
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSize dependent effect of new organometallic triptycene tectons on the dimensions of self-assembled macrocycles†
Sourav
Chakraborty
a,
Sourav
Bhowmick
a,
Jianqiu
Ma
b,
Hongwei
Tan
b and
Neeladri
Das
*a
aDepartment of Chemistry, Indian Institute of Technology Patna, Patna 800 013, Bihar, India. E-mail: neeladri2002@yahoo.co.in; neeladri@iitp.ac.in; Fax: +91 612 2277383; Tel: +91 612 2552023
bCollege of Chemistry, Beijing Normal University, Beijing 100875, People's Republic of China
First published on 23rd January 2015
Abstract
The design, synthesis and characterization of two new triptycene-containing ditopic Pt(II) organometallic complexes is being reported. These complexes comprise of two peripheral bis(trans-trialkylphosphine)platinum units either directly σ-bonded to the central triptycene moiety or connected via a bridging ethynyl spacer linkage. The potential utility of these organometallic complexes as ditopic acceptor building blocks for the construction of neutral metallasupramolecular macrocycles containing the triptycene motif is explored. Triptycene motif containing supramolecules were characterized using multinuclear NMR (including 1H DOSY), mass spectrometry (MALDI-TOF-MS) and elemental analyses. While the self-assembly of a longer acceptor linker with a terephthalate group results in the formation of a [3 + 3] self-assembled macrocycle, the use of a relatively shorter acceptor linker yields the corresponding [2 + 2] supramolecular framework. The shapes and dimensions of these supramolecular structures were also predicted by geometry optimization using PM6 semiempirical molecular orbital methods and the results corroborate well with the experimental observations. These two self-assembled macrocycles are unique examples of triptycene-based neutral “platinamacrocycles” reported in the literature to date. An investigation as to how the shape and size of the resulting discrete supramolecular framework is affected upon changing the length and rigidity of the triptycene-based acceptor linkers is discussed.
Introduction
Design and self-assembly of finite nanoscopic supramolecular coordination complexes (SCCs) via a coordination driven self-assembly approach is an important area of research in supramolecular chemistry.1 In the last two decades, a considerable number of 2D metallacycles and 3D metallacages have been reported.2 The geometrical shape and size of these supramolecules can be predicted prior to self-assembly based on the ‘bite angle’ of the organic donor and metal-containing acceptor tecton.2a,b Several structural motifs [such as ferrocene, carboranes, cavitands, highly aromatic systems (such as anthracene, phenanthrene, perylene, pyrene, etc.), dendrons and others] have been incorporated in these self-assembled supramolecules.3 This has been achieved by the design of specific modular subunits containing these structural motifs and subsequently using them as tectons in the self-assembly protocol. Additionally, the shape of the supramolecules has been effectively tailored using building blocks of various dimensions.Triptycene is the smallest member in the class of compounds called iptycenes. Triptycene has a rigid three dimensional framework decorated with three benzene rings in a paddlewheel orientation. Owing to the high level of symmetry associated with its rigid framework, triptycene derivatives have found application in supramolecular chemistry,4 material science4 and polymer chemistry.5 In this context, the use of triptycene motifs as tectons for discrete metallasupramolecular architectures is at a very nascent stage.6 A detailed literature search has revealed that only a handful of triptycene-based organometallic complexes have been reported so far.7 Consequently, their use in the design of self-assembled metallasupramolecular frameworks has not been explored. Recently, we have reported (for first time) the use of organometallic triptycene derivatives as building blocks in the design of ionic three dimensional discrete supramolecular cages.7b
In a continuation of our research efforts towards the synthesis of new triptycene-based donor/acceptor linkers and their use as modified subunits in supramolecular self-assembly,7a,b herein we report the synthesis of two new triptycene-containing ditopic organoplatinum complexes. Complex 1 was synthesized by the double oxidative-addition of Pt(PEt3)3 with dibromotriptycene.8 Using the σ-acetylide synthetic methodology,9 the organometallic complex 2 was obtained upon reaction of diethynyltriptycene with trans-bis(triethylphosphine)platinum(II). The halogen atoms (bromine/iodine) in these newly synthesized organometallic complexes (1 and 2) were subsequently exchanged with more labile nitrate anions through a salt metathesis reaction with silver nitrate (3 and 4 respectively). All of the new organoplatinum complexes (1–4) have been fully characterized using FT-IR and multinuclear NMR spectroscopies, mass spectrometry and elemental analyses. These ditopic triptycene-based organometallic linkers are structurally rigid with predefined bite angles and thus they have the potential to act as metal-containing acceptor tectons for the construction of discrete metallasupramolecular architectures. To illustrate this point, herein we report the self-assembly of two new platinum(II)-based neutral supramolecular macrocycles (5 and 6) using the triptycene-based organometallic linkers 3 and 4 as acceptor units and ditopic angular carboxylates as bridging ligands. Macrocycles 5 and 6 represent the first examples of triptycene-based neutral metallasupramolecular architectures. These newly synthesized metallasupramolecular macrocycles were completely characterized using multinuclear NMR spectroscopy including 1H DOSY, MALDI-TOF mass spectrometry and elemental analyses. Further insight into the shape and size of the neutral macrocycles was obtained via molecular simulations, employing a PM6 semiempirical molecular orbital method. Theoretical calculations suggest that the dimensions of the metallasupramolecules are in the nanoscalar range.
Results and discussion
Synthesis and characterization of triptycene-based ditopic organoplatinum acceptor linkers
Halogenated arenes are one of the most popular classes of synthons utilized in the design and synthesis of metal-containing acceptor building blocks for subsequent use in coordination driven self-assembly processes.10 The desired triptycene organoplatinum complex 1 was efficiently synthesized by the double oxidative addition8 of Pt(0) to the corresponding 2,6-dibromotriptycene11 in 66% yield as shown in Scheme 1.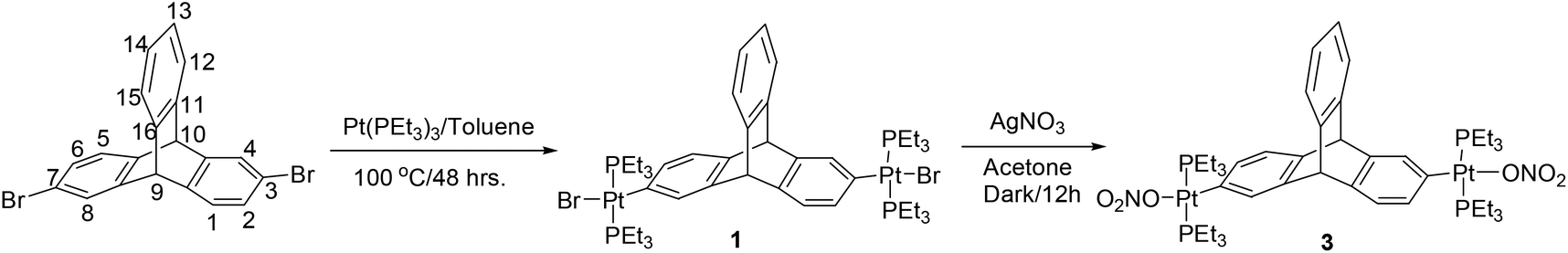 | ||
| Scheme 1 Synthesis of the triptycene-based organoplatinum complexes 1 and 3. | ||
In the past decade, the design and synthesis of organoplatinum acceptor complexes containing an ethynyl group as a spacer (bridging the central core motif and the peripheral metal center) has attracted the research attention of supramolecular chemists.12 Introduction of an ethynyl group imparts rigidity to the resulting linker and simultaneously alters its length, shape and directionality (bite angle). Subsequently, supramolecular tectons/building blocks are derived from these molecules with multiple ethynyl spacers, which in turn are utilized for the self-assembly of exciting nanoscalar macrocycles and cages.12
Through the reaction of an appropriate 2,6-diethynyltriptycene5d with trans-PtI2(PEt3)2 in a mixture of toluene (solvent) and triethylamine (base) in the presence of cuprous(I) iodide catalyst, the corresponding di-substituted metal complex 2 was obtained in a good yield (Scheme 2).
