
Syntheses, structures and magnetism of mixed-valence Mn19 and Mn21 complexes supported by alkylamine-based alkoxo-bridging ligands†
Hiromichi
Ida
,
Takuya
Shiga
,
Graham N.
Newton
and
Hiroki
Oshio
*
Department of Chemistry, Graduate School of Pure and Applied Sciences, University of Tsukuba, Tennodai 1-1-1, Tsukuba 305-8571, Japan. E-mail: oshio@chem.tsukuba.ac.jp; Fax: +81 29 853 4238; Tel: +81 29 853 4238
First published on 8th April 2015
Abstract
Two new high-nuclearity mixed-valence Mn clusters, Mn19 and Mn21, were synthesised utilising an alkylamine-based flexible ligand with five alkoxo-bridging groups, 1,3-bis-diethanolamino-2-propanol (H5bdp). They were obtained from the reaction of Mn(O2CPh)2·2H2O with H5bdp·2HCl·H2O and Et3N in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:7 molar ratio in CH3CN/CH3OH mixed solution. The X-ray crystal structures revealed {MnII8MnIII10MnIV} and {MnII6MnIII15} mixed-valence cores composed around {Mn7O8} corner-shared double cubanes. Magnetic susceptibility measurements on both complexes indicated dominant intramolecular antiferromagnetic interactions between Mn ions. No clear SMM behaviour was observed above 1.8 K due to the non-parallel alignment of magnetic anisotropy axes of the MnIII ions.
:1:7 molar ratio in CH3CN/CH3OH mixed solution. The X-ray crystal structures revealed {MnII8MnIII10MnIV} and {MnII6MnIII15} mixed-valence cores composed around {Mn7O8} corner-shared double cubanes. Magnetic susceptibility measurements on both complexes indicated dominant intramolecular antiferromagnetic interactions between Mn ions. No clear SMM behaviour was observed above 1.8 K due to the non-parallel alignment of magnetic anisotropy axes of the MnIII ions.
Introduction
Polynuclear transition metal complexes attract substantial attention due to their physical properties arising from the synergistic effects between metal ions.1 Such complexes are nano-sized materials, commonly synthesised through a combination of informed molecular design and serendipitous self-assembly, and exhibit significantly different physico-chemical properties from bulk materials or nanoparticles. Furthermore, control of their structural topology and the oxidation states of their constituent metal ions can be attained by chemical modification of the supporting organic ligands.In the study of transition metal complexes, nuclearity is one of the most important metrics, as it can directly influence the magnetic and electronic properties of the cluster.2 Similarly important is the manner in which metal centres are connected, as the properties of the cluster depend upon the degree to which the electronic or magnetic states of neighbouring metal centres are correlated.3 Small ionic ligands can mediate strong electronic or magnetic cooperativity between metal centres. For example, cyanide bridges can allow stimuli-responsive electron transfers in heterometal systems,4 and oxo bridges can mediate spin dependent electron delocalization in manganese clusters.5 While the directional nature of linear or 1D bridging ligands such as cyanide, azide and pyrazole allows their use in a controlled, or building block-type approach to the synthesis of high nuclearity clusters, the monoatomic nature of the oxo dianion ensures that the targeted generation of oxo-bridged clusters remains a challenging goal.
With the exception of polyoxometalates6 (POMs; discrete anionic metal–oxide species based on high valent V, Mo and W), which have been reported with nuclearities up to [Mo368],7 the synthesis of molecular metal oxide species is hampered by the harsh conditions required in such reactions, which can often yield bulk metal oxide and/or amorphous metal hydroxide. For this reason, the chemistry of transition metal oxide clusters is relatively under-developed, although in recent years there have been an increasing number of publications on their syntheses.8
Polynuclear Mn oxide clusters, consisting of Mn ions in their di-, tri- and tetravalent states, are a particularly attractive group of metal oxide clusters owing to their fascinating electrochemical9 and catalytic properties,10 and their propensity to display single molecule magnet (SMM) behaviour.11 Alterations to their synthetic conditions and the nature of the capping ligands used can allow the isolation of Mn oxide clusters with a wide range of nuclearities, geometries and electronic (oxidation) states. To date, Mn oxide clusters8a,12 with nuclearities of up to Mn84,12a have been reported.
