
Synthesis of diaryldifluoromethanes by Pd-catalyzed difluoroalkylation of arylboronic acids†
Ji-Wei
Gu
,
Wen-Hao
Guo
and
Xingang
Zhang
*
Key Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: xgzhang@mail.sioc.ac.cn; Fax: (+86)-21-6416-6128; Tel: (+86)-21-5492-5333
First published on 14th November 2014
Abstract
Diaryldifluoromethanes constitute a distinct class of fluorinated compounds in medicinal chemistry. But only limited methods to access this structural motif have been reported. Herein, we demonstrate the first example of Pd-catalyzed aryldifluoromethylation of arylboronic acids with readily available aryldifluoromethyl bromides. Because of its high efficiency, excellent functional group compatibility, and mild reaction conditions, this protocol provides a facile access to diaryldifluoromethanes. Preliminary mechanistic studies revealed that a Pd(0)Ln-initiated single electron transfer (SET) pathway is involved in the catalytic cycle.
Fluorinated arenes are an important class of structural motif that is found in many pharmaceuticals, agrochemicals, and advanced materials due to the unique properties of the fluorine atom and/or the carbon–fluorine bond.1 As a consequence, substitution of the hydrogen atom(s) in the parent molecules with the fluorine atom(s) is now a common strategy in drug discovery and development.2 Among the fluorinated arenes, diaryldifluoromethanes, in which the methylene group (CH2) is replaced by a difluoromethylene group (CF2), represent an appealing type compounds because of their important applications in medicinal chemistry. For instance, Ledipasvir, a diaryldifluoromethane containing molecule, is an oral NS5A inhibitor for the treatment of hepatitis C virus (HCV) infection,3 and has been filed for approval by the FDA. To date, however, only limited methods have been reported to access diaryldifluoromethanes. The most common approach to such a fluorinated structural motif relies on deoxyfluorination of a carbonyl with dialkylaminosulfur trifluorides (i.e. DAST or DeoxoFluor).4 Despite the importance of such a process, to directly introduce a functionalized CF2 group into organic molecules catalyzed by a transition metal would be an attractive alternative, as C–C bond formation catalyzed by transition-metals is a powerful strategy due to its broad substrate scope, high efficiency, and excellent functional-group compatibility. However, compared to transition-metal-catalyzed trifluoromethylation and fluorination of arenes,5 the difluoroalkylation of arenes through a similar strategy has been less studied,6,7 although the CF2 group plays an equally important role in medicinal chemistry and materials science.8 To the best of knowledge, there is no successful example that has been reported to prepare diaryldifluoromethanes catalyzed by a transition-metal. Therefore, it is highly desirable to develop an efficient reaction to access such a valuable structural motif.
As part of a systematic study on transition-metal-catalyzed reactions for the direct introduction of fluorinated moieties into organic molecules,7,9 herein we describe the first example of synthesis of diaryldifluoromethanes through Pd-catalyzed cross-coupling between readily available aryldifluoromethyl bromides and arylboronic acids. The notable advantages of this protocol are its mild reaction conditions, excellent functional group compatibility, and operational simplicity, thus providing a facile access to a range of diaryldifluoromethanes of interest in drug discovery and development. Furthermore, to demonstrate the usefulness of this method, the late-stage aryldifluoromethylation of bioactive molecules has also been performed.
