
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploiting nucleobase-containing materials – from monomers to complex morphologies using RAFT dispersion polymerization†
Yan
Kang
a,
Anaïs
Pitto-Barry
 a,
Helen
Willcock
a,
Wen-Dong
Quan
a,
Nigel
Kirby
b,
Ana M.
Sanchez
c and
Rachel K.
O'Reilly
*a
a,
Helen
Willcock
a,
Wen-Dong
Quan
a,
Nigel
Kirby
b,
Ana M.
Sanchez
c and
Rachel K.
O'Reilly
*a
aDepartment of Chemistry, University of Warwick, Library Rd., Coventry, CV4 7AL, UK. E-mail: Rachel.oreilly@warwick.ac.uk
bAustralian Synchrotron, 800 Blackburn Road, Clayton, VIC 3168, Australia
cDepartment of Physics, University of Warwick, Library Rd., Coventry, CV4 7AL, UK
First published on 11th September 2014
Abstract
The synthesis of nucleobase-containing polymers was successfully performed by RAFT dispersion polymerization in both chloroform and 1,4-dioxane and self-assembly was induced by the polymerizations. A combination of scattering and microscopy techniques were used to characterize the morphologies. It is found that the morphologies of self-assembled nucleobase-containing polymers are solvent dependent. By varying the DP of the core-forming block, only spherical micelles with internal structures were obtained in chloroform when using only adenine-containing methacrylate or a mixture of adenine-containing methacrylate and thymine-containing methacrylate as monomers. However, higher order structures and morphology transitions were observed in 1,4-dioxane. A sphere-rod-lamella-twisted bilayer transition was observed in this study. Moreover, the kinetics of the dispersion polymerizations were studied in both solvents, suggesting a different formation mechanism in these systems.
Introduction
The specific hydrogen bonding interactions between nucleobase pairs play a key role in nature for precise biosynthesis and stereospecific molecular assembly. Inspired by nature, nucleobases have been employed in synthetic polymer chemistry to control polymer tacticity,1 to mediate polymer composition or sequence2–4 and to template polymerizations.5–7 Moreover, nucleobase interactions have also been applied for driving self-assembly8–12 and for achieving a biomimetic segregation/templating approach.13 This pioneering work has expanded the window for further investigation into nucleobase materials.14 However, to our knowledge, although various morphologies, including large vesicles,8 rods9 and spheres,13 have been obtained, there is still very little research into the systematic study of the self-assembly of nucleobase-containing polymers. This might result from the poor solubility of the nucleobase-containing copolymers, which are only fully soluble in polar organic solvents. Therefore, nucleobase-containing polymers with a relatively high degree of polymerization (DP) have to be synthesized in polar solvents (e.g., DMSO, DMF) to avoid precipitation or to achieve good control; self-assembly is then achieved by post-polymerization processing. These multiple steps limit the synthesis, self-assembly, and other associated studies of nucleobase-containing polymers. Thus, a facile approach to make well-controlled nucleobase-containing polymers and to prepare their corresponding self-assemblies is worthy of investigation.Heterogeneous polymerizations (including dispersion, emulsion, suspension and precipitation) are easily performed and widely used in industry.15 In particular, heterogeneous polymerizations using controlled/living radical polymerization (CRP) techniques are of great interest recently,16–18 as they have compiled the advantages of heterogeneous polymerizations with the living nature of CRP. This approach has been exploited for the synthesis of block copolymers by reversible addition–fragmentation chain-transfer (RAFT) polymerization,16,19–21 various forms of atom-transfer radical polymerization (ATRP),22,23 nitroxide-mediated polymerizations (NMP),24,25 and other CRP techniques.26–28 Moreover, some of these processes have been developed for design of systematic ‘nanoobjects’ simply by varying the polymerization conditions.29–40 For example, RAFT dispersion polymerization has been used to grow a solvent-insoluble new block from solvent-soluble/miscible monomers in the presence of a solvent-soluble macromolecular chain transfer agent (macro-CTA) in both aqueous and organic media.29,30,41 Block copolymers with narrow size distributions have been obtained. Moreover, self-assemblies with controlled size and morphology are formed during the polymerization process without further steps. Spherical micelles, wormlike micelles and vesicles were usually obtained sequentially by increasing the length of the insoluble block.29,41 However, in some cases (e.g., when a relatively long macro-CTA was used30) only spherical micelles with different sizes were observed. Furthermore, a few novel morphologies (e.g., lumpy rod,42 framboidal morphology,43 or concentric vesicle44) or morphology transitions were observed on introduction of a special monomer or condition. In general, heterogeneous CRP has been demonstrated as a facile approach to make common polymers and their corresponding nanostructures. However, little work has been exploited for the synthesis of nucleobase-containing polymers and nanostructures by heterogeneous CRP, which is challenged by the interacting nature of nucleobases, leading to a complex and unpredictable system. Therefore, in this work, we have firstly taken advantage of heterogeneous CRP to prepare nucleobase-containing polymers and meanwhile produce their corresponding self-assemblies directly without other steps.
The nucleobases used in this study, adenine and thymine, are known as complementary base pairs in DNA. In synthetic chemistry, it has been proven that hydrogen bonding interactions still exist between modified adenine and thymine as long as the interaction sites of purine and pyrimidine functionalities are not trapped.13,14 Methacryloyl-type monomers containing adenine and thymine were previously synthesized and polymerized in our group.4 Moreover, it has been found that both chloroform (CHCl3) and 1,4-dioxane are good solvents to solubilize these monomers and more importantly support the nucleobase interactions (the association constants between monomers was measured as, Kasso = 20 M−1 in chloroform at 60 °C,4 and Kasso = 12 M−1 in 1,4-dioxane at 60 °C). According to our previous report, driven by the appearance of monomer interactions, an alternating copolymer has a tendency to be formed in CHCl3 when monomers containing adenine and thymine are copolymerized. Similar tendency is also expected in 1,4-dioxane.2 However, the copolymer is insoluble in both CHCl3 and 1,4-dioxane due to the presence of the strong hydrogen bonding interactions among copolymers. Therefore, both CHCl3 and 1,4-dioxane are ideal solvents for RAFT dispersion polymerization of these nucleobase-containing monomers. Poly(methyl methacrylate) (PMMA) has been selected to be the macro-CTA, as it is readily soluble in both CHCl3 and 1,4-dioxane and can be synthesized with good control by RAFT polymerization using 2-cyano-2-propyl dodecyl trithiocarbonate (CPDT) as a chain transfer agent (CTA).45
Experimental
Materials
Methyl methacrylate (MMA) was bought from Aldrich and passed through a column of neutral alumina to remove the inhibitor. 2,2-Azo-bis(isobutyronitrile) (AIBN) was purchased from Molekula and recrystallized from methanol. 2-Cyano-2-propyl dodecyl trithiocarbonate (CPDT) was synthesized according to published method.45 The synthesis of monomers 2-(2-(adenine-9-yl)acetoxyl) ethyl methacrylate (AMA) and 2-(2-(thymine-1-yl)acetoxyl) ethyl methacrylate (TMA) is based on our previous reference.4 1,4-Dioxane, CHCl3, and other solvents were used as received from Fisher Scientific. Deuterated solvents were purchased from Apollo Scientific.Polymerization of methyl methacrylate (MMA)
The typical procedure of RAFT homopolymerization of MMA was as follows: MMA (x), CPDT (1 eq.), and AIBN (0.1 eq.) were dissolved in toluene. The mixture was thoroughly degassed via 4 freeze–pump–thaw cycles, filled with oxygen-free nitrogen and then immersed into an oil bath at 60 °C for 5 hours. The reaction was quenched by putting into a liquid nitrogen bath and exposing to air. The mixture was precipitated in MeOH and filtered. The solid was dissolved in THF and precipitated again. The precipitation procedure was repeated 3 times in total. The light yellow polymers were dried in the vacuum oven overnight and characterized by 1H NMR spectroscopy and DMF size-exclusion chromatography (SEC) (PMMA standards). The DP of the block was varied by adding different amounts of the monomer.Synthesis of block polymers using PMMA as macro-CTA
The typical procedure was follows: PMMA (1 eq.), AMA (x), TMA (y), and AIBN (0.1 eq.) were dissolved in 1,4-dioxane or CHCl3. The mixture was thoroughly degassed via 4 freeze–pump–thaw cycles, filled with oxygen-free nitrogen and then immersed into an oil bath at 60 °C for 24 hours. The reaction was quenched by exposing to air and cooling down. The mixture was precipitated in MeOH and washed with MeOH several times. The light yellow polymers were dried in the vacuum oven overnight and characterized by 1H NMR spectroscopy and DMF SEC (PMMA standards). The DP of the block was varied by adding different amounts of the monomer.Kinetics of the dispersion polymerization for a target diblock copolymer PMMA70-b-(PAMA0.5-co-PTMA0.5)50
PMMA70 macro-CTA (35 mg, 0.005 mmol), AMA (38 mg, 0.125 mmol), TMA (37 mg, 0.125 mmol), and AIBN (0.1 mg, 0.0005 mmol) were dissolved in 6 mL of CHCl3 or 1,4-dioxane. The mixture was thoroughly degassed via 4 freeze–pump–thaw cycles, filled with oxygen-free nitrogen and then immersed into an oil bath at 60 °C. An aliquot of the polymerization solution was taken at designated times for 1H NMR, SEC, dynamic light scattering (DLS) and transmission electron microscopy (TEM) characterization.Characterization
1H NMR spectra were recorded on a Bruker DPX-300 or DPX-400 spectrometer with DMSO-d6 or deuterated chloroform (CDCl3) as the solvent. The chemical shifts of protons were reported relative to tetramethylsilane at δ = 0 ppm when using CHCl3 or solvent residues (DMSO 1H: 2.50 ppm).SEC was obtained in HPLC-grade DMF containing 5 mM NH3BF4 at 50 °C, with a flow rate of 1.0 mL per minute, on a set of two PLgel 5 μm Mixed-D columns, plus one guard column. SEC data was analyzed with Cirrus SEC software calibrated using polymethyl methacrylate (PMMA) standards. The SEC was equipped with both refractive index (RI) and UV detectors.