Previous studies in our group have illustrated the potential for both flexible9,13 and rigid ligands14 to stabilize polynuclear complexes of various transition metals, while flexible multidentate ligands with alkoxo-bridging groups have been shown to be able supports for Mn oxide clusters.12b,15 In this study, we use an alkylamine based ligand with a large number of alkoxo bridging groups to aid in the isolation of high nuclearity Mn oxide clusters. Herein, we report the syntheses, crystal structures and magnetic properties of two Mn oxide clusters, Mn19 and Mn21, supported by the alkylamine based flexible ligands, H5bdp and H4bdp*, which have five and four alkoxo-bridging moieties, respectively (Scheme 1).
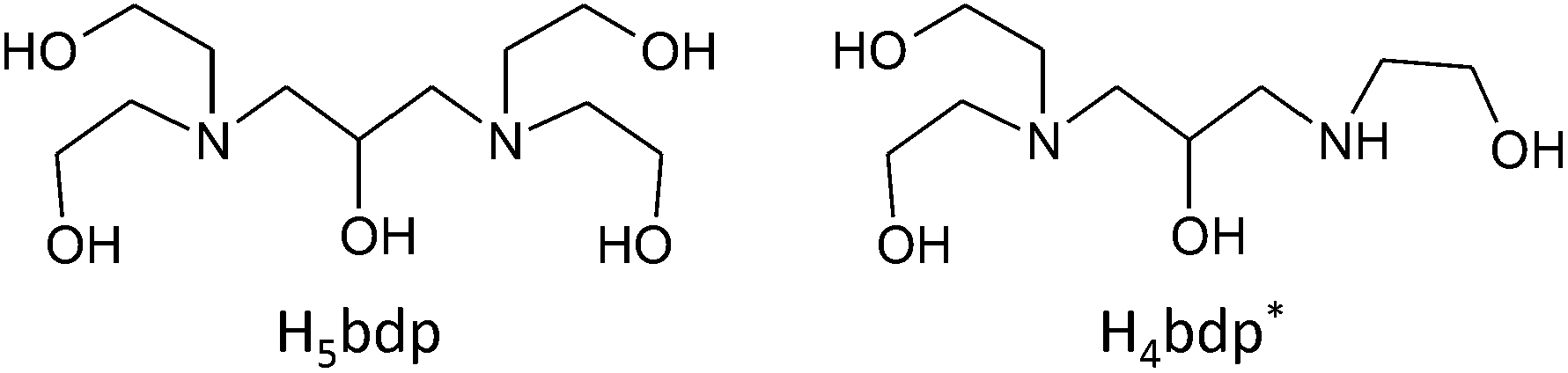 | ||
| Scheme 1 Molecular structures of the ligand H5bdp and its decomposition product H4bdp*. | ||
Experimental
Materials and physical measurements
All reagents and solvents were purchased from commercial suppliers and used without further purification. All experiments were carried out under ambient conditions. Mn(O2CPh)2·2H2O16 and the ligand 1,3-bis-diethanolamino-2-propanol (H5bdp·2HCl·H2O)17 were prepared according to the literature procedures. Elemental analyses were performed using a Perkin Elmer 2400 elemental analyser. Magnetic data were measured with a MPMS XL5 SQUID susceptometer (Quantum Design). DC magnetic susceptibilities were measured in a 1.8–300 K temperature range under an applied magnetic field of 500 Oe. The data were corrected for diamagnetic contributions using Pascal's constants. Ac magnetic susceptibilities were measured in a 1.8–5.0 K temperature range under an ac magnetic field of 3 Oe and zero dc field in the frequency range of 1–1000 Hz.Syntheses
X-ray crystallography
Diffraction data were collected using a Bruker SMART APEX II diffractometer equipped with a CCD type area detector with graphite monochromated MoKα (λ = 0.71073 Å) radiation. An empirical absorption correction was applied using SADABS. The structures were solved using direct methods and refined by the full-matrix least squares method using the SHELXTL package. Non-hydrogen atoms were refined using anisotropic thermal parameters. The SQUEEZE program was used to remove the contribution of the highly disordered solvent molecules from the structural calculations. Hydrogen atoms were included in calculated positions and refined with isotropic thermal parameters riding on those of the parent atoms. A summary of the crystallographic parameters and data is given in Table 1.| 1 | 2 | |