Initial studies focused on the Pd-catalyzed cross-coupling reaction of phenylboronic acid 2a with the readily available 4-(bromodifluoromethyl)-1,1′-biphenyl 1a10 in the presence of a variety of phosphine ligands (Table 1, entries 1–8). It was found that the choice of phosphine ligand was very critical to the reaction efficiency, and the bulky ligand cataCXium AHI [PAd2(n-Bu)·HI]11 was the optimal one, providing difluoromethylenated product 3a in 83% yield (determined by 19F NMR, Table 1, entry 6). This is probably because of formation of an active T-shaped palladium complex (Ph)(CF2Ar)Pd[PAd2(n-Bu)] that benefits the reductive elimination.12 However, only a poor yield (27%) of 3a was obtained when XantPhos, which was previously demonstrated to be an excellent bisphosphine ligand for reductive elimination from fluoroalkyl palladium complexes,7c,13 was employed (Table 1, entry 8). Encouraged by these results, several reaction parameters were examined to further optimize the reaction conditions (Table 1, entries 9–15, for details, see ESI†). It was revealed that K2CO3, 1,4-dioxane, and Pd(OAc)2 were the optimal choices, leading to compound 3a in the best yield (91% upon isolation) when the ratio of ligand/Pd was decreased from 2/1 to 1/1 (Table 1, entry 10). Other bases, solvents, or palladium catalysts either afforded lower yields or showed no activity. The absence of palladium catalyst or ligand failed to provide the desired product, thus demonstrating that a Pd(0/II) catalytic cycle is involved in the reaction (Table 1, entries 16 and 17).
|
|
|||
|---|---|---|---|
| Entry | [Pd] (x) | L (y) | 3a, Yieldb (%) |
| a Reaction conditions (unless otherwise specified): 1a (0.2 mmol, 1.0 equiv.), 2a (0.3 mmol, 1.5 equiv.), K2CO3 (2.0 equiv.), 1,4-dioxane (2 mL), 8 h. b Determined by 19F NMR using PhCF3 as an internal standard and number in parentheses is isolated yield. | |||
| 1 | Pd(OAc)2 (5) | PPh3 (10) | 27 |
| 2 | Pd(OAc)2 (5) | P(o-OMePh)3 (10) | 4 |
| 3 | Pd(OAc)2 (5) | PCy3·HBF4 (10) | 39 |
| 4 | Pd(OAc)2 (5) | Pt-Bu3·HBF4 (10) | 15 |
| 5 | Pd(OAc)2 (5) | CyJohnPhos (10) | 8 |
| 6 | Pd(OAc)2 (5) | PAd2(n-Bu)·HI (10) | 83 |
| 7 | Pd(OAc)2 (5) | DPPE (10) | 8 |
| 8 | Pd(OAc)2 (5) | XantPhos (10) | 27 |
| 9 | Pd(OAc)2 (5) | PAd2(n-Bu)·HI (7.5) | 89 |
| 10 | Pd(OAc)2 (5) | PAd2(n-Bu)·HI (5) | 97(91) |
| 11 | Pd(PPh3)4 (5) | PAd2(n-Bu)·HI (5) | Trace |
| 12 | Pd2(dba)3 (2.5) | PAd2(n-Bu)·HI (5) | 34 |
| 13 | Pd2(Allyl)2Cl2 (2.5) | PAd2(n-Bu)·HI (5) | 56 |
| 14 | Pd(PPh3)2Cl2 (2.5) | PAd2(n-Bu)·HI (5) | Trace |
| 15 | PdCl2(dppp) (2.5) | PAd2(n-Bu)·HI (5) | 14 |
| 16 | Pd(OAc)2 (5) | none | NR |
| 17 | None | PAd2(n-Bu)·HI(5) | NR |
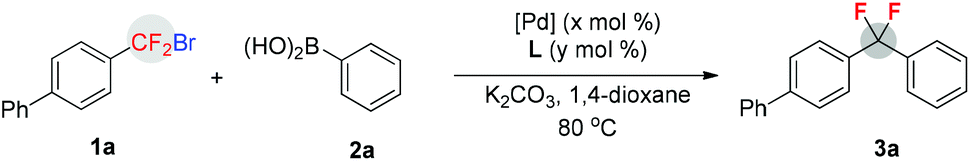
Upon identification of viable reaction conditions, the substrate scope of this transformation was tested and representative results are illustrated in Table 2. Overall, aromatic boronic acids with either electron-rich or electron-deficient substituents underwent the present cross-coupling process with aryldifluoromethyl bromide 1a in moderate to high yields. A variety of important functional groups, such as the formyl, alkoxycarbonyl, enolizable ketone, and trimethylsilyl groups, were tolerated quite well (3l–p). The sterically hindered ortho-tolylboronic acid and its derivative were excellent substrates, and the cross-coupling with 1a proceeded with high efficiency (3d and 3e). Furthermore, other aryldifluoromethyl bromides were also applicable to the reaction and moderate yields were obtained (3r–u). However, it should be mentioned that in the cases of reactions of phenyldifluoromethyl bromide 1b with arylboronic acids, a significant amount of PhCF2H was observed under standard conditions. We supposed that the formation of PhCF2H could be ascribed to the reaction of solvent with PhCF2˙ radical that was generated from 1b with Pd–PAd2(n-Bu)–base. To our delight, when 1.0 equiv. of H2O was added to the reaction, moderate yields of 3r and 3s were obtained. However, the role of H2O in this reaction remains uncertain. Notably, aryl chloride was also compatible with the reaction conditions, thus providing a good opportunity for downstream transformations (3u).
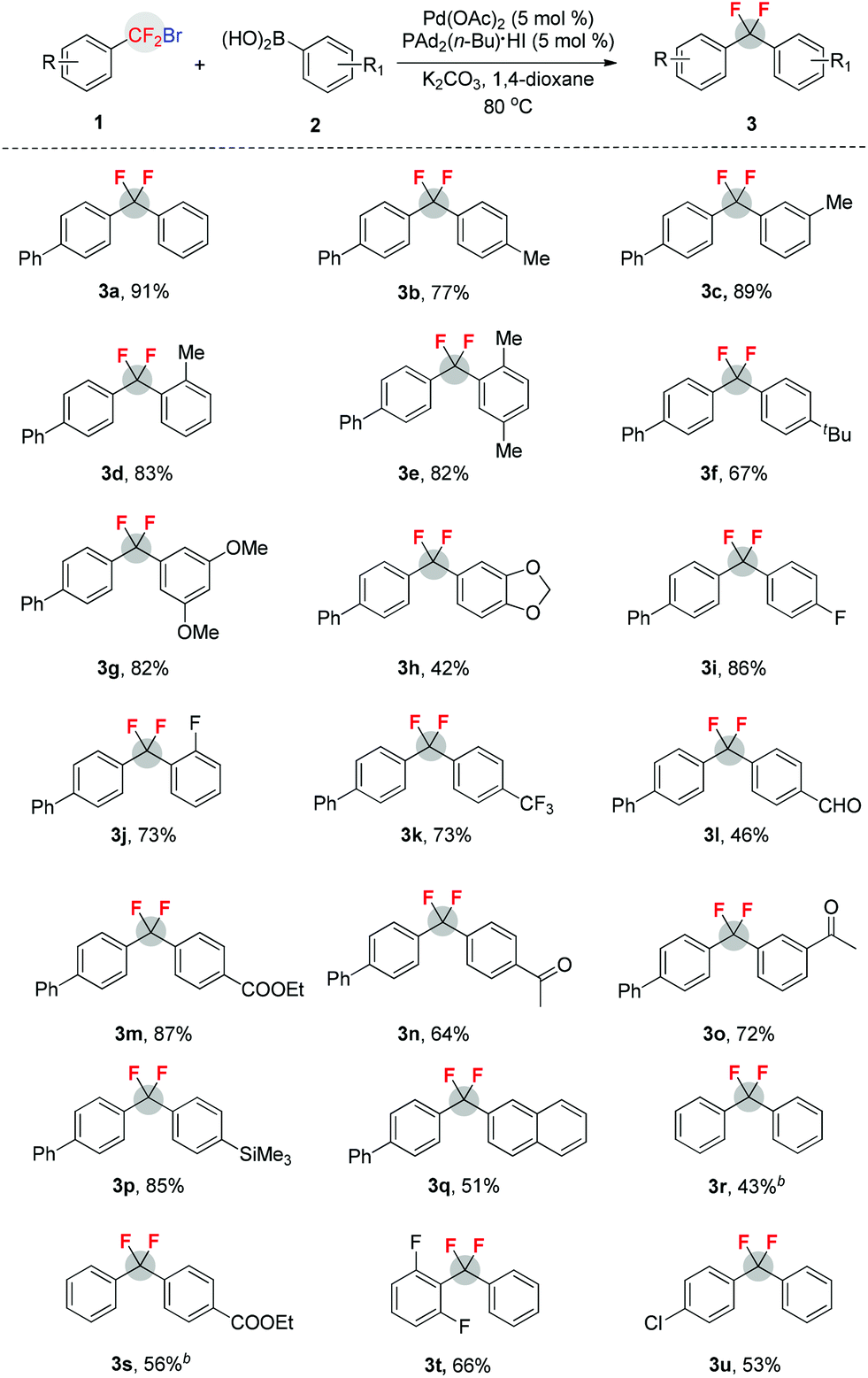
The importance and usefulness of this method can also be highlighted by late-stage aryldifluoromethylation of bioactive molecules. As shown in Scheme 1, treatment of estrone-derived arylboronic acid 4 with the difluoroalkyl bromide 1a afforded difluoroalkylated compound 5 with high efficiency (79%). Similarly, the flavanone-derived arylboronic acid 6 also led to its corresponding difluoroalkylated product 7 in high yield (82%). In view of the fact that CF2 can functionalize as a bioisostere of the oxygen atom or carbonyl group,14 and leads to profound changes in physical and biological properties of CF2 containing molecules,8 we view these transformations as a highly valuable opportunity for application in drug discovery and development.
 | ||
| Scheme 1 Late-stage functionalization for synthesis of biologically active molecules. | ||
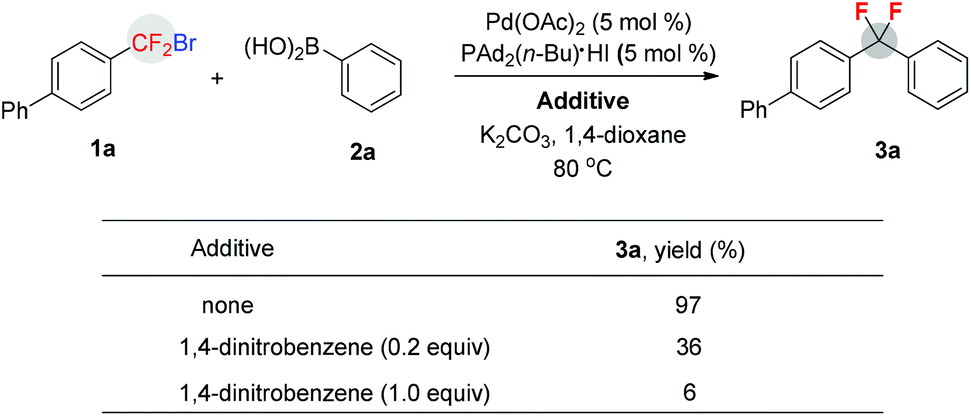
To gain some mechanistic insight into the present reaction, radical inhibition experiments were conducted (Scheme 2). A significant decrease of the yields were observed when an electron transfer (ET) inhibitor (1,4-dinitrobenzene) was added to the reaction mixture of 1a and 2a in the presence of Pd(OAc)2 (5 mol%), PAd2(n-Bu)·HI (5 mol%), and K2CO3 in dioxane.15 Thus, these preliminary studies demonstrate that a single electron transfer (SET) pathway via an aryldifluoromethyl radical is involved in the catalytic cycle.
 | ||
| Scheme 2 Radical inhibition experiments. | ||
To confirm that a free aryldifluoromethyl radical was generated during the reaction, a radical clock experiment was conducted (Scheme 3). When compound 816 was treated with 1a under standard conditions, a ring-expanded product 9 was formed in 7% yield (determined by 19F NMR) along with 3% yield of protonated product 10 and 78% of starting material 1a unreacted. This finding demonstrated that a free radical was indeed generated during the reaction.
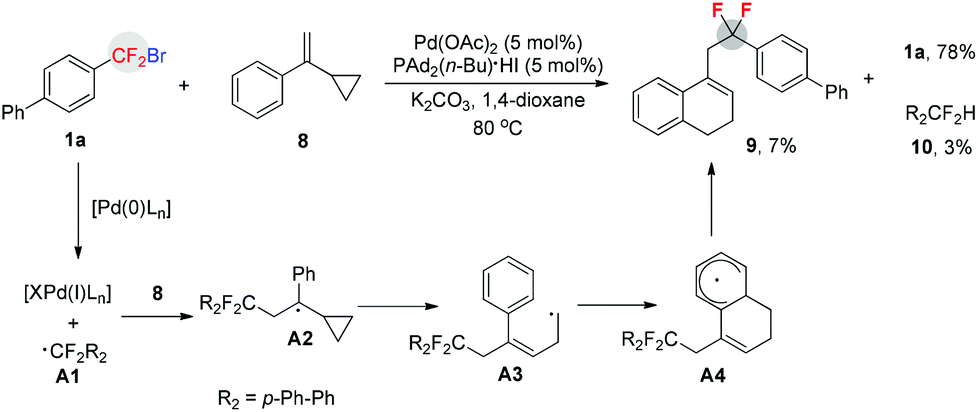 | ||
| Scheme 3 Radical clock experiment. | ||
On the basis of these results, a plausible reaction mechanism was proposed (Scheme 4). The reaction is initiated by a [PdLn(0)]-promoted SET pathway to generate aryldifluoroalkyl radical (ArCF2˙) A. A subsequently reacts with LnPd(I)Br to produce Pd(II)-complex B. After transmetalation, a key intermediate C would be generated. As the final step of the catalytic cycle, reductive elimination of C affords diaryldifluoromethanes 3. However, for the detailed reaction mechanism remains a subject to be studied.
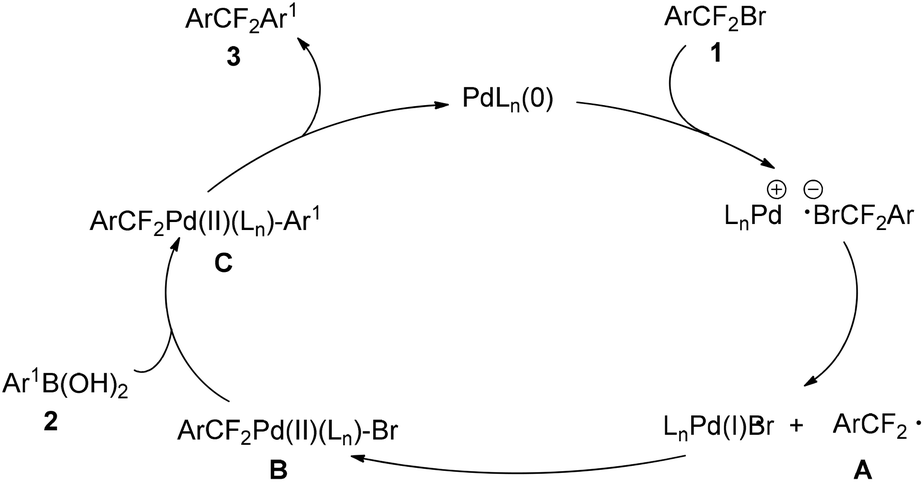 | ||
| Scheme 4 Proposed reaction mechanism. | ||
Conclusions
In conclusion, we have demonstrated the first example of palladium-catalyzed aryldifluoromethylation of arylboronic acids with readily available aryldifluoromethyl bromides. The utility of PAd2(n-Bu)·HI is critical for the reaction efficiency. The reaction proceeds under mild reaction conditions with excellent functional group compatibility. Thus, these transformations provide a facile route for application in medicinal chemistry. Mechanistic studies revealed that a [Pd(0)Ln] promoted SET pathway via a free fluoroalkyl radical is involved in the reaction. Further studies to uncover the reaction mechanism as well as other derivative reactions are now in progress in our laboratory.Acknowledgements
The National Basic Research Program of China (973 program (no. 2015CB931900)), the NSFC (nos 21425208, 21421002, 21172242, and 21332010), and SIOC are greatly acknowledged for funding this work.Notes and references
- For selected reviews, see: (a) B. E. Smart, J. Fluorine Chem., 2001, 109, 3 CrossRef CAS; (b) P. Maienfisch and R. G. Hall, Chimia, 2004, 58, 93 CrossRef CAS; (c) Special issue on “Fluorine in the Life Sciences”, ChemBioChem, 2004, 5, 557;; (d) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC.
- For selected reviews, see: (a) K. L. Kirk, J. Fluorine Chem., 2006, 127, 1013 CrossRef CAS PubMed; (b) J.-P. Begue and D. Bonnet-Delpon, J. Fluorine Chem., 2006, 127, 992 CrossRef CAS PubMed; (c) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (d) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed; (e) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC.
- J. O. Link, J. G. Taylor, L. Xu, M. Mitchell, H. Guo, H. Liu, D. Kato, T. Kirschberg, J. Sun, N. Squires, J. Parrish, T. Keller, Z.-Y. Yang, C. Yang, M. Matles, Y. Wang, K. Wang, G. Cheng, Y. Tian, E. Mogalian, E. Mondou, M. Cornpropst, J. Perry and M. C. Desai, J. Med. Chem., 2014, 57, 2033 CrossRef CAS PubMed.
- For selected examples of difluorination of carbonyl groups with fluorinated reagents, see: (a) W. J. Middleton and E. M. Bingham, J. Org. Chem., 1980, 45, 2883 CrossRef CAS; (b) G. S. Lal, G. P. Pez, R. J. Pesaresi, F. M. Prozonic and H. Cheng, J. Org. Chem., 1999, 64, 7048 CrossRef CAS; (c) D. Solas, R. L. Hale and D. V. Patel, J. Org. Chem., 1996, 61, 1537 CrossRef CAS; (d) S. Kobayashi, A. Yoneda, T. Fukuhara and S. Hara, Tetrahedron, 2004, 60, 6923 CrossRef CAS PubMed; (e) T. Umemoto, R. P. Singh, Y. Xu and N. Saito, J. Am. Chem. Soc., 2010, 132, 18199 CrossRef CAS PubMed; (f) A. L'Heureux, F. Beaulieu, C. Bennett, D. R. S. Clayton, F. LaFlamme, M. Mirmehrabi, S. Tadayon, D. Tovell and M. Couturier, J. Org. Chem., 2010, 75, 3401 CrossRef PubMed.
- For selected reviews, see: (a) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed; (b) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475 CrossRef CAS PubMed; (c) C. Hollingworth and V. Gouverneur, Chem. Commun., 2012, 48, 2929 RSC; (d) T. Besset, C. Schneider and D. Cahard, Angew. Chem., Int. Ed., 2012, 51, 5048 CrossRef CAS PubMed; (e) F.-L. Qing, Chin. J. Org. Chem., 2012, 32, 815 CrossRef CAS; (f) X. Wang, Y. Zhang and J. Wang, Sci. Sin.: Chim., 2012, 42, 1417 CrossRef CAS PubMed.
- For transition-metal-catalyzed difluoroalkylation, see: (a) K. Araki and M. Inoue, Tetrahedron, 2013, 69, 3913 CrossRef CAS PubMed; (b) S. Ge, W. Chaladj and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 4149 CrossRef CAS PubMed.
- For our contribution to transition-metal-catalyzed difluoroalkylation, see: (a) Z. Feng, F. Chen and X. Zhang, Org. Lett., 2012, 14, 1938 CrossRef CAS PubMed; (b) Z. Feng, Y.-L. Xiao and X. Zhang, Org. Chem. Front., 2014, 1, 113 RSC; (c) Z. Feng, Q.-Q. Min, Y.-L. Xiao, B. Zhang and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 1669 CrossRef CAS PubMed; (d) Q.-Q. Min, Z. Yin, Z. Feng, W.-H. Guo and X. Zhang, J. Am. Chem. Soc., 2014, 136, 1230 CrossRef CAS PubMed; (e) Y.-L. Xiao, W.-H. Guo, G.-Z. He, Q. Pan and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 9909 CrossRef CAS PubMed; (f) Y.-B. Yu, G.-Z. He and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 10457 CrossRef CAS PubMed; (g) Y.-L. Xiao, B. Zhang, Z. Feng and X. Zhang, Org. Lett., 2014, 16, 4822 CrossRef CAS PubMed.
- For selected reviews, see: (a) X.-L. Qiu, X.-H. Xu and F.-L. Qing, Tetrahedron, 2010, 66, 789 CrossRef CAS PubMed; (b) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529 CrossRef CAS PubMed; For selected papers, see: (c) C. S. Burgey, K. A. Robinson, T. A. Lyle, P. E. J. Sanderson, S. Dale Lewis, B. J. Lucas, J. A. Krueger, R. Singh, C. Miller-Stein, R. B. White, B. Wong, E. A. Lyle, P. D. Williams, C. A. Coburn, B. D. Dorsey, J. C. Barrow, M. T. Stranieri, M. A. Holahan, G. R. Sitko, J. J. Cook, D. R. McMasters, C. M. McDonough, W. M. Sanders, A. A. Wallace, F. C. Clayton, D. Bohn, Y. M. Leonard, T. J. Detwiler Jr., J. J. Lynch Jr., Y. Yan, Z. Chen, L. Kuo, S. J. Gardell, J. A. Shafer and J. P. Vacca, J. Med. Chem., 2003, 46, 461 CrossRef CAS PubMed; (d) J. Li, S. Y. Chen, B. J. Murphy, N. Flynn, R. Seethala, D. Slusarchyk, M. Yan, P. Sleph, H. Zhang, W. G. Humphreys, W. R. Ewing, J. A. Robl, D. Gordon and J. A. Tino, Bioorg. Med. Chem. Lett., 2008, 18, 4072 CrossRef CAS PubMed; (e) C. L. Lynch, C. A. Willoughby, J. J. Hale, E. J. Holson, R. J. Budhu, A. L. Gentry, K. G. Rosauer, C. G. Caldwell, P. Chen, S. G. Mills, M. MacCoss, S. Berk, L. Chen, K. T. Chapman, L. Malkowitz, M. S. Springer, S. L. Gould, J. A. DeMartino, S. J. Siciliano, M. A. Cascieri, A. Carella, G. Carver, K. Holmes, W. A. Schleif, R. Danzeisen, D. Hazuda, J. Kessler, J. Lineberger, M. Miller and E. A. Eminic, Bioorg. Med. Chem. Lett., 2003, 13, 119 CrossRef.
- (a) C.-Y. He, S. Fan and X. Zhang, J. Am. Chem. Soc., 2010, 132, 12850 CrossRef CAS PubMed; (b) X. Zhang, S. Fan, C.-Y. He, X. Wan, Q.-Q. Min, J. Yang and Z.-X. Jiang, J. Am. Chem. Soc., 2010, 132, 4506 CrossRef CAS PubMed; (c) S. Fan, F. Chen and X. Zhang, Angew. Chem., Int. Ed., 2011, 50, 5918 CrossRef CAS PubMed; (d) Z. Chen, C.-Y. He, Z. Yin, L. Chen and X. Zhang, Angew. Chem., Int. Ed., 2013, 52, 5813 CrossRef CAS PubMed.
- J. He and C. U. Pittman, Synth. Commun., 1999, 29, 855 CrossRef CAS.
- (a) A. Ehrentraut, A. Zapf and M. Beller, Synlett, 2000, 1589 CAS; (b) A. Zapf, A. Ehrentraut and M. Beller, Angew. Chem., Int. Ed., 2000, 39, 4153 CrossRef CAS.
- A. G. Sergeev, A. Spannenberg and M. Beller, J. Am. Chem. Soc., 2008, 130, 15549 CrossRef CAS PubMed.
- (a) V. V. Grushin and W. J. Marshall, J. Am. Chem. Soc., 2006, 128, 12644 CrossRef CAS PubMed; (b) V. I. Bakhmutov, F. Bozoglian, K. Gómez, G. González, V. V. Grushin, S. A. Macgregor, E. