
Novel, fast-processed crystalline and amorphous manganese oxide nanoparticles for stem cell labeling†
Jessica M.
Rosenholm
*abc,
Riikka M.
Korpi
de,
Eveliina
Lammentausta
d,
Siri
Lehtonen
f,
Petri
Lehenkari
g,
Rasmus
Niemi
hi,
Wangchuan
Xiao
b,
Jixi
Zhang
bj,
Desiré
Lindberg
c,
Hongchen
Gu
b,
Cecilia
Sahlgren‡
hik and
Roberto
Blanco Sequeiros‡
*dl
aPharmaceutical Sciences Laboratory, Faculty of Science and Engineering, Åbo Akademi University, Turku, Finland. E-mail: jerosenh@abo.fi; Tel: +358-2-215 3255
bNano Biomedical Research Center, Med-X Research Institute and School for Biomedical Engineering, Shanghai Jiao Tong University, Shanghai, P.R. China
cLaboratory for Physical Chemistry, Faculty of Science and Engineering, Åbo Akademi University, Turku, Finland
dDepartment of Diagnostic Radiology, Oulu University Hospital, Oulu, Finland
eDepartment of Radiology, Helsinki University Hospital, Helsinki, Finland
fUniversity of Oulu, Department of Anatomy and Cell Biology and Oulu University Hospital, Medical Research Center and Respiratory Research Unit, Finland
gDepartment of Anatomy and Clinical Research Center, University of Oulu, Oulu, Finland
hCell Biology, Faculty of Science and Engineering, Åbo Akademi University, Turku, Finland
iTurku Centre for Biotechnology, University of Turku and Åbo Akademi University, Turku, Finland
jChongqing University, College of Bioengineering, Chongqing, China
kDepartment of Biomedical Engineering, Technical University of Eindhoven, The Netherlands. E-mail: C.M.Sahlgren@tue.nl
lSouth West Finland Imaging Centre, Turku University Hospital, Turku, Finland. E-mail: roberto.blanco@tyks.fi; Tel: +358-2 313 1975
First published on 11th May 2015
Abstract
Magnetic resonance (MR) imaging, with its inherent good spatial resolution and without tissue penetration depth limitations associated with other cell tracking techniques, is considered a valuable tool to assess the effects of cellular therapy. However, in order to allow in vivo tracking of transplanted cells with MRI, the cells must be labeled with contrast agents, usually in the form of magnetic or paramagnetic nanoparticles. Typically these are iron oxides, which are associated with a number of drawbacks related e.g. to the generation of hypointensities of the MR image due to signal loss. In this study, two chemically distinct manganese oxide-based nanostructures were developed and their feasibility as labels for human mesenchymal stem cells (hMSCs) was investigated. The ability to monitor the produced particles alone or within the labeled cells in vitro using MR imaging was further evaluated. Two novel synthetic approaches, the polyol process and microwave digestion, were combined to yield a “green”, and extremely rapid means of producing water-dispersible crystalline (MnO) and amorphous (MnOx) manganese oxides. To increase their water dispersability, capping agents in the form of organic polymers, poly(vinyl pyrrolidone) (PVP) and poly(acrylic acid) (PAA), were added to the synthesis process. Crystalline MnO was not formed when PAA was used as the capping agent, since Mn ions (Mn2+) cannot be hydrolyzed to Mn(OH)2, which is an intermediate step in the formation of MnO, under the acidic conditions provided by PAA. PVP, on the other hand, served to induce a spherical shape to the formed nanocrystals. Remarkably, the relaxation times of the as-prepared amorphous MnOx were significantly shorter than those of their crystalline counterparts, and the biocompatibility was also higher for MnOx. To the best of our knowledge, this is the first report which describes the use of an amorphous MnOx as a cell label for MR imaging.
Introduction
Exogenous or autologous cell transplantation is regarded as a potential therapeutic method for a large variety of diseases. In the transplantation process, the ability to target and monitor transplanted cells is of critical importance. Magnetic resonance (MR) imaging offers non-invasive in vivo monitoring of cells labeled with suitable contrast agents with a high spatial resolution, additional anatomical and pathological details, and no exposure to ionizing radiation. The contrast agents serve to enhance the contrast of cellular targets in MR images by decreasing the relaxation times T1 (signal increase on the MR image) and/or T2 (signal loss on the MR image) to improve the image quality, acquisition time as well as enable enlargements of detectable organs/targets. For cellular labeling purposes, magnetic nanoparticles are of particular interest.1–3To date, small/ultra-small superparamagnetic iron oxide nanoparticles (SPIO/USPIO) have been the most widely used as contrast agents for cellular MR imaging.3–5 Traditionally, these are prepared via the conventional coprecipitation method, which is difficult to control in terms of size distribution, crystallinity, saturation magnetization values as well as particle aggregation and dispersability in aqueous solutions.6,7 To overcome these drawbacks, new synthesis approaches have been developed, such as high-temperature decomposition of iron precursors in nonpolar solvents, which produces high-quality monodisperse iron oxide nanoparticles with a tightly controlled size distribution and high crystallinity.7–9 These particles are inherently hydrophobic, and need to be either coated by a hydrophilic layer via surface functionalization procedures and/or phase transferred into water before they are suitable for biomedical applications. The addition of appropriate capping agents, such as carboxylic acids, in the synthesis process – resulting in hydrophilic groups on the formed particles has been an important development.10,11 Another method for producing intrinsically hydrophilic metal or metal oxide nanostructures with controlled properties is the polyol process, i.e. the reduction of metal salts in polyalcohols.6–8,10–17 When combined with microwave digestion, which is a well-established route in organic synthesis,18 this synthesis strategy may provide a simple, rapid (within minutes), effective, low energy-consuming, and environmentally friendly “green” synthesis method for producing inorganic nanostructures with controllable characteristics as well.8,14,18,19
Gadolinium-based complexes, such as gadopentate dimeglumine, and manganese (Mn) both accelerate longitudinal T1 relaxation increasing signal intensity on MR images as opposed to iron oxides. However widely used in drug administration applications,20 gadopentate dimeglumine is associated with nephrogenic systemic fibrosis21,22 and is therefore a less favorable agent for clinical applications. The molecular size of gadopentate dimeglumine is also an issue. Whereas free manganese ions (Mn2+) are also toxic and thus are mainly applicable for animal manganese-enhanced MR imaging studies,23 manganese oxide (MnO) in its crystalline form has a tolerable cellular toxicity range and is considered a promising agent for longitudinal cell tracking, as it is easy to deliver and maintain good image quality.23–25 However, it is still controversial how the labeling with MnO affects cell function and cell differentiation capacity26 and thus, there are only few studies reporting the use of MnO as a contrast agent for cellular tracking in vivo. Still, the obvious advantage of Mn-based substances related to the positive T1 contrast, which generates a signal increase that is opposite to iron oxide particles, opens up a multitude of opportunities in anatomical and functional imaging that can be used to augment tissue imaging. The effect could be especially beneficial in detecting metabolic events and adverse or pathological anatomy in tissues that naturally have low signal in MRI.24 Various advantages thus justify developing manganese-based nanoparticles for future clinical applications within molecular and cellular imaging.
The purpose of this study is to produce and characterize two chemically distinct types of manganese oxide, crystalline and amorphous, using the combined advantages of the polyol process and the microwave technique. We demonstrate the production of novel, water-dispersible (hydrophilic), fast-processed manganese oxide nanoparticles and assess the feasibility of these nanoparticles for the labeling of human mesenchymal stem cells as well as MR imaging.