|---|---|---|
| Formula | C150H182N4Cl2Mn19O80 | C140H224N9Mn21O95 |
| M W | 4435.75 | 4707.01 |
| T [K] | 100 | 100 |
| Crystal system | Monoclinic | Triclinic |
| Space group | C2/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a [Å] | 35.738(7) | 15.472(5) |
| b [Å] | 19.551(4) | 19.315(6) |
| c [Å] | 25.253(5) | 33.584(10) |
| α [°] | 90 | 96.546(5) |
| β [°] | 99.033(3) | 102.138(4) |
| γ [°] | 90 | 109.674(4) |
| V [Å3] | 17426(6) |
9051(5) |
| Z | 4 | 2 |
| ρ calc [g cm−3] | 1.691 | 1.782 |
| μ [mm−1] | 1.452 | 1.512 |
| GOF on F2 | 1.029 | 0.877 |
| R 1 [I > 2σ(I)] | 0.0760 | 0.0723 |
| wR2 (all data) | 0.1975 | 0.1642 |
Results & discussion
The flexible multidentate ligand 1,3-bis(diethanolamino)-2-propanol (H5bdp; Scheme 1 left) was obtained according to the previously reported method17 from the one-pot reaction of epichlorohydrine and diethanolamine, and isolated as the hydrochloride salt H5bdp·2HCl·H2O. Addition of base to solutions of H5bdp with manganese salts under basic conditions was expected to lead to deprotonation of the ligand hydroxyl groups, enabling it to interact with multiple metal centres, and stabilize high nuclearity molecular manganese oxide clusters.The reaction of Mn(O2CPh)2·2H2O, H5bdp·2HCl·H2O and Et3N in a 4:1:7 molar ratio in MeOH/MeCN (1:4) mixed solution under aerobic condition afforded a brown precipitate and a dark-brown solution. The precipitate was collected by vacuum filtration and dissolved in 1,2-dichloroethane, the slow evaporation of which gave single crystals of a nonadecanuclear mixed-valence complex, [MnII8MnIII10MnIVO10(OH)4(Hbdp)2(O2CPh)18]·4H2O (1·4H2O) after a few days. Slow evaporation of the dark brown mother liquor yielded single crystals of a heneicosanuclear mixed-valence complex, [MnII6MnIII15O12(OH)4(bdp)2(Hbdp*)(H2bdp*)(O2CPh)14]·7H2O (2·7H2O), after approximately 1 month, during which time partial decomposition of the ligand H5bdp occurred through elimination of one hydroxyethyl group to give H4bdp* (Scheme 1, right).