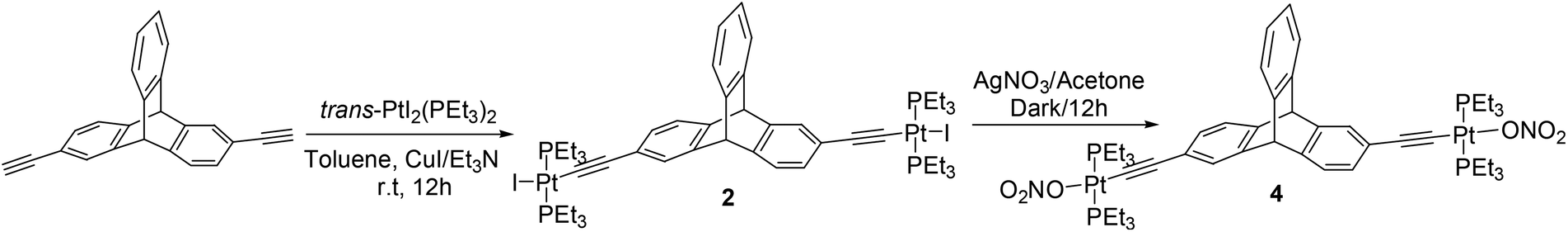 | ||
| Scheme 2 Synthesis of the triptycene-based organoplatinum complexes 2 and 4. | ||
The halogen atoms (bromine/iodine) in these newly synthesized organometallic complexes (1 and 2) were subsequently exchanged with more labile nitrate anions through a salt metathesis reaction with silver nitrate. The nitrate salts (3 and 4) were obtained in excellent yields (Schemes 1 and 2). These triptycene-organoplatinum complexes are white coloured solids that are stable in air/moisture with a high solubility in common organic solvents. The FTIR spectra of complexes 2 and 4 exhibit intense peaks at 2111 cm−1 and 2121 cm−1 respectively, due to the ethynyl functional group. The 1H NMR spectra also suggest the formation of the desired organometallic complexes due to the appearance of a singlet peak between 5.06 and 5.31 ppm corresponding to the bridgehead protons in complexes 1–4 (ESI†). As expected, the appearance of peaks in the range of δ = 0.95 to 2.20 ppm for complexes 1–4 is attributed to the ethyl group of the PEt3 units bound to the Pt(II) centers. Organometallic complexes were also characterized using 31P{1H} NMR spectroscopy (ESI†). The 31P{1H} NMR spectra of both of the halogenated complexes 1 and 2 show a sharp singlet at 12.39 and 8.56 ppm respectively with concomitant 195Pt satellites (1JPPt = 1383 Hz for 1 and 1166 Hz for 2). In the case of the nitrate salts 3 and 4, the 31P resonance due to the phosphine group shows a downfield shift in its peak position compared to those of the singlets observed for the corresponding halo analogues, appearing at 18.23 ppm (1JPPt = 1453 Hz) and 20.19 ppm (1JPPt = 1244 Hz) respectively. These chemical shifts observed in the 31P{1H} NMR are in the expected range for a trans-PtP2 system connected to arene8 and ethynyl moieties.9 In the 31P NMR of complexes 1 and 2, the appearance of a single sharp singlet also suggests that the phosphine groups are chemically equivalent with a trans orientation of the phosphine groups at the square planar Pt(II) centre. The formation of complexes 1–4 was further confirmed from mass spectrometry (ESI-MS) analyses and elemental analyses (ESI†).
Self assembly of 2D macrocycles
The triptycene-based organoplatinum complexes 3 and 4 are ditopic (having two reactive sites) moieties and hence they are potential acceptor tectons that may be reacted with appropriate ditopic donor ligands to yield metallamacrocycles via a coordination driven self-assembly reaction. In order to test this hypothesis, an acetone solution of 3 was reacted with an aqueous solution of disodium terephthalate in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio at ambient temperature (Scheme 3). This resulted in the gradual precipitation of the neutral assembly 5 as a white solid, which upon washing (with water) and recrystallization (from chloroform) yielded a white microcrystalline solid (81% isolated yield). The product obtained from this self-assembly reaction is highly soluble in common organic solvents such as DCM, chloroform, ethanol, methanol and THF.
:1 stoichiometric ratio at ambient temperature (Scheme 3). This resulted in the gradual precipitation of the neutral assembly 5 as a white solid, which upon washing (with water) and recrystallization (from chloroform) yielded a white microcrystalline solid (81% isolated yield). The product obtained from this self-assembly reaction is highly soluble in common organic solvents such as DCM, chloroform, ethanol, methanol and THF.
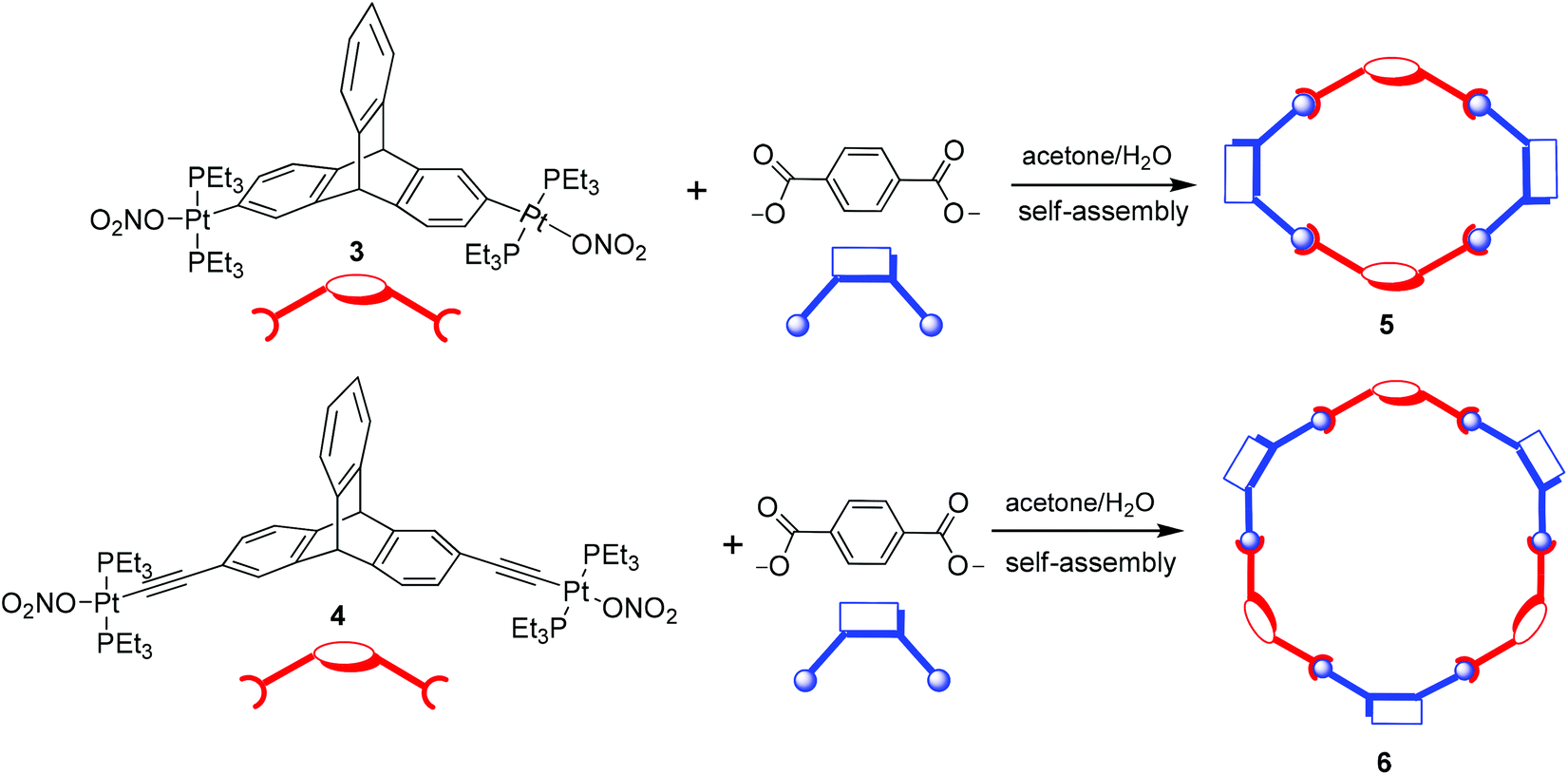 | ||
| Scheme 3 Synthesis of the molecular macrocycles 5 and 6. | ||
The product thus obtained was subjected to NMR spectroscopic analysis to ascertain its composition. The 31P{1H} spectrum of the product recorded in CDCl3, exhibited a sharp singlet (δ 16.14 ppm) with accompanying 195Pt satellites (1JPPt = 1459 Hz) shifted upfield relative to the precursor acceptor linker 3 by 2 ppm, suggesting the formation of a highly symmetrical self-assembled species in which all of the phosphorous atoms are chemically equivalent (Fig. 1a). The shift in the peak position of the 31P NMR signal is a clear indication of a new metal–ligand coordination. The 1H NMR spectrum of the product (Fig. 1b) also suggested the incorporation of both terephthalate (the sharp singlet at 7.96 ppm is assigned to protons on the phenyl ring of the terephthalate unit) and triptycene motifs (peaks at 5.07 ppm due to the bridgehead proton). Integration of the 1H peaks corresponding to terephthalate and that due to the triptycene motif suggests the presence of these two tectons in a 1:1 stoichiometric ratio. Thus coordination between a triptycene containing PtII2 unit and a terephthalate is confirmed from the multinuclear NMR spectra of the product. Additionally, this organometallic macrocycle (5) possesses a single trace in the 1H DOSY NMR (ESI†), indicating the formation of a single product and thereby ruling out the presence of additional species such as other macrocycles or oligomers in solution.
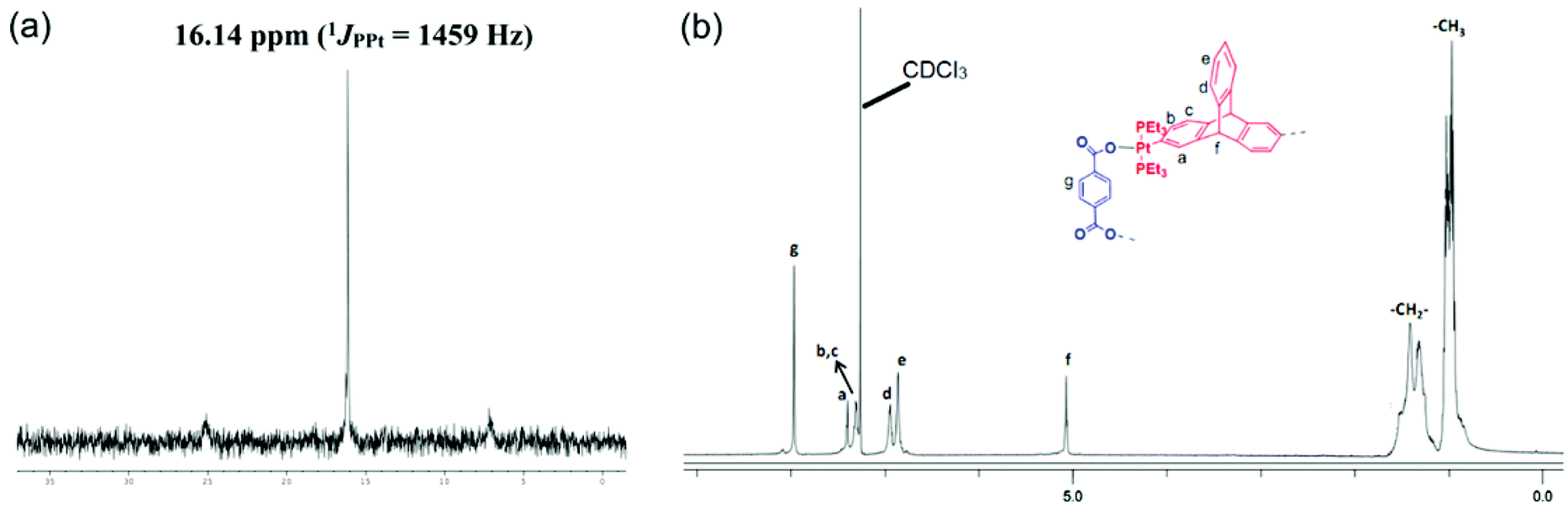 | ||
| Fig. 1 (a) The 31P{1H} NMR and (b) 1H NMR spectra of macrocycle 5 in CDCl3. | ||
However, the NMR spectroscopy technique does not predict the number of each unit (organometallic acceptor/organic donor) present in the self-assembled product. In such a situation, mass spectrometry is utilized as a powerful tool to interpret the composition of the macrocyclic product. In the present case, MALDI-TOF-MS spectrometric analysis confirmed the actual composition of macrocycle 5 (ESI†). The MALDI-TOF-MS spectrum of 5 showed two peaks at m/z = 2560.9 and m/z = 2582.9 corresponding to the [2 + 2 + H]+ and [2 + 2 + Na]+ species respectively, assuming a [2 + 2] self-assembly between two units each of a triptycene-containing PtII2 unit and a terephthalate. These signals were isotopically resolved (Fig. 2a) and these are in good agreement with theoretically calculated isotopic distribution patterns, assuming a [2 + 2] self-assembled macrocycle.
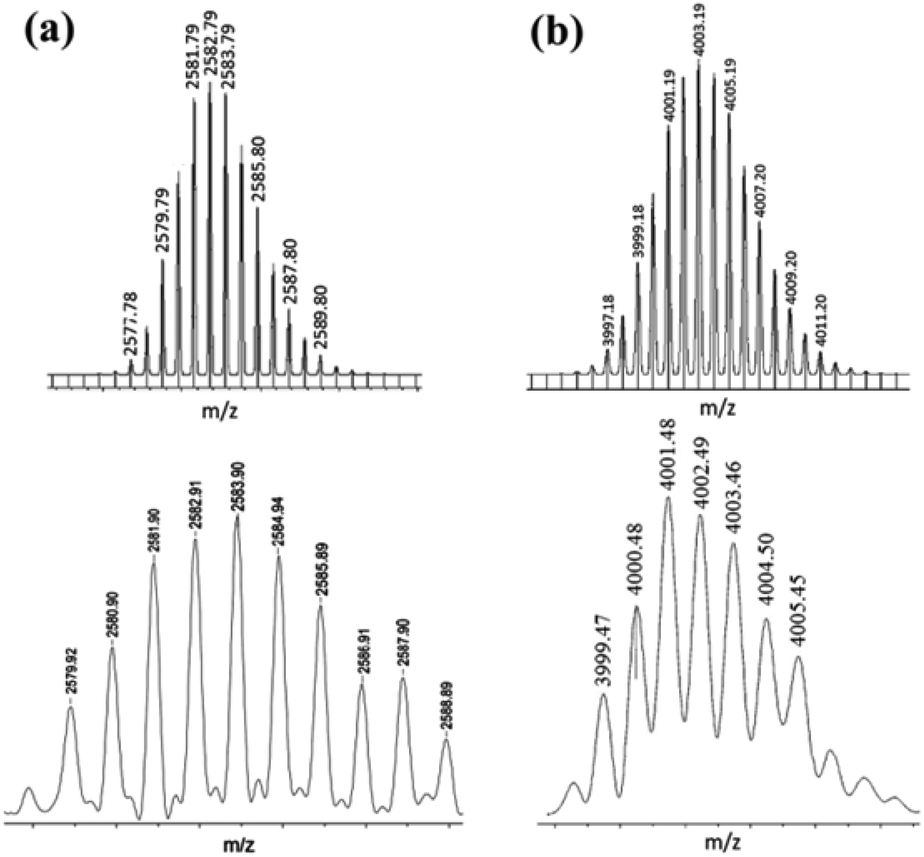 | ||
| Fig. 2 Theoretical (top) and experimental (bottom) MALDI-TOF-MS spectra of (a) macrocycle 5 and (b) macrocycle 6. | ||
Both organometallic complexes 3 and 4 have a central triptycene core and contain two Pt(II) acceptor centers (with the same predefined bite angle). Moreover, in both 3 and 4, the same phosphine ligands are attached to the Pt(II) center. However, the latter is longer in length than the former, because of the presence of the ethynyl spacer groups in 4 that bridge the triptycene core and the peripheral Pt(II) centers. Reaction of the acceptor linker 4 with disodium terephthalate proceeded in a similar manner to that described for 3, yielding a white microcrystalline solid (6, isolated yield 78%). 1H and 31P{1H} NMR (ESI†) of the product suggested the incorporation of both of the subunits (the organometallic triptycene-based acceptor unit and the terephthalate) as well as the formation of a highly symmetrical product (a sharp 31P singlet at 18.30 ppm with concomitant Pt satellites; 1JPPt = 1258 Hz). Furthermore, a single diffusion coefficient in the 1H DOSY NMR spectrum points towards the formation of a single product and rules out the possibility of an oligomeric species in the solution (ESI†). In this case also, the product obtained was subjected to MALDI-TOF-MS analysis to identify its composition. MALDI-TOF-MS analysis of 6 exhibited the formation of a [3 + 3] macrocycle due the presence of a peak at m/z = 4001.4 corresponding to a [3 + 3 + Na]+ species. This peak (m/z = 4001.4) was also isotopically resolved and was found to be in good agreement with its theoretical distribution (Fig. 2b). Interestingly, in this case, evidence for the formation of the [2 + 2] self-assembled product was not observed from MALDI-TOF-MS spectrometric studies. This is evident from the absence of molecular ion peaks for the [2 + 2] adducts at m/z = 2653.8 [2 + 2 + H]+ and 2675.7 [2 + 2 + Na]+ from MALDI-TOF-MS analysis. Thus, mass spectrometric studies indicated that the use of a longer acceptor tecton (4) results in the formation of a [3 + 3] self-assembled metallamacrocycle, while a relatively smaller acceptor tecton (3) yields a [2 + 2] supramolecular framework (Scheme 3).
All of our attempts to obtain X-ray quality single crystals in the case of the self-assembled macrocycles 5 and 6 were unsuccessful. However, to explain the observed difference in composition of the self-assembled products upon the use of acceptor linkers of different sizes, thermodynamic parameters of the dimeric and the trimeric adducts were calculated using a PM6 semiempirical molecular orbital method.13 On comparing the [2 + 2] and [3 + 3] metallacycles, it is known that the formation of the [3 + 3] adduct is entropically disfavored, as a smaller number of molecular ensembles would result compared to the number of [2 + 2] macrocycles that can be generated using the same number of building blocks.14 Therefore, in the case of linker 4, the anomalous formation of the [3 + 3] self-assembled macrocycle 6 over the entropically more preferable [2 + 2] product can be explained by comparing the stabilization energy which is brought by a single monomer of the adduct. The energy (heat of formation) optimization, calculated using the PM6 semiempirical molecular orbital method, reveals that for linker 4, the stabilization energy for a single monomer of the [3 + 3] adduct is E[3+3]/3 = (−1656.64/3) = −552.21 kJ mol−1. On the other hand, in the case of the [2 + 2] adduct, the stabilization energy of the single monomer is E[2+2]/2 = (−981.50/2) = −490.75 kJ mol−1. Therefore in case of the [3 + 3] adduct, the stabilization energy which is brought by a single monomer of the adduct is higher than that of the [2 + 2] adduct by 61.46 kJ mol−1. This energy calculation strongly supports the observed formation of a [3 + 3] macrocycle over a [2 + 2] structure in the case of linker 4 as indicated from MALDI-TOF-MS analysis. Additionally, the geometries of the macrocycles 5 and 6 were also optimized using PM6 molecular orbital methods. The internal dimensions of the macrocycles 5 and 6 are depicted in Fig. 3. The geometry optimization of macrocycles 5 and 6 reveals that in both cases, the platinum centers display slightly distorted square planar geometries resulting from the coordination of oxygen atoms of the dicarboxylate ligands.
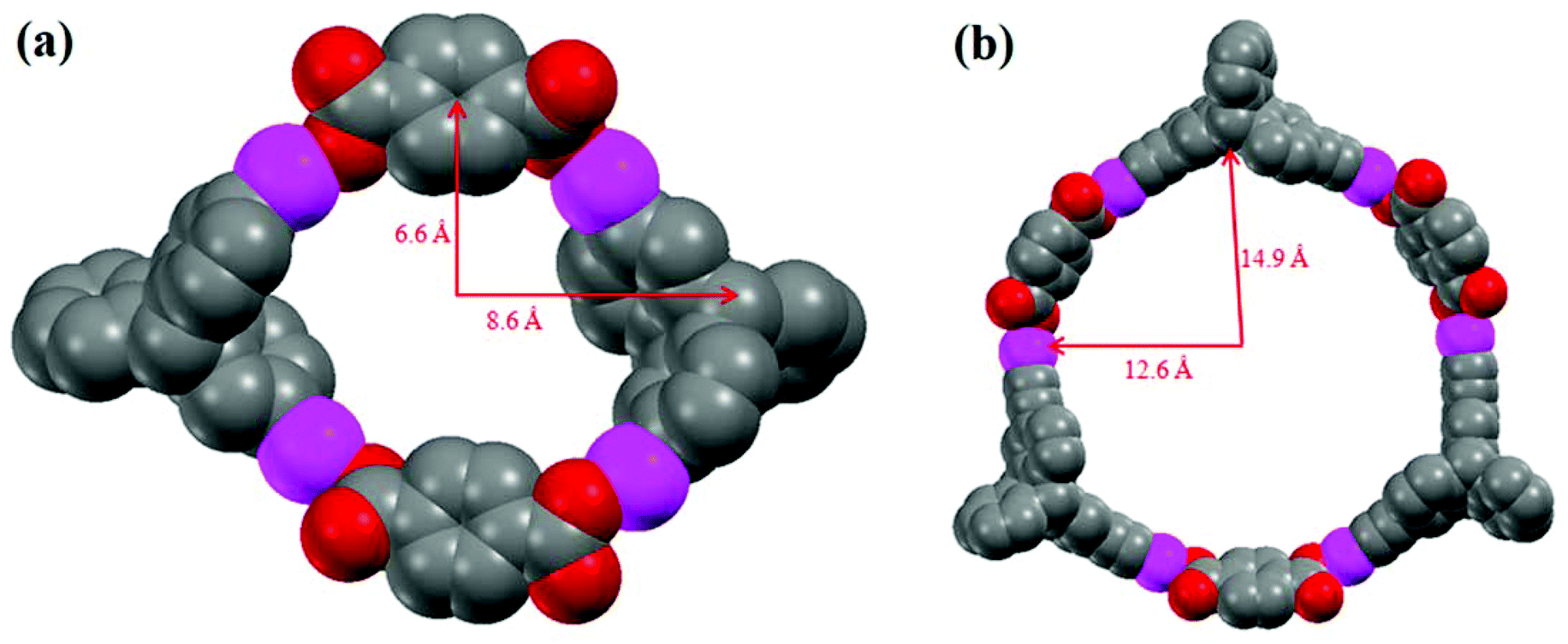 | ||
| Fig. 3 Simulated spacefill molecular models of (a) macrocycle 5 and (b) macrocycle 6 optimized using PM6 semiempirical molecular orbital methods. The phosphine ligands and H atoms are omitted for clarity (grey: C, red: O, pink: Pt). | ||
Conclusions
In summary, we report the synthesis of two new triptycene-based organoplatinum complexes containing two Pt(II) centers in reasonably good yields. In these complexes, the triptycene core is either σ-bonded to the peripheral bis(trans-trialkylphosphine)platinum units directly or via a bridging ethynyl spacer unit. These triptycene-containing ditopic PtII2 organometallic complexes and their corresponding nitrate salts were characterized using FT-IR, multinuclear NMR spectroscopy, mass spectrometry and elemental analyses. The presence of a triptycene or ethynyltriptycene backbone imparts rigidity to these organometallic complexes. Moreover, the separation of the Pt(II) centers has been spatially tuned by the inclusion or exclusion of ethynyl groups. Further, the potential of these organometallic tectons as supramolecular acceptor synthons in coordination driven self-assembly protocols has been tested/explored. In this regard, two neutral nanoscalar metallamacrocycles (5 and 6) have been constructed using these triptycene-based organoplatinum complexes in conjugation with a dicarboxylate anion (terephthalate). Multinuclear NMR spectroscopy, including 1H DOSY, of these supramolecules (5 and 6) ruled out the formation of multiple oligomeric species and suggested the formation of single highly symmetrical discrete moieties. The chemical compositions of these macrocycles were determined from mass spectrometric (MALDI-TOF-MS) analysis, which indicated the formation of a [3 + 3] self-assembled metallamacrocycle (6) in cases where the longer acceptor linker 4 was used, while the use of the relatively short linker 3 yields the [2 + 2] supramolecular framework 5. The formation of the entropically disfavoured [3 + 3] self-assembled adduct in the case of linker 4 was further supported by results obtained from computational calculations. The comparison of the stabilization energies, optimized using the PM6 semiempirical molecular orbital method, suggests the preferential formation of the [3 + 3] adduct over the corresponding [2 + 2] adduct in the case of linker 4 as experimentally observed from MALDI-TOF-MS analysis. The shapes and dimensions of these nanoscalar frameworks were also predicted from molecular simulations using PM6 semiempirical molecular orbital methods. To the best of our knowledge, the self-assembled macrocycles 5 and 6 are unique examples of triptycene-based neutral “platinamacrocycles” to date. To summarize, a facile and efficient synthetic protocol for the construction of neutral metallasupramolecular frameworks containing a triptycene motif has been described. Additionally, the effect of changing the dimension of the triptycene-based acceptor building blocks on the shape and size of the resulting supramolecular framework was studied. Triptycene-based organometallic acceptors, as described in this manuscript, have immense potential for the design of new self-assembled Supramolecular Coordination Complexes (SCCs) and Metal–Organic Frameworks (MOFs). The platinamacrocycles reported in the literature are known to be very stable and have several applications in terms of host–guest chemistry. Due to the presence of the large void space in their cavities, the newly synthesized platinamacrocycles reported herein may also have interesting host–guest properties. Studies are currently being undertaken in our laboratory in this direction.Experimental section
General details
All of the chemicals and anhydrous solvents used in this work were purchased from commercial sources and were used without further purification. 2,6-Dibromotriptycene,11 2,6-diethynyltriptycene5d and tris(triethylphosphine)platinum(0)15 were prepared by following reported literature procedures. FTIR spectra were recorded using a PerkinElmer Spectrum 400 FT-IR spectrophotometer. 1H and 31P NMR spectra were recorded using Bruker 400 or 500 MHz spectrometers. Elemental analyses were carried out using a Thermo Scientific Flash 2000 Organic Elemental Analyzer. MALDI-TOF-MS spectra of the compounds were recorded using a Bruker UltrafleXtreme™ MALDI-TOF/TOF mass spectrometer. DOSY NMR measurements were performed using a Bruker AV 500 NMR spectrometer with a 5 mm gradient probe at 298 K. DOSY experiments were done using a standard Bruker pulse sequence (ledbpgp2s) with a longitudinal eddy current delay.:1, up to 9:1, v/v) affording the pure organometallic complex 1 as a white solid.
