Mastrantonio, J. Antimicrob. Chemother., 2011, 66, 2227–2234 CrossRef CAS PubMed.
- I. Lowy, D. C. Molrine, B. A. Leav, B. M. Blair, R. Baxter, D. N. Gerding, G. Nichol, W. D. Thomas, M. Leney, S. Sloan, C. A. Hay and D. M. Ambrosino, N. Engl. J. Med., 2010, 362, 197–205 CrossRef CAS PubMed.
- G. S. Tillotson and J. Tillotson, F1000 Medicine Reports, 2011, vol. 3, (1 March 2011).
- K. Weiss, Int. J. Antimicrob. Agents, 2009, 33, 4–7 CrossRef CAS PubMed.
- S. J. Abdeen, R. J. Swett and A. L. Feig, ACS Chem. Biol., 2010, 5, 1097–1103 CrossRef CAS PubMed.
- A. W. Puri, P. J. Lupardus, E. Deu, V. E. Albrow, K. C. Garcia, M. Bogyo and A. Shen, Chem. Biol., 2010, 17, 1201–1211 CrossRef CAS PubMed.
- A. Greco, J. G. S. Ho, S. J. Lin, M. M. Palcic, M. Rupnik and K. K. S. Ng, Nat. Struct. Mol. Biol., 2006, 13, 460–461 CAS.
- N. M. O'Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, J. Chem. Inf., 2011, 3(33), 1–14 Search PubMed.
- O. Trott and A. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- D. H. Joziasse, J. H. Shaper, D. H. Vandeneijnden, A. J. Vantunen and N. L. Shaper, J. Biol. Chem., 1989, 264, 14290–14297 CAS.
- S. Bernatchez, C. M. Szymanski, N. Ishiyama, J. J. Li, H. C. Jarrell, P. C. Lau, A. M. Berghuis, N. M. Young and W. W. Wakarchuk, J. Biol. Chem., 2005, 280, 4792–4802 CrossRef CAS PubMed.
- P. Reddy and S. Baskaran, Tetrahedron Lett., 2002, 43, 1919–1922 CrossRef CAS.
- W. Brandt, T. Herberg and L. Wessjohann, Biopolymers, 2011, 96, 651–668 CrossRef CAS.
- D. M. Schnur, Y. H. Yuh and D. R. Dalton, J. Org. Chem., 1989, 54, 3779–3785 CrossRef CAS.
- N. H. Shah, G. L. Butterfoss, K. Nguyen, B. Yoo, R. Bonneau, D. L. Rabenstein and K. Kirshenbaum, J. Am. Chem. Soc., 2008, 130, 16622–16632 CrossRef CAS PubMed.
- E. N. Kitova, A. El-Hawiet, P. D. Schnier and J. S. Klassen, J. Am. Soc. Mass Spectrom., 2012, 23, 431–441 CrossRef CAS PubMed.
- P. Strazzolini, A. G. Giumanini and S. Cauci, Tetrahedron, 1990, 46, 1–2 CrossRef.
- J. P. Praly and R. U. Lemieux, Can. J. Chem., 1987, 65 Search PubMed; R. U. Lemieux and K. Bock, Arch. Biochem. Biophys., 1983, 221, 213–223 CrossRef.
- M. R. Wormald, A. J. Petrescu, Y. L. Pao, A. Glithero, T. Elliott and R. A. Dwek, Chem. Rev., 2002, 102, 371–386 CrossRef CAS PubMed.
- D. Adlercreutz, J. T. Weadge, B. O. Petersen, J. Ø. Duus, N. J. Dovichi and M. M. Palcic, Carbohydr. Res., 2010, 345, 1384–1388 CrossRef CAS PubMed.
- S. Bernatchez, S. Bernatchez, C. M. Szymanski, N. Ishiyama, J. Li, H. C. Jarrell, P. C. Lau, A. M. Berghuis, N. M. Young and W. W. Wakarchuk, J. Biol. Chem., 2005, 280, 4792–4802 CrossRef CAS PubMed.
- F. González Núñez, M. T. Campos Valdes, E. Aruca, R. R. Schmidt, V. V. Bencomo, E. Aruca, M. T. Valdes, R. R. Schmidt and V. V. Bencomo, J. Carbohydr. Chem., 2003, 22, 1–13 CrossRef PubMed.
- J. X. Sun, E. N. Kitova, W. J. Wang and J. S. Klassen, Anal. Chem., 2006, 78, 3010–3018 CrossRef CAS PubMed; W. J. Wang, E. N. Kitova and J. S. Klassen, Anal. Chem., 2003, 75, 4945–4955 CrossRef.
- J. G. S. Ho, A. Greco, M. Rupnik and K. K. S. Ng, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18373–18378 CrossRef CAS PubMed.
- H. Lin, E. N. Kitova, M. A. Johnson, L. Eugenio, K. K. Ng and J. S. Klassen, J. Am. Soc. Mass Spectrom., 2012, 23, 2122–2131 CrossRef CAS PubMed.
- W. Kabsch, J. Appl. Crystallogr., 1993, 26, 795–800 CrossRef CAS.
- G. N. Murshudov, A. A. Vagin and E. J. Dodson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1997, 53, 240–255 CrossRef CAS PubMed.
- P. Emsley and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2126–2132 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Mass and NMR spectra, Python scripts for virtual library screening. See DOI: 10.1039/c4ob01838a |
| This journal is © The Royal Society of Chemistry 2015 |