TEM observations were performed on a JEOL 2000FX electron microscope at an acceleration voltage of 200 kV. High magnification TEM images were obtained from a JEOL 2100FX electron microscope at an acceleration voltage of 200 kV. All TEM samples were prepared on graphene oxide (GO)-coated carbon grids (Quantifoil R2/2), which allows high contrast TEM images to be acquired without staining.46 Generally, a drop of sample (20 μL) was pipetted on a grid, blotted immediately and left to air dry.
Hydrodynamic diameters (Dh, app) and size distributions of the self-assemblies were determined by DLS. The DLS instrumentation consisted of a Malvern Zetasizer NanoS instrument operating at 25 °C with a 4 mW He–Ne 633 nm laser module. Measurements were made at a detection angle of 173° (back scattering), and Malvern DTS 5.02 software was used to analyze the data. Static light scattering (SLS) measurement was conducted with an ALV CGS3 (λ = 632 nm) at both 20 °C and 50 °C. The data were collected from 30° up to 150° with an interval of 10°, calibrated with filtered toluene and filtered CHCl3 or 1,4-dioxane as backgrounds. The RI of the polymer 3 in CHCl3 was measured to be 0.053 mL g−1 and polymer 10 in 1,4-dioxane was measured to be 0.033 mL g−1.
Atomic force microscopy (AFM) images were taken in tapping mode on a Multimode AFM with Nanoscope IIIA Controller with Quadrex. Silicon AFM tips were used with nominal spring constant and resonance frequency of 3.5 Nm−1 and 75 kHz (MikroMasch NSC18). Samples were imaged either on the same quantifoil Cu/GO grids used in TEM analysis or onto freshly cleaved mica discs (Agar Scientific, G250-2). Data were analyzed using Gwyddion software.
Sonication experiments were carried out with a Sonopuls (Bandelin) ultrasonic homogenizer HD 2200 equipped with a MS72 microtip at a frequency of 2 kHz.
Small-angle neutron scattering (SANS) experiments were recorded on the ISIS neutron beam facility, sans2d instrument at the Rutherford Appleton Laboratory, Oxford. Sample 3 was measured at 20 mg mL−1 in CDCl3, which provides a high contrast in scattering length to the polymer. Small-angle X-ray scattering (SAXS) were carried out on the SAXS-WAXS beamline at the Australian Synchrotron facility at a photon energy of 15 keV. The samples were prepared in 1,4-dioxane and were run using 1.5 mm diameter quartz capillaries. The measurements were collected at 25 °C with a sample to detector distance of 7.160 m to give a q range of 0.0015 to 0.08 Å−1, where q is the scattering vector and is related to the scattering angle (2θ) and the photon wavelength (λ) by the following equation:
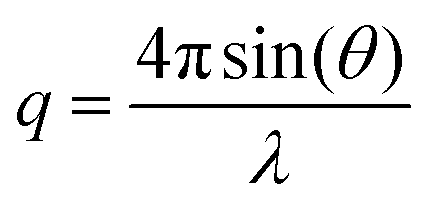 | (1) |
All patterns were normalized to a fixed transmitted flux using a quantitative beam stop detector. The scattering from a blank (1,4-dioxane) was measured in the same location as sample collection and was subtracted for each measurement. The two-dimensional SAXS images were converted in one-dimensional SAXS profile (I(q) versus q) by circular averaging, where I(q) is the scattering intensity. ScatterBrain and NCNR Data Analysis IGOR PRO software were used to plot and analyze SAXS and SANS data.47 The scattering length density of the solvents and monomers were calculated using the “Scattering Length Density Calculator” provided by NIST Center for Neutron Research.
Results
There are a few reports on the self-assembly of nucleobase-containing polymers.14 Spherical micelles,13 giant vesicles,8 and rod morphologies9 were observed upon assembly of single or mixtures of nucleobase-containing polymers. However, there has been little research into the morphology transition of nanostructures constructed by nucleobase-containing polymers. A possible reason for the difficulty in studying morphology transition is the poor solubility of the nucleobase-containing polymers, which are only fully soluble in organic solvents with high polarity (e.g., DMF, DMSO). Therefore, it is not easy to make controlled nucleobase-containing polymers with a high DP and self-assembly must be achieved by post-polymerization processing.The formation of a range of diblock copolymer nanostructures in either aqueous or organic media by heterogeneous CRP has been demonstrated in previous reports.19,29–40 This approach is easy to perform and also results in polymers with good control. However, as far as we know, this robust method hasn't been applied to synthesize nucleobase-containing polymers or to provide their corresponding nanostructures. Herein, we demonstrate the synthesis of well-controlled nucleobase-containing polymers and a range of their corresponding nanostructures by RAFT dispersion polymerization. Moreover, new morphologies have been observed in this system.
Nucleobase-containing monomers
Nucleobase-containing monomers, AMA and TMA, were synthesized and both homopolymerized and copolymerized in our previous work.4 The results showed that the hydrogen bonding interactions between AMA and TMA were solvent-dependent. Monomer interactions occurred in CHCl3, but were suppressed in DMF. Moreover, the apparent reactivity ratios of copolymerizations of AMA and TMA were investigated. The results indicated that the presence and absence of monomer interactions could result in copolymers with different compositions (alternating copolymers tended to be formed in CHCl3 and statistical copolymers were most likely to be synthesized in DMF). It has been reported that both CHCl3 and 1,4-dioxane are good solvents to support nucleobase interactions among polymers.48,49 Moreover, both AMA and TMA are soluble in these two solvents. Therefore, similar observations on polymerization of the two monomers were expected in 1,4-dioxane to those in CHCl3.Synthesis of the macro-CTA
The synthesis route for macro-CTA PMMA is shown in Scheme 1. PMMA was synthesized using CPDT as the CTA, AIBN as the initiator, and toluene as the solvent. The polymerization was stopped at a relatively low conversion (∼36%) to ensure good end-group fidelity. The DP of the PMMA was determined by 1H NMR spectroscopy by comparing the integration of the backbone signals with those of the end group from CPDT (Fig. S1†). The resulting DP of the PMMA is ca. 70. Furthermore, SEC (DMF as eluent, PMMA standards) was used to determine the molecular weight and the molecular weight distribution. The molecular weight from SEC is 7.4 kDa, which is also consistent with the result from 1H NMR spectroscopy. Additionally, RI and UV (309 nm, from the trithiocarbonate end group) overlap well, indicating good end group fidelity. The characterization data of the PMMA macro-CTA is shown in Table 1. | ||
| Scheme 1 Synthesis route for the macro-CTA (PMMA, 1) and the nucleobase-containing diblock copolymers using RAFT polymerization. | ||
| Solvent | Polymer/polymerization | Conv. (%) | M n, th (kDa) | M n, NMR (kDa) | M n, SEC (kDa) | Đ M |
|---|---|---|---|---|---|---|
| Toluene | PMMA701 | 36 | 4.5 | 7.0 | 7.4 | 1.24 |
| CHCl3 | PMMA70-b-PAMA502 | 92 | 20.5 | 21.4 | 24.7 | 1.46 |
| CHCl3 | PMMA70-b-(PAMA0.5-co-PTMA0.5)503 | 95 | 21.3 | 26.8 | 26.1 | 1.32 |
| CHCl3 | PMMA70-b-PAMA1004 | 95 | 37.0 | 40.3 | 41.1 | 1.20 |
| CHCl3 | PMMA70-b-(PAMA0.5-co-PTMA0.5)1005 | 98 | 37.0 | 44.8 | 38.2 | 1.76 |
| 1,4-Dioxane | PMMA70-b-PAMA506 | 92 | 20.5 | 21.4 | 25.2 | 1.39 |
| 1,4-Dioxane | PMMA70-b-(PAMA0.5-co-PTMA0.5)507 | 90 | 20.5 | 23.8 | 25.6 | 1.25 |
| 1,4-Dioxane | PMMA70-b-PAMA1008 | 98 | 37.0 | 40.3 | 32.0 | 1.21 |
| 1,4-Dioxane | PMMA70-b-(PAMA0.5-co-PTMA0.5)1009 | 90 | 37.0 | 44.8 | 30.5 | 1.36 |
| 1,4-Dioxane | PMMA70-b-PAMA15010 | 99 | 52.0 | 55.6 | 38.6 | 1.09 |
| 1,4-Dioxane | PMMA70-b-(PAMA0.5-co-PTMA0.5)15011 | 90 | 52.0 | 54.7 | 38.4 | 1.50 |
Synthesis of diblock copolymers
To synthesize diblock copolymers, RAFT polymerizations were performed in both CHCl3 and 1,4-dioxane, first using exclusively AMA and then using a mixture of AMA and TMA as monomers. The DP of the nucleobase block was varied by adding different amounts of the monomer. It should be noted that AMA and TMA are both soluble in CHCl3 and 1,4-dioxane, but form insoluble homopolymers or copolymers in these two solvents. This suggests that RAFT dispersion polymerization would proceed and self-assembly would be induced by the polymerizations.30The characterization data of all polymers are shown in Table 1. High conversion (≥90%) was attained for each chain extension polymerization. SEC traces of macro-CTA and synthesized diblock copolymers were all overlapped and summarized (Fig. S2 and S3†). These studies indicated that well-defined diblock copolymers were obtained with relatively high blocking efficiencies and low macro-CTA contamination. Compared to previously reported systems where monodisperse diblock copolymers (ĐM < 1.40) were produced,29,30 this system is generally as good as the published system although some polymers have slightly higher dispersities. The nature of nucleobases leads to potential interactions among monomers and polymers and hence results in a more complex system of polymerization.