Materials & methods
Preparation of manganese oxide nanoparticles
The manganese oxide syntheses were performed in a microwave reactor with a controllable temperature/heating and time program of a focused microwave synthesis system (Discover SClass, CEM, USA). In a typical synthesis, 12 mmol of sodium acetate was dissolved in 30 ml ethylene glycol by careful heating, ultrasonication and stirring. When dissolved, 4 mmol of MnCl2·4H2O was added to the mixture and dissolved accordingly. The synthesis mixture was either used as such, or capping agents poly(acrylic acid), PAA (Mw 5000) or poly(vinyl pyrrolidone), PVP (Mw 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) at an amount of Mn2+: PAA/PVP = 1:2 (i.e. 8 mmol capping agent). The different reaction mixtures were then digested in the microwave at different temperatures (200 °C, 220 °C, and 240 °C) for different reaction times (5–50 min). After the reaction, quick cooling to room temperature was realized by high pressure air flow. The formed particles were precipitated by addition of 15 ml ethyl acetate and 3 ml absolute ethanol, and separated by centrifugation at 7000 rpm for 10 min. The final particles were harvested by subsequent washing three times in ethanol, and redispersed in ethanol until further use. For ICP measurements, 100 μl of such suspension was dissolved into concentrated HNO3 and diluted to 5 ml with ddH2O.
000) at an amount of Mn2+: PAA/PVP = 1:2 (i.e. 8 mmol capping agent). The different reaction mixtures were then digested in the microwave at different temperatures (200 °C, 220 °C, and 240 °C) for different reaction times (5–50 min). After the reaction, quick cooling to room temperature was realized by high pressure air flow. The formed particles were precipitated by addition of 15 ml ethyl acetate and 3 ml absolute ethanol, and separated by centrifugation at 7000 rpm for 10 min. The final particles were harvested by subsequent washing three times in ethanol, and redispersed in ethanol until further use. For ICP measurements, 100 μl of such suspension was dissolved into concentrated HNO3 and diluted to 5 ml with ddH2O.
Characterization
Transmission electron microscopy (TEM, JEOL 2010, at the Instrument Centre of Shanghai Jiao Tong University) was used for observing the morphology of the obtained nanoparticles. The hydrodynamic size was measured on either a high performance particle sizer (HPPS 5001, Malvern Instruments Ltd., Worcestershire, UK) or a Malvern ZetaSizer (Malvern ZetaSizer NanoZS, Malvern Instruments Ltd, Worcestershire, UK) of which the latter also was used for zeta potential measurements. X-ray diffraction (XRD) was recorded on a Rigaku D/max 2250 VB/PC with CuKα radiation (1.54056 Å) at 40 kV, 200 mA to confirm the nanocrystalline phase. Manganese concentrations were determined by inductively coupled plasma emission spectroscopy (ICP, ICA P6300, Thermo Fischer, USA) after dissolution into concentrated HNO3 with subsequent dilution in ddH2O.Relaxivity measurements
The relaxivity properties of the as-prepared particle suspensions were measured on an NMR spectrometer (Minispec, mq60, Bruker, Germany). The proton resonance frequency was 60 MHz to assess their feasibility for MR imaging. The Carr–Purcell–Meiboom–Gill (CPMG) spin echo sequence was used for T2 measurements and saturation recovery sequence was used for T1 measurements. Known concentrations of particles, as determined from ICP, was diluted in water and kept at 37 °C prior to measurements. The transverse and longitudinal relaxation times in water of the as-prepared nanoparticles were measured at different Mn concentrations until the same value was provided by three consecutive measurements recorded.Cell viability
Viability was determined by the WST-1 assay: human mesenchymal stem cells were transferred to 96-well plate (9000 cells per well) and allowed to attach and grow. After 24 h, the medium was removed and replaced with 100 μl medium containing different concentrations of particles (10, 20 and 40 μg ml−1). After incubation for 2 h, the medium was removed and replaced with a particle free medium. At various time points after particle treatments 10 μl of WST-1 (Roche Applied Science) reagent was added to cells and further incubated for 90 min, after which the 96-well plate was analyzed at 430 nm wavelength using a Varioskan plate reader to determine the cell viability. Cell media without particles were used as a control.MR imaging
For MR imaging measurements, crystalline (prepared at 240 °C for 30 min without additives) and amorphous (prepared at 240 °C for 20 min with PAA as an additive) manganese oxide suspensions were diluted in agar at concentrations of 0.1–1 mM in 0.1 mM increments, and for more accurate relaxivity characterization, at concentrations of 0.01–0.1 mM in 0.01 mM increments were prepared and imaged with an MR scanner. The same suspensions in water were also pre-evaluated on a Minispec instrument.For MR imaging measurements of particle treated MSC cells, 125000 MSCs were incubated with 20 and 40 μg ml−1 particles for 2 h. Subsequently, the medium was replaced and the cells were allowed to grow for 24 h after which the cells were harvested and pelleted in a 3% agar in Eppendorf tubes.
T1 and T2 relaxation times of all the samples were measured using a 3T clinical MR scanner (Siemens Magnetom Skyra, Siemens Healthcare, Erlangen, Germany) and an 8-channel receive-only small animal coil (RAPID Biomedical GmbH, Rimpar, Germany). The T1 relaxation time was measured using a single slice inversion recovery fast spin echo sequence (TR 10000 ms, TE 8.6 ms, ten TIs between 50 and 9500 ms, FOV 12 cm, matrix 256 × 256 yielding an in-plane resolution of 0.47 mm, slice thickness 3 mm, ETL 8, NEX 1). The T2 relaxation time was measured using a multi-slice multi echo spin echo sequence (TR 1680 ms, 12 TEs between 11.5 and 138 ms, ETL 5; FOV, matrix and slice thickness unchanged). Slices were positioned along the cross-section of the test tubes. Circular ROIs were manually segmented into each test tube, and the relaxation times were calculated pixelwise with non-linear fitting and assuming monoexponential relaxation using the in-house MATLAB application (The MathWorks Inc., Natick, MA, USA). The mean value and standard deviation were calculated for each ROI.
