
A 2-D coordination polymer incorporating cobalt(II), 2-sulfoterephthalate and the flexible bridging ligand 1,3-di(4-pyridyl)propane†
Amitabha
Datta
*a,
Kuheli
Das
b,
Chiara
Massera
c,
Jack K.
Clegg
*d,
Michael C.
Pfrunder
d,
Eugenio
Garribba
e,
Jui-Hsien
Huang
*a,
Chittaranjan
Sinha
*b,
Tapas Kumar
Maji
f,
Takashiro
Akitsu
g and
Shingo
Orita
g
aDepartment of Chemistry, National Changhua University of Education, Changhua 50058, Taiwan. E-mail: amitd_ju@yahoo.co.in; juihuang@cc.ncue.edu.tw
bDepartment of Chemistry, Jadavpur University, Kolkata – 700032, India. E-mail: c_r_sinha@yahoo.com
cDipartimento di Chimica, Università degli Studi di Parma, Viale delle Scienze 17/A, 43124 Parma, Italy
dSchool of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane St Lucia, QLD 4072, Australia. E-mail: j.clegg@uq.edu.au
eDipartimento di Chimica e Farmacia, and Centro Interdisciplinare per lo Sviluppo della Ricerca Biotecnologica e per lo Studio della Biodiversita della Sardegna, Universitá di Sassari, Via Vienna 2, I-07100 Sassari, Italy
fMolecular Materials Laboratory, Chemistry and Physics of Materials Unit, Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR), Jakkur, Bangalore 560064, India
gDepartment of Chemistry, Faculty of Science, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan
First published on 15th December 2014
Abstract
The cobalt derivative {[Co3(2-stp)2(dpp)4]·2H2O}n (1) has been hydro(solvo)thermally synthesised by combining sodium 2-sulfoterephthalic acid (2-stpH2Na) and divalent cobalt nitrate in presence of the flexible N-donor co-ligand dpp (1,3-di(4-pyridyl)propane). Single-crystal X-ray diffraction shows that 1 is a two-dimensional coordination polymer with carboxylate and sulfonate oxygens and pyridyl nitrogens coordinated to the two symmetry independent octahedral cobalt(II) centres. UV-Vis, magnetic and EPR studies show that the cobalt(II) centres are high-spin. Compound 1 proved to be robust to the removal of the solvent water molecules, which was achieved by heating the sample to yield the desolvated complex {[Co3(2-stp)2(dpp)4]}n (2). 2 was also characterised by single-crystal X-ray diffraction at 331 K. Gas sorption studies on this material showed only surface binding of N2 and modest uptake for CO2. The crystallographic analysis also shows that upon desolvation, the flexible propyl groups of the dpp ligand adjust their positions, reducing the potentially accessible void volumes and accounting for the gas adsorption results.
Introduction
The preparation of coordination polymers has received significant interest due to both their diverse network topologies1 and the wide number of potential applications in catalysis, storage, separation and sensing, along with their many interesting physical properties including unusual chirality, luminescence and magnetism.2 While it is possible to design new coordination polymers with the help of reticular methods3 and, to some extent, predict their properties using computational methods,4 the applicability of these methods are often limited to systems in which rigid symmetric ligands is to be employed. Accordingly, the most used building blocks in the construction of coordination properties have been the bipyridine and imidazole-type ligands containing N donor groups, and the versatile aromatic carboxylic ligands. The incorporation of flexible bridging ligands into coordination polymers has received significantly less attention due to the unpredictability of the connectivity of the final assembly.5,6 Despite this limitation, the use of flexible components has the potential to yield materials with structures and functions that are inaccessible when rigid components alone are employed.5,7With the aim of producing materials with either unusual topologies or physical properties, we have recently investigated the formation of new metal–organic frameworks incorporating the rigid but non-symmetrical 2-sulfoterephthalate (2-stp) ligand.8 This ligand presents a diverse number of potential coordination and bridging modes, along with multiple accessible protonation states due to the presence of both the carboxylate and the sulfonate groups. In addition, the aromatic spacer provides π-surfaces for intermolecular interactions. When 2-stp was used in combination with the rigid linear 4,4′-bipyridine (bipy) building block both a zinc-containing pseudorotaxane and a 3D copper coordination polymer could be prepared.8
As an extension of these studies, in the present investigation we have employed the flexible 1,3-di(4-pyridyl)propane (dpp) ligand in combination with 2-stp and cobalt(II) to generate a new 2D-coordination polymer {[Co3(2-stp)2(dpp)4]·2H2O}n (1) and its desolvated analogue, {[Co3(2-stp)2(dpp)4]}n (2). Synthesis, X-ray diffraction analysis, magnetic investigations and gas-sorption data are reported.