Kinetics of RAFT dispersion polymerization in CHCl3
The kinetics of the RAFT dispersion polymerization in CHCl3 was studied by chain-extending the PMMA macro-CTA using a mixture of AMA and TMA at 60 °C. Monomer conversion data obtained by 1H NMR are shown in Fig. S4† for a target diblock copolymer of PMMA70-b-(PAMA0.5-co-PTMA0.5)50, 3. It should be noted that conversions of each monomer were very similar during the whole polymerization. High conversion (≥85%) was obtained after 10 hours. The evolution of SEC traces with monomer conversion is shown in Fig. 1. Interestingly, obvious bimodal peaks were observed at low conversions (3 hours and 5 hours). The SEC traces after 3 hours and 5 hours show that the high molecular weight polymer traces stay the same, but the peaks for the low molecular weight polymers shift and drop. As the polymerization proceeds, the low molecular weight polymer trace decreases in intensity when the peaks for the high molecular weight polymer are normalized. This observation indicates that the macro-CTAs were not all chain-extended at the same time or at the same rate. However, close to monodisperse diblocks were obtained at high conversions. Three separate polymerizations with the same starting mixture were also performed and stopped at designated reaction times (4 hours, 10 hours and 16 hours). The monomer conversion data and SEC traces are shown in Fig. S5.† Similar results were observed, demonstrating that this phenomenon is reproducible and repeatable. This observation is different to the previously reported results in the literature, where the molecular weight increases linearly with conversion, indicating that an unusual RAFT dispersion polymerization is occurring in this system.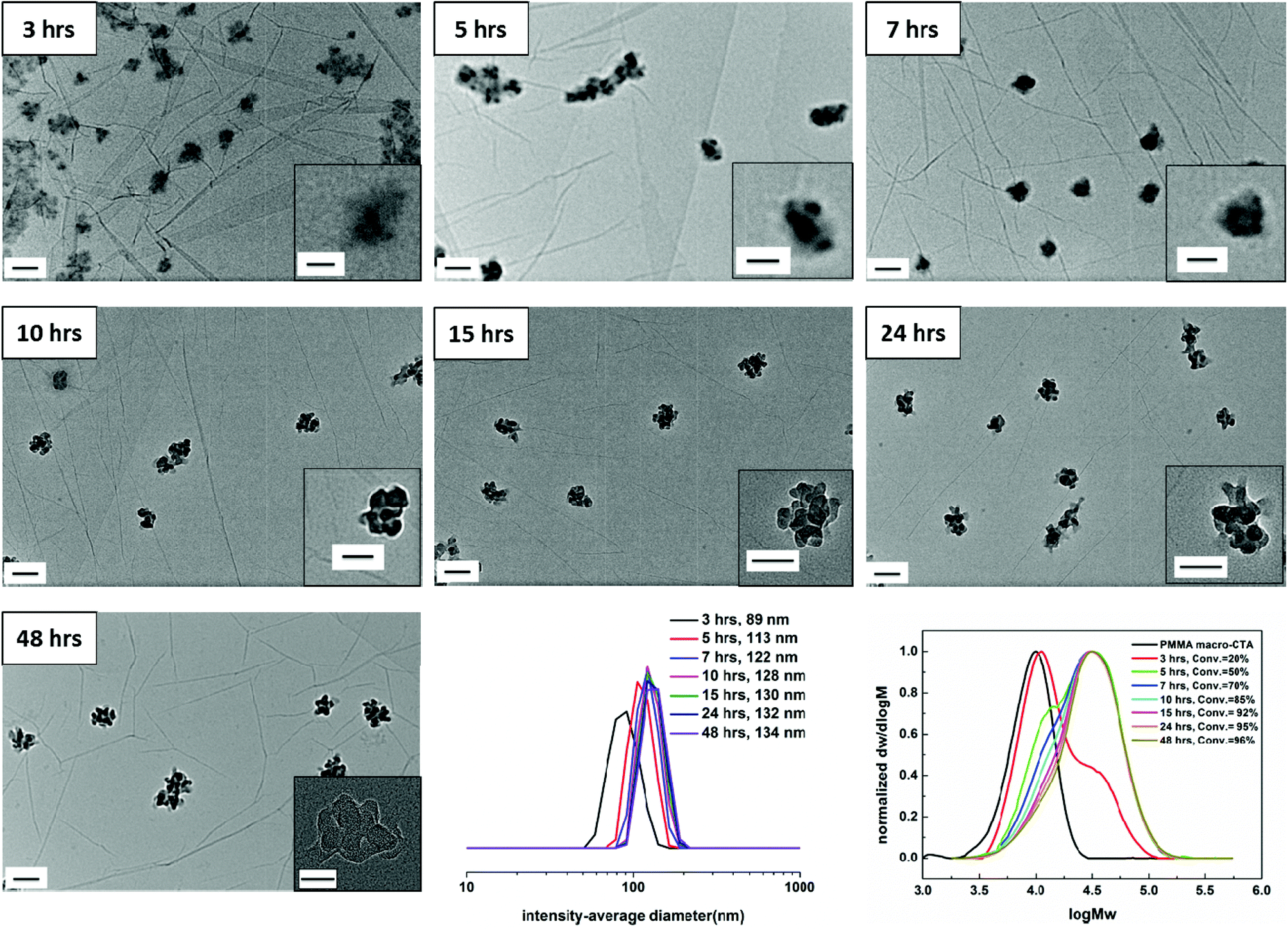 | ||
| Fig. 1 Evolution of the self-assembly by unstained TEM analysis, intensity-average diameter as determined by DLS and SEC traces (DMF as eluent, PMMA standards) with monomer conversion using a PMMA70 macro-CTA for the RAFT dispersion polymerization of a mixture of AMA and TMA in CHCl3 at 60 °C. The target diblock composition was PMMA70-b-(PAMA0.5-co-PTMA0.5)50, 3. Scale bar: 100 nm (inset: 50 nm). The inset image of 48 hours was taken by TEM at a higher magnification (150k). | ||
Morphologies induced by polymerization in CHCl3
To assess the evolution of morphology and size distribution with monomer conversion in this RAFT dispersion polymerization in CHCl3, the polymerization solution 3 was also periodically sampled for unstained dry-state TEM on graphene oxide (GO)46 and DLS analysis,50 the results of which are shown in Fig. 1. During the polymerization the mixture became translucent after approximately 3 hours (20% conversion) and as the polymerization proceeded, the mixture became more opalescent, but no precipitation was observed. It should be noted that no obvious color change was observed by visual inspection after 10 hours, which is also consistent with the high monomer conversion obtained after 10 hours. These observations suggest that self-assembly was induced in this system. Moreover, a small-angle neutron scattering (SANS) experiment was conducted at the end of the polymerization, which allows access to all morphologies available in solution.When monomer conversion was about 20% and a bimodal SEC trace is obtained (Fig. 1, SEC trace of 3 hours), close to spherical micelles with some evidence of internal structure (tiny dark dots inside) were observed with a hydrodynamic diameter (Dh) of 89 nm (Fig. 1, TEM and DLS of 3 hours). Increasing the conversion to 50% (Fig. 1, SEC trace of 5 hours) also resulted in spherical micelles with internal structure being formed (Fig. 1, TEM of 5 hours). It should be noted that at this conversion (50%) a bimodal SEC trace was still observed, but compared to the copolymers obtained at 3 hours, the relative ratio of the two populations was reversed (more polymers with high molecular weight were obtained at 5 hours). Larger spherical micelles were produced from the 7 hour sample and 70% monomer conversion was achieved (Fig. 1, 7 hours). Moreover, as mentioned above, the low molecular weight trace reduced further (Fig. 1, SEC trace of 7 hours). Once the conversion increased to 85%, when monodisperse diblocks were generally obtained (Fig. 1, SEC trace of 10 hours), popcorn-like structures were observed in the TEM (Fig. 1, TEM of 10 hours). There was no obvious morphology change observed with further increasing monomer conversion (Fig. 1, 15 hours, 24 hours, and 48 hours). The sample from 48 hours was also characterized by TEM at a higher magnification (150k), which is shown in the inset of Fig. 1, 48 hours. Internal structure is clearly observed and thus popcorn-like micelles were the final structure attained in this case. Moreover, the particles were treated with sonication for 1 hour to test their stability. No obvious size and morphology changes were observed, suggesting a high stability of the particles (Fig. S6†). SANS was conducted on the very concentrated liquid sample (20 mg mL−1), which was directly obtained after polymerization without further dilution. Unfortunately, models including a Guinier-Porod, a ‘Raspberry’, a ‘Fuzzy sphere’ and a ‘Polycore shell’ were unsuccessfully fitted. A fractal model with some dispersity for the subunits was used to determine some dimensions of the assembly (Fig. S7†).51 For a fractal object of fractal dimension d (i.e. its mass M scales with its size R according to M ∝ Rd), the scattering cross-section follows I(q) ∝ q−d. Thus it is possible to determine the fractal dimension at low q with a power law when plotting log(I(q)) versus log(q).52 A fractal dimension of 2.9 was found which correlates with the mass fractal and indicates a 3D-object was afforded.
To further study the morphologies induced by polymerization in CHCl3, a range of diblock copolymers PMMA70-b-(PAMAx-co-PTMAy)n (Table 1, polymer 2, 3, 4, and 5) were analyzed by TEM to assess their morphologies. Only spherical structures were observed in most cases when using CHCl3 as the solvent (Fig. 2). When chain extended with exclusively AMA, irregular but almost spherical micelles were observed (Fig. 2, 2 and 4). TEM images reveal that the sizes of particles increase significantly by performing higher targeted DP values for the core-forming block (Fig. 2, 2, DP is 50; Fig. 2, 4, DP is 100). This observation was also confirmed by DLS studies. It shows that the intensity-average diameters of particles were increased from 81 nm to 178 nm when the targeted DP of polymer of AMA (PAMA) block went from 50 to 100 (Fig. 2). No additional morphologies were obtained when further increasing the DP of PAMA block to 150 and 200 (Table S1 and Fig. S8 and S9†). However, the sizes of the spherical particles increased and the periphery of the particles became smoother with increasing length of PAMA blocks. Moreover, it is noticed that the sizes obtained from DLS are larger than that observed by TEM, which is due to the collapsed structure of the polymers in the dry-state. In the case of a mixture of AMA and TMA as monomers, spherical structures with rough surfaces were obtained (Fig. 2, 3 and 5). The particles grew bigger when a higher DP of the core-forming block was targeted. This size change was again witnessed by a combination of TEM and DLS. Particularly, the morphology of particles prepared by polymerization 4 and 5 is very similar to the structure, staggered lamella, which has been observed previously.53 In the reference this morphology is formed due to the strong π–π stacking interactions between side groups. In our study, there are strong hydrogen bonding interactions and π–π stacking interactions between nucleobase side groups, which might contribute to the formation of similar structures.
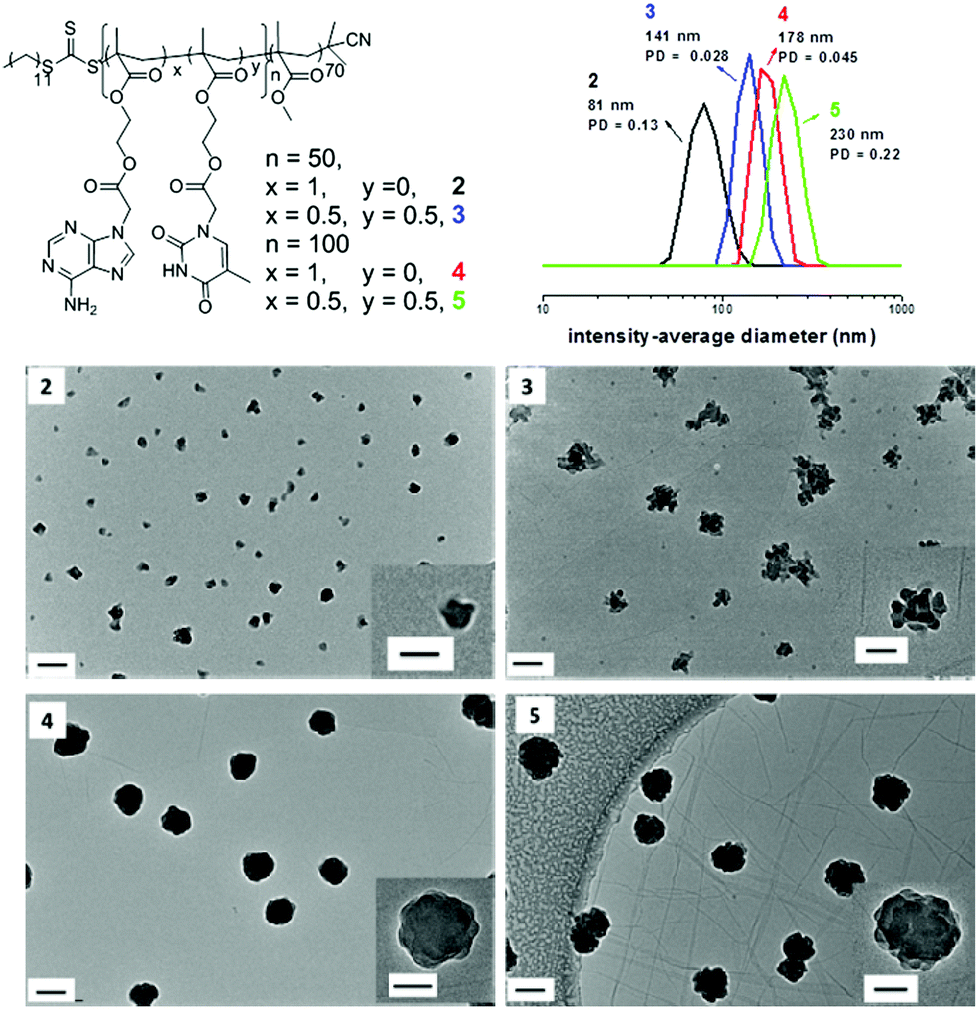 | ||
| Fig. 2 Representative TEM images of self-assemblies prepared by RAFT dispersion polymerization in CHCl3, their corresponding polymer structures and DLS particle size distributions of 2, 3, 4 and 5. Scale bar 100 nm (inset 50 nm). | ||
To further study the formation of this morphology, polymerizations were also performed in the presence of an aromatic solvent, anisole. Anisole is not a good solvent for the monomer AMA, therefore, mixtures of anisole and chloroform were selected as the solvents in this study. For the homopolymerization of AMA, the optimized conditions were a chloroform–anisole ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with a monomer concentration being the same as the one used in the polymerization of 4 (Fig. S10†). By TEM analysis, it was observed that staggered lamellae were still the major morphologies, however, a few small micelles were also observed. This observation indicates that the formation of staggered lamellae was interrupted by the addition of the aromatic solvent. This is to be expected as the aromatic solvent can competitively interact with adenine and thus affect the π–π stacking interactions between nucleobases (anisole is known to have an effect on interchain π–π stacking between conjugated polymers).54
:1 with a monomer concentration being the same as the one used in the polymerization of 4 (Fig. S10†). By TEM analysis, it was observed that staggered lamellae were still the major morphologies, however, a few small micelles were also observed. This observation indicates that the formation of staggered lamellae was interrupted by the addition of the aromatic solvent. This is to be expected as the aromatic solvent can competitively interact with adenine and thus affect the π–π stacking interactions between nucleobases (anisole is known to have an effect on interchain π–π stacking between conjugated polymers).54
In polymerizations where a mixture of AMA and TMA were used, mixtures of chloroform and anisole at different ratios were investigated (Table S2†). Precipitation instead of self-assembly was obtained when the ratio of chloroform to anisole was 1:1. However, when the ratio of chloroform to anisole was changed to 2:1, a mixture of aggregates of staggered lamellae and small sheet-like micelles were observed (Fig. S11†). The aggregates may be formed due to the poorer solubility of polymers in the presence of anisole and the formation of small sheet-like micelles may result from the interruption caused by anisole. Further variation of the ratio of chloroform to anisole to 5:1 also led to aggregates of staggered lamellae with some small micelles observed by TEM analysis (Fig. S12†). It was also noticed that there were less small micelles at a higher chloroform–anisole ratios. These results indicate that the formation of staggered lamellae can be affected by the presence of aromatic solvent, this further suggests that π–π stacking interactions between nucleobases could be one of the driving forces for the formation of staggered lamellae in this study. However, the exact reason for this behavior needs further investigation.
As observed above, in terms of a mixture of AMA and TMA as monomers, the micelles, prepared by polymerization 3, appear to be ‘popcorn-like’ structures. To further study the structures, AFM was carried out on the same grid which was used in TEM microscopy. From AFM images, ‘popcorn-like’ structures along with a few small spheres are clearly observed (Fig. S13†). Moreover, the heights of the large particles were measured using AFM to give a value around 80 nm (Fig. S13†), which fits well with the results shown from TEM and is close to the value from DLS. To eliminate the possibility that the popcorn-like structures were formed by simply aggregation or fusion of smaller micelles, which may result from the cooling process or a drying-effect, variable-temperature SLS and DLS were utilized to obtain more details about the popcorn structures. As these techniques are conducted on a liquid sample, no artifacts from TEM grid preparation can occur.
