
Phosphine-free ruthenium-arene complex for low temperature one-pot catalytic conversion of aldehydes to primary amides in water†
Deepika
Tyagi‡
,
Rohit K.
Rai‡
,
Ambikesh D.
Dwivedi
,
Shaikh M.
Mobin
and
Sanjay K.
Singh
*
Discipline of Chemistry, School of Basic Sciences, Indian Institute of Technology (IIT) Indore, Khandwa Road, Indore 452 017, India. E-mail: sksingh@iiti.ac.in; Tel: +91 731 2438 730
First published on 11th November 2014
Abstract
A highly active phosphine-free ruthenium-arene complex, [(η6-C6H6)RuCl2(C6H5NH2)], exhibits excellent catalytic performance for a one-pot conversion of aldehydes to primary amides at low temperature (60 °C), in water and without any inert gas protection. The reported catalyst performed exceptionally well for a huge range of aldehydes, including aromatic, heteroaromatic, aliphatic and conjugated systems, with a high tolerance for other functional groups. The development of such highly active catalysts using simple reagents will offer new opportunities for the development of improved phosphine-free catalytic systems for this and other related catalytic reactions.
Introduction
Amide bonds are not only abundantly present in nature, as a major component of proteins and natural products, but they are also key functional groups in synthetic organic chemistry owing to their numerous applications in pharmaceuticals and other industries.1–3 The classical method for amide synthesis is remarkably general, where an amide linkage is formed by coupling carboxylic acids and their derivatives with amines.4 However, these reactions are regarded as an expensive and wasteful method due to the use of toxic and/or expensive reagents, poor functional group tolerance, and the generation of waste which may cause potential environmental threats.4 Not surprisingly, the development of an atom efficient and waste-free process for amide synthesis using environment friendly components is considered as the top challenge for organic chemistry.2 Therefore, improved methodologies for amide bond formation are in great demand. In this context, aldehydes, which are easily available from nature, have been identified as important starting materials. Significant efforts have been made to develop a one-pot methodology for the synthesis of amides from aldehydes, via metal-catalysed rearrangement of an aldoxime.3 In recent years, several such catalytic reactions have been accomplished using various catalytic systems, e.g. Ru, Rh, Ir, Pd, Cu, Fe, Zn, In and Sc, that facilitate an atom-economical approach to amide formation.5–11Moreover, the ability to perform organic reactions in water, which is cheap, safe and most importantly environment friendly, is in high demand and essentially required.12 Notably, two recent reports have demonstrated a synthesis of amides from aldoxime rearrangements in water, using metal-arene complexes [(η6-C6Me6)RuCl2{P(NMe2)3}] and [(η5-C5Me5)Ir(H2O)3][OTf3]2 (shown in Fig. 1).5,8 Cadierno et al. demonstrated that the complex [(η6-C6Me6)RuCl2{P(NMe2)3}] provides high yields of amides, through aldoxime rearrangement in water, but requires a high reaction temperature (100 °C) and protection by an inert atmosphere.5 However, the iridium complex, [(η5-C5Me5)Ir(H2O)3][OTf3]2, reported by Li et al. exhibited high activity for amide formation in water without inert gas protection, but it also operates only at a very high temperature (110 °C).8
 | ||
| Fig. 1 Metal-arene complex-based active catalysts for amide synthesis from aldoxime rearrangement in water. | ||
Therefore, in light of the above, we focused our efforts on developing a highly efficient catalyst without phosphine ligands for a one-pot synthesis of primary amides from aldehydes at a lower reaction temperature, under aqueous-aerobic conditions. Here, we report a highly efficient and selective catalyst based on an arene-ruthenium complex with a readily available aniline ligand, [(η6-arene)RuCl2(C6H5NH2)] (η6-arene = C6H6 ([Ru]-2a) and C10H14 ([Ru]-2b)) (Fig. 1), for the one-pot conversion of aldehydes with a broad range of aromatic, heteroaromatic, aliphatic and conjugated systems to amides in water, without inert gas protection, and most importantly at a lower reaction temperature (60 °C).
Experimental
Materials and instrumentation
All reactions were performed without inert gas protection, using chemicals of high purity purchased from Aldrich and Alfa Aesar. Ruthenium arene precursors [{(η6-C6H6)RuCl2}2] and [{(η6-C10H14)RuCl2}2] were synthesized according to the literature procedures.13,14 The catalytic reactions were monitored using a thin layer chromatography (TLC) method. 1H NMR (400 MHz), 13C NMR (100 MHz), and 19F NMR (376.5 MHz) spectra were recorded at 298 K using CDCl3 or DMSO-d6 as the solvent on a Bruker Avance 400 spectrometer. Tetramethylsilane (TMS) was used as an external standard and the chemical shifts in ppm are reported relative to the center of the singlet at 7.26 ppm for CDCl3 and 2.50 ppm for DMSO-d6 in 1H NMR, and to the center of the triplet at 77.0 ppm for CDCl3 and 39.50 ppm for DMSO-d6 in 13C NMR. Coupling constants, J values, are reported in Hertz (Hz), and the splitting patterns are designated as follows: s, singlet; d, doublet; t, triplet; m, multiplet; br., broad. Single-crystal X-ray structural studies of complex [Ru]-2a were carried out using Agilent Technologies Supernova CCD system. Elemental analysis was carried out using a Thermo Scientific FLASH 200 elemental analyzer. High-resolution mass spectra (HRMS) were recorded on a micrOTF-Q II mass spectrometer.General procedure for the synthesis of the complexes
General procedure for the catalytic synthesis of amides from aldehydes