Results
Synthesis and characterization of manganese oxides
When no capping agents were used in the synthesis protocols, 30 min at 240 °C proved to be sufficient for obtaining of a pale green synthesis mixture, characteristic of the formation of the crystalline MnO phase23 as illustrated in Fig. 1a. After purification and redispersion into ethanol, the obtained stable suspension was black in color and further TEM analysis revealed the presence of nanocrystals (Fig. 1b). Based on this observation, together with ICP analysis (yield) the reaction temperature was set to 240 °C and different reaction times (10, 20, 30 min) were evaluated for the manganese oxide syntheses together with the capping agents PAA and PVP (Table 1). Interestingly, the use of PVP resulted in similar black suspensions after ethanol purification, whereas for the PAA-addition resulted in white suspensions (Fig. 1c). TEM analysis of such a suspension suggested that an amorphous structure was formed (Fig. 1d). The distinction in the two different reaction systems is as follows. MnO (crystalline) was formed in two steps. Firstly, Mn(OH)2 was formed by the hydrolysis of Mn2+ in the presence of CH3COONa as the alkalinity provider. The second step included the dehydration of Mn(OH)2 at high temperature:| Ac− + H2O = HAc + OH− | (1) |
| Mn2+ + OH− = Mn(OH)2 | (2) |
| Mn(OH)2 = MnO + H2O | (3) |
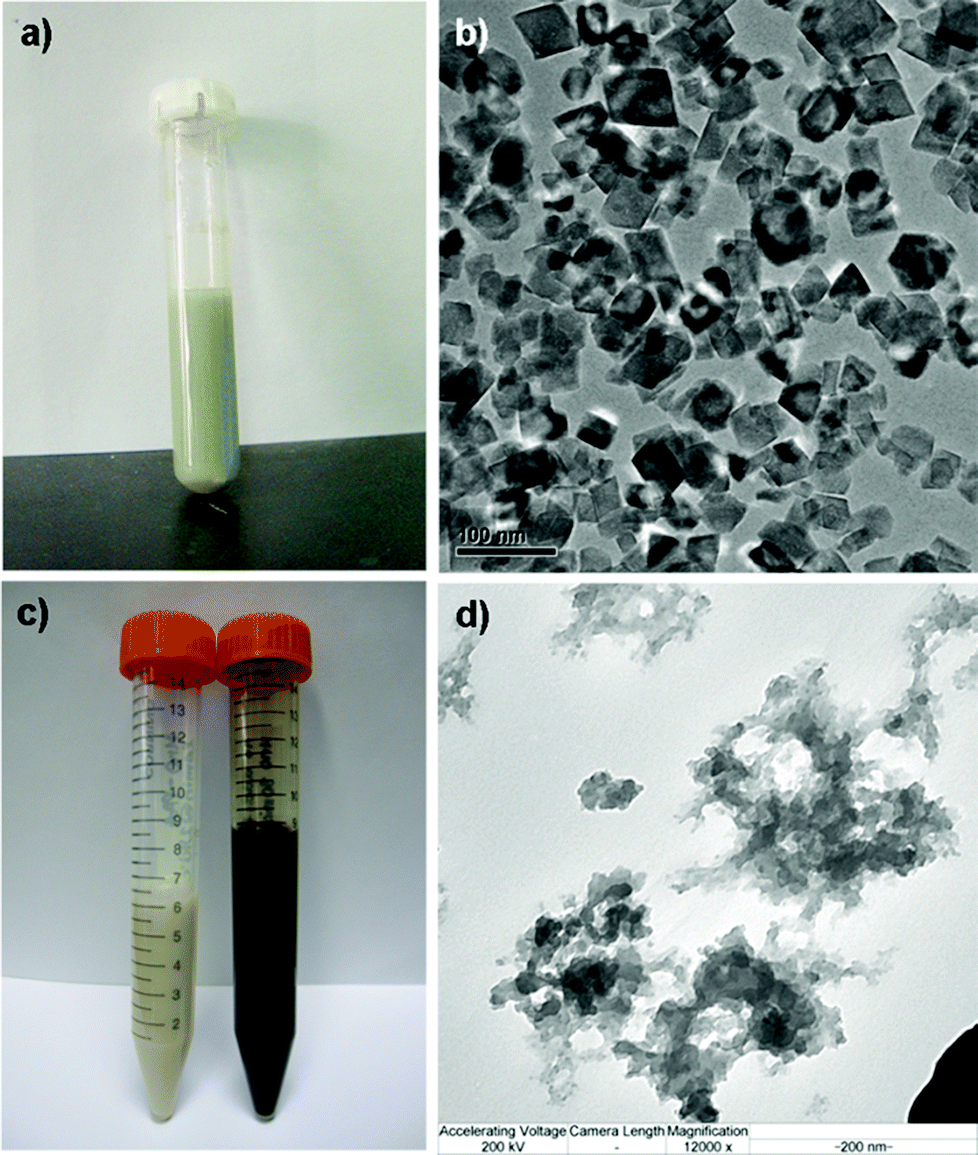 | ||
| Fig. 1 Image characterization of the formed manganese oxides. (a) The synthesis mixture directly after microwave digestion when no additives were used; (b) TEM image of (a) after washing and dispersing into ethanol; (c) ethanol suspensions of amorphous (white) and crystalline (black) manganese oxide; (d) TEM image of the “white” suspension (using PAA as additive) indicating that an amorphous structure had been formed. | ||
| Sample (reaction conditions) | Hydrodynamic diameter in EtOH | Hydrodynamic diameter in H2O |
|---|---|---|
| PVP 10 min@240 °C | 129 ± 9 nm | 383 ± 84 nm |
| PVP 20 min@240 °C | 204 ± 8 nm | 349 ± 42 nm |
| PVP 30 min@240 °C | 156 ± 22 nm | 411 ± 191 nm |
| PAA 10 min@240 °C | N/A | 105 ± 11 nm |
| PAA 20 min@240 °C | N/A | 178 ± 42 nm |
| PAA 30 min@240 °C | N/A | 113 ± 11 nm |
Therefore, MnO was not formed when PAA was used as the capping agent, since Mn2+ cannot be hydrolyzed to Mn(OH)2 under the acidic conditions provided by PAA (a polyacid). Moreover, the reaction of Mn2+ to MnO indicates that there is no oxidation or reduction reaction taking place in this system.
To determine the behavior in solution of the formed materials, whereby electron microscopy only provides information in dry state, the water/buffer suspensions of the amorphous and crystalline manganese oxides were investigated. Their suspension behavior especially in aqueous solvent is crucial for successful applicability, as the utilization of the produced materials, such as cellular labeling, will naturally take place under aqueous conditions. Thus, their hydrodynamic size and net surface charge (zeta potential) at neutral pH were investigated with dynamic light scattering and electrokinetic measurements (Fig. 2). For the crystalline sample, a hydrodynamic diameter of 99.17 ± 0.17 nm (z-average) was found (Fig. 2a), with a low polydispersity index (PdI = 0.076), indicative of well-dispersed particles with a narrow (hydrodynamic) size distribution. For the amorphous sample, the particles were not as discrete and well-defined, as also evident from their structure from TEM (Fig. 1) but they could also be readily dispersed in an aqueous solvent and provided better hydrodynamic characteristics than their crystalline counterparts under the same conditions (Table 1). However, under ethanolic (parent/storage solvent) conditions, also the crystalline MnO exhibited narrow hydrodynamic size distributions (Table 1), indicative of discrete particulates. Whereas the amorphous MnOx net surface charge, as measured by the ζ-potential in HEPES buffer at pH 7.2 yielded a negative surface charge −25.7 ± 1.47 mV due to the carboxylic acid groups resulting from the PAA used as the capping agent, the crystalline sample exhibited a net neutral charge at neutral pH (Fig. 2c), the characteristic isoelectric point of Mn(OH)2;27 further corroborated by considering that PVP does not contain chargeable groups. The net neutral charge at neutral pH is also reflected in the “worse” hydrodynamic size results in ddH2O of these samples, due to the absence of strong repulsive forces as in the case of the negatively charged MnOx. The overall characteristics of the PVP and PAA capped suspensions have been summarized in Table 1.
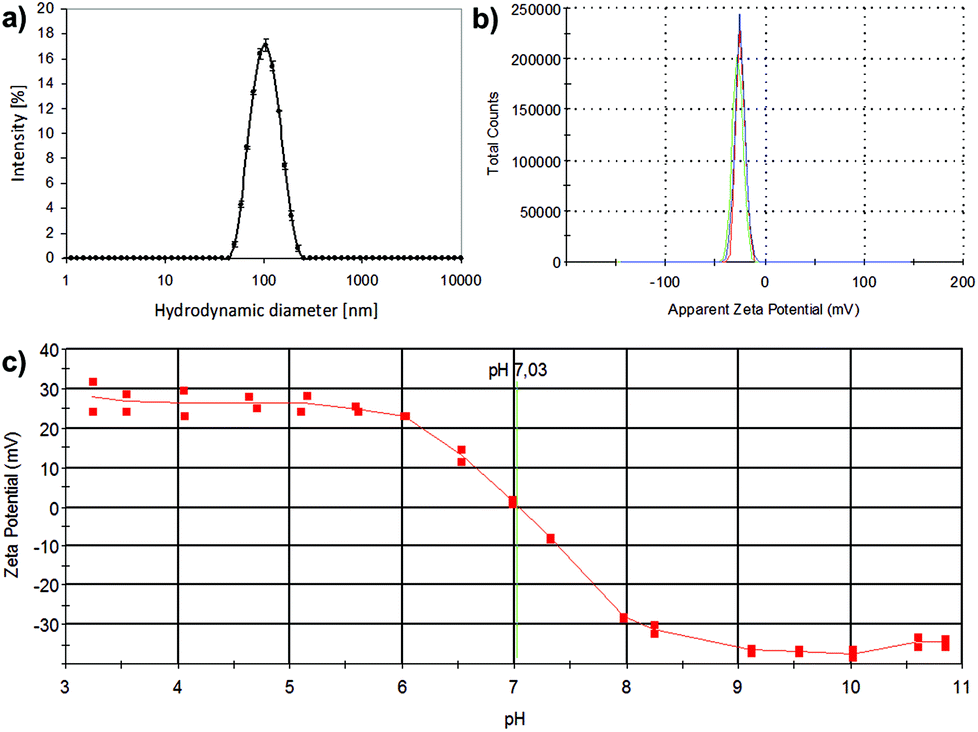 | ||
| Fig. 2 Characterization of manganese oxide suspensions. (a) Dynamic light scattering (intensity plot) of crystalline MnO prepared without capping polymers (30 min@240 °C) in ddH2O; (b) zeta-potential of an amorphous MnOx (20 min@240 °C) in HEPES buffer (25 mM, pH 7.2); (c) isoelectric titration graph of sample (a) yielding a net neutral charge at physiological pH. | ||
Based on dynamic light scattering data (Table 1), 20 min was concluded to be sufficient for obtaining manganese oxide particles when stabilized by polymeric agents PVP or PAA. For confirmation of the crystalline phase (or absence thereof), selected samples were analyzed by XRD (Fig. 3).
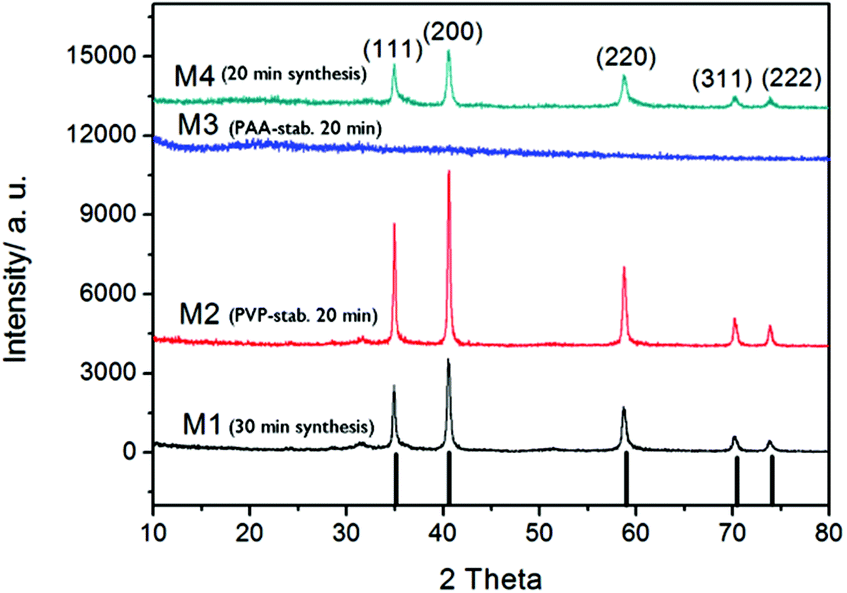 | ||
| Fig. 3 XRD patterns for selected manganese oxide samples. | ||
The diffraction patterns of samples produced via microwave digestion for 20 or 30 min at 240 °C without any additional polymer, showed clearly resolved peaks that could be indexed to a crystalline MnO phase.28 The same pattern was observed for the PVP-stabilized sample digested for 20 min at 240 °C, whereas for the corresponding sample prepared with capping agent PAA, no distinctive XRD patterns could be found, indicative of an amorphous phase and in accordance with what was deduced from TEM characterization.
TEM analysis of the above samples (Fig. 4) revealed some differences in size and morphology, also suggesting that 30 min provides for more uniform morphology than 20 min, whereas the addition of PVP seems to facilitate some additional morphology/size control inducing a more uniform and spherical shape rather than crystal-like particulates; whereby 20 min microwave digestion is sufficient for the formation of ∼10 nm MnO nanoparticles.
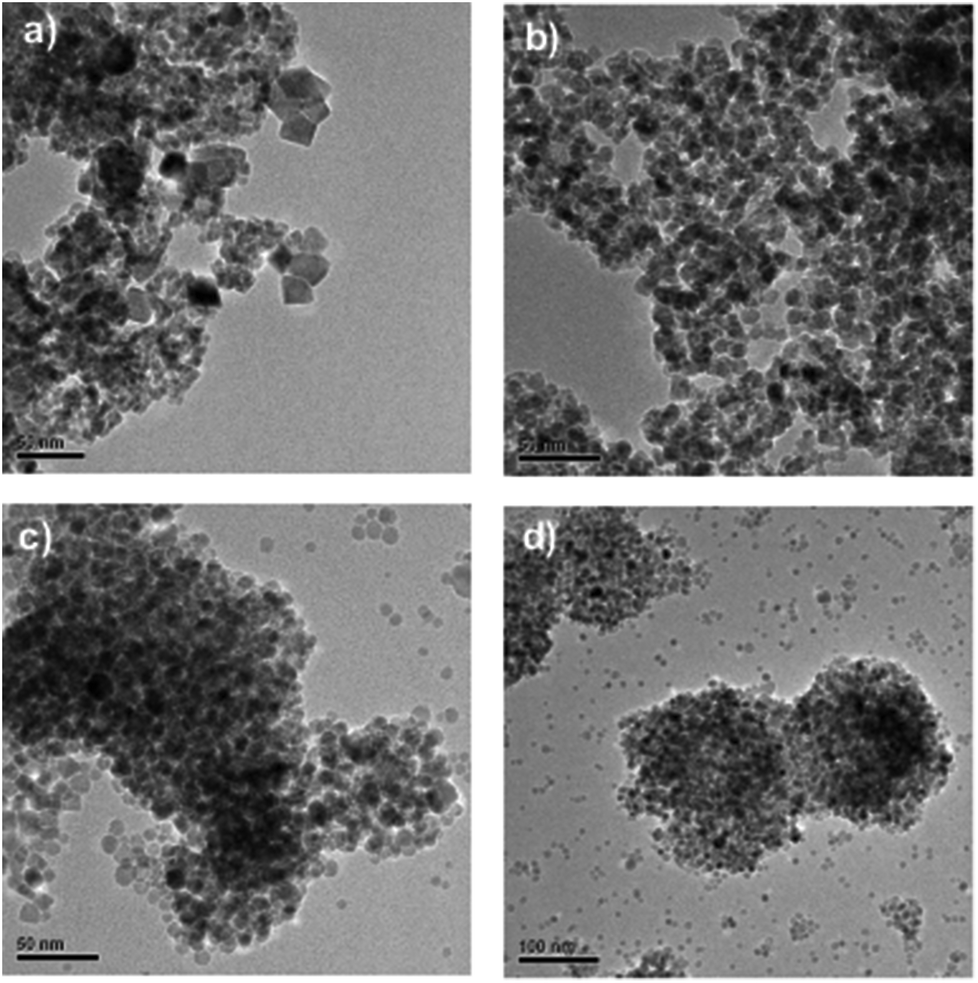 | ||
| Fig. 4 Morphology investigation through TEM revealed more spherical-like particles for PVP-stabilized samples. MnO crystals formed without addition of polymers: (a) 20 min reaction time & (b) 30 min reaction time; MnO crystals formed with addition of PVP; (c) 20 min reaction time & (d) 30 min reaction time. | ||
As can be deduced from these TEM images, especially when PVP is used as the capping agent, there seems to be a coexistence of nanoclusters together with independent nanoparticles, which may explain why the individual particle diameter is about 10 nm but the hydrodynamic size (by DLS) is exceeding 200 nm (Table 1). In this case, the PVP thus seems to function primarily as a morphology inducer and/or growth regulator rather than an effective dispersion agent. Clearly, separate and almost spherical nanoparticles with diameters less than 10 nm are formed (Fig. 4c and d), but to keep them properly separated also in the final suspension, either the PVP concentration would need to be optimized, or a second dispersion agent would need to be added, or possibly some other suspension media could be used. It should be noted, however, that particle aggregation would also readily take place upon drying on the grids used for electron microscopy, due to the large surface energy of such small particles. Upon application in biological systems, further organic coating of the nanoparticles would also commonly come in question.3
Relaxometric properties
To assess the suitability of the produced manganese oxides for MR imaging, the relaxometric properties of the as-prepared materials, in terms of their relaxation times T1 and T2 as a function of Mn concentration, was first determined using a minispec NMR spectrometer (Fig. 5). T1 and T2 describe the longitudinal and transverse relaxations of excited protons, respectively; and a shorter relaxation time is associated with a brighter (T1) or darker (T2) image intensity.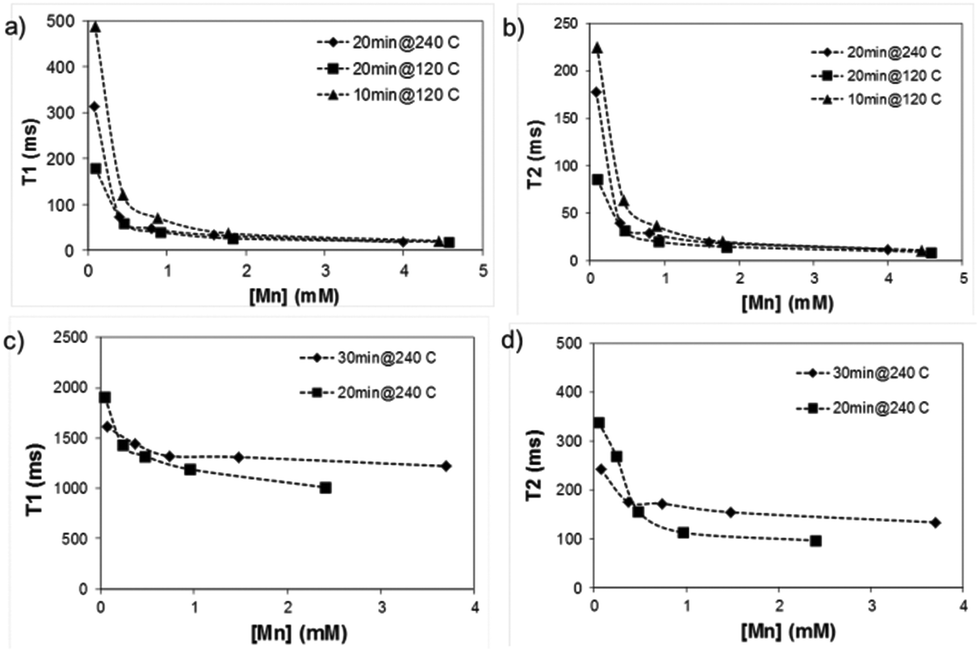 | ||
| Fig. 5 Relaxivity measurements of as-prepared manganese oxides in water. (a) & (b) amorphous and (c) & (d) crystalline MnO. Measurements were performed on a minispec table-top NMR instrument. | ||
Remarkably, the relaxation times of the amorphous samples (Fig. 5a and b) were significantly shorter than those of the crystalline particles down to extremely low values (T1 below 20 ms and T2 below 10 ms at higher concentrations of Mn). The corresponding relaxivity plots (Fig. 6) revealed r1,2 values of r1 = 15.6, 26.7, 24.9 mM−1 s−1 and r2 = 30.8, 52.2, 39.1 mM−1 s−1 for the amorphous MnOx. The corresponding relaxivity values for the crystalline MnO (ESI Fig. 1a and b†) are r1 = 0.3, 0.09 mM−1 s−1 and r2 = 6.7, 1.4 mM−1 s−1, i.e. approximately an order of magnitude lower. These striking differences in relaxometric properties can be due to many reasons, related to different material characteristics such as saturation magnetization, particle size and surface coating as well as dispersability in aqueous solvent.29–33 For instance, when superparamagnetic particles are very small, their 1/T2 is shown to be proportional to particle size; whereas when the particles are large, 1/T2 is proportional to the inverse particle size.29 The slight deviations from linearity observed for most of the samples may also be well related to the aggregation in solution, which, in turn, may very likely also partly be a consequence of time waiting dependence.32,33 Time dependence in the T2 relaxation time has been investigated for iron oxides, and found to be due to large particles generally having a high saturation magnetization value and poor stability in water, which, in combination, may lead to aggregation under the 1.4T magnetic field used for relaxivity measurements of suspensions; whereby the relaxation time changes with time.32 We have also recently shown the importance of an optimized surface functionalization for preventing aggregation of magnetic particles under a magnetic field.34 These notions point to the fact that careful surface engineering could be facilitative also from a relaxometric point of view.