Complex 1 crystallised in the monoclinic space group C2/c, and the molecule lies on a crystallographic inversion centre (Fig. 1). The structure of 1 consists of a mixed-valent {Mn19} core comprising one MnIV, ten MnIII and eight MnII ions, which are bridged by eight μ4-oxo, two μ3-oxo and four μ3-hydroxo ligands. The core is further capped by two partially deprotonated Hbdp4− ligands in μ7:η3:η1:η1:η2:η1:η3:η2 bridging mode (Scheme 2 top) and eighteen benzoate groups, and, as a result, the complex molecule has an overall neutral charge. The oxidation states of the Mn ions were assigned according to their bond lengths, coordination environments and bond valence sum (BVS) calculations (Table S1†).18 The degree of protonation of the inorganic O atoms was also confirmed by BVS calculations (Table S2†). The molecules are connected by intermolecular hydrogen bonds to form a columnar structure in the crystal lattice (Fig. S1†), in which the shortest Mn⋯Mn distance is ∼6.9 Å.
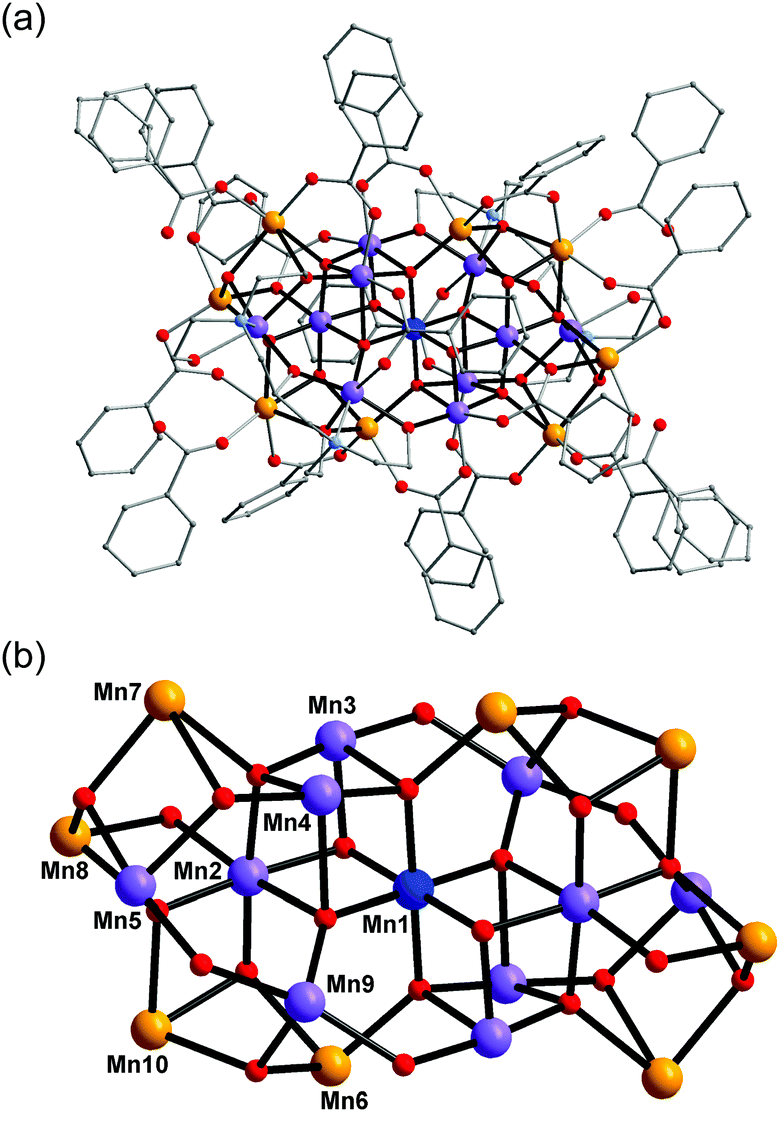 | ||
| Fig. 1 Structure of complex 1 (a) and its core (b). Colour scheme: MnII orange; MnIII purple; MnIV blue; O red; N light blue; C grey. | ||
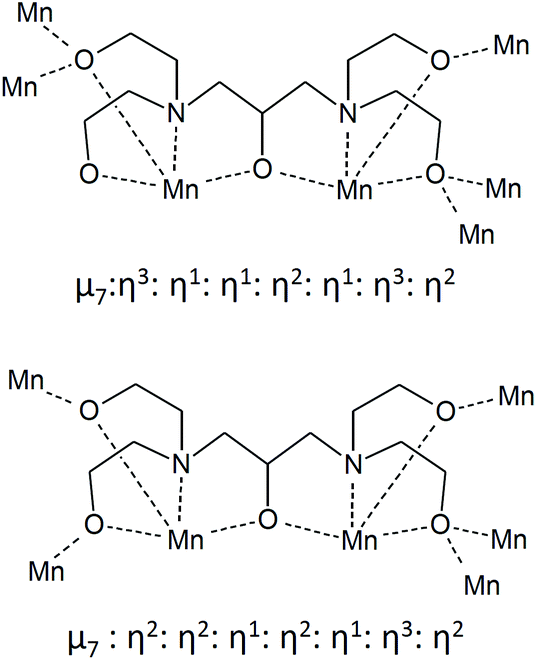 | ||
| Scheme 2 The coordination modes of deprotonated H5bdp ligands in 1 (top) and 2 (bottom). | ||
Complex 2 crystallised in the triclinic space group P (Fig. 2). In contrast to 1, the complex molecule 2 displays no crystallographic symmetry. The structure of 2 consists of a mixed-valence {Mn21} core comprising fifteen MnIII and six MnII ions, which are bridged by eight μ4-oxo, four μ3-oxo, three μ3-hydroxo and one μ2-hydroxo ligands. The core is further capped by two fully deprotonated bdp5− in η2:η2:η1:η2:η1:η3:η2:μ7 bridging mode (Scheme 2 bottom), two partially deprotonated Hbdp*4−/H2bdp*3− ligands and fourteen benzoate groups, as a result the complex molecule has an overall neutral charge. The oxidation states of the Mn ions were assigned according to their bond lengths, coordination environments and bond valence sum (BVS) calculations (Table S3†). The degree of protonation of the inorganic O atoms was also confirmed by BVS calculations (Table S4†). The molecules are linked by hydrogen bonds to form a supramolecular columns in the crystal lattice (Fig. S2†), with a minimum intermolecular Mn⋯Mn distance of ∼8.1 Å.
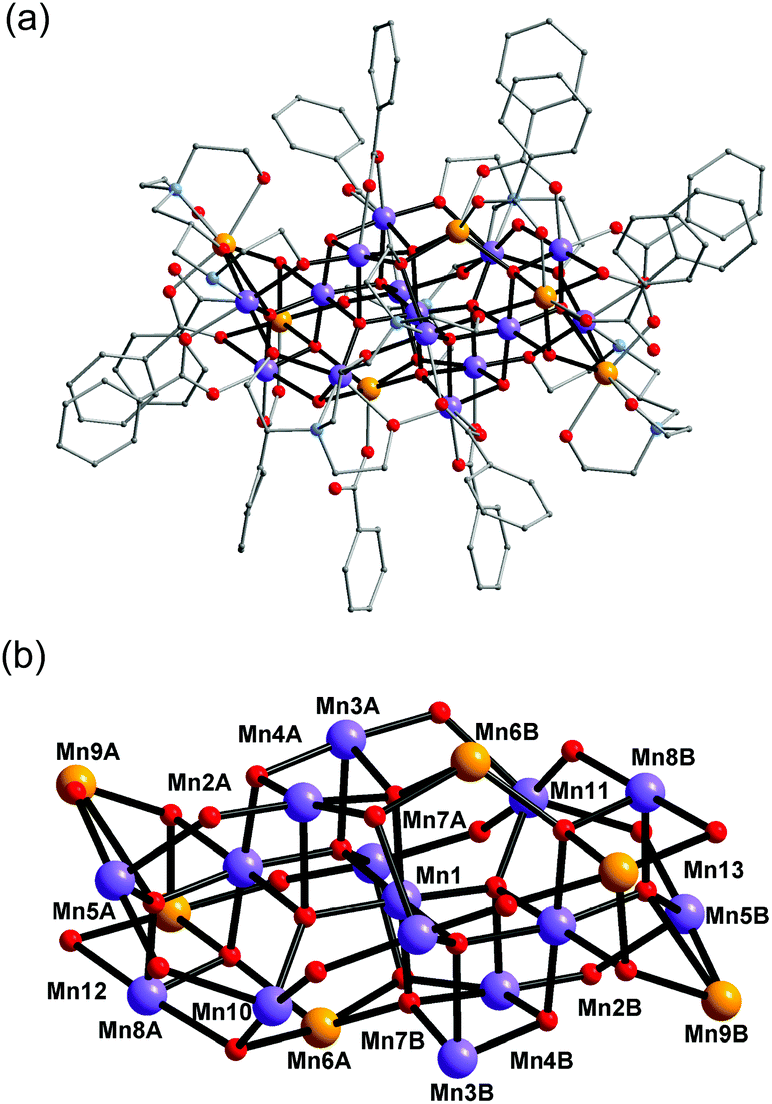 | ||
| Fig. 2 Structure of complex 2 (a) and its core (b). Colour scheme is given in Fig. 1. | ||
In both 1 and 2, the Mn oxide core is based around a corner-sharing double cubane, which consists of seven Mn atoms, six μ4-oxo and two μ3-oxo groups (Fig. 3). It is interesting to note that while cubane and double-cubane units are common structural motifs in manganese cluster chemistry, the previous examples of 19- and 21-nuclear Mn clusters have generally been based on open or defective cubane-type topologies.8a In 1, the central octahedral Mn1 atom of the double cubane is MnIV and is coordinated by six bridging oxo ions with an average bond distance of 1.915(4) Å. The remaining Mn ions that constitute the double cubane (Mn2, Mn3 and Mn4) are trivalent and exhibit Jahn–Teller distorted octahedral coordination environments with axial (Mn–Oax) and equatorial (Mn–Oeq) Mn–O bond distances in the range of 2.114(4)–2.326(4) Å and 1.901(4)–1.985(4) Å, respectively. In contrast, all Mn atoms in the double cubane at the core of complex 2 (Mn1–Mn4B) are trivalent and exhibit Jahn–Teller distorted octahedral coordination environments with Mn–Oax and Mn–Oeq bond distances in the range of 2.116(5)–2.440(5) Å and 1.853(4)–2.053(5) Å, respectively. The double cubanes of both 1 and 2 are capped by MnIII/MnII mixed-valence shells through a combination of inorganic oxo and hydroxo ions, the alkoxo groups of the capping ligands and the carboxylate moieties of the benzoate ions. In addition to these ligands, the two H4bdp* ligands formed during the reaction period act as a bridge between the double cubane core and the mixed-valence shell in complex 2.