To a stirred solution of ruthenium(II) complex [(η6-C10H14)RuCl2(C6H5NH2)] [Ru]-2a (0.017 g, 0.05 mmol, 5 mol%, dissolved in 3 ml of water) in a round bottom flask, was added a primary aldehyde (1 mmol), hydroxylamine hydrochloride (0.090 g, 1.3 mmol), NaHCO3 (0.109 g, 1.3 mmol) and 5 ml of water (or a water–methanol mixture with a ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) in the case of ferrocenecarboxaldehyde) under open air conditions, and the reaction mixture was stirred at 60 °C for 5–24 hours. After this time, the reaction mixture was extracted using dichloromethane at least 5–6 times, the organic layer from the extraction was dried with sodium sulfate to remove moisture and then the solvent was evaporated under reduced pressure, which gave the corresponding amide. The conversion and selectivity for the corresponding amide were determined using the 1H, 13C and 19F NMR spectra.
:1 (v/v) in the case of ferrocenecarboxaldehyde) under open air conditions, and the reaction mixture was stirred at 60 °C for 5–24 hours. After this time, the reaction mixture was extracted using dichloromethane at least 5–6 times, the organic layer from the extraction was dried with sodium sulfate to remove moisture and then the solvent was evaporated under reduced pressure, which gave the corresponding amide. The conversion and selectivity for the corresponding amide were determined using the 1H, 13C and 19F NMR spectra.
Single-crystal X-ray diffraction studies
Single-crystal X-ray structural studies of [Ru]-2a were performed on a CCD Agilent Technologies (Oxford Diffraction) SUPER NOVA diffractometer. Data were collected at 150(2) K using graphite-monochromated Mo Kα radiation (λα = 0.71073 Å). The strategy for the data collection was evaluated using the CrysAlisPro CCD software. The data were collected using the standard ‘phi-omega’ scan techniques, and were scaled and reduced using CrysAlisPro RED software. The structures were solved by direct methods using SHELXS-97, and refined using full matrix least-squares with SHELXL-97, refining on F2.15 The positions of all the atoms were obtained by direct methods. All non-hydrogen atoms were refined anisotropically, except for the solvent molecules (O111 and O222) which were refined isotropically. The remaining hydrogen atoms were placed in geometrically constrained positions, and refined with isotropic temperature factors, generally 1.2Ueq of their parent atoms. The crystal and refinement data are summarized in Table 1. The bond lengths and bond angles are summarized in Tables S1 and S2 (ESI†). The file with CCDC number 995119 contains the supplementary crystallographic data for [Ru]-2a.| Chemical formula | C24H26Cl4N2O2Ru2 |
| Fw | 718.41 |
| T (K) | 150(2) |
| Wavelength (Å) | 0.71073 |
| Cryst syst, space group | Triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| Cryst size (mm) | 0.33 × 0.26 × 0.21 |
| a (Å) | 8.0036(3) |
| b (Å) | 8.7265(3) |
| c (Å) | 18.6255(7) |
| α (°) | 76.921(3) |
| β (°) | 85.191(3) |
| γ (°) | 74.312(3) |
| V (Å3) | 1219.56(8) |
| Z | 2 |
| ρ calcd (g cm−3) | 1.956 |
| μ (mm−1) | 11.704 |
| F(000) | 712 |
| θ range (°) | 2.97–25.00 |
| Index ranges | −9 ≤ h ≤ 9; −10 ≤ k ≤ 10;−22 ≤ l ≤ 22 |
| Completeness to θmax | 99.8% |
| No. of data collected/unique data | 11690/4285 [Rint = 0.1943] |
| Absorption correction | Semi-empirical from equivalents |
| No. of params/restraints | 297/0 |
| Refinement method | Full-matrix least-squares on F2 |
| Goodness of fit on F2 | 1.047 |
| R 1 [I > 2σ(I)] | 0.0829 |
| wR2 [I > 2σ(I)] | 0.2091 |
| Largest diff peak and hole (e Å−3) | 2.989 and −4.032 |
Results and discussion
Synthesis of the complexes and their crystal structure
Mononuclear complexes [(η6-C6H6)RuCl2(C6H5NH2)] ([Ru]-2a) and [(η6-C10H14)RuCl2(C6H5NH2)] ([Ru]-2b) were obtained in high yields by treating methanol solutions of the respective ruthenium-arene dimer precursors, [{(η6-arene)RuCl2}2] (η6-arene = C6H6 ([Ru]-1a), C10H14 ([Ru]-1b)), with aniline under reflux conditions (Scheme 1). The isolated complexes, [Ru]-2a and [Ru]-2b, are soluble in polar solvents such as dichloromethane, chloroform, methanol and water, whereas insoluble in petroleum ethers and hexane. The identity of the complexes has been established using NMR, elemental analysis and ESI mass spectrometry (details are given in the Experimental section and ESI†). In the 1H NMR of complex [Ru]-2a, η6-C6H6 resonates at 5.35 ppm as a sharp singlet. The aromatic protons of C6H5NH2 were significantly down-shielded and resonate as a multiplet at 7.45–7.36 ppm for the five protons of the phenyl ring of aniline, whereas the NH2 protons show a broad singlet at 4.87 ppm. In the 13C NMR of complex [Ru]-2a, η6-C6H6 resonates at 87.68 ppm (DMSO-d6).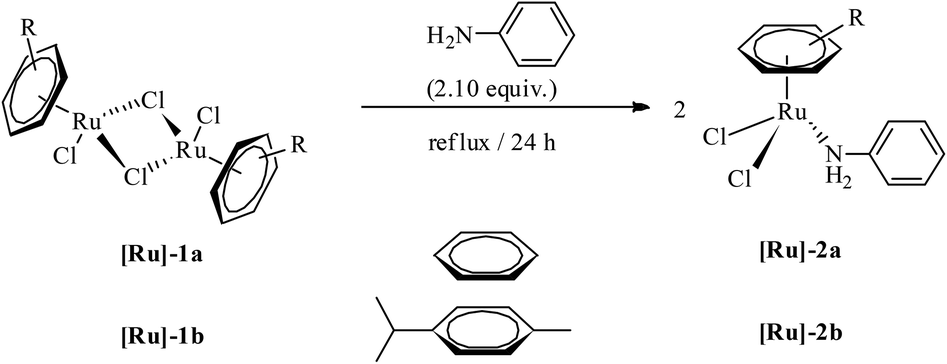 | ||
| Scheme 1 The synthesis of ruthenium-arene complexes [(η6-arene)RuCl2(C6H5NH2)] ([Ru]-2a and [Ru]-2b). | ||
The structure of the complex [(η6-C6H6)RuCl2(C6H5NH2)] ([Ru]-2a) was further confirmed by single-crystal X-ray diffraction analysis of suitable crystals of [Ru]-2a, grown by slow evaporation of a solution of [Ru]-2a in a methanol–dichloromethane (2:1) mixture. The complex [Ru]-2a crystallizes in the triclinic P space group, where two crystallographically independent molecules have been observed in the unit cell of [Ru]-2a (Fig. 2, Fig. S1,†Table 1, and Tables S1 and S2 in the ESI†). An ORTEP view of a single molecule of the complex [Ru]-2a and selected bond lengths and angles are given in Fig. 2. The Ru centre in complex [Ru]-2a exhibits the usual piano-stool geometry, with the nitrogen atom of aniline and two chloride ligands as the legs. The η6-coordinated benzene ring in the complex [Ru]-2a is almost planar and the ruthenium centre is displaced by 1.662 Å from the centroid of the benzene ring. Both the Ru–nitrogen bond distances (2.158 Å) and the Ru–chlorine bond distances (2.431 Å) were within the expected bonding distances for ruthenium arene complexes.16 The angles between the legs, Cl–Ru–Cl (88.48°) and Cl–Ru–N (82.87°), and the angles between the legs and the centroid of the η6-C6H6 ring (CCt) (126.5°–133.7°) are comparable with other similar complexes.16 Moreover, the Ru1–N1–C7 and Ru2–N2–C19 angles of 117.7(4)° and 117.8(5)°, respectively, indicated an away placement of the phenyl ring of aniline from the Ru centre.
 | ||
| Fig. 2 An ORTEP view of the complex [Ru]-2a (50% probability thermal ellipsoids). Hydrogen atoms are omitted for clarity. Selected bond lengths (Å): Ru1–Cav 2.1758, Ru1–Cl1 2.4242(15), Ru1–Cl2 2.4329(18), Ru1–N1 2.162(7), N1–C7 1.448(9); and bond angles (°): Cl1–Ru1–Cl2 88.35(6), Cl1–Ru1–N1 82.26(15), Cl2–Ru1–N1 82.99(18), Ru1–N1–C7 117.7(4). | ||
Catalytic activities of the complexes
The catalytic activity of complexes [Ru]-2a and [Ru]-2b for the synthesis of amides from aldehydes in water was evaluated using benzaldehyde as a model substrate and hydroxylamine hydrochloride as the NH2 source (Table 2). In recent reports, analogous catalytic reactions were performed at ≥100 °C using NaHCO3 or Na2CO3 base in water (Table 2, entries 14–15) or in other organic solvents (Table 2, entries 16–18).5,6b,e,8,10 We thought to bring down the reaction temperature, and treated benzaldehyde with NH2OH·HCl in water at 80 °C, under N2 atmosphere and in the presence of a [Ru]-2a catalyst. Surprisingly, complex [Ru]-2a provided a high yield (>99%) of benzamide (Table 2, entry 5). It is worth noting the superior catalytic performance of the complex [Ru]-2a, as previously reported active catalyst [(η6-C6H6)RuCl2{P(NMe2)3}] exhibits a drastic reduction in the catalytic yield to ∼55% with a minor decrease in the reaction temperature to 80 °C.5 The above results led us to further evaluate the catalytic performance of complex [Ru]-2a for the synthesis of amides from aldehydes at lower temperatures (<80 °C). Interestingly, the catalytic synthesis of benzamide from benzaldehyde proceeds well even at a reaction temperature as low as 40 °C, however, with a little loss in yield. The average reaction yields for the conversion of benzaldehyde to benzamide were >99% at 80 °C (Table 2, entry 5), >99% at 60 °C (Table 2, entries 1 and 6), 95% at 50 °C (Table 2, entry 7) and 86% at 40 °C (Table 2, entry 8). At the optimized reaction temperature of 60 °C, the catalytic performances of other analogues of [Ru]-2a, such as [Ru]-2b (Table 2, entry 2), [Ru]-1a (Table 2, entry 3), and [Ru]-1b (Table 2, entry 4), were also examined, but the complex [Ru]-2a outperforms the others. Moreover, compared to other bases used for this catalytic reaction, NaHCO3 promoted the performance of the catalysts (Table 2, entries 9–12). Moreover, the catalytic activity of the [Ru]-2a catalyst was also evaluated by lowering the catalyst loading to 2.5 mol%, and an appreciable high catalytic performance was observed (after 5 h of reaction, TON = 25.3 and TOF = 5.1 h−1) for the conversion of aldehydes to primary amides (Table 2, entry 13).|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Base | T (°C)/time (h) | Conv./sel. (%)b |
| a Conditions: benzaldehyde (1.0 mmol), NH2OH·HCl (1.3 equiv.), [Ru] (5 mol%), NaHCO3 (1.3 equiv.), H2O (8 mL). b Determined by 1H NMR. c Reactions were performed under a N2 atmosphere. d [Ru] (2.5 mol%), TON 25.3 (5 h) (in parentheses: selectivity at 24 h). e Ref. 5. f Ref. 8. g Cat. (1.5 mol%), with p-chlorobenzaldehyde as the substrate. h Ref. 6b. i Reaction was performed in toluene. j Ref. 6e. k Ref. 10. l Reaction was performed in DMSO–H2O. m Isolated yields. | ||||
| 1 | [(η6-C6H6)RuCl2(C6H5NH2)] ([Ru]-2a) | NaHCO3 | 60/5 | 99/99 |
| 2 | [(η6-C10H14)RuCl2(C6H5NH2)] ([Ru]-2b) | NaHCO3 | 60/5 | 99/65 |
| 3 | [{(η6-C6H6)RuCl2}2] ([Ru]-1a) | NaHCO3 | 60/5 | 99/54 |
| 4 | [{(η6-C10H14)RuCl2}2] ([Ru]-1b) | NaHCO3 | 60/5 | 99/84 |
| 5c | [(η6-C6H6)RuCl2(C6H5NH2)] ([Ru]-2a) | NaHCO3 | 80/4 | 99/99 |
| 6c | [(η6-C6H6)RuCl2(C6H5NH2)] ([Ru]-2a) | NaHCO3 | 60/5 | 99/99 |
| 7 | [(η6-C6H6)RuCl2(C6H5NH2)] ([Ru]-2a) | NaHCO3 | 50/5 | 96/99 |
| 8 | [(η6-C6H6)RuCl2(C6H5NH2)] ([Ru]-2a) | NaHCO3 | 40/5 | 94/92 |
| 9 | [(η6-C6H6)RuCl2(C6H5NH2)] ([Ru]-2a) | K2CO3 | 60/5 | 99/91 |
| 10 | [(η6-C6H6)RuCl2(C6H5NH2)] ([Ru]-2a) | NaOH | 60/5 | 98/98 |
| 11 | [(η6-C6H6)RuCl2(C6H5NH2)] ([Ru]-2a) | KOH | 60/5 | 95/85 |
| 12 | [(η6-C6H6)RuCl2(C6H5NH2)] ([Ru]-2a) | Na2CO3 | 60/5 | 92/77 |
| 13d | [(η6-C6H6)RuCl2(C6H5NH2)] ([Ru]-2a) | NaHCO3 | 60/5 | 99/64(83) |
| 14e | [(η6-C6Me6)RuCl2{P(NMe2)3}] | NaHCO3 | 100/7 | 88m |
| 15f,g | [(η5-C5Me5)Ir(H2O)3][OTf3]2 | Na2CO3 | 110/12 | 85m |
| 16h,i | [terpyRu(PPh3)Cl2] | NaHCO3 | Reflux/17 | 88m |
| 17i,j | [Ru(DMSO)4Cl2] | NaHCO3 | Reflux/6 | 24m |
| 18k,l | Pd(OAc)2 | Cs2CO3 | 100/15 | 98m |
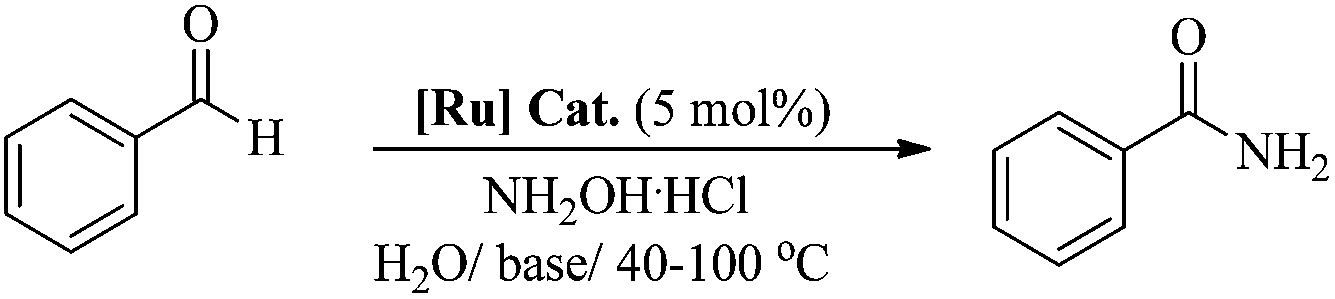
Most importantly, the complex [Ru]-2a not only exhibits higher catalytic performance even at a lower temperature (60 °C) in comparison to other catalysts, it also exhibits excellent stability in an aerobic environment. It is worth mentioning here that removal of the N2 gas protection, does not exhibit any adverse effect on the catalytic performances of the studied complexes (Table 2, entries 1 and 6). Use of inert gas was considered in previous reports to be essential for the catalytic conversion of aldehydes to amides.5 Therefore, we performed all of the catalytic reactions under aerobic conditions without any inert gas protection. The optimal reaction conditions for the catalytic conversion of aldehydes to amides are: a substrate/catalyst/NH2OH·HCl/NaHCO3 ratio of 100/5/130/130; H2O as the solvent; a reaction temperature 60 °C; and without any inert gas protection. The generality and scope of complex [Ru]-2a were then explored using a wide range of aldehydes (entries 1–18, Table 3 and entries 1–7, Table 4), including aromatic, heteroaromatic, aliphatic and conjugated systems, and the results obtained are summarized in Tables 3 and 4.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Product | Time (h) | Conv./sel. (%) | TONd/TOFe |
|
a Conditions: aldehyde (1.0 mmol), NH2OH·HCl (1.3 equiv.), [Ru]-2a (5 mol%), NaHCO3 (1.3 equiv.), H2O (8 mL).
b Conversion and selectivity were determined by 1H NMR.
c Reaction was performed in a H2O–methanol mixture (10:1 v/v).
d TON = turnover number.
e TOF = turnover frequency (h−1).
|
|||||
| 1 |

|
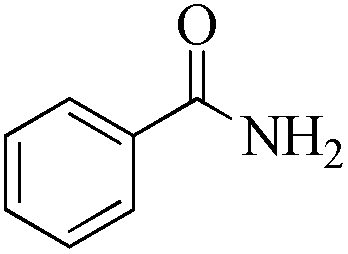
|
5 | 99/99 | 19.60/3.92 |
| 2 |

|
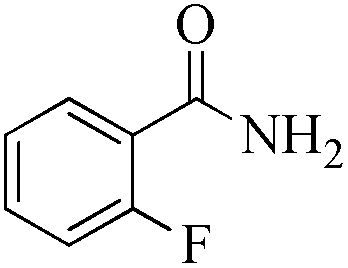
|
5 | 99/60 | 11.88/2.37 |
| 3 |
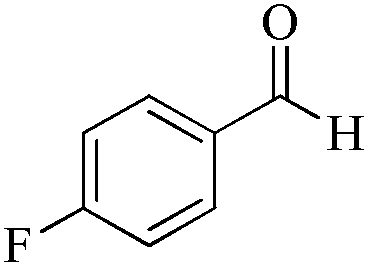
|
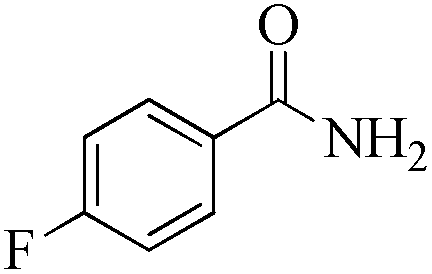
|
5 | 99/84 | 16.63/3.32 |
| 4 |
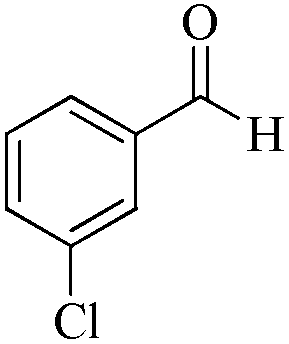
|

|
5 | 99/99 | 19.60/3.92 |
| 5 |

|
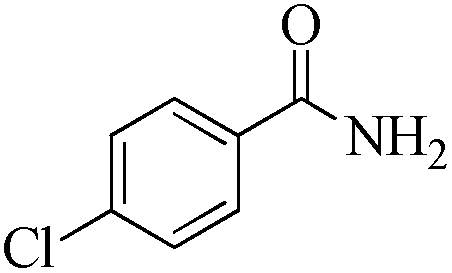
|
5 | 99/86 | 17.02/3.40 |
| 6 |

|

|
5 | 99/56 | 11.08/2.21 |
| 7 |
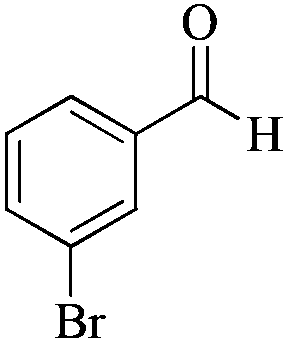
|
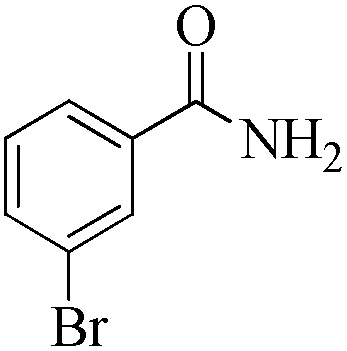
|
5 | 99/55 | 10.89/2.17 |
| 8 |
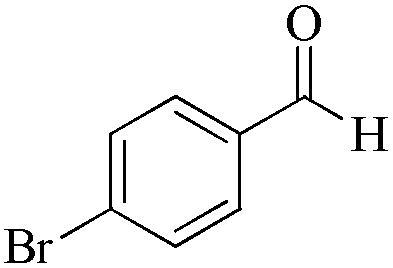
|
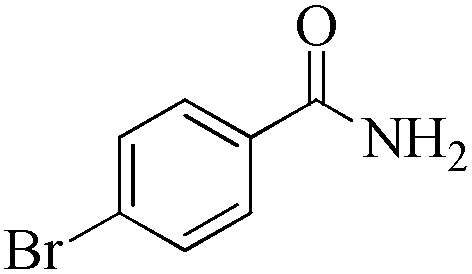
|
5 | 99/90 | 17.82/3.56 |
| 9 |
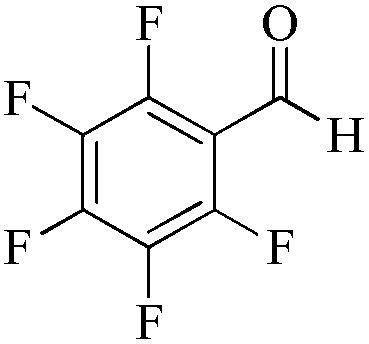
|
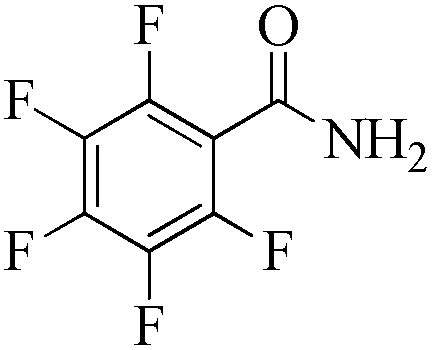
|
5 | 99/91 | 18.01/3.60 |
| 10 |
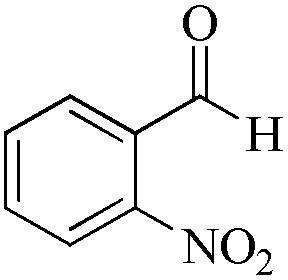
|
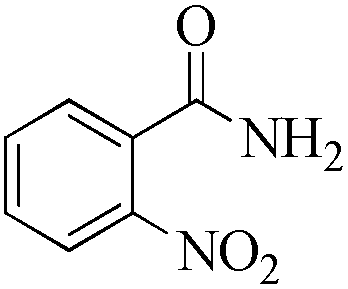
|
5 | 99/45 | 8.91/1.78 |
| 11 |

|
|
5 | 99/73 | 14.45/2.89 |
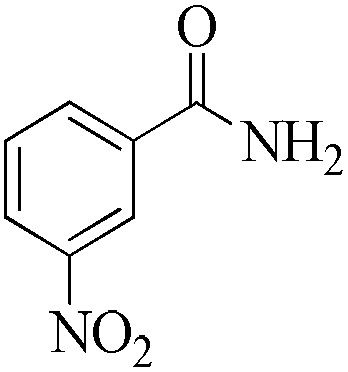
| 12 |
|

|
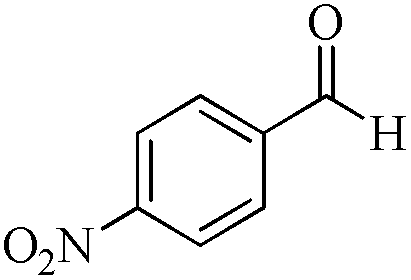
5 | 99/23 | 4.55/0.91 |
| 24 | 99/80 | 15.84/0.66 | |||
| 13 |

|
|
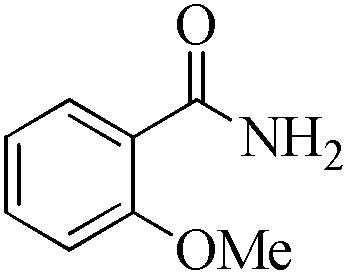
24 | 99/93 | 18.41/0.76 |
| 14 |
|

|
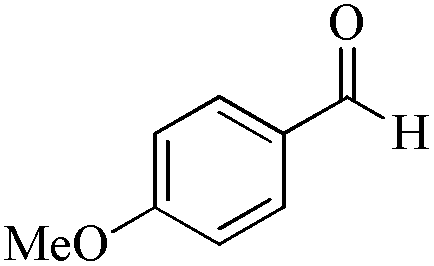
5 | 99/60 | 11.88/2.38 |
| 24 | 99/90 | 17.82/0.74 | |||
| 15 |
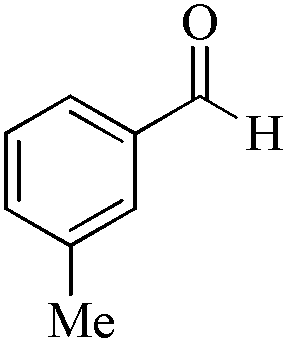
|
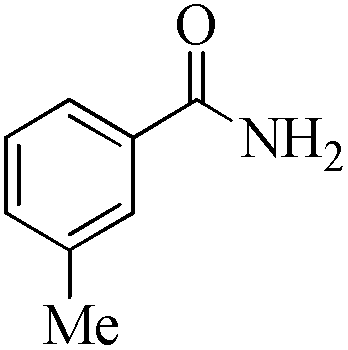
|
5 | 99/90 | 17.82/3.56 |
| 16 |

|
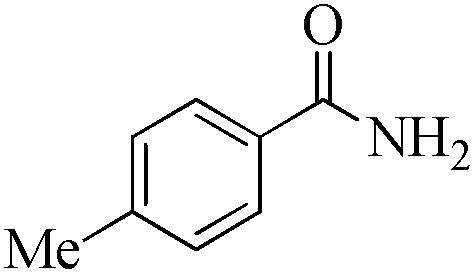
|
5 | 99/92 | 18.21/3.64 |
| 17 |
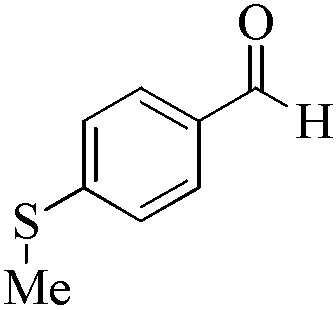
|
|
24 | 99/99 | 19.60/0.81 |
| 18c |

|
|
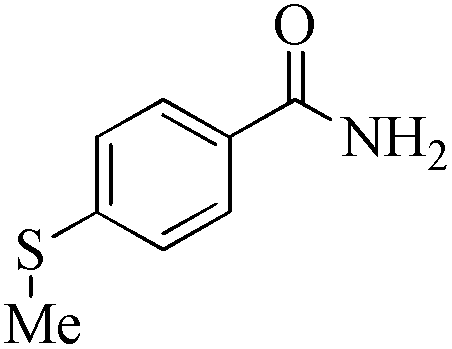
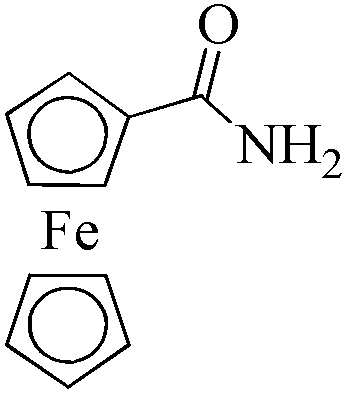
24 | 99/99 | 19.60/0.81 |