Experimental
2-Sulfoterephthalic acid monosodium salt (2-stpH2Na) (Acros Organics), 1,3-di(4-pyridyl)propane (dpp) and cobalt nitrate hexahydrate (Aldrich) were used as received without further purification. Elemental analysis was performed on crushed sample that had been dried for 24 hours over P2O5 using a Thermo Flash 2000 CHN-O elemental analyzer. Infrared spectra were recorded on a Perkin-Elmer 883-Infrared spectrophotometer as KBr pellets. Emission spectra were recorded with a LS 55 Perkin–Elmer spectrofluorimeter at room temperature (298 K) in CH3CN solution. N2 (77 K) and CO2 (195 K) absorption measurements were performed using an AUTOSORB IQ2 instrument. Prior to the measurement of the isotherms, the sample was de-solvated for 72 h under high vacuum conditions (<0.1 Pa) at 130 °C. EPR spectra were recorded from 0 to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Gauss in the temperature range 77–298 K with an X-band (9.15 GHz) Varian E-9 spectrometer. Magnetic properties were investigated with a Quantum Design MPMS-XL superconducting quantum interference device magnetometer (SQUID) at an applied field 0.5 T in a temperature range 5–300 K and at 5 K in a field range −2.5 − +2.5 T. Thermogravimetric analysis was carried out at a heating rate of 10 °C min−1 with a Mettler-Toledo Star TGA/SDTA-851e thermal analyzer system in a dynamic atmosphere of N2 (flow rate 30 mL min−1) in an alumina crucible for the range 25–350 °C. Powder X-ray measurements were performed using a Shimadzu Lab-X 6000 diffractometer. Calculations were performed using SPARTAN'149 at the DFT level employing the B3LYP/6-31G* basis set.
000 Gauss in the temperature range 77–298 K with an X-band (9.15 GHz) Varian E-9 spectrometer. Magnetic properties were investigated with a Quantum Design MPMS-XL superconducting quantum interference device magnetometer (SQUID) at an applied field 0.5 T in a temperature range 5–300 K and at 5 K in a field range −2.5 − +2.5 T. Thermogravimetric analysis was carried out at a heating rate of 10 °C min−1 with a Mettler-Toledo Star TGA/SDTA-851e thermal analyzer system in a dynamic atmosphere of N2 (flow rate 30 mL min−1) in an alumina crucible for the range 25–350 °C. Powder X-ray measurements were performed using a Shimadzu Lab-X 6000 diffractometer. Calculations were performed using SPARTAN'149 at the DFT level employing the B3LYP/6-31G* basis set.
Crystallography
Data for 1 were recorded at 190(2) K on a Bruker APEX II diffractometer employing graphite-monochromated Mo-Kα radiation generated from a sealed tube. Data integration and reduction were undertaken with SAINT and XPREP.10 Multi-scan empirical absorption corrections were applied to the data using the program SADABS.11 Data for 2 at were collected at 331(2) K on an Oxford Diffraction Gemini Ultra diffractometer using graphite-monochromated Mo-Kα radiation generated from a sealed tube. Data integration, reduction and multi-scan empirical absorption corrections were performed using CrysAlisPro.12 Subsequent computations were carried out using the WinGX-32 graphical user interface.13 The structures of 1 and 2 were solved by direct methods using the SIR97 program14 then refined and extended with SHELXL-97.15 Carbon-bound hydrogen atoms were included in idealised positions and refined using a riding model. Hydrogen atoms bound to solvent water molecules that could not be located in the difference Fourier map were not included in the model. In the structure of 1, all non-hydrogen atoms were refined with anisotropic atomic displacements. Positional disorder in the dpp ligands in structure 2 prevented anisotropic refinement of the associated atoms. CCDC 936366 and 1003710.![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 10.616(5) Å, b = 11.949(4) Å, c = 14.908(6) Å, α = 80.973(6)°, β = 83.402(6)°, γ = 73.213(5)°, V = 1783(1) Å3, Dc = 1.390 g cm−3, Z = 1, crystal dimensions = 0.15 × 0.10 × 0.08, dark pink prism, 190(2) K, λ(Mo-Kα) = 0.71073, μ(Mo-Kα) 0.818 mm−1, T(empirical)min,max = 0.598, 0.746, 2θmax = 30.90, hkl range = −15 15, −17 17, −21 21, N = 28971, Nind = 10960 (Rmerge 0.0519), Nobs = 7740 (I > 2σ(I)), Nvar = 439, residuals R1 (F, 2σ) = 0.0476, wR2(F2 all) = 0.1308, GoF(all) = 1.013, Δρmin,max = −0.348, 0.893, e− Å−3. The refinement residuals are defined as ∑||Fo| − |Fc||/∑|Fo| for Fo > 2σ(Fo) and wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fc2)2]}1/2 where w = 1/[σ2(Fo2) + (0.0704 P)2 + 0.1155P], P = (Fo2 + 2Fc2)/3.
, a = 10.6094(6) Å, b = 11.9098(6) Å, c = 14.7412(7) Å, α = 81.495(4)°, β = 83.114(4)°, γ = 72.479(5), V = 1751(1) Å3, Dc = 1.381 g cm−3, Z = 1, crystal dimensions = 0.32 × 0.24 × 0.07, dark pink prism, 331(2) K, λ(Mo-Kα) = 0.71073, μ(Mo-Kα) 0.830 mm−1, T(empirical)min,max = 0.908, 1, 2θmax = 57.68, hkl range = −13 14, −16 15, −19 19, N = 12842, Nind = 7787 (Rmerge 0.0360), Nobs = 5378 (I > 2σ(I)), Nvar = 520, residuals R1 (F, 2σ) = 0.0482, wR2(F2 all) = 0.0966, GoF(all) = 1.008, Δρmin,max = −0.397, 0.467 e− Å−3. The refinement residuals are defined as ∑||Fo| − |Fc||/∑|Fo| for Fo > 2σ(Fo) and wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fc2)2]}1/2 where w = 1/[σ2(Fo2) + (0.03 P)2], P = (Fo2 + 2Fc2)/3.