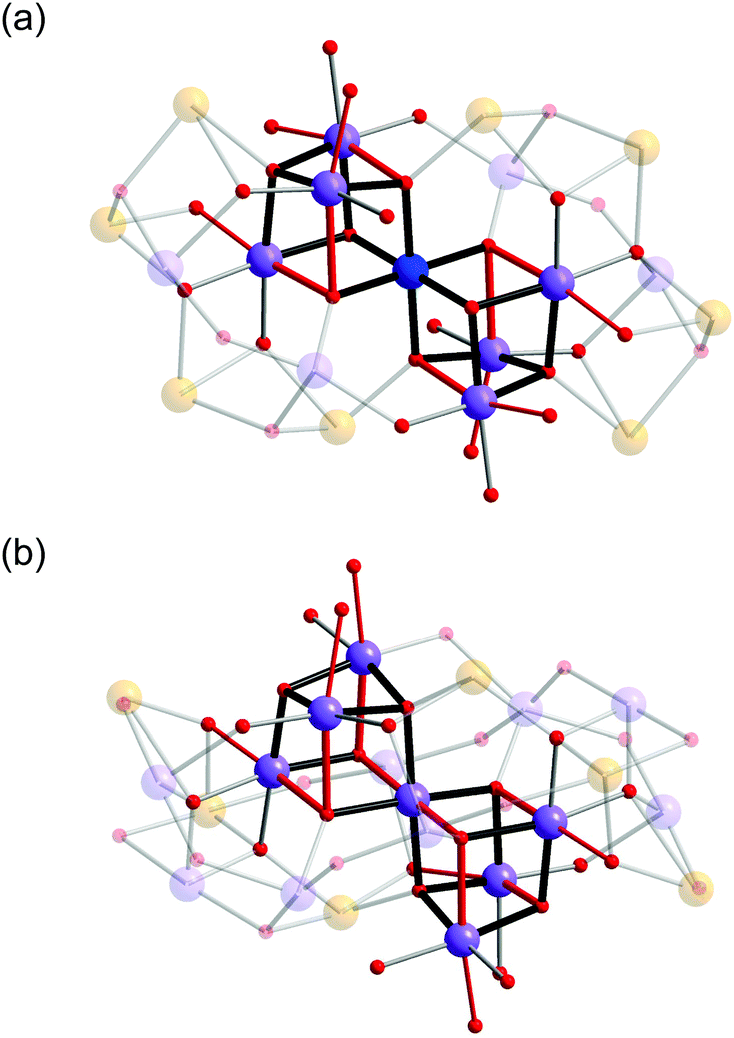 | ||
| Fig. 3 Details of {Mn7O8} double cubanes in the core of 1 (top) and 2 (bottom). The red coloured bonds indicate elongated Jahn–Teller axes of MnIII ions. Colour scheme is given in Fig. 1. | ||
In 1, all six MnIII ions in the mixed-valence shell (Mn5 and Mn9) have Jahn–Teller distorted octahedral coordination environments with {O5N} coordination spheres in which the N atoms of the Hbdp4− ligands occupy their equatorial positions. Of the eight MnII ions in the shell, the Mn10 ion has a distorted octahedral coordination environment, while the Mn7 ion exists in an {O6} coordination sphere which includes one elongated Mn–O interaction with a distance of 2.469(4) Å; the five remaining Mn–O interactions are in the range of 2.083(4)–2.299(4) Å. The remaining MnII atoms, Mn6 and Mn8, have highly distorted five coordinated geometries.
In 2, all eight MnIII ions in the shell (Mn5A, Mn5B, Mn7A, Mn7B, Mn8A, Mn8B, Mn10 and Mn11) have Jahn–Teller distorted octahedral coordination environments. Two (Mn8A and Mn8B) are in {O6} ligand fields, while the remainder (Mn5A, Mn5B, Mn7A, Mn7B, Mn10 and Mn11) have {O5N} coordination spheres in which the N atoms of the bdp5− and the Hbdp*3−/H2bdp*2− ligands occupy equatorial positions. Of the six MnII ions in the shell, four exhibit distorted octahedral coordination environments and two (Mn6A and Mn6B) have highly distorted five coordinated geometries.