|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Product | Time (h) | Conv./Sel. (%) | TONc/TOFd |
| a Conditions: aldehyde (1.0 mmol), NH2OH·HCl (1.3 equiv.), [Ru]-2a (5 mol%), NaHCO3 (1.3 equiv.), H2O (8 mL). b Conversion and selectivity were determined by 1H NMR. c TON = turnover number. d TOF = turnover frequency (h−1). | |||||
| 1 |

|
|
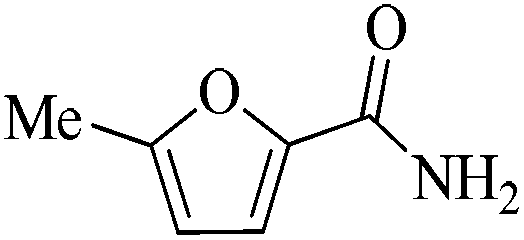
24 | 99/65 | 12.87/0.536 |
| 2 |

|
|
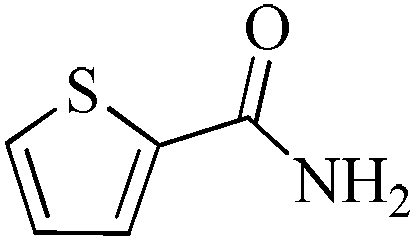
24 | 99/74 | 14.65/0.61 |
| 3 |
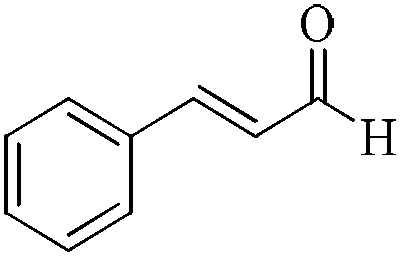
|

|
5 | 99/99 | 19.60/3.92 |
| 4 |
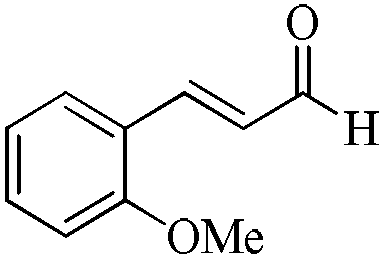
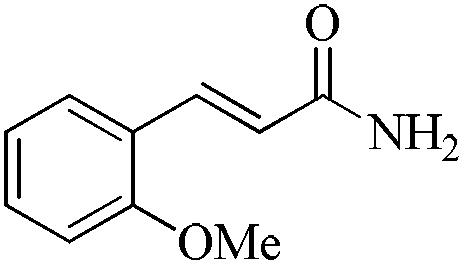
|
|
5 | 99/92 | 18.81/3.76 |
| 5 |

