
Gold-catalyzed highly efficient benzylation of alcohols with N-Cbz-N-benzyl-propargylamine†
Jing-Rui
Zhao
,
Xiaolong
Yuan
,
Zhaoyan
Wang
,
Shiwu
Chen
,
Zhan-Xin
Zhang
* and
Weihua
Xue
*
School of Pharmacy, Lanzhou University, Lanzhou, 730000, China. E-mail: xuewh@lzu.edu.cn; zhangzhx@lzu.edu.cn; Fax: +86(931)8915686
First published on 12th November 2014
Abstract
N-Cbz-N-benzyl-propargylamine is easily synthesized and described as a benzylating reagent. The treatment of the novel reagent with various alcohols in the presence of the IPrAuNTf2 catalyst affords benzyl ethers in high yields. The protocol derives practical benefits from the mild conditions and the high functional group tolerance, and also eliminates the need for excess base additives or moisture-sensitive promoters.
Benzyl ethers play significant roles in synthetic chemistry. Above all, the O-benzyl group is a useful protective group which is stable over a wide range of conditions in the total synthesis of complex natural products.1 Secondly, some benzylated glycosyl donors for the construction of glycosides prove importantly to be more reactive than acylated analogues.2 Typically, benzyl ethers are prepared by reacting alcohols with a benzyl halide in the presence of an excess of a strong base.3 Alternatively, the benzylation of alcohols can be accomplished using benzyl trichloroacetimidate or its mutants in the presence of an acid promoter.4 Benzyl diphenylphosphinite,5 2-benzyloxy-1-methylpyridinium triflate,6 and 4-(4,6-diphenoxy-1,3,5-triazin-2-yl)-4-benzylmorpholinium trifluoromethanesulfonate7 are recently known as effective benzylating agents of hydroxy groups under neutral conditions. However, these methods are still plagued with problems including the poor compatibility with functional groups,3,4 the use of hygroscopic promoters,4 the requirement of a long reaction time5,6 and excess additives.5–7 Another interesting report is that Asao described gold-catalyzed etherification of alcohols using a designed benzyl ortho-alkynylbenzate as an effective benzylating agent.8 Although the reactions proceed under mild conditions, with low catalyst loadings, to give the products in good to high yields, there are still drawbacks such as the utility of the environmentally unfriendly solvent as well as the moisture-sensitive silver salt in the known procedure. The development of new alternative methods for the synthesis of benzyl ethers is therefore of considerable synthetic importance. A topic of current interest is the catalytic activation for coupling reactions using gold.9 We are particularly attracted to the potential of gold catalysts. Adrio's team found that N-Boc-N-benzyl propargylamine activated by a gold complex catalyst underwent a cyclization to N-benzyl 2-oxazolidinones.10 Based on their observation, we envisioned that N-Cbz-N-benzyl propargylamine might behave as an electrophilic benzylating reagent when it is treated with a nucleophilic alcohol in the presence of a gold catalyst, the reaction would proceed through the intramolecular construction of N-benzyl 2-oxazolidinones as a leaving group and subsequent nucleophilic attack of alcohols. In this work, a new compound, N-Cbz-N-benzyl propargylamine, was reported as a benzylating reagent, which enables the synthesis of benzyl ethers under the catalysis of gold.
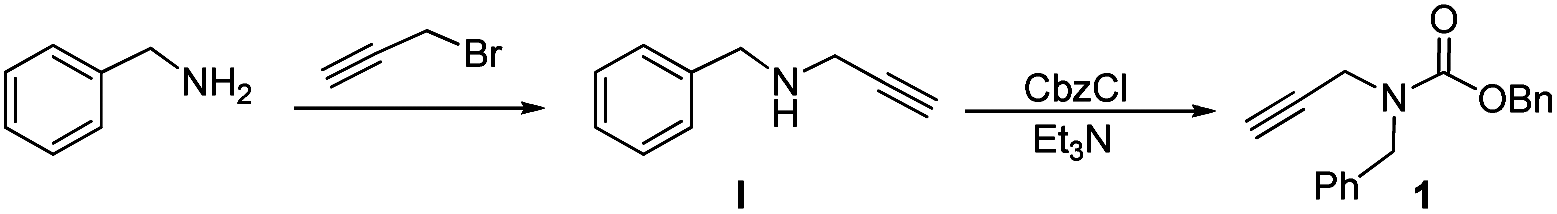
On the basis of Adrio's study, N-Boc-N-benzyl-propargylamine cyclises quickly when catalyzed by gold, so we decided to examine the reaction conditions initially using its analogue N-Cbz-N-benzyl-propargylamine (1) as a potential benzylation reagent. The carbamate was readily prepared as shown in Scheme 1.