Yield: 84 mg, 71%, mp 237–238 °C; 1H NMR (CDCl3, 400 MHz): δ 1.13–1.22 (m, 36H, –CH3), 1.48–1.57 (m, 24H, –CH2–), 5.06 (s, 2H, –CH), 6.82–6.93 (m, 6H, Ar–H), 7.26–7.29 (m, 2H, Ar–H), 7.32–7.33 (m, 2H, Ar–H). 31P NMR (CDCl3, 162 MHz): δ 12.39 (1JPPt = 1383 Hz). IR (KBr): 2961, 2925, 2876, 2852, 1585, 1450, 1413, 1378, 1255, 1240, 1168, 1035, 848, 764, 742 cm−1. Anal. Calcd For C44H72Br2P4Pt2: C, 41.45; H, 5.69. Found: C, 41.57; H, 5.81. ESI-MS: m/z calcd for [M − Br]+: 1193.3, found 1194.29.
Yield: 83 mg, 64%, mp 240–242 °C; 1H NMR (CDCl3, 400 MHz): δ 1.09–1.73 (m, 36H, –CH3), 2.14–2.20 (m, 24H, –CH2–), 5.29 (s, 2H, –CH), 6.90 (dd, J = 7.6, J = 1.2, 2H, Ar–H), 6.96–6.99 (m, 2H, Ar–H), 7.20–7.22 (d, J = 7.6, 2H, Ar–H), 7.25 (s, 2H, Ar–H), 7.34–7.36 (m, 2H, Ar–H). 31P NMR (CDCl3, 162 MHz): δ 8.56 (1JPPt = 1166 Hz). IR (KBr): 3061, 2961, 2930, 2874, 2111, 1610, 1465, 1409, 1376, 1251, 1031, 767, 742 cm−1. Anal. Calcd For C48H72I2P4Pt2: C, 40.69; H, 5.12. Found: C, 40.82; H, 5.26. ESI-MS: m/z calcd for [M − I]+: 1289.28, found 1289.00.
Complex 3. Yield: 35 mg, 94%, mp 234–236 °C; 1H NMR (CDCl3, 400 MHz): δ 0.95–1.09 (m, 36H, –CH3), 1.24–1.46 (m, 24H, –CH2–), 5.10 (s, 2H, –CH), 6.78 (d, J = 7.6 Hz, 2H, Ar–H), 6.86 (d, J = 7.2, 2H, Ar–H), 6.95–6.98 (m, 2H, Ar–H), 7.30–7.32 (m, 4H, Ar–H). 31P NMR (CDCl3, 162 MHz): δ 18.23 (1JPPt = 1453 Hz). IR (KBr): 2962, 2925, 2875, 2853, 1585, 1465, 1444, 1376, 1276, 1237, 1034, 997, 765, 738 cm−1. Anal. Calcd For C44H72N2O6P4Pt2: C, 44.79; H, 5.64; N, 2.18. Found: C, 44.92; H, 5.74; N, 2.25. ESI-MS: m/z Calcd for [M − NO3]+: 1176.37, Found: 1176.13.
Complex 4. Yield: 36 mg, 93%, mp 204–206 °C; 1H NMR (CDCl3, 500 MHz): δ 1.15–1.21 (m, 36H, –CH3), 1.87–1.93 (m, 24H, –CH2–), 5.31 (s, 2H, –CH), 6.87 (d, J = 7.2 Hz, 2H, Ar–H), 6.99–7.00 (m, 2H, Ar–H), 7.21–7.23 (m, 4H, Ar–H), 7.35–7.37 (m, 2H, Ar–H). 31P NMR (CDCl3, 202 MHz): δ 20.19 (1JPPt = 1244 Hz). IR (KBr): 2966, 2932, 2877, 2121, 1476, 1467, 1384, 1277, 1036, 989, 768, 743 cm−1. Anal. Calcd For C48H72N2O6P4Pt2: C, 41.29; H, 5.64; N, 2.33. Found: C, 41.37; H, 5.71; N, 2.4. ESI-MS: m/z Calcd for [M − NO3]+: 1224.37, Found: 1224.40.
Macrocycle 5. Yield 13.7 mg, 81%; 1H NMR (CDCl3, 500 MHz): δ 0.95–1.04 (m, 72H, –CH3), 1.30–1.53 (m, 48H, –CH2–), 5.07 (s, 4H, –CH), 6.86 (s, 4H, Ar–H), 6.94–6.95 (m, 8H, Ar–H), 7.30–7.31 (m, 4H, Ar–H), 7.39 (s, 4H, Ar–H), 7.96 (s, 8H, Ar–H). 31P NMR (CDCl3, 162 MHz): δ 16.14 (1JPPt = 1459 Hz). Anal. Calcd For C104H152O8P8Pt4: C, 48.82; H, 5.99. Found: C, 48.65; H, 6.13. IR (KBr): 2963, 2929, 2877, 1611, 1454, 1413, 1378, 1330, 1253, 1034, 821, 766, 743 cm−1. MALDI-TOF-MS: m/z Calcd for [M + H]+: 2560.81, Found: 2560.9 and Calcd for [M + Na]+: 2582.79, Found: 2582.9.