|

|
5 | 99/86 | 17.03/3.40 |
| 6 |
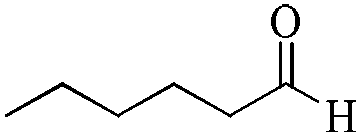
|

|
5 | 99/99 | 19.60/3.92 |
| 7 |
|

|
5 | 99/99 | 19.60/3.92 |
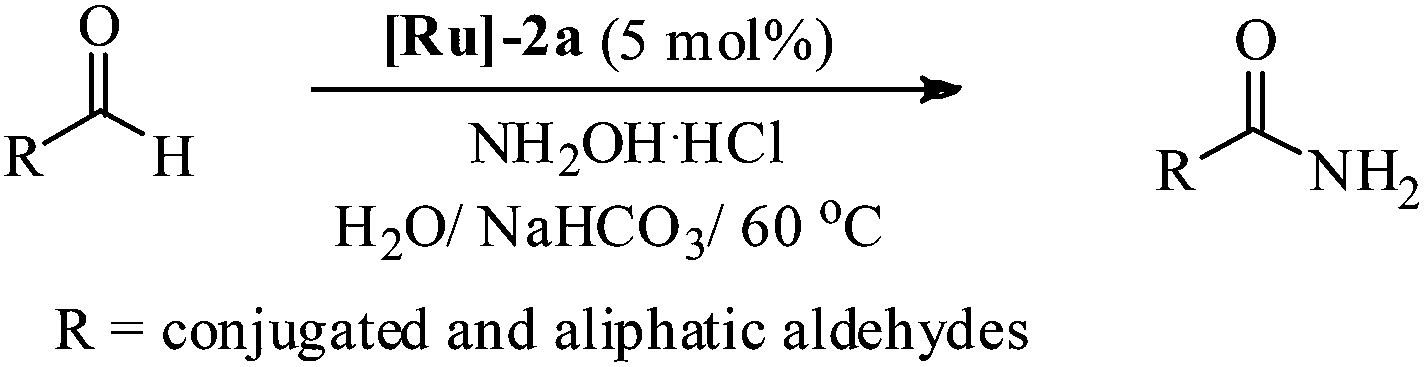
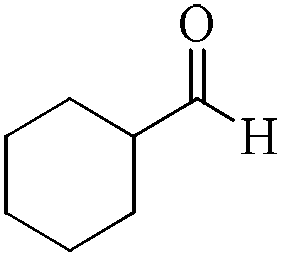
As observed for benzaldehyde, complex [Ru]-2a exhibits a high catalytic performance, >99% conversion and good selectivity, for almost all of the substituted aromatic aldehydes used to synthesize the corresponding primary amides, regardless of the position and electronic behavior of the substituents on the phenyl ring (Table 3). However, aromatic aldehydes with electron donating substituents (entries 13–17, Table 3) displayed a high selectivity in comparison to those with electron withdrawing substituents (entries 2–12, Table 3). Among the substrates with electron withdrawing substituents, the –NO2 substituted aromatic aldehydes exhibited very low selectivity in the amide formation due to the strong electron withdrawing nature of the –NO2 group, which retards the rearrangement of the aldoxime intermediate into the corresponding amide (Table 3, entry 12).11d Moreover, as expected, the ortho-substituted (entries 2,6, Table 3) aromatic aldehydes showed a lower activity in comparison to the corresponding meta- and para-substituted aromatic aldehydes, presumably due to steric effects (entries 3–5,7–8, Table 3). In comparison to –Me substituted aromatic aldehydes (Table 3, entries 15–16), with the 2-OMe substituted aromatic aldehyde (Table 3, entry 13) there is a possibility of coordination of the resulting aldoxime, as O,N chelation, to the metal centre thereby preventing the rearrangement of the aldoxime to the corresponding amide, which may be responsible for the long reaction period used (24 h) to generate 2-methoxybenzamide.19 As an extreme case, aromatic (e.g. salicylaldehyde) and heteroaromatic (e.g. pyridine-2-carboxaldehyde) aldehydes which are highly coordinating in nature, remain completely unreactive.17 It is worth mentioning here that the complex [Ru]-2a exhibits a high tolerance towards various labile functional groups, such as halides (entries 2–9, Table 3), nitro groups (entries 10–12, Table 3) and thioethers (entry 17, Table 3). The tolerance towards sulfur containing substrates is worth noting as sulfur species can poison homogeneous catalysts. Moreover, the complex [Ru]-2a could be advantageously used to synthesize ferrocenecarboxamide (entry 18, Table 3) from ferrocenecarboxaldehyde in higher yields and at a lower temperature (60 °C), when compared to the classical two-step method for its synthesis which uses harmful reagents such as thionyl chloride.18 In addition, heteroaromatic aldehydes, furyl (entry 1, Table 4) and thienyl (entry 2, Table 4) aldehydes, and α,β-unsaturated aldehydes (entries 3–4, Table 4) having conjugated olefin-carbonyl conjugated bonds, can also be converted to the corresponding primary amides. Further exploration with aliphatic aldehydes (CH3(CH2)nCHO, n = 2,4) (entries 5–6, Table 4) and cyclohexanecarboxaldehyde (entry 7, Table 4) revealed that [Ru]-2a exhibits an exceptionally high catalytic activity (selectivities 86%–99%) for the transformation of these aldehyde substrates into the corresponding amides. Almost all of the aldehydes exhibit high conversions and selectivities in the transformation to the corresponding amides, the only byproduct observed is aldoxime, which is also an intermediate for the aldehyde to amide conversion,7 for those aldehyde substrates having a poor selectivity. Moreover, pure crystals of the amides, in most of the cases, could be isolated by cooling the reaction mixture to 0 °C after completion of the reaction, and the identity of the isolated amides were analyzed by NMR spectroscopy (Fig. S2, ESI†).
To elucidate the reaction mechanism, we monitored the catalytic reaction by 1H NMR spectroscopy using benzaldehyde as the representative aldehyde substrate. As shown in Fig. 3, the benzaldoxime formed in the early stages of the catalytic cycle is slowly consumed to generate benzamide. To our surprise, benzonitrile was not observed, which was considered to be an important intermediate in the aldoxime rearrangement process. So the classical pathway of hydration of a benzonitrile intermediate by water can be discarded, instead aldoxime assisted hydration of benzonitrile may be the plausible pathway.8,19 To further demonstrate the above assumption, a reaction of benzonitrile was attempted in water at 60 °C in the presence of the [Ru]-2a catalyst, but no conversion was observed. However, under analogous reaction conditions, hydration of benzonitrile can be facilitated in the presence of butylaldoxime to yield benzamide, which further supports the alternative pathway for the hydration of benzonitrile (Schemes 2 and S1, ESI†).
 | ||
| Fig. 3 Plot of the time-dependent reaction progress for the catalytic conversion of benzaldehyde to benzamide in the presence of [Ru]-2a, in water at 60 °C. | ||
 | ||
| Scheme 2 Plausible mechanism for the catalytic conversion of an aldehyde to a primary amide. | ||
Dissociation of the ancillary ligands from the metal complex has previously been reported to generate a catalytically active species.20 Therefore, the possible structural changes in the [Ru]-2a catalyst, such as the dissociation of aniline, during the catalytic reaction were studied by 1H NMR and mass spectrometry, using the substrate 3-chlorobenzaldehyde at a substrate to catalyst ratio (S/C) of 6:1. While characterizing the catalyst recovered after the catalytic reaction using 1H NMR, we observed that the peaks corresponding to the ruthenium bound aniline were missing. Moreover, the mass spectrum of the recovered catalyst exhibits peaks corresponding to only [(η6-C6H6)RuCl(OH2)] (m/z 232.9285) and [(η6-C6H6)RuCl] (m/z 214.9190), whereas those corresponding to [(η6-C6H6)RuCl(C6H5NH2)] were absent (Fig. S3, ESI†). The observed 1H NMR and mass spectral data indicates that, presumably, replacement of the aniline ligand is the first step in the catalytic reaction, to provide a vacant coordination site for subsequent coordination of aldoxime to the ruthenium metal centre (Scheme 2).20 Comparing the catalytic performance of [(η6-C6H6)RuCl2(C6H5NH2)] ([Ru]-2a) with the corresponding arene-ruthenium dimer [{(η6-C6H6)RuCl2}2], it is easy to understand that the presence of aniline has a positive effect on the observed enhanced catalytic performance of the [(η6-C6H6)RuCl2(C6H5NH2)] ([Ru]-2a) catalyst, and presumably this is accounted for by the easy replacement of aniline with aldoxime. To further address the question of whether aniline is working as a base or a co-catalyst, we performed a reaction in the absence of base and observed >99% conversion but with only 94% selectivity for the amide. These results indicated that presumably the free aniline may act as a base or co-catalyst without coordination to the ruthenium centre. However, using aniline as an additive may retard the catalytic reaction due to it undergoing condensation with the aldehyde and preventing the formation of the crucial aldoxime intermediate. Moreover, surprisingly, free aniline in the crude reaction mixture could not be detected by either 1H NMR or mass spectrometry.