 | ||
| Scheme 1 Preparation of N-Cbz-N-benzyl-propargylamine. | ||
Treatment of benzylamine with propargyl bromide generated secondary amine I,11 which was then reacted with benzyl chloroformate in CH2Cl2 to form 1 as a colorless oil in 52% overall yield at a low cost. The compound shows high solubility in chloroform, dichloromethane and toluene. It is stable in both air and moisture and did not deteriorate after storage on the bench for more than one month at ambient temperature.
Menthol and N-Cbz-N-benzyl-propargylamine were selected as prototypical partners for benzylation reactions. The desired transformation was examined using a variety of commercially available gold complexes in CH2Cl2 at room temperature (Table 1). While Ph3PAuSbF6 proved to be extremely efficient in the procedure reported by Adrio and co-workers, poor results were obtained in our case with 10 mol% of this catalyst (Table 1, entry 1). The longer reaction time or employment of related gold phosphine catalysts led to no significant improvement in conversion (Table 1, entries 2–5). Imidazolium carbene complexes with gold have recently found many applications as catalysts for cyclization12 and carbene-transfer13 reactions. In particular, these findings also showed that the use of strongly σ-donating N-heterocyclic carbene (NHC) ligands could be beneficial in cases where the use of phosphine led to a poor catalytic activity of the gold(I) complexes.14 Therefore, we decided to test NHC as the ligand for gold-catalyzed benzylation. To our delight, when 10 mol% of IMesAuNTf2 was used, after 2 h TLC indicated consumption of the starting materials and the final product was isolated in 85% yield (Table 1, entry 6). Further optimization revealed that IPrAuNTf2 was more effective to catalyze the same transformation. When we observed the reaction for 2 h using 10 mol% IPrAuNTf23a was obtained in 94% yield (Table 1, entry 7) together with 2-oxazolindinoe in 90% yield. IPrAuNTf2 loading to 5 mol% led to an apparent loss in yield (78%) and a longer reaction time (10 h) was observed (Table 1, entry 8). IPrAuNTf2-catalyzed benzylation afforded exclusively the ether 3a without any undesirable byproduct being detected. We also observed that the change of the N-substituent from benzyl to butyl, phenyl, trifluorophenyl did not render higher reactivity to the benzylation reagent. Additionally, noteworthy is the fact that IPrAuNTf2 in a solid form is not hygroscopic and can be easily stored and weighed. This is particularly convenient when required for anhydrous reactions.