, a = 10.616(5) Å, b = 11.949(4) Å, c = 14.908(6) Å, α = 80.973(6)°, β = 83.402(6)°, γ = 73.213(5)°, V = 1783(1) Å3, Dc = 1.390 g cm−3, Z = 1, crystal dimensions = 0.15 × 0.10 × 0.08, dark pink prism, 190(2) K, λ(Mo-Kα) = 0.71073, μ(Mo-Kα) 0.818 mm−1, T(empirical)min,max = 0.598, 0.746, 2θmax = 30.90, hkl range = −15 15, −17 17, −21 21, N = 28971, Nind = 10960 (Rmerge 0.0519), Nobs = 7740 (I > 2σ(I)), Nvar = 439, residuals R1 (F, 2σ) = 0.0476, wR2(F2 all) = 0.1308, GoF(all) = 1.013, Δρmin,max = −0.348, 0.893, e− Å−3. The refinement residuals are defined as ∑||Fo| − |Fc||/∑|Fo| for Fo > 2σ(Fo) and wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fc2)2]}1/2 where w = 1/[σ2(Fo2) + (0.0704 P)2 + 0.1155P], P = (Fo2 + 2Fc2)/3.
, a = 10.6094(6) Å, b = 11.9098(6) Å, c = 14.7412(7) Å, α = 81.495(4)°, β = 83.114(4)°, γ = 72.479(5), V = 1751(1) Å3, Dc = 1.381 g cm−3, Z = 1, crystal dimensions = 0.32 × 0.24 × 0.07, dark pink prism, 331(2) K, λ(Mo-Kα) = 0.71073, μ(Mo-Kα) 0.830 mm−1, T(empirical)min,max = 0.908, 1, 2θmax = 57.68, hkl range = −13 14, −16 15, −19 19, N = 12842, Nind = 7787 (Rmerge 0.0360), Nobs = 5378 (I > 2σ(I)), Nvar = 520, residuals R1 (F, 2σ) = 0.0482, wR2(F2 all) = 0.0966, GoF(all) = 1.008, Δρmin,max = −0.397, 0.467 e− Å−3. The refinement residuals are defined as ∑||Fo| − |Fc||/∑|Fo| for Fo > 2σ(Fo) and wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fc2)2]}1/2 where w = 1/[σ2(Fo2) + (0.03 P)2], P = (Fo2 + 2Fc2)/3.
Results and discussion
The use of flexible bridging ligands in the formation of coordination polymers and discrete supramolecular assemblies has the potential to produce a degree of uncertainty in the connectivity of products resulting from self-assembly reactions.5,6 This uncertainty stems from the wide range of potential coordination vectors and distances between donor atoms that result from the ligand adopting various conformers. In the case of dpp, there are a total of nine calculated low energy conformers (five of which are degenerate) in each of which the propyl group is in a staggered conformation. The calculated gas-phase structures9 of the four equal energy but non-degenerate conformers (A–D) are given in Fig. 1. In these structures the nitrogen–nitrogen distances, which approximate the length of the ligand, range from 5.00–9.68 Å and their coordination vectors range from 31°−113°, highlighting the difficulties in predicting the likely connectivity of a coordination polymer incorporating this ligand and providing the potential for significant topological diversity. It should also be noted, that these structures only represent the geometries that the dpp ligand is likely to adopt in its lowest energy forms – there are many thousands on higher energy structures that could be adopted, for example, with eclipsed or partially eclipsed arrangements that would result in an even larger set of potential coordination vectors and donor atom distances.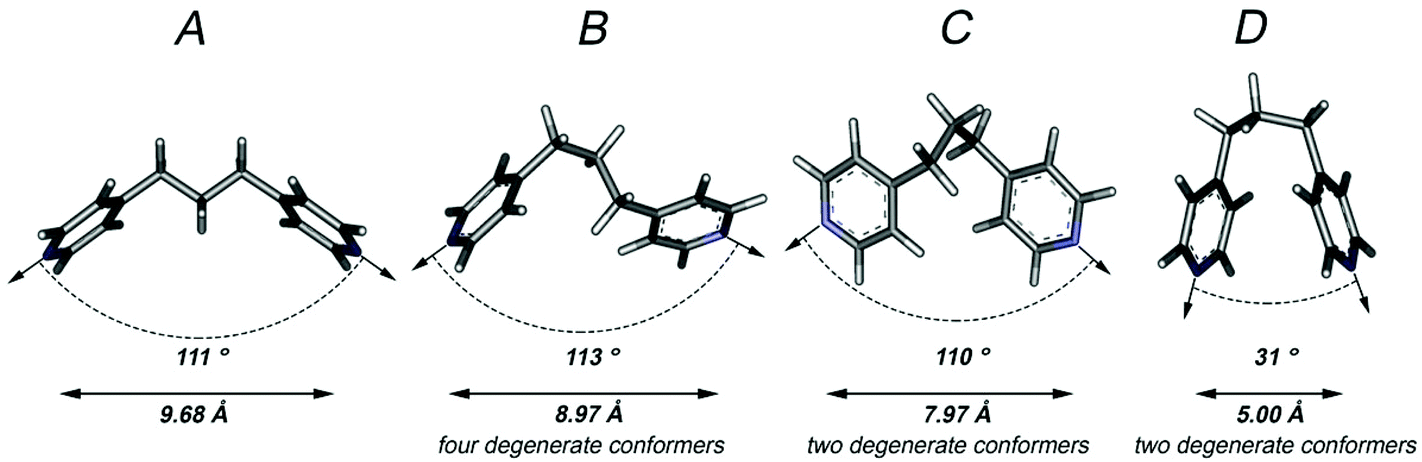 | ||
| Fig. 1 The four calculated equal energy conformers A–D of dpp. Distances are measured between nitrogen atoms and angles represent approximate coordination vectors. The coordination vector angles in A and D were approximated as the angle of intersection between the two mean pyridyl planes; the coordination vectors in B and C were approximated by measuring the nitrogen–methylene–methylene–nitrogen torsion angles. | ||
The hydrothermal reaction of NaH22-stp, dpp and cobalt(II) nitrate hexahydrate in a 1:2:30 ratio resulted in the formation of pink crystals of {[Co3(2-stp)2(dpp)4]·2H2O}n, which PXRD (Fig. S6†) indicated to be a single phase. The single crystal X-ray structure showed that 1 is a two-dimensional coordination polymer (Fig. 2).