Conclusions
In conclusion, we demonstrated a new highly efficient catalytic system based on a phosphine-free ruthenium-arene complex, [(η6-C6H6)RuCl2(C6H5NH2)] ([Ru]-2a), for a one-pot primary amide synthesis from aldehydes at a remarkably lower temperature (as low as 60 °C, superior to other related reports operated at 100–110 °C). Moreover, the reported method is environmentally sustainable too, as all the reactions are performed in water and without inert gas protection. Advantageously, the reported complex displayed a wide generality and tolerance for reactive/unprotected functional groups on the aldehyde substrates. The current report will offer new opportunities for the development of improved phosphine-free catalytic systems for this and other related catalytic reactions.Acknowledgements
Financial support from IIT Indore, CSIR, New Delhi and SERB (DST), New Delhi is acknowledged. SIC IIT Indore is acknowledged for instrumentation facilities. D.T. and R.K.R. thank UGC, New Delhi and CSIR, New Delhi, respectively, for their fellowships. A.D.D. thanks CSIR, New Delhi [01(2722)/13/EMR-II] for his fellowship.Notes and references
- (a) G. Arture, The Amide Linkage: Selected Structural Aspects in Chemistry, Biochemistry, and Materials Science, Wiley-Interscience, 2000 Search PubMed; (b) J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 RSC; (c) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC.
- (a) V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed; (b) R. García-Álvarez, P. Crochet and V. Cadierno, Green Chem., 2013, 15, 46–66 RSC; (c) C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405–3415 RSC.
- B. S. Jursic and Z. Zdravkovdki, Synth. Commun., 1993, 23, 2761–2770 CrossRef CAS.
- R. García-Álvarez, A. E. Díaz-Álvarez, P. Crochet and V. Cadierno, RSC Adv., 2013, 3, 5889–5894 RSC.
- (a) R. García-Álvarez, M. Zablocka, P. Crochet, C. Duhayon, J. P. Majural and V. Cadierno, Green Chem., 2013, 15, 2447–2456 RSC; (b) D. Gnanamgan and R. H. Crabtree, Organometallics, 2009, 28, 922–924 CrossRef; (c) N. A. Owston, A. J. Parker and J. M. J. Williams, Org. Lett., 2007, 9, 3599–3601 CrossRef CAS PubMed; (d) N. Raja, M. V. Raja and R. Ramesh, Inorg. Chem. Commun., 2012, 19, 51–54 CrossRef CAS PubMed; (e) J. F. Hull, S. T. Hilton and R. H. Crabtree, Inorg. Chim. Acta, 2010, 363, 1243–1245 CrossRef CAS PubMed; (f) R. N. Prabhu and R. Ramesh, RSC Adv., 2012, 2, 4515–4524 RSC; (g) T. Naota and S.-I. Murahasi, Synlett, 1991, 693–694 CrossRef CAS; (h) S. Muthaiah, S. C. Ghosh, J.-E. Jee, C. Chen, J. Zhang and S. H. Hong, J. Org. Chem., 2010, 75, 3002–3006 CrossRef CAS PubMed.
- (a) H. Fujiwara, Y. Ogasawara, M. Kotani, K. Yamaguchi and N. Mizuno, Chem. – Asian J., 2008, 3, 1715–1721 CrossRef CAS PubMed; (b) H. Fujiwara, Y. Ogasawara, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2007, 46, 5202–5205 CrossRef CAS PubMed.
- C. Sun, P. Qu and F. Li, Catal. Sci. Technol., 2014, 4, 988–996 CAS.
- N. A. Owston, A. J. Parker and J. M. J. Williams, Org. Lett., 2007, 9, 73–75 CrossRef CAS PubMed.
- M. A. Ali and T. Punniyamurthy, Adv. Synth. Catal., 2010, 352, 288–292 CrossRef CAS.
- (a) A. Martínez-Asencio, M. Yus and D. J. Ramón, Tetrahedron, 2012, 68, 3948–3951 CrossRef PubMed; (b) N. C. Ganguly, S. Roy and P. Mondal, Tetrahedron Lett., 2012, 53, 1413–1416 CrossRef CAS PubMed; (c) S. K. Sharma, S. D. Bisshop, C. L. Allen, R. Lawrence, M. J. Bamford, A. A. Lapkin, P. Plucinski, R. J. Watson and J. M. J. Williams, Tetrahedron Lett., 2011, 52, 4252–4255 CrossRef CAS PubMed; (d) R. R. Gowda and D. Chakrabarty, Eur. J. Org. Chem., 2011, 2226–2229 CrossRef CAS; (e) C. L. Allen and J. M. J. Williams, Tetrahedron Lett., 2010, 51, 2724–2726 CrossRef CAS PubMed; (f) B. K. Allam and K. N. Singh, Tetrahedron Lett., 2011, 52, 5851–5854 CrossRef CAS PubMed.
- (a) C. J. Li and T. H. Chan, in Comprehensive Organic Reactions in Aqueous Media, John Wiley & Sons, Hoboken, NJ, 2007 Search PubMed; (b) Organic Reactions in Water Principles, Strategies and Applications, ed. Lindstrom, Blackwell Publishing Ltd., Oxford, UK, 2007 Search PubMed; (c) Water in Organic Synthesis, ed. S. Kobayashi, Thieme-Verlag, Stuttgart, Germany, 2012 Search PubMed.
- M. A. Bennett, T. N. Huang, T. W. Matheson and A. K. Smith, Inorg. Synth., 1982, 21, 74–78 CrossRef CAS.
- R. A. Zelonka and M. C. Baird, Can. J. Chem., 1972, 50, 3063–3072 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed; Program for Crystal Structure Solution and Refinement, University of Goettingen, Goettingen, Germany, 1997 Search PubMed.
- (a) S. K. Singh, M. Trivedi, M. Chandra, A. N. Sahay and D. S. Pandey, Inorg. Chem., 2004, 43, 8600–8608 CrossRef CAS PubMed; (b) R. García-Álvarez, J. Díez, P. Crochet and V. Cadierno, Organometallics, 2011, 30, 5442–5451 CrossRef; (c) J. G. Malecki, M. Jawarska and R. Kruszynski, Polyhedron, 2006, 25, 2519–2524 CrossRef CAS PubMed.
- (a) S. K. Singh, M. Chandra and D. S. Pandey, J. Organomet. Chem., 2004, 689, 2073–2079 CrossRef CAS PubMed; (b) S. K. Singh, M. Chandra, D. S. Pandey, M. C. Puerta and P. Valerga, J. Organomet. Chem., 2004, 689, 3612–3620 CrossRef CAS PubMed; (c) K. F. Konidaris, C. D. Polyzou, G. E. Kostakis, A. J. Tasiopoulos, O. Roubeau, S. J. Teat, E. Manessi-Zoupa, A. K. Powell and S. P. Perlepes, Dalton Trans., 2012, 41, 2862–2865 RSC; (d) M. A. M. Abu-Youssef, S. M. Soliman, V. Langer, Y. M. Gohar, A. A. Hasanen, M. A. Makhyoun, A. H. Zaky and L. R. Öhrström, Inorg. Chem., 2010, 49, 9788–9797 CrossRef CAS PubMed.
- T. H. Galow, J. Rodrigo, K. Cleary, G. Cooke and V. M. Rotello, J. Org. Chem., 1999, 64, 3745–3746 CrossRef CAS.
- R. García-Álvarez, A. E. Díaz-Álvarez, J. Borge, P. Crochet and V. Cadierno, Organometallics, 2012, 31, 6482–6490 CrossRef.
- W.-C. Lee and B. J. Frost, Green Chem., 2012, 14, 62–66 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and spectral data for the amides. Structural data for [Ru]-2a. CCDC 995119. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qi00115j |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2015 |