|
|
|||
|---|---|---|---|
| Entry | Catalyst (equiv.) | Time (h) | Yieldb (%) |
| a Reaction conditions: 1 (0.22 mmol), 2a (0.2 mmol), and [Au] (0.02 mmol) in CH2Cl2 (4 mL) at room temperature. b Isolated yield. c AuPPh3SbF6 was generated in situ from AuPPh3Cl and AgSbF6. | |||
| 1 | AuPPh3SbF6c (0.1) | 1 h | 5.0 |
| 2 | AuPPh3SbF6 (0.1) | 24 h | 7.0 |
| 3 | AuPPh3(CH3CN)SbF6 (0.1) | 24 h | 8.8 |
| 4 | MePhosAuNTf2 (0.1) | 24 h | 12 |
| 5 | XPhosAuNTf2 (0.1) | 24 h | 10 |
| 6 | IMsAuNTf2 (0.1) | 2 h | 85 |
| 7 | IPrAuNTf2 (0.1) | 2 h | 94 |
| 8 | IPrAuNTf2 (0.05) | 10 h | 78 |
|
|||
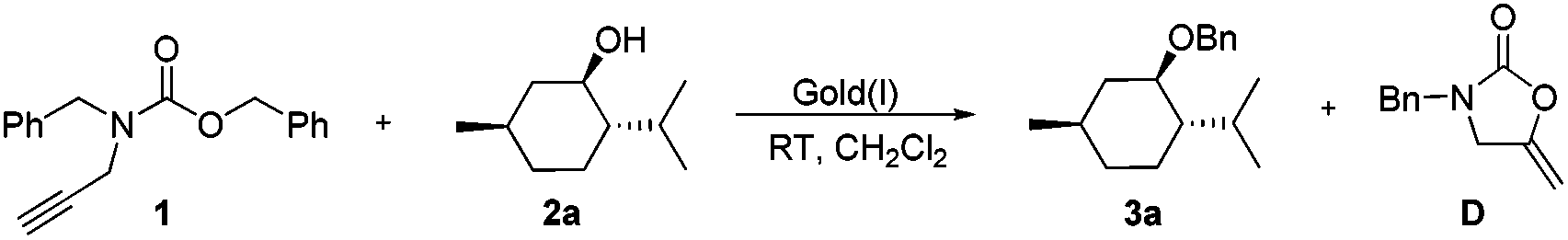
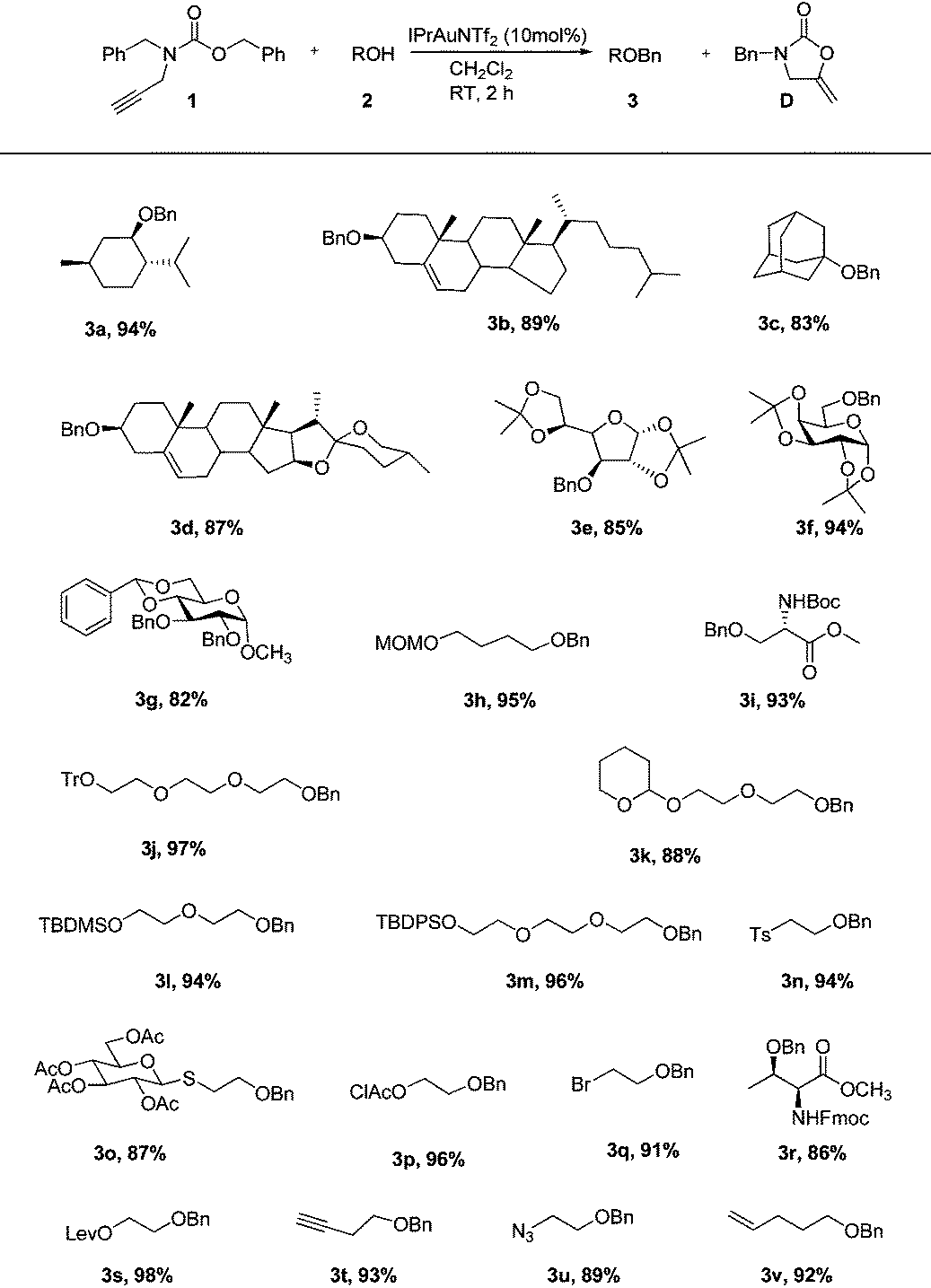
Under the optimal reaction conditions (Table 1, entry 7), various alcohol substrates were tested to examine the generality of the current protocol. As shown in Table 2, all of the substrates including primary, secondary and tertiary alcohols provided the corresponding benzyl ethers within 2 h in good to excellent yields (3a–v). In particular, the compatibility of functionalities with the present new reaction conditions was well confirmed. It should be emphasized that these reactions well proceeded with substrates possessing acid-sensitive functional groups and gave the functionalized products (3e–m) which are difficult to obtain via acid-promoted benzylation reactions. These cases fully indicate that the reaction conditions are relatively mild and not sufficiently acidic to remove the acid-labile protective groups. On the other hand, substrates which are usually not easily benzylated under standard basic conditions underwent smooth cyclization to yield the corresponding protected products (3n–s) without β-elimination or cleavage of protective groups. Noticeably, 3-butyn-1-ol was not intramolecularly cyclized into enol ether under the catalysis of gold, which was consistent with the observation by Pale,15 but was smoothly converted into the desired product (3t). The example pithily demonstrated the higher reactivity of the benzylating reagent bearing an alkyne moiety than the terminal alkynyl alcohol, probably due to the more nucleophilic nature of the carbonyl group toward the intramolecularly activated acetylenic bond. In the latter cases other functionalities such as azide (3u) and olefin (3v) did not also affect the reactions. Hence, the main characteristic of this approach lies in selectively masking alcohols in the presence of other functionalities. Such chemoselectivity is a greatly desirable feature in complex total synthesis. It is noteworthy that no epimerization occurred during the benzylation of chiral alcohols, which rationalized from the following plausible mechanism.
| a Isolated yields. |
|---|
|
A probable mechanism for the benzylation reaction is proposed in Scheme 2. The nucleophilic attack of the carbamate carbonyl group on the activated Au(I)-alkyne complex A is likely to involve the participation of the nitrogen lone pair leading to the formation of 2-(benzyloxy)oxazolinium B which could be an actual active species. The oxazolinium can be degraded into intermediate C and benzyl cationic species.
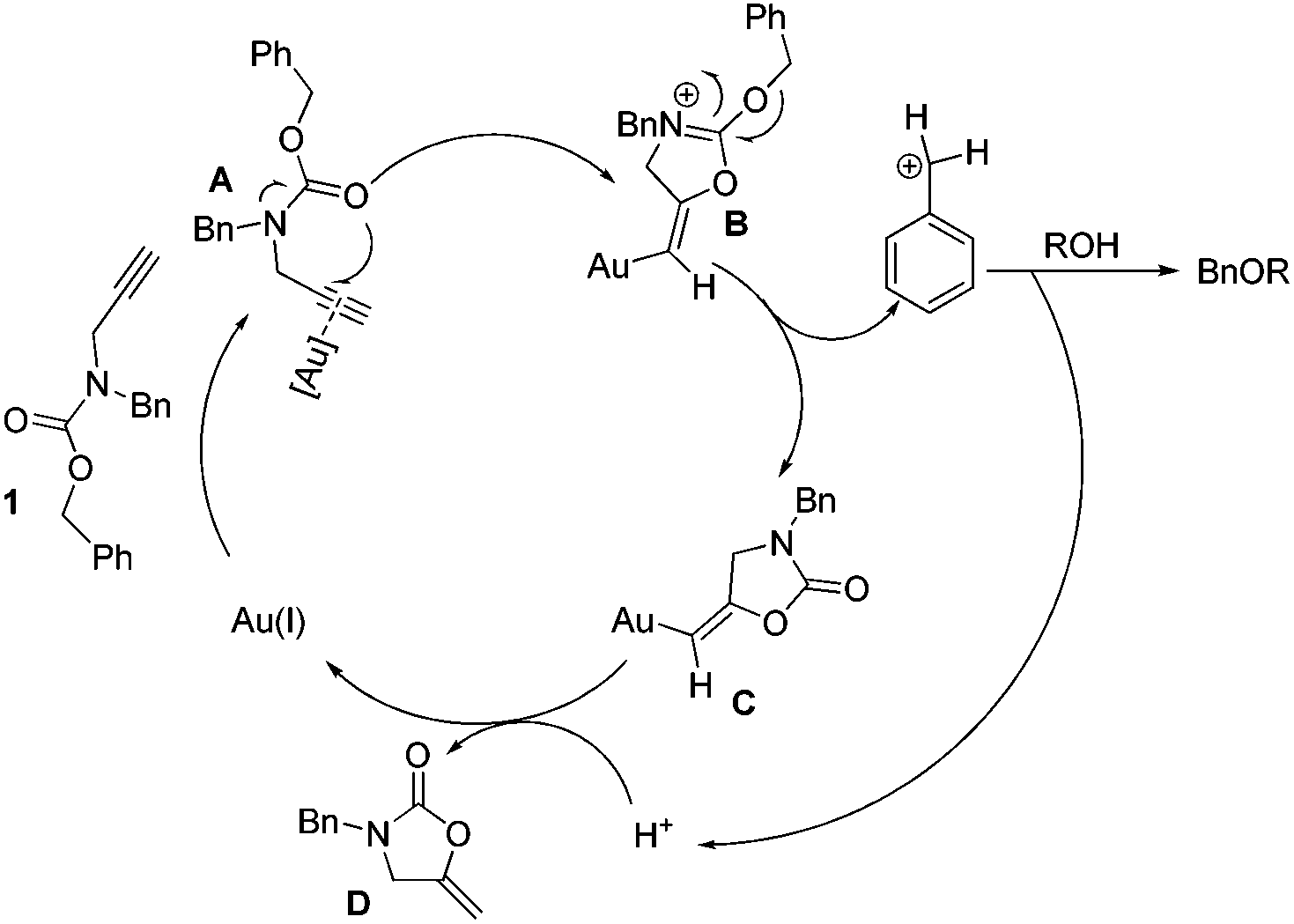 | ||
| Scheme 2 Plausible reaction mechanism. | ||
Then the latter is swiftly attacked by a nucleophilic alcohol to give the corresponding ether and the proton. Subsequently protodeauration of C affords the 2-oxazolidinone D and releases the gold catalyst in the next catalytic cycle. The isolation and characterization of intermediate D were performed, which helped us to gain insight into the detailed catalytic cycle.
To further probe the reaction mechanism we performed benzylation using π-rich heteroaromatics such as 2-methylfuran which is difficult to alkylate under traditional Friedel–Crafts conditions (Scheme 3). We were pleased to find that reaction in CH2Cl2 at room temperature after 2 h afforded 5-benzyl-2-methylfuran F2 in 69% yield. No other side product was observed from the reaction as previously described by Asao. On the basis of the above results, we presume that IPrAuNTf2 was able to catalyze the benzylation of 2-methylfuran with 1 through a Friedel–Crafts process, which indicates that an electrophilic benzyl cationic species might be involved in the reaction and argues in favor of a more SN1-like pathway.