Temperature dependent dc magnetic susceptibilities were measured for 1·4H2O and 2·7H2O in the temperature range of 1.8–300 K, and the results are plotted in Fig. 4. At 300 K, the χT value of 1·4H2O is 49.7 emu K mol−1, which is much smaller than the spin only value of 66.9 emu K mol−1 expected for the uncorrelated spins of eight MnII, ten MnIII and one MnIV ion. As the temperature is lowered, the χT value decreases gradually at first, and then rapidly to reach a minimum value of 1.53 emu K mol−1 at 1.8 K, indicating dominant antiferromagnetic interactions are operative between the metal centres. For 2·7H2O, the χT value is 38.8 emu K mol−1 at 300 K, which is much smaller than the spin only value of 71.3 emu K mol−1 expected for the uncorrelated spins of six MnII and fifteen MnIII ions. The χT value decreases with decreasing temperature, reaching a plateau at about 20 K with a value of ∼11.6 emu K mol−1 (corresponding to a S = 10/2 or 8/2 ground spin state, depend on the g value). On further cooling below 5 K, the χT value again decreases to a minimum value of 10.5 emu K mol−1 due to zero-field splitting and/or weak intermolecular antiferromagnetic interactions (the shortest intermolecular Mn⋯Mn distance is ∼8.1 Å). Although both complexes have Mn–oxide cores based around a similar corner-shared double cubane, {Mn7O8}, the magnetic behaviour of the two complexes is different, suggesting that different magnetic interactions are operative between Mn ions. Such magnetic pathways are very sensitive to bridging bond angles and electronic configuration. Furthermore, the relative positions of the Jahn–Teller axes of the MnIII ions are a key factor in the determination of the magnetic interaction pathways.19 As shown in Fig. 3, the distortion of the double cubane and the relative alignments of the Jahn–Teller axes of the MnIII ions are significantly altered by the inclusion of the MnIV ion in 1 in place of the MnIII ion in 2. Hence, there is a substantial difference in the magnetic interactions in 1 and 2. The magnetization curves of both complexes, neither of which reach saturation up to 5 T, are shown in Fig. S3.† Low temperature ac magnetic measurements indicate no clear SMM behaviour above 1.8 K for either 1·4H2O or 2·7H2O (Fig. S4 and S5†), possibly due to the small molecular magnetic anisotropy, D, resulting from the non-parallel alignment of the Jahn–Teller axes of the MnIII ions.
 | ||
| Fig. 4 χT vs. T plots for 1·4H2O (●) and 2·7H2O (○). | ||
Conclusions
High nuclearity mixed-valence Mn oxide clusters, [MnII8MnIII10MnIVO10(OH)4(Hbdp)2(O2CPh)18] (1) and [MnII6MnIII15O12(OH)4(bdp)2(Hbdp*)(H2bdp*)(O2CPh)14] (2) were synthesised utilizing alkylamine based multidentate ligands with multiple alkoxo bridging groups. The cores of both complexes were constructed similar corner-shared double cubanes {Mn7O8} with the only difference in the oxidation state of the central Mn ions. 1 and 2 show dominant antiferromagnetic interactions between Mn ions and no SMM behaviour was observed above 1.8 K due to the non-parallel alignment of MnIII magnetic anisotropy axes.Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers 25248014 and 26410065.Notes and references
- (a) T. Ito, T. Hamaguchi, H. Nagino, T. Yamaguchi, J. Washington and C. P. Kubiak, Science, 1997, 277, 660 CrossRef; (b) V. Balzani, A. Juris, M. Venturi, S. Campagna and S. Serroni, Chem. Rev., 1996, 96, 759 CrossRef PubMed.
- (a) L. K. Thompson, O. Waldmann and Z. Xu, Coord. Chem. Rev., 2005, 249, 2677 CrossRef PubMed; (b) M. Ruben, J. Rojo, F. J. Romero-Salguero, L. H. Uppadine and J.-M. Lehn, Angew. Chem., Int. Ed., 2004, 43, 3644 CrossRef PubMed.
- O. Kahn, Molecular Magnetism, VCH Publishers, Weinheim, 1993 Search PubMed.
- (a) A. Bleuzen, V. Marvaud, C. Mathoniere, B. Sieklucka and M. Verdaguer, Inorg. Chem., 2009, 48, 3453 CrossRef PubMed; (b) J. M. Herrera, V. Marvaud, M. Verdageur, J. Marrot, M. Kalisz and C. Mathonière, Angew. Chem., Int. Ed., 2004, 41, 5468 CrossRef PubMed; (c) N. Shimamoto, S. Ohkoshi, O. Sato and K. Hashimoto, Inorg. Chem., 2002, 41, 678 CrossRef PubMed; (d) G. N. Newton, M. Nihei and H. Oshio, Eur. J. Inorg. Chem., 2011, 2011, 3031 CrossRef PubMed.
- P. W. Anderson and H. Hasegawa, Phys. Rev., 1955, 100, 675–681 CrossRef CAS.
- D.-L. Long, R. Tsunashima and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 1736 CrossRef PubMed.
- A. Müller, E. Bechmann, H. Bögge, M. Schmidtmann and A. Dress, Angew. Chem., Int. Ed., 2002, 41, 1162 CrossRef.
- (a) G. E. Kostakis, A. M. Ako and A. K. Powell, Chem. Soc. Rev., 2010, 39, 2238 RSC; (b) D. Gatteschi, M. Fittipaldi, C. Sangregorio and L. Sorace, Angew. Chem., Int. Ed., 2012, 51, 4792 CrossRef PubMed.