There was no size change observed by variable-temperature DLS (Fig. S14†). The number-average diameters of ‘popcorn-like’ particles were measured at different concentrations (0.032 mg mL−1 to 1 mg mL−1) and at variable temperatures (20 °C to 60 °C). It was found that at one temperature there was no obvious size change upon dilution. Meanwhile, no change in size was observed when a sample was heated from 20 °C to 60 °C and then cooled down from 60 °C to 20 °C. Furthermore, the aggregation numbers were carefully measured by SLS at both 20 °C and 50 °C, which are close to the temperatures used for microscopy sample preparation and polymerization, respectively. The concentrations of the sample were from 0.25 mg mL−1 to 0.031 mg mL−1. It was determined that there was no significant change on aggregation number observed from the results of SLS (Fig. S15 and S16,†Nagg = 29,000 at 20 °C and Nagg = 33,000 at 50 °C). P-ratio (Rg/Rh, ratio between radius of gyration and hydrodynamic radius) at both temperatures were calculated to be smaller than 1 (Rg/Rh = 0.81 at 20 °C and Rg/Rh = 0.79 at 50 °C), which indicates that the particle was a solid sphere at both temperatures. However, it should be noted that the calculated large aggregation numbers indicate the formation of complex spheres rather than simple core–shell structures. These results prove that the particles don't tend to fall apart when heating from room temperature to 50 °C and thus suggests that the ‘popcorn-like’ structure was formed during the polymerization and was not induced by cooling down or a drying effect upon analysis.
To further investigate the stabilities of the irregular particles, sizes of self-assemblies prepared by polymerization 2 and 5 were also measured at different temperatures by DLS (Fig. S17†). No size change was detected. This suggests that the particles formed in CHCl3 are very stable upon heating, although they appear to be irregular in shape as determined by TEM analysis. The stability of staggered lamellae 5 was also studied by sonication. No obvious size and morphology changes were observed upon sonication for 1 hour (Fig. S18†), showing that the staggered lamellae are highly stable due to the strong hydrogen bonding interactions. A few small sheetlike nanostructures were observed when the staggered lamellae were treated with sonication for 8 hours (Fig. S19†), which is consistent with previous observation in the literature.53
Kinetics of RAFT dispersion polymerization in 1,4-dioxane
The kinetics of RAFT dispersion polymerizations were also investigated in 1,4-dioxane. Monomer conversion data obtained by 1H NMR spectroscopy is shown in Fig. S20† for a target diblock copolymer 7, PMMA70-b-(PAMA0.5-co-PTMA0.5)50. Both monomers were incorporated at the same rate. It should be noted that polymerization conditions used for polymerization 7 were identical to polymerization 3 except for the choice of solvent. 80% of monomer conversion was obtained after 15 hours and conversion reached 90% after 24 hours. Compared to the result in CHCl3, polymerization in 1,4-dioxane is slightly slower (85% of monomer conversion after 10 hours in CHCl3). The evolution of SEC traces with monomer conversion is shown in Fig. 3. The molecular weight generally increases linearly with conversion and the dispersities remain narrow. Compared to the results obtained in CHCl3, monodisperse diblock copolymers were obtained at each conversion, which is consistent with previously reported systems in the literature.30 These results indicate that the RAFT dispersion polymerization of a mixture of AMA and TMA in 1,4-dioxane differs significantly to that in CHCl3.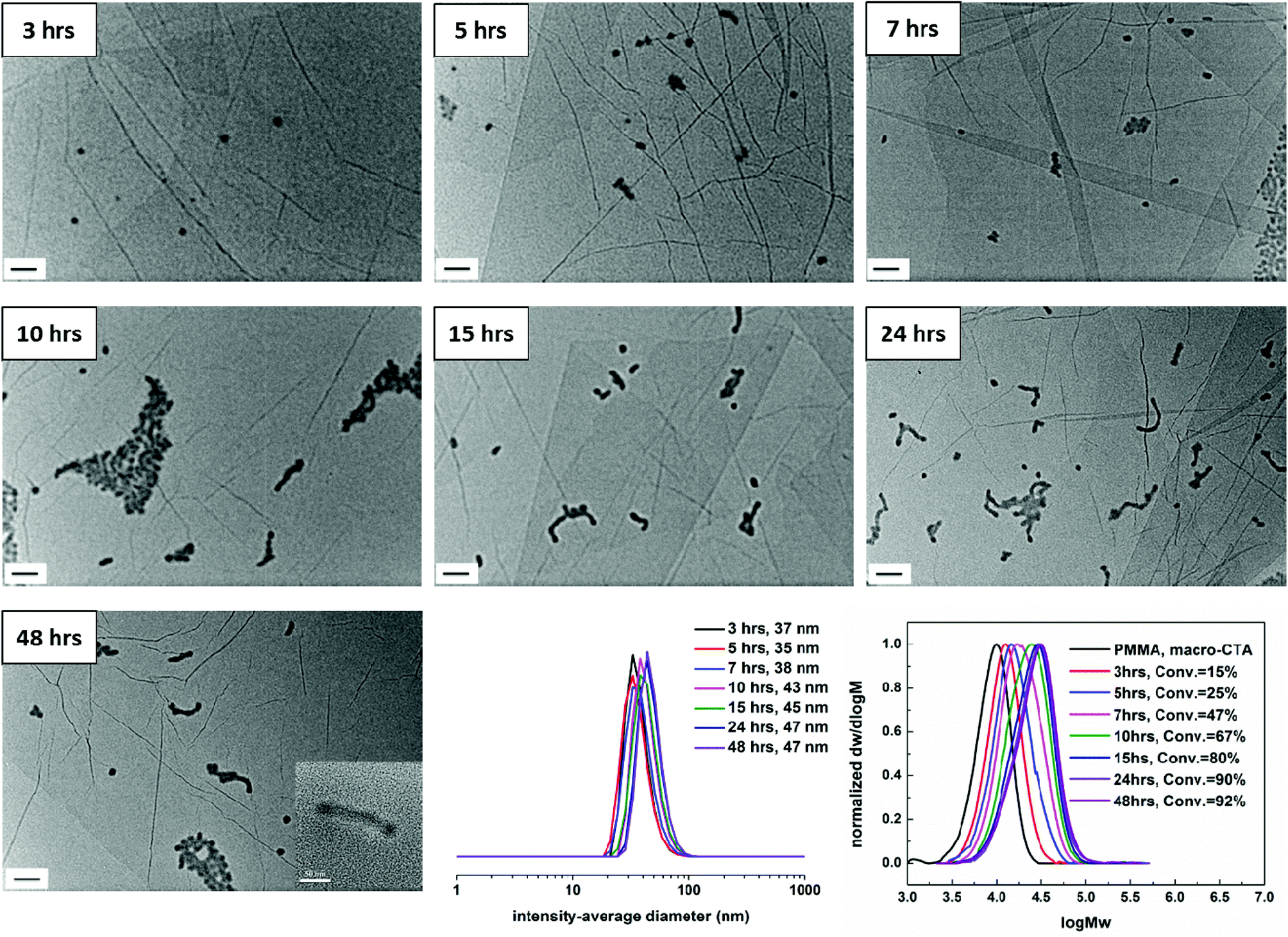 | ||
| Fig. 3 Evolution of the self-assembly by unstained TEM analysis, intensity-average diameter as determined by DLS and SEC traces (DMF as eluent, PMMA standards) with monomer conversion using a PMMA70 macro-CTA for the RAFT dispersion polymerization of a mixture of AMA and TMA in 1,4-dioxane at 60 °C. The target diblock composition was polymer 7, PMMA70-b-(PAMA-co-PTMA)50. Scale bar: 100 nm. The inset image of 48 hours was taken by TEM at a higher magnification (60k). | ||
Morphologies induced by polymerization in 1,4-dioxane
The polymerization mixture of 7 appeared clear until 7 hours (47% conversion), but became more opalescent as the polymerization proceeded. There was no obvious difference in color after 15 hours which was also consistent with monomer conversion (high conversion attained after 15 hours). To monitor the evolution of the morphology as the polymerization progressed, samples were also prepared for TEM analysis. The representative TEM images and their corresponding size distributions are shown in Fig. 3. At low monomer conversion when the polymerization solution looked clear (3 hours, 5 hours, and 7 hours), exclusively spherical micelles were observed (Fig. 3, 3 hours, 5 hours, and 7 hours). The hydrodynamic diameter of micelles was less than 40 nm by DLS. Further increase of the monomer conversion (10 hours, 67% conversion) led to a mixture of remaining spherical micelles and short wormlike micelles (Fig. 3, 10 hours). When high conversion was reached (80%, 15 hours), a mixture of some spherical micelles remained and longer wormlike micelles were formed (Fig. 3, 15 hours). There was no significant morphology change after 15 hours. A phase made of spherical and wormlike micelles was the final structure for polymerization 7 (Fig. 3, 24 hours and 48 hours). Small-angle X-ray scattering (SAXS) was performed on this sample to provide a global view of the resultant morphologies. The Guinier-Porod model was used to provide information on the shape of the scattering objects (Rg and anisotropy).55 High-q range was not taken into account for this model as it does not reproduce oscillations characteristic of form factors for monodisperse scattering objects. A dimension parameter of 0.52 was obtained, which suggests the presence of slightly elongated objects, or a mixture of spherical and elongated particles (Table S3 and Fig. S21†). From TEM analysis, the second option is more plausible. More complex structural models based on shape form factors of scattering objects were applied for SAXS analysis: a model for spherical objects with some dispersity (Polycore model56) and a model for cylindrical particles (Cylinder model57). The particles were assumed to have a uniform density for both models. Both the cylinder and the sphere models did not provide high quality fits, thus a linear combination of these two models (named the Sum model) was created to take into account the two morphologies present in solution as seen by TEM (Fig. S21†). These results thus confirm the hypothesis we made about the results of the Guinier-Porod fit: both spherical and cylindrical micelles coexist in solution. The spheres have a similar radius to the cylinders (Table S4†). Owing to the fact that the SAXS data were normalized, the scale parameter given by the models is equal to the volume fraction, and thus it is possible to estimate the number ratio between cylinders and spheres (Table S4†). It was determined that the volume fraction of cylindrical micelles is bigger than that of the spherical objects but by number there are more spheres than cylinders in solution. The evolution of morphology also indicates that the wormlike micelles observed were formed by the fusion of the spherical micelles, which is consistent with the reported mechanism of sphere-to-worm transitions.29 The possible reason for this formation is that the relatively high polymer curvature results in a lower stabilizer chain density and thus less effective steric stabilization at the cylinder-ends, which further induces the fusion. Furthermore, compared to polymerization 3 which was performed in CHCl3, the self-assembled structures formed in 1,4-dioxane were different (popcorn-like structures were formed in CHCl3). This further suggests that the RAFT dispersion polymerization of a mixture of AMA and TMA in 1,4-dioxane is significantly different to the same polymerization in CHCl3. In other words, solvent choice plays an important role on morphologies formed by RAFT dispersion polymerization of nucleobase-containing monomers.To further study the morphologies induced by polymerization in 1,4-dioxane, a range of diblock copolymers PMMA70-b-(PAMAx-co-PTMAy)n (Table 1, polymer 6–11) prepared in 1,4-dioxane were analyzed by TEM to assess their morphologies. Representative TEM images are shown in Fig. 4. Higher order morphologies were observed in this system than in CHCl3. AFM was also utilized to further assess these high order structures. In particular, height information of these structures could be attained accurately as well as insights regarding the morphologies. SAXS experiments were also performed on some samples to provide more details on the resultant morphologies in solution.