 | ||
| Fig. 2 The symmetry-generated trinuclear unit building up the molecular structure of {[Co3(2-stp)2(dpp)4]·2H2O}n. The atoms labelled in red belong to adjacent units and are included to show the coordination environment around the metal centres. Hydrogen atoms and lattice water molecules have been omitted for clarity. | ||
There are two symmetry independent octahedral cobalt(II) centres in the asymmetric unit, each of them bridged by both 2-stp and dpp ligands. Co1 has an N3O3 coordination sphere, with three nitrogen atoms from three dpp ligands in a meridonal plane and the remaining positions occupied by three carboxylate oxygens atoms from two different 2-stp ligands; one of the carboxylate groups is a bidentate chelate and the third carboxylate oxygen is donated from an adjacent 2-stp ligand. Co2, which lies on an inversion centre, has an N2O4 coordination sphere. The nitrogen atoms from two dpp ligands are located axially and the four equatorial oxygens come from 2-stp ligands; two of the oxygens are from monodentate sulfonate groups and two from monodentate (but bridging) carboxylate groups. Each 2-stp ligand eptly bridges three metal ions forming metallocyclic loops that produce infinite [1 0 0] chains of formula [Co3(2-stp)2]n (Fig. 3a).16 These chains are connected by dpp units (Fig. 3b) in the direction of the crystallographic b-axis forming polymeric metallocyclic [Co6(dpp)6]n rings. Combined, these two sets of polymers result in a two-dimensional motif of thick sheets that stack parallel to the crystallographic ab-plane (Fig. 3c). If Co1 is considered a 5-connected node, Co2 as a 4-c node, the 2-stp ligand as 3-c node and the dpp ligand as two-fold connector, the topology of 1 can be described as a (3, 4, 5)-c trinodal net with point symbol17 {4·62}2{42·63·8}{42·66·82}2 as calculated with TOPOS.18 A schematic representation of the resulting network is shown in Fig. 4.
 | ||
| Fig. 3 (a) Perspective view of the [Co3(2-stp)2]n chains propagating along the a axis of the unit cell; the repetitive cyclic unit is highlighted in green. Colour code: cobalt, blue; oxygen, red; sulfur, yellow; carbon, grey; hydrogen, white. (b) Perspective view of the connectivity of the Co ions through dpp ligands; the repetitive cyclic unit is highlighted in orange. (c) Perspective view of part of a thick sheet parallel to the ab plane; a [Co3(2-stp)2]n chain is highlighted in green and a cyclic [Co6(dpp)6] unit is highlighted in orange. H atoms and water molecules are omitted for clarity. | ||
 | ||
| Fig. 4 Schematic view of the (3,4,5)-c trinodal net. Co1, Co2 and 2-stp are represented as pink, green and blue spheres, respectively. | ||
Each dpp ligand is present in the same conformer, which is very closely related to the calculated structure B (Fig. 5). Indeed, both the coordination vector (113°) and nitrogen separation (8.97 Å) are identical with the only marked difference a 45° rotation around one methylene–pyridine bond in the crystallised conformer resulting in an insignificant difference in energy between the two.
 | ||
| Fig. 5 Overlay of the calculated ligand in conformer B (red) and the ligand present in the crystal structure of {[Co3(2-stp)2(dpp)4]·2H2O}n (blue). | ||
The arrangement of the ligands resulting in metallocyclic units produces small cavities within each two-dimensional sheet of approximately 100 Å3 (which represents ∼6% of total void unit cell volume).19 The solvent water molecules are located within these cavities and hydrogen bond with an uncoordinated sulfonate oxygen that points into the cavity.
In the solid state, 1 exhibits some fluorescence behaviour. Upon excitation between 280–360 nm a strong emission band is observed at 408 nm with a shoulder at 486 nm (Fig. S3†). As the 2-stp ligand itself fluoresces between 473 and 515 nm,20 the emission is probably ligand centred with some possible contributions from LMCT, MLCT or intra-ligand charge transfer.