- G. N. Newton, S. Yamashita, K. Hasumi, J. Matsuno, N. Yoshida, M. Nihei, T. Shiga, M. Nakano, H. Nojiri, W. Wernsdorfer and H. Oshio, Angew. Chem., Int. Ed., 2011, 50, 5716 CrossRef PubMed.
- G. C. Dismukes, R. Brimblecombe, G. A. N. Felton, R. S. Pryadun, J. E. Sheats, L. Spiccia and G. F. Swiegers, Acc. Chem. Res., 2009, 42, 1935 CrossRef CAS PubMed.
- (a) G. Christou, D. Gatteschi, D. N. Hendrickson and R. Sessoli, MRS Bull., 2000, 25, 66 CrossRef; (b) D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, Oxford University Press, New York, 2006 and references therein Search PubMed.
- (a) A. J. Tasiopoulos, A. Vinslava, W. Wernsdorfer, K. A. Abboud and G. Christou, Angew. Chem., Int. Ed., 2004, 43, 2117 CrossRef PubMed; (b) E. E. Moushi, C. Lampropoulos, W. Wernsdorfer, V. Nastopoulos, G. Christou and A. J. Tasiopoulos, J. Am. Chem. Soc., 2010, 132, 16146 CrossRef CAS PubMed; (c) M. Manoli, R. Inglis, M. J. Manos, V. Nastopoulos, W. Wernsdorfer, E. K. Brechin and A. J. Tasiopoulos, Angew. Chem., Int. Ed., 2011, 50, 4441 CrossRef CAS PubMed; (d) S. K. Langley, R. A. Stott, N. F. Chilton, B. Moubaraki and K. S. Murray, Chem. Commun., 2011, 47, 6281 RSC.
- S. Yamashita, T. Shiga, M. Kurashina, M. Nihei, H. Nojiri, H. Sawa, T. Kakiuchi and H. Oshio, Inorg. Chem., 2007, 46, 3810 CrossRef CAS PubMed.
- (a) T. Matsumoto, G. N. Newton, T. Shiga, S. Hayami, Y. Matsui, H. Okamoto, R. Kumai, Y. Murakami and H. Oshio, Nat. Commun., 2014, 5, 3865 CAS; (b) G. N. Newton, T. Onuki, T. Shiga, M. Noguchi, T. Matsumoto, J. S. Mathieson, M. Nihei, M. Nakano, L. Cronin and H. Oshio, Angew. Chem., Int. Ed., 2011, 50, 4844 CrossRef CAS PubMed.
- (a) A.-J. Zhou, J.-D. Leng, J.-S. Hu and M.-L. Tong, Dalton Trans., 2013, 42, 9428 RSC; (b) A. Saha, K. A. Abboud and G. Christou, Inorg. Chem., 2011, 50, 12774 CrossRef CAS PubMed; (c) S. Nayak, M. Evangelisti, A. K. Powell and J. Reedijk, Chem. – Eur. J., 2010, 16, 12865 CrossRef CAS PubMed; (d) M. Manoli, A. Collins, S. Parsons, A. Candini, M. Evangelisti and E. K. Brechin, J. Am. Chem. Soc., 2008, 130, 11129 CrossRef CAS PubMed; (e) R. Bagai, K. A. Abboud and G. Christou, Inorg. Chem., 2008, 47, 621 CrossRef CAS PubMed; (f) A.-J. Zhou, L.-J. Qin, C. C. Beedle, S. Ding, M. Nakano, J.-D. Leng, M.-L. Tong and D. N. Hendrickson, Inorg. Chem., 2007, 46, 8111 CrossRef CAS PubMed.
- M. W. Wemple, H.-L. Tsai, S. Wang, J. P. Claude, W. E. Streib, J. C. Huffman, D. N. Hendrickson and G. Christou, Inorg. Chem., 1996, 35, 6437 CrossRef CAS PubMed.
- J. S. Pierce and J. Wotiz, J. Am. Chem. Soc., 1944, 66, 879 CrossRef CAS.
- (a) I. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244 CrossRef; (b) W. Liu and H. H. Thorp, Inorg. Chem., 1993, 32, 4102 CrossRef CAS.
- (a) O. Kahn, Molecular Magnetism, VHC, New York, 1991 Search PubMed; (b) T. Matsumoto, T. Shiga, M. Noguchi, T. Onuki, G. N. Newton, N. Hoshino, M. Nakano and H. Oshio, Inorg. Chem., 2010, 49, 368 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional structural and magnetic data. CCDC 1043428 (1) and 1043429 (2). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qi00013k |
| This journal is © the Partner Organisations 2015 |