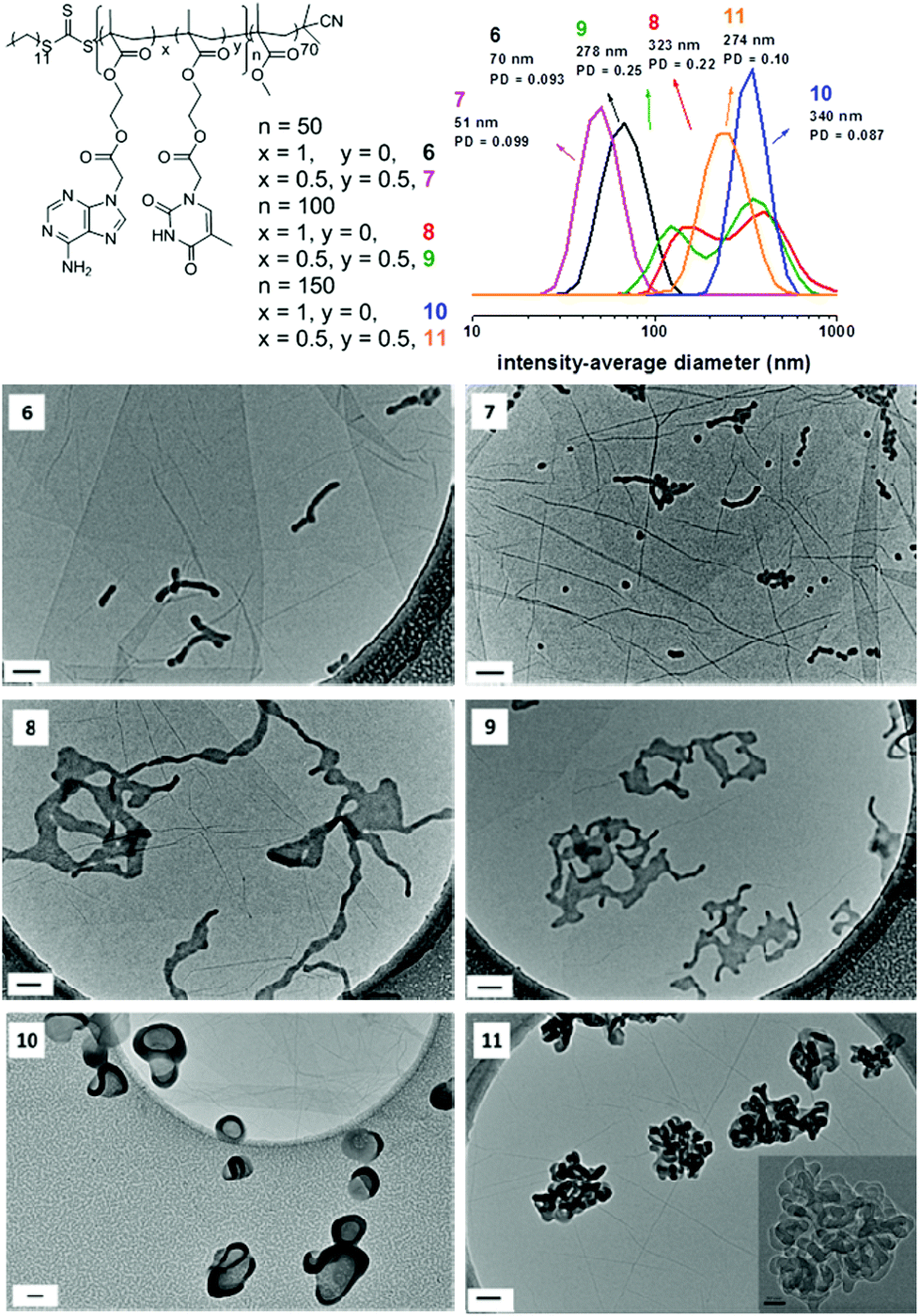 | ||
| Fig. 4 Representative TEM images of self-assemblies prepared by RAFT dispersion polymerization in 1,4-dioxane, their corresponding polymer structures and DLS particle size distributions of 6, 7, 8, 9, 10 and 11. Scale bar: 100 nm (inset 50 nm). The inset of 11 was taken by TEM with a higher magnification of 50k. | ||
When only AMA was used as the monomer, cylinders along with some remaining spherical micelles were observed by TEM in the case of nucleobase-containing block with a target DP of 50 (Fig. 4, 6). The lengths of the cylinders are about 150 nm as estimated from TEM images. Moreover, the widths of cylinders are very close to the diameter of the spheres. On close inspection of these TEM images, the cylinders growth in these polymerizations appears to result from the fusion of spherical micelles. This mechanism of cylinder formation has been previously reported as mentioned above.29,30 These morphologies could also be clearly observed by AFM. The heights of cylinders are about 15 nm for polymerization 6, which is also very consistent with the heights of the spheres formed in this system (Fig. S22†). SAXS was also performed on this sample and the analysis was conducted in the same way as for polymer 7, which further confirmed the results obtained by TEM and AFM: a mixed phase of cylinders (length of 158 nm, radius of 15 nm) and spheres (radius of 18 nm) with a bigger volume fraction of cylinders compared to the spheres (Tables S3, S4 and Fig. S23†). This observation further suggests that cylinders were formed by the fusion of spherical micelles. Targeting a DP of 100 led to lamellae along with ‘tentacles’. This morphology has been previously reported and called the octopus-like morphology (Fig. 4, 8).29 The ‘main-body’ of the octopus (lamellae) looks relatively flat when observed by TEM. AFM was also conducted to allow access to more details about this structure. The heights of the structures were measured to give a value of 15 nm (Fig. S24†). Furthermore, the ‘main-bodies’ of the octopus appear to be of similar height, which further proves the formation of flat lamellae. This octopus-like morphology is considered as an intrinsic intermediate morphology between worms and bilayers during the process of polymerization.29 Therefore, ‘jellyfish’ morphology or vesicles would be expected to be developed as a result of further increase of the length of the nucleobase-containing blocks. However, different morphologies were unexpectedly observed when the targeted DP of the core-forming block was increased to 150. The morphologies were firstly observed by TEM analysis. The octopus ‘tentacles’ underwent fusion and completely disappeared. Lamellae which were either partially wrapped-up or slightly twisted were observed (Fig. 4, 10). It seems that a vesicle phase started to form but was slightly disrupted. AFM was also performed to characterize the new morphologies. For polymerization 10, the height was estimated to be about 100 nm to 250 nm (Fig. S25†). It is much higher than the flat lamellae which were observed above (Fig. S24,† the height is ca. 15 nm). The analysis of heights of the aggregations suggests that the lamellae are no longer flat. SLS was conducted on this sample to further study its morphology in solution. As the scatterers are very large (q × R > 1 over the whole q-range investigated), the Zimm formulation is not applicable. Therefore, the shape of scatters is determined by the plot of q-dependency of R/Kc.58 The particle fractal dimension determined from the log–log plot of R/Kc as a function of q is 2.5 (Fig. S26†), which is not a reported value (2 is for thin disk and 3 is for 3D-objects with smooth surfaces), which indicates a novel structure formed. It further suggests that structures might be partially enclosed.