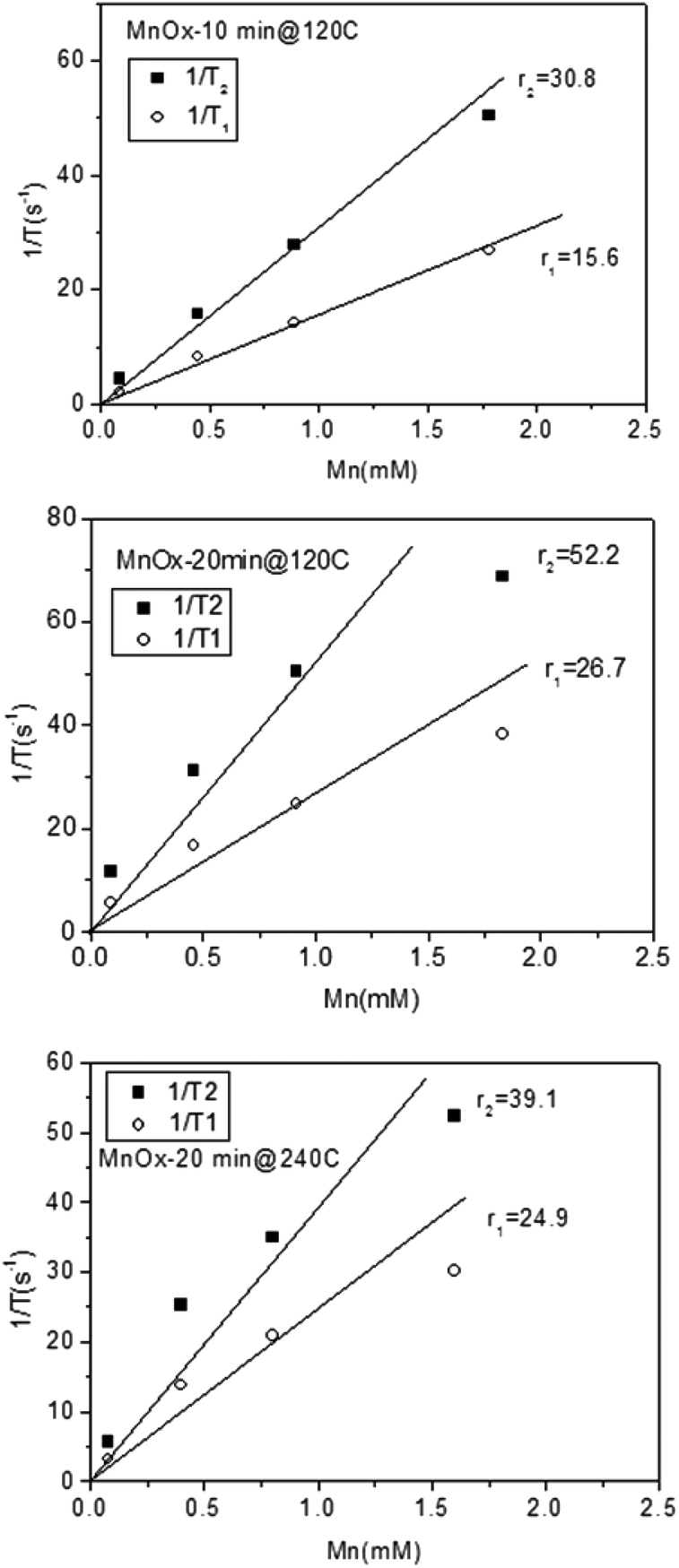 | ||
| Fig. 6 Relaxivity plots for the as-prepared amorphous MnOx prepared at different reaction times and temperatures. | ||
Cytocompatibility
To ensure the feasibility of the produced particles for cellular labeling also in terms of cytocompatibility, we tested the effect on cell viability by labeling hMSCs for 2 h with 10, 20, and 40 μg ml−1 of amorphous (PAA, 20 min@240 °C) or crystalline (no additives, 30 min@240 °C) particle samples. Viability/proliferation was measured up to 24 h after labeling. After 2 h, there were no significant changes in viability, however a reduction of 20% versus 50% (non-treated cells) in viability was observed at 6 h and 24 h after labeling respectively (Fig. 7). As there was no significant decrease in viability over time within the individual treatments, it indicated that cells did not die but instead the proliferation was halted. When the results were plotted as a proliferation index, it became evident that proliferation was affected in a dose dependent manner. The cells treated with 40 μg ml−1 ceased to proliferate, while the cells labeled with 10 μg ml−1 continued to proliferate albeit slightly slower than non-treated cells. Amorphous particles demonstrated higher cytocompatibility than crystalline particles, and cells treated with 10 μg ml−1 amorphous particles continued to proliferate.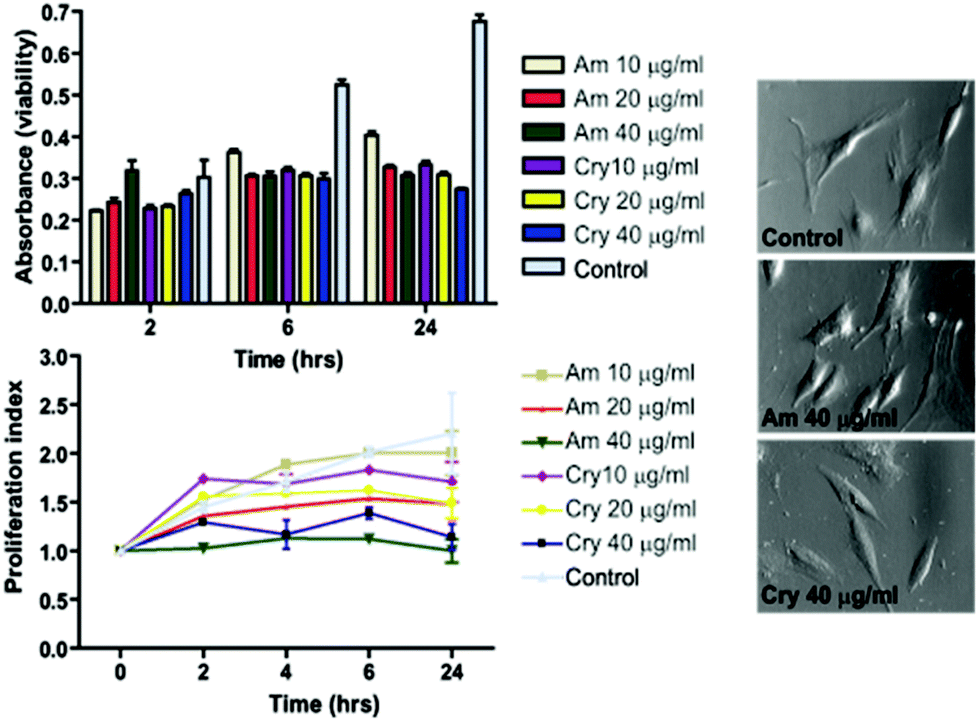 | ||
| Fig. 7 Viability and proliferation of hMSC labeled with Mn-oxide particles. | ||
MR imaging
MR imaging measurements showed similar results as those aquired with the NMR spectrometer, where the amorphous particles displayed slightly higher relaxivity, indicated by shorter relaxation times at low concentrations (Fig. 8). However, when examining the relaxometric properties of amorphous vs. crystalline MnO(x) in agar, measured on the clinical 3T MRI scanner, the differences between the amorphous and crystalline samples are not that remarkable as when measured as aqueous suspensions (Fig. 5). The corresponding relaxivity values for agar-immobilized MnO(x) particles measured at 3T where r1 = 30.4 mM−1 s−1, r2 = 65.7 mM−1 s−1 for the amorphous MnOx, and r1 = 10.0 mM−1 s−1, r2 = 65.0 mM−1 s−1 for the crystalline MnO. This indicates that the differences in relaxivity observed in the aqueous suspension measurements may be chiefly related to the colloidal stability in water, whereby aggregation of particles will lead to a decreased relaxivity. Indeed, the crystalline samples exhibited poorer dispersability properties (Table 1) as a result of the net neutral charge under these conditions (Fig. 2c) as compared to the amorphous MnOx, which were electrostatically stabilized in water (Fig. 2b). The corresponding MR image (Fig. 9) of the amorphous MnOx particles in agar demonstrates the apparent differences in the signal intensity profile related to particle concentration (Fig. 8). The relaxation times for labeled hMSCs are shown in Table 2. It is interesting to note that the reduction in T2 seemed more proficient than T1, even though MnO has to date mainly been put forward as a T1 contrast agent.23,24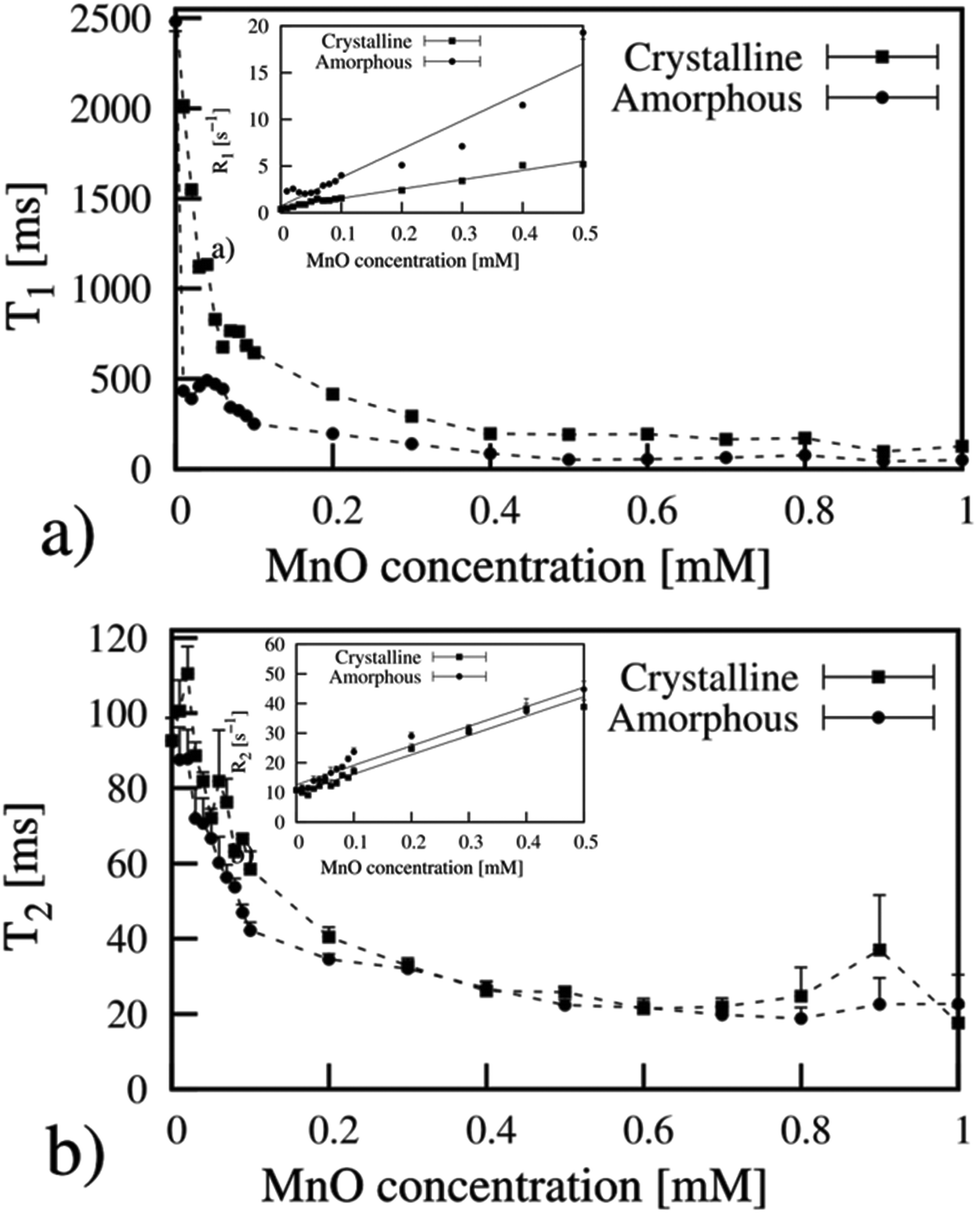 | ||
| Fig. 8 Relaxation time measurements on manganese oxide particles in agar measured with a 3T MR-scanner. Insets show the corresponding relaxivity plots, resulting in relaxivity values of (a) r1 = 30.4 mM−1 s−1 (amorphous) and r1 = 10.0 mM−1 s−1 (crystalline); (b) r2 = 65.7 mM−1 s−1 and r2 = 65.0 mM−1 s−1. | ||
 | ||
| Fig. 9 3T MR image illustrating relaxation measurements. Slice was set to form a circular cross-section of the test tubes. Each test tube contains a certain concentration of amorphous MnOx (bottom left corner: 0 mM, increasing from left to right and from bottom to top with an interval of 0.01 mM up to 0.1 mM) in agar. | ||