EPR spectra of the polycrystalline sample of 1 were recorded at 77 and 298 K. At room temperature, no spectrum is observed (Fig. 6a). At 77 K a very weak signal appears (Fig. 6b), but the sum of many scans (40) was necessary to obtain an interpretable spectrum (see ESI†). In the literature, few EPR spectra of high-spin cobalt(II) complexes are observed at 77 K.21 The short spin-lattice relaxation time for high-spin Co(II) species and, probably, the presence of two different chemical environments around the Co1 and Co2 in 1 cause the broad absorptions in the region 0–4000 Gauss and the poor resolution. No hyperfine coupling between the unpaired electrons and the nucleus of 59Co isotope (100% abundance, I = 7/2) is revealed; this suggests that, under these conditions, the line width is larger than the value of the hyperfine constant A.22 However, even if the hyperfine structure is absent, resonances at g1 ∼ 5.5, g2 ∼ 4.0 and g3 ∼ 2.1 are detected. Values of g larger than 4.0 are typical of high-spin Co(II) compounds,23 and has been measured for other hexa-coordinated species with tetragonal symmetry.24 The spectrum can be associated with the Kramer doublet ±1/2 arising from the S = 3/2 spin state,25 for which two signals approximately at g2 ∼ 2g3 are expected. Overall, EPR results confirm that the ground state of 1 is high-spin S = 3/2 (as indicated also by χMT vs. T plot, see Fig. 7) and are consistent with the magnetic susceptibility measurements.
 | ||
| Fig. 6 EPR spectra on the polycrystalline powder of complex 1 at: (a) 298 K and (b) 77 K. The two spectra were recorded using the same instrumental gain, but the spectrum in the trace b was obtained summing 40 different scans. | ||
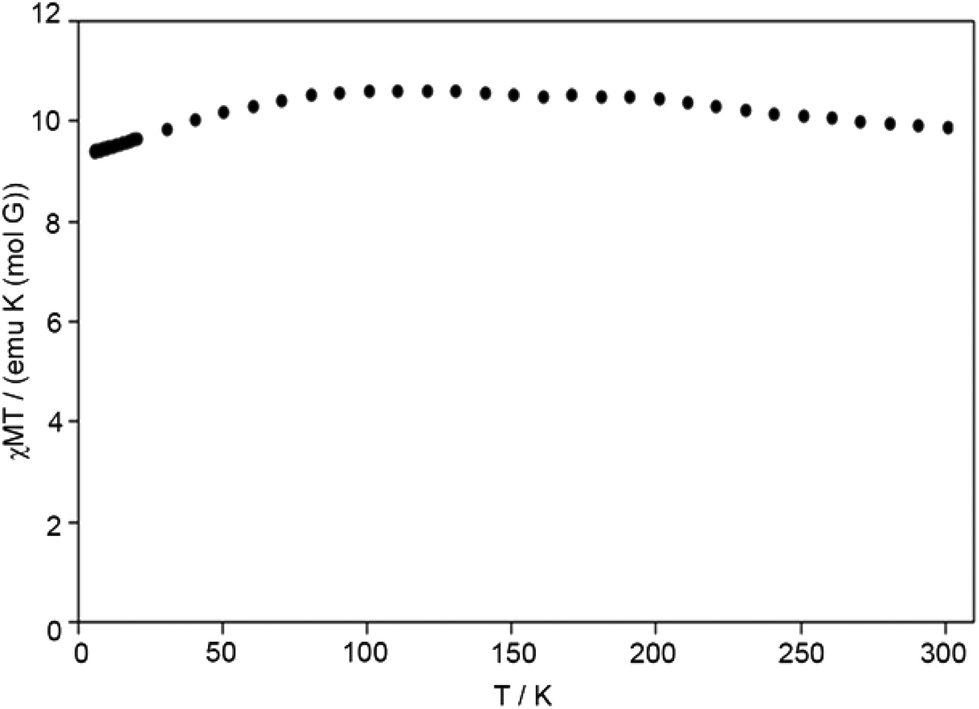 | ||
| Fig. 7 The χMT vs. T plot under 0.5 T for 1. | ||
Both VT magnetic susceptibility and EPR investigations of 1 were undertaken. Fig. 7 and S4† depict the χMT vs. T plot and the M vs. H plot for 1, respectively. At room temperature the χMT of complex 1 reaches the value of 9.90 emu K mol−1. No hysteresis width in the M vs. H plot and no sharp increasing (almost constant values) of susceptibility in the low temperature region suggest that complex 1 is paramagnetic for all temperatures (Fig. 7). The susceptibility and the resulting effective magnetic moment are consistent with the presence of high-spin octahedral cobalt(II) centres.
Thermogravimetric analysis (Fig. S1†) showed that the solvent water molecule could be removed by heating the sample above 100 °C and that the desolvated complex was stable to ca. 300 °C. Indeed, upon heating to 100 °C in an oven or in situ on the diffractometer to 58 °C, desolvation occurred without the loss of crystallinity producing the desolvated analogue {[Co3(2-stp)2(dpp)4]}n, 2, whose structure was confirmed by crystal structure determination at 331(2) K; 1 is therefore a rare example of a two-dimensional coordination polymer that maintains its single crystallinity upon desolvation.26 Indeed, immersion of the desolvated samples in water rapidly yielded the resolvated product without loss of crystallinity. The desolvation/resolvation process could be repeated many times, with the structural change confirmed by single crystal unit cell analysis after each cycle.