When a mixture of AMA and TMA were used as monomers, the sample exhibited a mixed sphere/cylinder phase (Fig. 4, 7) when the target DP was 50, the morphology of which was also observed in the kinetics study (Fig. 3). The lengths of the cylinders are about 100 nm and the diameters of spheres are around 20 nm. Estimated by AFM, the cylinders’ heights are around 15 nm (Fig. S27†), which is also consistent with the size of the spheres and further proves the fusion mechanism of sphere-to-worm transition. Similar to the case when exclusively AMA was used, targeting a DP of 100 generated lamellae along with ‘tentacles’ (Fig. 4, 9). AFM was also conducted to allow access to more details about this structure. The heights of the structures were measured to give a value of 15 nm (Fig. S28†). Furthermore, the ‘main-bodies’ of the octopuses were also confirmed to be flat by analyzing the AFM height image. SAXS analysis confirmed the formation of lamellae with “tentacles”: a Guinier-Porod fit gave a dimension variable value of 1.59, which indicates either the presence of morphologies between rods and plates or the presence of a mixture of these two morphologies (Table S3 and Fig. S29†). Detailed modeling was then carried out to obtain more information by using a linear sum of a dilute lamellar form factor model59,60 with various cylinder models: no dispersity, dispersity on the length or dispersity on the radius57 (Fig. S29 and Table S5† for the three sum models). Better data fit was obtained with the model having some dispersity on the radius, as previously observed for cylindrical structures.61,62 The sum model resulted in a mixture of lamellae with a thickness of 20 nm (slightly bigger than by TEM and AFM, which is often the case as SAXS is performed in solution and TEM and AFM in a dried state) and cylinders of 80 nm of length and 12 nm of radius (Fig. S29†). Further increasing the target DP to 150 led to a new structure. High resolution TEM was used to obtain a clear image of this structure (Fig. 4, inset of 11). Compared to the observation in the case of the homopolymerization of AMA, deeply twisted lamellas/cylinders were obtained. A ‘jellyfish’ morphology or pure vesicle phase was also not produced in this system. AFM was also performed to characterize the new morphologies. The heights of aggregations prepared by polymerization 11 were analyzed and the average height is about 150 nm, which is higher than the mono-lamella. Knot-like structures were also observed (Fig. S30†). SAXS analysis performed on this sample did not allow a proper Guinier-Porod fit which could indicate the presence of too many different morphologies or morphologies which are not recognized by the Guinier-Porod model. A fractal model with disperse cylinders as building blocks was used to fit the experimental data (Fig. S31†). A fractal dimension of 3.3 was found, which indicates that a 3D-object was formed. Unfortunately, this model does not reflect the twisted cylinders. Generally speaking, this observation suggests that more complex and higher order structures were generated in these systems.
DLS was also carried out to analyze the morphologies. The results of intensity-average diameters were summarized (Fig. 4). They show that by increasing the DP of the core-forming blocks, the sizes of the self-assemblies were significantly increased. It should be noted that due to the high order morphologies obtained in these systems, the sizes obtained from DLS are relative values (for DLS, the assumption that the particles are spherical is made).63 However, as CONTIN analysis is able to detect multiple populations in solution and obtain polydispersity information, the results from DLS are still valuable even if the structures are not spherical.
Discussion
As mentioned in the monomer study section above, both CHCl3 and 1,4-dioxane are low polarity solvents and support nucleobase interactions among polymers.48,49 However, different polymerization and self-assembly behavior were observed in these two media when using nucleobase-containing monomers to undergo RAFT dispersion polymerization. Moreover, a diversity of morphologies was observed in this nucleobase-containing system. We hypothesize that a combination of complementary nucleobases interactions and solubility (determined by a balance of intermolecular forces between solvent and solute) leads to this novel behavior.Although H-bonding interactions between adenine and thymine exist in both solvents as reported,48,49 the difference in solubility of the nucleobases also needs to be taken into account. In other words, there is a competition of nucleobase-nucleobase interactions and intermolecular forces between nucleobases and solvent (nucleobase solubility). Moreover, we observe that the solubility of nucleobases in 1,4-dioxane is relatively higher than in CHCl3, which indicates a stronger intermolecular force between the nucleobase and 1,4-dioxane than CHCl3. This is supported by the observation that monomers (particularly AMA) have better solubility in 1,4-dioxane than in CHCl3, which is assessed by the visual inspection that at room temperature AMA is fully soluble in 1,4-dioxane at 20 mg mL−1, but not in CHCl3. Another observation is that the homopolymer of TMA with a low DP (ca. 20) is soluble in 1,4-dioxane but not in CHCl3. Therefore, in CHCl3 the nucleobase interactions are much stronger and the intermolecular force between nucleobases and solvent (solubility of nucleobase) can be ignored. However, in 1,4-dioxane, the effect of intermolecular force between nucleobases and solvent (solubility of the nucleobase) in 1,4-dioxane can't be ignored, and even overtakes the importance of nucleobase interactions for monomers or polymer with a low DP due to the relatively low concentration of nucleobase compared to the amount of solvent. However, with further formation of polymers or increase of the local concentration of nucleobases, the nucleobase interactions become more and more important.
Based on the observations and discussion above, we hypothesize that compared to 1,4-dioxane, the solubility of nucleobase-containing polymers in CHCl3 is poorer and thus leads to a lower critical micelle degree of polymerization (CMDP), which is a key parameter in the process of dispersion polymerization and can be influenced greatly by many factors, including the solvent used.31,64–66 Moreover, as nucleobase interactions are the main interactions among monomers and polymers in CHCl3, frozen core-forming nucleobase-containing blocks are induced above the CMDP. Therefore, the chain propagation can be affected, which results in different polymerization rates as observed in the kinetics study in CHCl3. Furthermore, as a result of the frozen core formed, morphology transitions can't occur during the process of dispersion polymerization and hence only spheres were observed in CHCl3.
In contrast, a higher CMDP and relatively mobile polymer growing chains are expected in 1,4-dioxane due to a better solubility of the core-forming polymers, including both homopolymers of AMA and copolymers of AMA and TMA. These could lead to a controlled chain extension as observed in the kinetics study and also allow access to ergodic aggregates during dispersion polymerization. However, with increasing length of the nucleobase-containing blocks or local concentration of the nucleobases, the nucleobase interactions appear to be the main interactions and the growing polymer chains are no longer mobile. As a result, the morphology transitions are hindered and hence twisted structures are generated instead of vesicles (Fig. 4, 10 and 11). Complementary nucleobase interactions between adenine and thymine are stronger than adenine–adenine interactions.67,68 Therefore, a slightly folded structure was observed for the polymerization of 10, while a deeply twisted structure was afforded in the polymerization of 11.
Conclusions
In conclusion, copolymers containing nucleobases were synthesized by RAFT dispersion polymerization and the obtained polymers were well-controlled in terms of molecular weight and molecular weight distributions. Moreover, self-assembly was induced during the RAFT dispersion polymerizations in CHCl3 and 1,4-dioxane, using monomers containing adenine and thymine. Different morphological evolutions were observed in these two media. Only spheres were observed in CHCl3 with increasing size upon increasing the length of the core-forming blocks. In contrast, morphology transitions from spheres through cylinders to lamellae were observed in 1,4-dioxane due to a better chain mobility. However, the transitions were disrupted by the presence of strong nucleobase interactions with increasing length of nucleobase-containing polymers. This study provides insights into understanding the properties of nucleobase-containing polymers and also offers a simple method to prepare self-assemblies containing nucleobases. By simply varying the corona-forming block into a biological compatible polymer, this route is expected to have potential applications in drug delivery and other biological aspects.Acknowledgements
The authors thank the University of Warwick, NSF and EPSRC for research funding. The SECs used in this research were obtained though Birmingham Science City: Innovative Uses for Advanced Materials in the Modern World (West Midlands Centre for Advanced Materials Project 2), with support from Advantage West Midlands (AWM), and partly funded by the European Regional Development Fund (ERDF). Mr Daniel Wright is acknowledged for help with refractive index measurements and helpful discussions about SLS analysis. Dr Stephen King (Rutherford Appleton Laboratory, Oxford, UK) is gratefully acknowledged for carrying out the SANS measurements. Dr Ana M. Sanchez thanks the Science City Research Alliance and the HEFCE Strategic Development Fund for funding support. Dr Neil Wilson and Mr Mark Skilbeck are thanked for assistance with AFM microscopy.Notes and references
- Y. Tao, K. Satoh and M. Kamigaito, Macromol. Rapid Commun., 2011, 32, 226–232 CrossRef CAS PubMed.
- K. Takemoto, M. Akashi and Y. Inaki, J. Polym. Sci., Polym. Chem. Ed., 1974, 12, 1861–1869 CrossRef CAS.
- M. Akashi, Y. Kita, Y. Inaki and K. Takemoto, J. Polym. Sci., Polym. Chem. Ed., 1979, 17, 301–312 CrossRef CAS.
- Y. Kang, A. Lu, A. Ellington, M. C. Jewett and R. K. O'Reilly, ACS Macro Lett., 2013, 2, 581–586 CrossRef CAS.
- A. Khan, D. M. Haddleton, M. J. Hannon, D. Kukulj and A. Marsh, Macromolecules, 1999, 32, 6560–6564 CrossRef CAS.
- C. R. South and M. Weck, Macromolecules, 2007, 40, 1386–1394 CrossRef CAS.
- P. K. Lo and H. F. Sleiman, J. Am. Chem. Soc., 2009, 131, 4182–4183 CrossRef CAS PubMed.