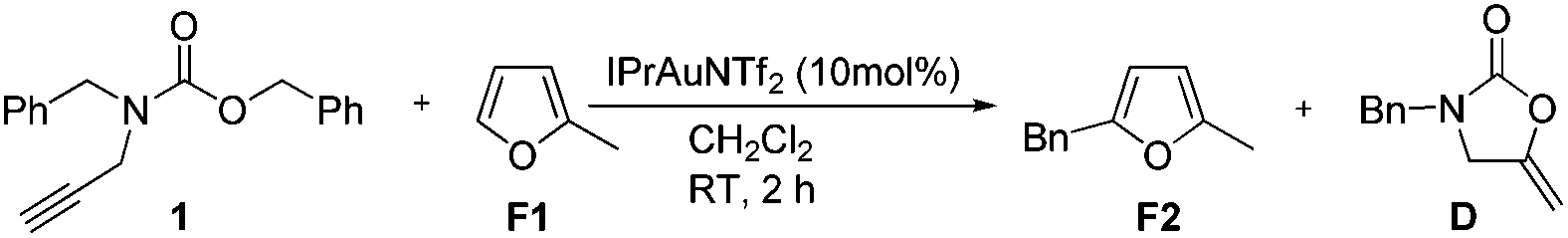 | ||
| Scheme 3 Insight into the potential reaction mechanism. | ||
In conclusion, a new, stable and easily handled benzylating reagent, N-Cbz-N-benzyl-propargylamine, was introduced. We have demonstrated that a series of benzyl ethers was synthesized via gold(I)-catalyzed etherification of the reagent with a diverse range of alcohols. The advantages of the present protocol are the shorter reaction times, the simplicity in operation, and the satisfactory yields of products. Furthermore, its good compatibility with functionalities allows us to believe that this procedure may provide a potential alternative to the existing reagents reported in the literature.
Acknowledgements
We gratefully acknowledge the financial support from the National Science Foundation of China (21372110, 21402075 and 21405067) and the Fundamental Research Funds for the Central Universities, Lanzhou University (lzujbky-2014-k20).Notes and references
- T. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, John Wiley and Sons, 3rd edn, New York, 1999 Search PubMed.
- (a) H. Paulsen, Angew. Chem., Int. Ed. Engl., 1982, 21, 155 CrossRef; (b) S. Hotha and S. J. Kashyap, Am. Chem. Soc., 2006, 128, 9620 CrossRef CAS PubMed.
- Selective examples: (a) W. J. Williamson, J. Chem. Soc., 1852, 4, 106 Search PubMed; (b) T. Iwashige and H. Saeki, Chem. Pharm. Bull., 1967, 15, 1803 CrossRef CAS; (c) S. Czemecki, C. Georgoulis and C. Provelenghiou, Tetrahedron Lett., 1976, 17, 3535 CrossRef; (d) R. Kuhn, I. Löw and H. Trischmann, Chem. Ber., 1957, 90, 203 CrossRef CAS; (e) M. M. El-Abadelah, Tetrahedron, 1973, 29, 589 CrossRef CAS; (f) J. M. Berry and L. D. Hall, Carbohydr. Res., 1976, 47, 307 CrossRef CAS; (g) R. A. W. Johnstone and M. E. Rose, Tetrahedron, 1979, 35, 2169 CrossRef CAS; (h) H.-O. Kalinowski, G. Crass and D. Seebach, Chem. Ber., 1981, 114, 477 CrossRef CAS.
- (a) T. Iversen and D. R. Bundle, J. Chem. Soc., Chem. Commun., 1981, 1240 RSC; (b) H.-P. Wessel, T. Iversen and D. R. Bundle, J. Chem. Soc., Perkin Trans. 1, 1985, 2247 RSC; (c) M. Onaka, M. Kawai and Y. Izumi, Bull. Chem. Soc. Jpn., 1986, 59, 1761 CrossRef CAS; (d) L. J. Liotta and B. Ganem, Tetrahedron Lett., 1989, 30, 4759 CrossRef CAS; (e) S. Hatakeyama, S. Mori, K. Kitano, H. Yamada and M. Nishizawa, Tetrahedron Lett., 1994, 35, 4367 CrossRef CAS; (f) M. Nakano, J. Matsuo and T. Mukaiyama, Chem. Lett., 2000, 29, 1352 CrossRef; (g) E. E. Dueno, F. Chu, S.-I. Kim and K. W. Jung, Tetrahedron Lett., 1999, 40, 1843 CrossRef CAS; (h) B. Boyer, E.-M. Keramane, J.-P. Roque and A. A. Pavia, Tetrahedron Lett., 2000, 41, 2891 CrossRef CAS; (i) M. Izumi and K. Fukase, Chem. Lett., 2005, 34, 594 CrossRef CAS; (j) H. Aoki and T. Mukaiyama, Chem. Lett., 2005, 34, 1016 CrossRef CAS; (k) H. Aoki and T. Mukaiyama, Bull. Chem. Soc. Jpn., 2006, 79, 1255 CrossRef CAS; (l) Y. Okada, M. Ohtsu, M. Bando and H. Yamada, Chem. Lett., 2007, 36, 992 CrossRef CAS; (m) K. Yamada, H. Fujita and M. Kunishima, Org. Lett., 2012, 14, 5026 CrossRef CAS PubMed.