Upon desolvation, the structure maintains its connectivity, remaining a two-dimensional polymer. The only significant differences between the structures of 1 and 2 (other than the loss of solvent water) is a decrease of the cell volume from 1783 Å3 to 1751 Å3 and the introduction of positional disorder in the flexible propyl sections of the dpp ligands; which maintain their overall conformation. A diagram showing the disorder present in the dpp ligands in the desolvated structure is provided as ESI (Fig. S2†).
To assess the propensity of compound 2 to accommodate other small guest molecules, dehydrated single crystals were immersed in dry methanol and dry acetone. Visual inspection of a crystal submerged in dry methanol indicated that the overall crystallinity was lost within seconds of contact with the solvent. A crystal placed in a capillary containing dry acetone retained a sufficient level of single-crystallinity for the collection of meaningful X-ray diffraction data, however, even after soaking for 12 hours the resulting electron density map did not provide evidence supporting the adsorption of acetone molecules into the lattice.
We then undertook preliminary studies into the gas sorption behaviour of 2 towards both N2 and CO2. The sorption profile for nitrogen at 77 K (Fig. S5†) showed typical type-II behaviour with a modest uptake of 7 mL g−1, suggesting that only surface adsorption of the gas had occurred, while with CO2 at 195 K a type I profile was observed (Fig. 8).
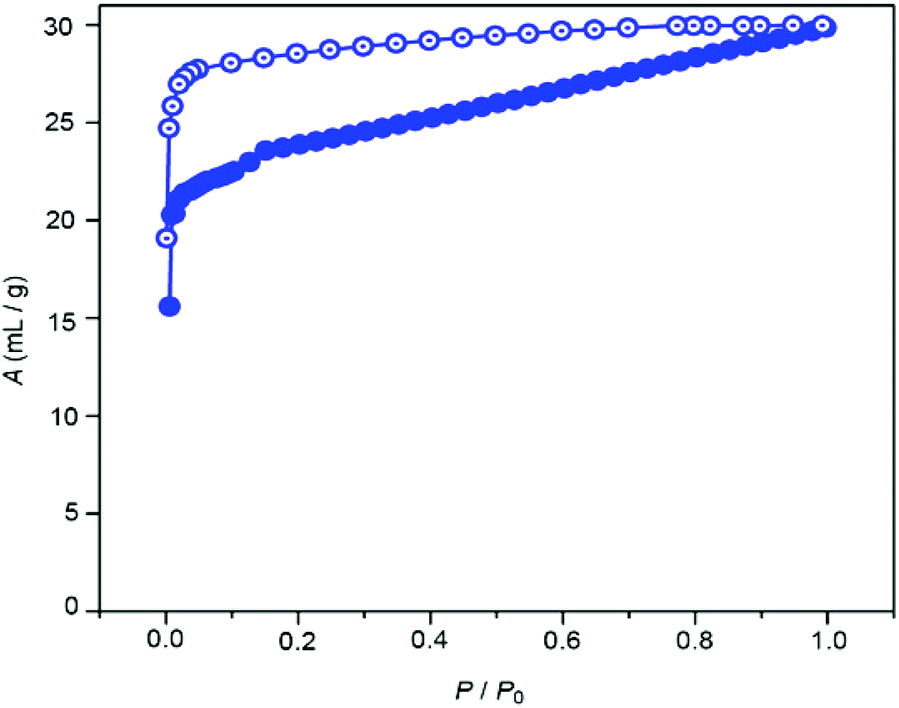 | ||
| Fig. 8 CO2 adsorption isotherm of desolvated 1 measured at 195 K. P0 = 680 mm of Hg. Closed symbols and open symbols correspond to adsorption and desorption, respectively. | ||
A steep uptake of CO2 was observed at very low pressure (P/P0 = 0.005), however this tapered off and a maximum loading of 27 mL g−1 was obtained. The Langmuir surface area calculated from the CO2 uptake profile was found to be 116 m2 g−1. The uptake behaviour of CO2, however, was found to display some hysteresis. Upon degassing, a residual quantity of gas (15 mL g−1 at P/P0 = 0.0009) was retained, suggesting a reasonably strong interaction (or kinetic trapping) between the pores of 1 and the CO2 molecules.
The relatively modest gas uptake behaviour presumably results from the reduction of the pore volume associated with the loss of the lattice water molecules. Close examination of the dehydrated crystal structure show that the disordered sections of the dpp ligands (which are not disordered in the solvated structure) occupy the cavities of the coordination polymer. Indeed, in addition to the decrease of the unit cell volume, the cavity volume decreases to 82 Å3.19 This reduction in solvent accessible void space is likely to impede guest uptake.