Macrocycle 6. Yield 13.76 mg, 78%; 1H NMR (CDCl3, 500 MHz): δ 1.14–1.19 (m, 108H, –CH3), 1.87–1.90 (m, 72H, –CH2–), 5.28 (s, 6H, –CH), 6.85 (d, J = 7.5 Hz, 6H, Ar–H), 6.96–6.97 (m, 6H, Ar–H), 7.20–7.21 (m, 12H, Ar–H), 7.33–7.35 (m, 6H, Ar–H), 7.95 (s, 12H, Ar–H). 31P NMR (CDCl3, 162 MHz): δ 18.30 (1JPPt = 1258 Hz). Anal. Calcd For C168H228O12P12Pt6: C, 50.68; H, 5.77. Found: C, 50.45; H, 5.91. IR (KBr): 2963, 2926, 2877, 2851, 2114, 1625, 1465, 1377, 1328, 1034, 822, 766, 742 cm−1. MALDI-TOF-MS: m/z Calcd for [M + Na]+: 4001.19, Found: 4001.4.
Authors’ contributions
N.D. and S.C. conceived the research idea. S.C. synthesized all of the new triptycene-based organometallic complexes and the neutral metallacycles reported in this manuscript. S.B. assisted in the synthesis of some of the literature reported triptycene precursors. J.M. and H.T. optimized the energy minimized geometry of the metallacycles 5 and 6 using the PM6 semiempirical molecular modeling method. All of the authors have contributed to compiling the manuscript and have approved the final manuscript.Acknowledgements
N.D. thanks the Department of Science and Technology, Govt. of India, New Delhi (DST no: SR/FT/CS-028/2009) and the Indian Institute of Technology Patna for financial support. The authors acknowledge Dr P.R. Rajamohanan, Central NMR Facility, CSIR-NCL, Pune for recording DOSY NMR spectra. The authors also acknowledge Dr Sivaramaiah Nallapeta (Bruker Daltonics India) for MALDI-TOF mass spectrometric data. S.C. and S.B. thank IIT Patna for an Institute Research Fellowship. The authors acknowledge SAIF-Panjab University and SID, IISc Bangalore for providing analytical facilities.References
- (a) J. W. Steed, D. R. Turner and K. J. Wallace, Core Concepts in Supramolecular Chemistry and Nano chemistry, Wiley, West Sussex, UK, 2007 Search PubMed; (b) J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4763 CrossRef CAS PubMed; (c) J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley & Sons, Ltd, United Kingdom, 2009 Search PubMed; (d) M. Fujita, D. Oguro, M. Miyazawa, H. Oka, K. Yamaguchi and K. Ogura, Nature, 1995, 378, 469 CrossRef CAS; (e) B. Olenyuk, J. A. Whiteford, A. Fechtenkotter and P. J. Stang, Nature, 1999, 398, 796 CrossRef CAS PubMed; (f) K. N. Raymond, Nature, 2009, 460, 585 CrossRef CAS.
- (a) T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734 CrossRef CAS PubMed; (b) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810 CrossRef CAS PubMed; (c) M. D. Pluth, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2009, 42, 1650 CrossRef CAS PubMed; (d) M. Yoshizawa and M. Fujita, Bull. Chem. Soc. Jpn., 2010, 83, 609 CrossRef CAS; (e) M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418 CrossRef CAS PubMed; (f) C. G. Oliveri, P. A. Ulmann, M. J. Wiester and C. A. Mirkin, Acc. Chem. Res., 2008, 41, 1618 CrossRef CAS PubMed; (g) D. Samanta and P. S. Mukherjee, Chem. Commun., 2014, 50, 1595 RSC; (h) N. J. Young and B. P. Hay, Chem. Commun., 2013, 49, 1354 RSC; (i) P. Thanasekaran, C.-C. Lee and K.-L. Lu, Acc. Chem. Res., 2012, 45, 1403 CrossRef CAS PubMed; (j) B. Therrien, Chem. – Eur. J., 2013, 19, 8378 CrossRef CAS PubMed; (k) T. R. Cook, V. Vajpayee, M. H. Lee, P. J. Stang and K.-W. Chi, Acc. Chem. Res., 2013, 46, 2464 CrossRef CAS PubMed; (l) J. K. Clegga, F. Li and L. F. Lindoy, Coord. Chem. Rev., 2013, 257, 2536 CrossRef PubMed; (m) M. L. Saha, S. Neogi and M. Schmittel, Dalton Trans., 2014, 43, 3815 RSC.
- (a) B. H. Northrop, H.-B. Yang and P. J. Stang, Chem. Commun., 2008, 5896 RSC and references therein; (b) M. Fujita, S.-Y. Yu, T. Kusukawa, H. Funaki, K. Ogura and K. Yamaguchi, Angew. Chem., Int. Ed., 1998, 37, 2082 CrossRef CAS; (c) L. Xu, L.-J. Chen and H.-B. Yang, Chem. Commun., 2014, 50, 5156 RSC and references therein; (d) N. Das, A. M. Arif, P. J. Stang, M. Sieger, B. Sarkar, W. Kaim and J. Fiedler, Inorg. Chem., 2005, 44, 57984 Search PubMed; (e) N. Das, P. J. Stang, A. M. Arif and C. F. Campana, J. Org. Chem., 2005, 70, 10440 CrossRef CAS PubMed.
- (a) C.-F. Chen, Chem. Commun., 2011, 47, 1674 RSC and references therein; (b) Y. Jiang and C.-F. Chen, Eur. J. Org. Chem., 2011, 6377 CrossRef CAS and references therein; (c) C.-F. Chen and Y.-X. Ma, Iptycene Chemistry: from Synthesis to Applications, Springer-Verlag, Berlin, 2013 Search PubMed; (d) J. H. Chong and M. J. MacLachlan, Chem. Soc. Rev., 2009, 38, 3301 RSC and references therein; (e) A. K. Crane, B. O. Patrick and M. J. MacLachlan, Dalton Trans., 2013, 42, 8026 RSC; (f) A. K. Crane, E. Y. L. Wong and M. J. MacLachlan, CrystEngComm, 2013, 15, 9811 RSC; (g) M. Mastalerz, Synlett, 2013, 781 CrossRef CAS PubMed and references therein; (h) L. Zhao, Z. Li and T. Wirth, Chem. Lett., 2010, 39, 658 CrossRef CAS; (i) S. Chakraborty, S. Mondal and N. Das, Inorg. Chim. Acta, 2014, 413, 214 CrossRef CAS PubMed; (j) Y. Han, Z. Meng, Y.-X. Ma and C.-F. Chen, Acc. Chem. Res., 2014, 47, 2026 CrossRef CAS PubMed and references therein.
- (a) T. M. Swager, Acc. Chem. Res., 2008, 41, 1181 CrossRef CAS PubMed; (b) B. VanVeller, D. Robinson and T. M. Swager, Angew. Chem., Int. Ed., 2012, 51, 1182 CrossRef CAS PubMed; (c) B. VanVeller, D. J. Schipper and T. M. Swager, J. Am. Chem. Soc., 2012, 134, 7282 CrossRef CAS PubMed; (d) S. Mondal, S. Chakraborty, S. Bhowmick and N. Das, Macromolecules, 2013, 46, 6824 CrossRef CAS; (e) C. Zhang, Y. Liu, B. Li, B. Tan, C.-F. Chen, H.-B. Xu and X.-L. Yang, ACS Macro Lett., 2012, 1, 190 CrossRef CAS; (f) C. Zhang, Z. Wang, J.-J. Wang, L. Tan, J.-M. Liu, B. Tan, X.-L. Yang and H.-B. Xu, Polymer, 2013, 54, 6942 CrossRef CAS PubMed; (g) C. Zhang, J.-J. Wang, Y. Liu, H. Ma, X.-L. Yang and H.-B. Xu, Chem. – Eur. J., 2013, 19, 5004 CrossRef CAS PubMed; (h) S. Mondal and N. Das, RSC Adv., 2014, 4, 61383 CAS.
- (a) C. Azerraf and D. Gelman, Organometallics, 2009, 28, 6578 CrossRef CAS; (b) C. Azerraf and D. Gelman, Chem. – Eur. J., 2008, 14, 10364 CrossRef CAS PubMed; (c) A. Ishii, N. Nakata, R. Uchiumi and K. Murakami, Angew. Chem., Int. Ed., 2008, 47, 2661 CrossRef CAS PubMed; (d) A. Ishii, H. Kamon, K. Murakami and N. Nakata, Eur. J. Org. Chem., 2010, 1653 CrossRef CAS; (e) N. Nakata, R. Uchiumi, T. Yoshino, T. Ikeda, H. Kamon and A. Ishii, Organometallics, 2009, 28, 1981 CrossRef CAS; (f) Y. Yamaguchi, N. Nakata and A. Ishii, Eur. J. Inorg. Chem., 2013, 30, 5233 CrossRef; (g) S. Toyota, H. Okuhara and M. Ōki, Organometallics, 1997, 16, 4012 CrossRef CAS; (h) H.-B. Yang, K. Ghosh, N. Das and P. J. Stang, Org. Lett., 2006, 8, 3991 CrossRef CAS PubMed.
- (a) S. Chakraborty, S. Mondal, Q. Li and N. Das, Tetrahedron Lett., 2013, 54, 1681 CrossRef CAS PubMed; (b) S. Chakraborty, S. Mondal, S. Bhowmick, J. Ma, H. Tan, S. Neogi and N. Das, Dalton Trans., 2014, 43, 13270 RSC; (c) A. Dubey, A. Mishra, J. W. Min, M. H. Lee, H. Kim, P. J. Stang and K.-W. Chi, Inorg. Chim. Acta, 2014, 423, 326 CrossRef CAS PubMed.
- J. Manna, C. J. Kuehl, J. A. Whiteford and P. J. Stang, Organometallics, 1997, 16, 1897 CrossRef CAS.
- S. Leininger and P. J. Stang, Organometallics, 1998, 17, 3981 CrossRef CAS.
- (a) S. Leininger, M. Schmitz and P. J. Stang, Org. Lett., 1999, 1, 1921 CrossRef CAS; (b) C. J. Kuehl, C. L. Mayne, A. M. Arif and P. J. Stang, Org. Lett., 2000, 2, 3727 CrossRef CAS PubMed; (c) Y. K. Kryschenko, S. R. Seidel, A. M. Arif and P. J. Stang, J. Am. Chem. Soc., 2003, 125, 5193 CrossRef CAS PubMed; (d) H.-B. Yang, A. M. Hawkridge, S. D. Huang, N. Das, S. D. Bunge, D. C. Muddiman and P. J. Stang, J. Am. Chem. Soc., 2007, 129, 2120 CrossRef CAS PubMed.
- Z. Chen and T. M. Swager, Macromolecules, 2008, 41, 6880 CrossRef CAS.
- (a) H.-B. Yang, K. Ghosh, N. Das and P. J. Stang, Org. Lett., 2006, 8, 3991 CrossRef CAS PubMed; (b) M.-C. K. Wong and V. W.-W. Yam, Acc. Chem. Res., 2011, 44, 424 CrossRef PubMed; (c) C.-H. Tao and V. W.-W. Yam, J. Photochem. Photobiol., C, 2009, 10, 130 CrossRef CAS PubMed; (d) S. Li, J. Huang, F. Zhou, T. R. Cook, X. Yan, Y. Ye, B. Zhu, B. Zheng and P. J. Stang, J. Am. Chem. Soc., 2014, 136, 5908 CrossRef CAS PubMed; (e) S. Shanmugaraju, H. Jadhav, Y. P. Patil and P. S. Mukherjee, Inorg. Chem., 2012, 51, 13072 CrossRef CAS PubMed; (f) S. Shanmugaraju, A. K. Bar, K.-W. Chi and P. S. Mukherjee, Organometallics, 2010, 29, 2971 CrossRef CAS; (g) J. Zhang, X.-D. Xu, L.-J. Chen, Q. Luo, N.-W. Wu, D.-X. Wang, X.-L. Zha and H.-B. Yang, Organometallics, 2011, 30, 4032 CrossRef CAS; (h) W. Wang and H.-B. Yang, Chem. Commun., 2014, 50, 5171 RSC and references therein; (i) D. Samanta and P. S. Mukherjee, Dalton Trans., 2013, 42, 16784 RSC.
- J. J. P. Stewart, J. Mol. Model, 2007, 13, 1173 CrossRef CAS PubMed.
- N. Das, A. Ghosh, A. M. Arif and P. J. Stang, Inorg. Chem., 2005, 44, 7130 CrossRef CAS PubMed.
- T. Yoshida, T. Matsuda, S. Otsuka, G. W. Parshall and W. G. Peet, Inorg. Synth., 1990, 28, 122 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 31P{1H} NMR spectra for complexes 1–4 and macrocycles 5 and 6. ESI-MS spectra of complexes 1–4. MALDI-TOF-MS spectra and 1H DOSY NMR spectra for macrocycles 5 and 6. See DOI: 10.1039/c4qi00211c |
| This journal is © the Partner Organisations 2015 |