| Labeled cell samples at 3T | ||||
|---|---|---|---|---|
| T 2 [ms] | T 1 [ms] | |||
| Mean | SD | Mean | SD | |
| Am 20 μg ml−1 | 3506 | 208 | 2040 | 5508 |
| Am 40 μg ml−1 | 3707 | 351 | 1986 | 4603 |
| Cry 20 μg ml−1 | 3451 | 149 | 1598 | 5341 |
| Cry 40 μg ml−1 | 3183 | 132 | 1257 | 4296 |
Discussion
To overcome the known drawbacks of excessive signal loss with ferrous agents, other nanoparticles and small-molecular compounds such as manganese and gadopentate dimeglumine are under research. The current study is based on the production of novel manganese oxide nanostructures for cell labeling. In general, the purpose of all cellular MR imaging techniques is to visualize the cells without interpretational challenges with subcellular structures or molecules. Recently, in the published literature, a few applications have pointed out the ability of MR imaging to detect MnO.23–26,35 However, heterogeneous sets of MnO solely or with adjunctive coating techniques were used, and therefore the results may not be comparable. Here, the feasibility of using amorphous and crystalline MnO as cellular labels for MR imaging was demonstrated on a 3T MRI scanner. Noteworthily, the amorphous MnOx exhibited promising relaxometric properties; and to the best of our knowledge, amorphous MnOx, which is traditionally used in catalysis and manganese oxide electrodes, has not been previously reported to be used in related applications. Due to the extraordinary relaxometric properties determined, a novel notion depicted on MR images both using agar and in vitro hMSCs, further studies using amorphous MnOx for cell labeling are justified.The polyol process has cleared the ground amongst inorganic nanoparticle syntheses especially when biomedical applications are aimed at, due to the inherently hydrophilic particles that are produced as a result of the hydroxyl (–OH) groups that are formed on the surface by a layer of polyol molecules.7 The polarity of the polyols moreover offer the ability to dissolve various inorganic salts, act as the reducing agent for many kinds of metal ions for the synthesis of metal and metal oxide nanoparticles, and furthermore, the high boiling point enables elevated temperature conditions which, in turn, may improve the reactivity of the used reactants.17 Synergistically, carrying out the synthesis with the aid of microwave irradiation offers a striking reduction of reaction times as opposed to conventional heating (oil bath or autoclave) from hours or days to minutes, allowing for a time- and energy-saving process.14 A superior colloidal stability under aqueous conditions could potentially still be achieved by the addition of a polyelectrolyte such as PAA, the chains of which can strongly coordinate to metal ions such as iron on a nanocrystal surface, while uncoordinated carboxylic acid groups can extend into the surrounding water.10 In the present case, a change in pH was exerted by the polyacid, inducing the formation of an amorphous phase over a crystalline one. Shifting PAA to the strong coordinating agent PVP, an amphiphilic, nonionic polymer, widely used to improve the colloidal stability of various particle types as it is known to adsorb well onto a broad range of different materials,36 produced crystalline materials as expected. Here, the PVP seemed to rather function as a morphology (shape) inducer and size regulator, leading to spherical nanocrystals of less than 10 nm in diameter. Further optimization of surface coating procedures will facilitate the application of the produced Mn-based nanostructures as cellular labels from a colloidal stability, cytocompatibility and thus, ultimately, also the relaxivity point of view.
Conclusions
In conclusion, novel, “green” and fast-processed manganese oxide nanostructures, both amorphous and crystalline, were developed using a combination of the “polyol” synthetic approach with microwave digestion. In both cases, a stable suspension of particles of nanometric dimensions was successfully produced. The amorphous material in particular exhibited prominent relaxometric properties, although both crystalline (MnO) and amorphous (MnOx) materials provided sufficient contrast enhancement on MR images. Thus we envision either of the techniques to be feasible and well suited for producing Mn-based nanostructures for cell labeling.Acknowledgements
The Instrument Centre of Shanghai Jiao Tong University, P.R. China, is acknowledged for instrument use. Natalie Råtts is acknowledged for assistance in work related to cell biology. Svenska Tekniska Vetenskapsakademien i Finland r.f., Tekniikan Edistämissäätiö, and the Academy of Finland (project #260599) are acknowledged for financial support (J. M. R.).Notes and references
- L. Ferreira, J. M. Karp, L. Nobre and R. Langer, Cell Stem Cell, 2008, 3, 136 CrossRef CAS PubMed.
- C. Sun, J. S. H. Lee and M. Zhang M, Adv. Drug Delivery Rev., 2008, 60, 1252 CrossRef CAS PubMed.
- A. Taylor, K. M. Wilson, P. Murray, D. G. Fernig and R. Lévy, Chem. Soc. Rev., 2012, 41, 2707 RSC.
- R. Weissleder, G. Elizondo, J. Wittenberg, C. A. Rabito, H. H. Bengele and L. Josephson, Radiology, 1990, 175, 489 CrossRef CAS PubMed.
- J. W. Bulte and D. L. Kraitchman, NMR Biomed., 2004, 17, 484 CrossRef CAS PubMed.
- Z. Li, Q. Sun and M. Gao, Angew. Chem., Int. Ed., 2005, 44, 123 CrossRef CAS PubMed.
- J. Wan, W. Cai, X. Meng and E. Liu, Chem. Commun., 2007, 5004 RSC.
- W. Xiao, H.-C. Gu, D. Li, D. Chen, X. Deng, Z. Jiao and J. Lin, J. Magn. Magn. Mater., 2012, 324, 488 CrossRef CAS PubMed.
- E. Wetterskog, M. Agthe, A. Mayence, J. Grins, D. Wang, S. Rana, A. Ahniyaz, G. Salazar-Alvarez and L. Bergström, Sci. Technol. Adv. Mater., 2014, 15, 055010 CrossRef.
- J. Ge, Y. Hu, M. Biasini, C. Dong, J. Guo, W. P. Beyermann and Y. Yin, Chem. – Eur. J., 2007, 13, 7153 CrossRef CAS PubMed.
- C. Cheng, Y. Wen, X. Xu and H.-C. Gu, J. Mater. Chem., 2009, 19, 8782 RSC.
- Y. Sun, Y. Yin, B. T. Mayers, T. Herricks and Y. Xia, Chem. Mater., 2002, 14, 4736 CrossRef CAS.
- S. H. Im, Y. T. Lee, B. Wiley and Y. Xia, Angew. Chem., Int. Ed., 2005, 44, 2154 CrossRef CAS PubMed.
- I. Bilecka, I. Djerdj and M. Niederberger, Chem. Commun., 2008, 886 RSC.
- S. E. Skrabalak, B. J. Wiley, M. Kim, E. V. Formo and Y. Xia, Nano Lett., 2008, 8, 2077 CrossRef CAS PubMed.
- X. Jia, D. Chen, X. Jiao and S. Zha, Chem. Commun., 2009, 968 RSC.
- C. Cheng, F. Xu and H.-C. Gu, New J. Chem., 2011, 35, 1072 RSC.
- I. Bilecka and M. Niederberger, Nanoscale, 2010, 2, 1358 RSC.
- W. Guan, W. Gu, L. Ye, C. Guo, S. Su, P. Xu and M. Xue, Int. J. Nanomed., 2014, 9, 5071 CAS.
- J. W. Bulte, AJR, Am. J. Roentgenol., 2009, 193, 314 CrossRef PubMed.
- H. S. Thomsen, Eur. J. Radiol., 2006, 16, 2619 CrossRef PubMed.
- J. Perez-Rodriguez, S. Lai, B. D. Ehst, D. M. Fine and D. A. Bluemke, Radiology, 2009, 250, 371 CrossRef PubMed.
- H. Na, J. Lee, K. An, Y. Park, M. Park, I. S. Lee, D.-H. Nam, S. T. Kim, S.-H. Kim, S.-W. Kim, K.-H. Lim, K.-S. Kim, S.-O. Kim and T. Hyeon, Angew. Chem., Int. Ed., 2007, 46, 5397 CrossRef CAS PubMed.
- A. Gilad, P. Walczak, M. McMahon, H. Na, J. Lee, K. An, T. Hyeon, P. van Zijl and J. Bulte, Magn. Reson. Med., 2008, 60, 1 CrossRef CAS PubMed.
- K. H. Bae, K. Lee, C. Kim and T. G. Park, Biomaterials, 2011, 32, 176 CrossRef CAS PubMed.
- T. Kim, E. Momin, J. Choi, K. Yuan, H. Zaidi, J. Kim, M. Park, N. Lee, M. T. McMahon, A. Quinones-Hinojosa, J. W. M. Bulte, T. Hyeon and A. A. Gilad, J. Am. Chem. Soc., 2011, 113, 2955 CrossRef PubMed.
- G. A. Parks, Chem. Rev., 1965, 65, 177 CrossRef CAS.
- F. Gao, J.-Y. Qu, Z.-B. Zhao, Y.-F. Dong, J. Yang, Q. Dong and J.-S. Qiu, New Res. Carbon Mater., 2014, 29, 316 CrossRef.
- D.-X. Chen, N. Sun and H.-C. Gu, J. Appl. Phys., 2009, 063906 CrossRef PubMed.
- L. E. W. LaConte, N. Nitin, O. Zurkiya, D. Caruntu, C. J. O'Connor, X. Hu and G. Bao, J. Magn. Reson. Imaging, 2007, 26, 1634 CrossRef PubMed.
- H. Duan, M. Kuang, X. Wang, Y. A. Wang, H. Mao and S. Nie, J. Phys. Chem. C, 2008, 112, 8127 CAS.
- N. Sun, D.-X. Chen, H.-C. Gu and X.-L. Wang, J. Magn. Magn. Mater., 2009, 321, 2971 CrossRef CAS PubMed.
- D.-X. Chen, N. Sun, Z.-J. Huang, C.-M. Cheng, H. Xub and H.-C. Gu, J. Magn. Magn. Mater., 2010, 322, 548 CrossRef CAS PubMed.
- T. Gulin-Sarfraz, J. Zhang, D. Desai, J. Teuho, J. Sarfraz, H. Jiang, C. Zhang, C. Sahlgren, M. Lindén, H. Gu and J. M. Rosenholm, Biomater. Sci., 2014, 2, 1750 RSC.
- C. Liang, C. Wang and Z. Liu, Part. Part. Syst. Charact., 2013, 30, 1006 CrossRef CAS PubMed.
- C. Graf, D. L. J. Vossen, A. Imhof and A. van Blaaderen, Langmuir, 2003, 19, 6693 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qi00033e |
| ‡ Equal contribution. |
| This journal is © the Partner Organisations 2015 |