- F. Ilhan, T. H. Galow, M. Gray, G. Clavier and V. M. Rotello, J. Am. Chem. Soc., 2000, 122, 5895–5896 CrossRef CAS.
- H. S. Bazzi and H. F. Sleiman, Macromolecules, 2002, 35, 9617–9620 CrossRef CAS.
- R. J. Thibault, P. J. Hotchkiss, M. Gray and V. M. Rotello, J. Am. Chem. Soc., 2003, 125, 11249–11252 CrossRef CAS PubMed.
- H. J. Spijker, A. J. Dirks and J. C. M. van Hest, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4242–4250 CrossRef CAS.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS.
- R. McHale, J. P. Patterson, P. B. Zetterlund and R. K. O'Reilly, Nat. Chem., 2012, 4, 491–497 CrossRef CAS PubMed.
- R. McHale and R. K. O'Reilly, Macromolecules, 2012, 45, 7665–7675 CrossRef CAS.
- J. K. Oh, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6983–7001 CrossRef CAS.
- J. Jennings, M. Beija, J. T. Kennon, H. Willcock, R. K. O'Reilly, S. Rimmer and S. M. Howdle, Macromolecules, 2013, 46, 6843–6851 CrossRef CAS.
- M. F. Cunningham, Prog. Polym. Sci., 2008, 33, 365–398 CrossRef CAS PubMed.
- P. B. Zetterlund, F. Aldabbagh and M. Okubo, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3711–3728 CrossRef CAS.
- V. Ladmiral, M. Semsarilar, I. Canton and S. P. Armes, J. Am. Chem. Soc., 2013, 135, 13574–13581 CrossRef CAS PubMed.
- X. W. Zhang, F. Boisson, O. Colombani, C. Chassenieux and B. Charleux, Macromolecules, 2014, 47, 51–60 CrossRef CAS.
- Y. Luo, X. Wang, Y. Zhu, B.-G. Li and S. Zhu, Macromolecules, 2010, 43, 7472–7481 CrossRef CAS.
- Y. Kagawa, H. Minami, M. Okubo and J. Zhou, Polymer, 2005, 46, 1045–1049 CrossRef CAS PubMed.
- Y. Kitayama, Y. Kagawa, H. Minami and M. Okubo, Langmuir, 2010, 26, 7029–7034 CrossRef CAS PubMed.
- J. Nicolas, A.-V. Ruzette, C. Farcet, P. Gérard, S. Magnet and B. Charleux, Polymer, 2007, 48, 7029–7040 CrossRef CAS PubMed.
- C. Farcet, B. Charleux and R. Pirri, Macromolecules, 2001, 34, 3823–3826 CrossRef CAS.
- Y. Kitayama, K. Kishida, H. Minami and M. Okubo, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1991–1996 CrossRef CAS.
- Y. Kitayama, M. Yorizane, H. Minami and M. Okubo, Polym. Chem., 2012, 3, 1394–1398 RSC.
- J. Tonnar, P. Lacroix-Desmazes and B. Boutevin, Macromolecules, 2007, 40, 6076–6081 CrossRef CAS.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed.
- L. A. Fielding, M. J. Derry, V. Ladmiral, J. Rosselgong, A. M. Rodrigues, L. P. D. Ratcliffe, S. Sugihara and S. P. Armes, Chem. Sci., 2013, 4, 2081–2087 RSC.
- J.-T. Sun, C.-Y. Hong and C.-Y. Pan, Soft Matter, 2012, 8, 7753–7767 RSC.
- C.-Q. Huang and C.-Y. Pan, Polymer, 2010, 51, 5115–5121 CrossRef CAS PubMed.
- X. Zhang, S. p. Boissé, W. Zhang, P. Beaunier, F. D'Agosto, J. Rieger and B. Charleux, Macromolecules, 2011, 44, 4149–4158 CrossRef CAS.
- W. Zhang, F. D'Agosto, P.-Y. Dugas, J. Rieger and B. Charleux, Polymer, 2013, 54, 2011–2019 CrossRef CAS PubMed.
- Z. Jia, V. A. Bobrin, N. P. Truong, M. Gillard and M. J. Monteiro, J. Am. Chem. Soc., 2014, 136, 5824–5827 CrossRef CAS PubMed.
- H. A. Zayas, N. P. Truong, D. Valade, Z. Jia and M. J. Monteiro, Polym. Chem., 2013, 4, 592–599 RSC.
- B. Karagoz, L. Esser, H. T. Duong, J. S. Basuki, C. Boyer and T. P. Davis, Polym. Chem., 2014, 5, 350–355 RSC.
- B. Karagoz, C. Boyer and T. P. Davis, Macromol. Rapid Commun., 2014, 35, 417–421 CrossRef CAS PubMed.
- Y. Su, X. Xiao, S. Li, M. Dan, X. Wang and W. Zhang, Polym. Chem., 2014, 5, 578–587 RSC.
- Y. Pei and A. B. Lowe, Polym. Chem., 2014, 5, 2342–2351 RSC.
- W. Cai, W. Wan, C. Hong, C. Huang and C. Pan, Soft Matter, 2010, 6, 5554–5561 RSC.
- S. Sugihara, S. P. Armes, A. Blanazs and A. L. Lewis, Soft Matter, 2011, 7, 10787–10793 RSC.
- P. Chambon, A. Blanazs, G. Battaglia and S. P. Armes, Macromolecules, 2012, 45, 5081–5090 CrossRef CAS.
- W.-J. Zhang, C.-Y. Hong and C.-Y. Pan, Macromolecules, 2014, 47, 1664–1671 CrossRef CAS.
- Y. K. Chong, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2007, 40, 4446–4455 CrossRef CAS.
- J. P. Patterson, A. M. Sanchez, N. Petzetakis, T. P. Smart, T. H. Epps III, I. Portman, N. R. Wilson and R. K. O'Reilly, Soft Matter, 2012, 8, 3322–3328 RSC.
- S. Kline, J. Appl. Crystallogr., 2006, 39, 895–900 CrossRef CAS.
- J.-F. Lutz, A. F. Thünemann and K. Rurack, Macromolecules, 2005, 38, 8124–8126 CrossRef CAS.
- J.-F. Lutz, A. F. Thünemann and R. Nehring, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4805–4818 CrossRef CAS.
- J. P. Patterson, M. P. Robin, C. Chassenieux, O. Colombani and R. K. O'Reilly, Chem. Soc. Rev., 2014, 43, 2412–2425 RSC.
- J. Teixeira, J. Appl. Crystallogr., 1988, 21, 781–785 CrossRef.
- G. Beaucage, J. Appl. Crystallogr., 1995, 28, 717–728 CrossRef CAS.
- X. Hu, J. Hu, J. Tian, Z. Ge, G. Zhang, K. Luo and S. Liu, J. Am. Chem. Soc., 2013, 135, 17617–17629 CrossRef CAS PubMed.
- M. He, L. Zhao, J. Wang, W. Han, Y. Yang, F. Qiu and Z. Lin, ACS Nano, 2010, 4, 3241–3247 CrossRef CAS PubMed.
- B. Hammouda, J. Appl. Crystallogr., 2010, 43, 716–719 CrossRef CAS.
- P. Bartlett and R. H. Ottewill, J. Chem. Phys., 1992, 96, 3306–3318 CrossRef CAS PubMed.
- A. Guinier and G. Fournet, Small-angle scattering of X-rays, Wiley, 1955 Search PubMed.
- S. Catrouillet, C. Fonteneau, L. Bouteiller, N. Delorme, E. Nicol, T. Nicolai, S. Pensec and O. Colombani, Macromolecules, 2013, 46, 7911–7919 CrossRef CAS.
- F. Nallet, R. Laversanne and D. Roux, J. Phys. II, 1993, 3, 487–502 CrossRef CAS.
- J. Berghausen, J. Zipfel, P. Lindner and W. Richtering, J. Phys. Chem. B, 2001, 105, 11081–11088 CrossRef CAS.
- L. Sun, N. Petzetakis, A. Pitto-Barry, T. L. Schiller, N. Kirby, D. J. Keddie, B. J. Boyd, R. K. O'Reilly and A. P. Dove, Macromolecules, 2013, 46, 9074–9082 CrossRef CAS.
- A. Pitto-Barry, N. Kirby, A. P. Dove and R. K. O'Reilly, Polym. Chem., 2014, 5, 1427–1436 RSC.
- N. Petzetakis, D. Walker, A. P. Dove and R. K. O'Reilly, Soft Matter, 2012, 8, 7408–7414 RSC.
- G. Zheng and C. Pan, Macromolecules, 2005, 39, 95–102 CrossRef.
- G. Zheng, Q. Zheng and C. Pan, Macromol. Chem. Phys., 2006, 207, 216–223 CrossRef CAS.
- W. Ji, J. Yan, E. Chen, Z. Li and D. Liang, Macromolecules, 2008, 41, 4914–4919 CrossRef CAS.
- B. D. Mather, M. B. Baker, F. L. Beyer, M. A. G. Berg, M. D. Green and T. E. Long, Macromolecules, 2007, 40, 6834–6845 CrossRef CAS.
- Y. Kyogoku, R. C. Lord and A. Rich, Proc. Natl. Acad. Sci. U. S. A., 1967, 57, 250–257 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra, kinetics experiments, SEC, AFM, SANS, SAXS, SLS data. See DOI: 10.1039/c4py01074d |
| This journal is © The Royal Society of Chemistry 2015 |