Conclusions
Complex 1 was synthesised and its structure determined through the use of single crystal X-ray diffraction. The compound was fully characterised using the usual methods. The Co(II) centres were found to be in the high spin state by both VT magnetic susceptibility and EPR spectroscopic measurements. Upon heating, crystals of 1 undergo reversible desolvation while retaining their single-crystallinity. The removal of the guest solvent molecules produced a smaller than expected pore volume due to an increase in disorder in the flexible components of the coordination polymer. The compound shows modest N2 and CO2 uptake, which is attributed to the collapse of the pore volume upon desolvation. These results highlight both the benefits and the potential problems with the use of flexible ligands for the construction of porous metal–organic framework as gas storage materials; flexibility in this case allows the crystal structure to re-adjust upon desolvation maintaining crystallinity while simultaneously reducing the accessible pore volume leading to limited gas sorption behaviour.Acknowledgements
AD and JHH gratefully acknowledge financial support from the National Science Council of Taiwan. KD and CS would like to thank the DST (Department of Science and Technology, New Delhi, India) for the grant (SR/S1/IC-31/2008). CM would like to thank Prof. D. Proserpio for fruitful discussion on topological analysis of MOFs. JKC and MP acknowledge the support of the Australian Research Council.Notes and references
- S. R. Batten, S. M. Neville and D. R. Turner, Coordination Polymers: Design, Analysis and Application, Royal Society of Chemistry, Cambridge, UK, 2009 CrossRef CAS PubMed; B. Moulton and M. J. Zarawotko, Chem. Rev., 2001, 101, 1629–1658 CrossRef CAS PubMed; J. L. Rowsell and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4670 CrossRef PubMed; J. J. Vittal, Coord. Chem. Rev., 2007, 251, 1781–1795 CrossRef PubMed; K. Biradha, C.-Y. Su and J. J. Vittal, Cryst. Growth Des., 2011, 11, 875–886 Search PubMed.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed; S. Zang, Y. Su, Y.-Z. Li, J. Lin, X. Duan, Q. Meng and S. Gao, CrystEngComm, 2009, 11, 122–129 RSC; A. Lan, K. Li, H. Wu, D. H. Olson, T. J. Emge, W. Ki, M. Hong and J. Li, Angew. Chem., Int. Ed., 2009, 48, 2334–2338 CrossRef PubMed; D. N. Dybtsev, A. L. Nuzhdin, H. Chun, K. P. Bryliakov, E. P. Talsi, V. P. Fedin and K. Kim, Angew. Chem., Int. Ed., 2006, 45, 916–920 CrossRef PubMed; Y.-F. Zeng, X. Hu, F.-C. Liu and X.-H. Bu, Chem. Soc. Rev., 2009, 38, 469–480 RSC; D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed; M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef PubMed; N. F. Sciortino, K. R. Scherl-Gruenwald, G. Chastanet, G. J. Halder, K. W. Chapman, J.-F. Letard and C. J. Kepert, Angew. Chem., Int. Ed., 2012, 51, 9944 CrossRef; L. J. Murray, M. Dinca and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC; M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330–1352 RSC; A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43, 58–67 CrossRef PubMed; C. Dey and R. Banerjee, Chem. Commun., 2013, 49, 6617–6619 RSC; W. M. Bloch, R. Babarao, M. R. Hill, C. J. Doonan and C. J. Sumby, J. Am. Chem. Soc., 2013, 135, 10441–10448 CrossRef PubMed; P. J. Saines, B. C. Melot, R. Seshadri and A. K. Cheetham, Chem. – Eur. J., 2010, 16, 7579–7585 CrossRef PubMed; G. J. Halder, C. J. Kepert, B. Moubaraki, K. S. Murray and J. D. Cashion, Science, 2002, 298, 1762–1765 CrossRef PubMed; Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- J. J. Perry IV, J. A. Perman and M. J. Zaworotko, Chem. Soc. Rev., 2009, 38, 1400–1417 RSC; D. J. Tranchemontagne, J. L. Mendoza-Cortes, M. O'Keeffe and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1257–1283 RSC; Q. Li, W. Zhang, O. S. Miljanic, C.-H. Sue, Y.-L. Zhao, L. Liu, C. B. Knobler, J. F. Stoddart and O. M. Yaghi, Science, 2009, 325, 855–859 CrossRef CAS PubMed; S. Hausdorf, J. r. Wagler, R. Moβig and F. O. R. L. Mertens, J. Phys. Chem. A, 2008, 112, 7567–7576 CrossRef PubMed; B. F. Abrahams, A. Hawley, M. G. Haywood, T. A. Hudson, R. Robson and D. A. Slizys, J. Am. Chem. Soc., 2004, 126, 2894–2904 CrossRef PubMed; B. F. Abrahams, N. J. FitzGerald and R. Robson, Angew. Chem., Int. Ed., 2007, 46, 8640–8643 CrossRef PubMed; O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef PubMed; O. K. Farha and J. T. Hupp, Acc. Chem. Res., 2010, 43, 1166–1175 CrossRef PubMed.
- O. K. Farha, Y. A. Oezguer, I. Eryazici, C. D. Malliakas, B. G. Hauser, M. G. Kanatzidis, S.-B. T. Nguyen, R. Q. Snurr and J. T. Hupp, Nat. Chem., 2010, 2, 944–948 CrossRef CAS PubMed; C. E. Wilmer, M. Leaf, C. Y. Lee, O. K. Farha, B. G. Hauser, J. T. Hupp and R. Q. Snurr, Nat. Chem., 2012, 4, 83–89 CrossRef PubMed; Y. J. Colon and R. Q. Snurr, Chem. Soc. Rev., 2014, 43, 5735–5749 RSC.
- W. M. Bloch, C. J. Doonan and C. J. Sumby, CrystEngComm, 2013, 15, 9663–9671 RSC.
- G. J. Halder and C. J. Kepert, J. Am. Chem. Soc., 2005, 127, 7891–7900 CrossRef CAS PubMed; J. Rabone, Y.-F. Yue, S. Y. Chong, K. C. Stylianou, J. Bacsa, D. Bradshaw, G. R. Darling, N. G. Berry, Y. Z. Khimyak, A. Y. Ganin, P. Wiper, J. B. Claridge and M. J. Rosseinsky, Science, 2010, 329, 1053–1057 CrossRef PubMed; C. R. K. Glasson, G. V. Meehan, C. A. Motti, J. K. Clegg, P. Turner, P. Jensen and L. F. Lindoy, Dalton Trans., 2011, 40, 12153–12159 RSC; J. K. Clegg, F. Li, K. A. Jolliffe, L. F. Lindoy, G. V. Meehan, S. Parsons, P. A. Tasker and F. J. White, Dalton Trans., 2013, 42, 14315–14323 RSC; T.-J. Won, J. K. Clegg, L. F. Lindoy and J. C. McMurtrie, Cryst. Growth Des., 2007, 7, 972–979 Search PubMed.
- J. E. Beves, B. A. Blight, C. J. Campbell, D. A. Leigh and R. T. McBurney, Angew. Chem., Int. Ed., 2011, 50, 9260–9327 CrossRef CAS PubMed; R. S. Forgan, J.-P. Sauvage and J. F. Stoddart, Chem. Rev., 2011, 111, 5434–5464 CrossRef PubMed; F. Li, J. K. Clegg, L. F. Lindoy, R. B. MacQuart and G. V. Meehan, Nat. Commun., 2011, 2, 205 CrossRef PubMed; A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC.
- A. Datta, K. Das, J.-Y. Lee, Y.-M. Jhou, C.-S. Hsiao, J.-H. Huang and H. M. Lee, CrystEngComm, 2011, 13, 2824–2827 RSC.
- SPARTAN'14 for Windows, Wavefunction Inc., 2014 Search PubMed.
- APEX v2.1, SAINT v.7 and XPREP v.6.14, Bruker AXS Inc., Madison, Wisconsin, USA, 2003 Search PubMed.
- SADABS: Empirical Absorption and Correction Software, University of Göttingen, 1996–2008 Search PubMed.
- CrysAlisPro, Agilent Technologies Ltd, 1.171.35.11, 2009–2011 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giocavazzo, A. Gaugliardi, G. C. Moliterni, G. Polidori and S. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. B. Deacon, C. M. Forsyth, O. Gazukin, P. C. Junk, G. Meyer, J. Sierau and D. R. Turner, Aust. J. Chem., 2014, 67, 1251–1256 CrossRef CAS.
- V. A. Blatov, M. O'Keeffe and D. M. Proserpio, CrystEngComm, 2010, 12, 44–48 RSC.
- V. A. Blatov, A. P. Shevchenko and V. N. Serezhkin, J. Appl. Crystallogr., 2000, 33, 1193 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- Y.-X. Ren, X.-J. Zheng and L.-P. Jin, CrystEngComm, 2011, 13, 5915–5923 RSC.
- K. Kojima, J. Matsuda, N. Kojima, T. Ban and I. Tsujikawa, Bull. Chem. Soc. Jpn., 1981, 60, 3213 CrossRef; S. Bellú, E. Hure, M. Trapé, C. Trossero, G. Molina, C. Drogo, P. A. M. Williams, A. M. Atria, J. C. Muñoz Acevedo, S. Zacchino, M. Sortino, D. Campagnoli and M. Rizzotto, Polyhedron, 2005, 24, 501–509 CrossRef PubMed; A. Dołęga, A. Pladzyk, K. Baranowska and J. Jezierska, Inorg. Chim. Acta, 2009, 362, 5085–5096 CrossRef PubMed.
- A. C. Rizzi, C. D. Brondino, R. Calvo, R. Baggio, M. T. Garland and R. E. Rapp, Inorg. Chem., 2003, 42, 4409–4416 CrossRef CAS PubMed.
- A. Abragam and M. H. L. Pryce, Proc. R. Soc. London, Ser. A, 1951, 206, 173–191 CrossRef CAS.
- A. Bencini, C. Benelli, D. Gatteschi and C. Zanchini, Inorg. Chem., 1980, 19, 1301–1304 CrossRef CAS; P. Thuery and J. Zarembowitch, Inorg. Chem., 1986, 25, 2001–2008 CrossRef; K. Sik Min, T. Weyhermuller and K. Wieghardt, Dalton Trans., 2004, 178–186 RSC.
- R. Vicente, A. Escuer and J. Ribas, Polyhedron, 1992, 11, 857–862 CrossRef CAS.
- M. W. Hosseini, Acc. Chem. Res., 2005, 38, 313–323 CrossRef CAS PubMed; W. M. Bloch and C. J. Sumby, Chem. Commun., 2012, 48, 2534–2536 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional details of the physical methods, additional figures and additional topological discussion, crystallographic data in CIF format. CCDC 936366 and 1003710. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qi00201f |
| This journal is © the Partner Organisations 2015 |