- T. Shintou, W. Kikuchi and T. Mukaiyama, Bull. Chem. Soc. Jpn., 2003, 76, 1645–1667 CrossRef CAS.
- (a) K. W. C. Poon, S. E. House and G. B. Dudley, Synlett, 2005, 3142 CAS; (b) K. W. C. Poon and G. B. Dudley, J. Org. Chem., 2006, 71, 3923 CrossRef CAS PubMed; (c) K. W. C. Poon, P. A. Albiniak and G. B. Dudley, Org. Synth., 2007, 84, 295 CrossRef CAS; (d) K. Yamada, Y. Karuo, M. Kitamura and M. Kunishima, Chem. – Eur. J., 2014, 20, 1 CrossRef PubMed.
- T.-W. Wang, T. Intaranukulkit, M. R. Rosana, R. Slegeris, J. Simon and G. B. Dudley, Org. Biomol. Chem., 2012, 10, 248 CAS.
- N. Asao, H. Aikawa, S. Tago and K. Umetsu, Org. Lett., 2007, 9, 4299 CrossRef CAS PubMed.
- For recent reviews on gold catalysis, see: (a) S. A. Hashmi and G. J. Huchings, Angew. Chem., Int. Ed., 2006, 45, 7896 CrossRef PubMed; (b) D. J. Gorin and F. D. Toste, Nature, 2007, 46, 395 CrossRef PubMed; (c) A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 2 CrossRef; (d) A. Stephen and K. Hashmi, Chem. Rev., 2007, 7, 3180 Search PubMed; (e) Z. Li, C. Brouwer and C. He, Chem. Rev., 2008, 8, 3239 CrossRef PubMed.
- R. Robles-Machin, J. Adrio and J. C. Carretero, J. Org. Chem., 2006, 71, 5023 CrossRef CAS PubMed.
- C. Monlinaro and T. F. Jamison, J. Am. Chem. Soc., 2003, 125, 8076 CrossRef PubMed.
- (a) N. D. Shapiro and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 4160 CrossRef CAS PubMed; (b) S. López, E. Herrero-Gomez, P. Perez-Galan, C. Nieto-Oberhuber and A. M. Echavarren, Angew. Chem., Int. Ed., 2006, 45, 6029 CrossRef PubMed; (c) M. R. Fructos, T. R. Belderrain, P. de Frémont, N. M. Scott, S. P. Nolan, M. M. Diaz-Requejo and P. J. Perez, Angew. Chem., Int. Ed., 2005, 44, 5284 CrossRef CAS PubMed; (d) P. De Frémont, E. D. Stevens, M. R. Fructos, M. M. Diaz-Requejo, P. J. Perez and S. P. Nolan, Chem. Commun., 2006, 2045 RSC.
- N. Marion, S. Diez-Gonzalez, P. De Frémont, A. R. Noble and S. P. Nolan, Angew. Chem., Int. Ed., 2006, 45, 3647 CrossRef CAS PubMed.
- L. Ricard and F. Gagosz, Organometallics, 2007, 26, 4704 CrossRef CAS.
- H. Harkat, J.-M. Weibel and P. Pale, Tetrahedron Lett., 2007, 48, 1439 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4qo00255e |
| This journal is © the Partner Organisations 2015 |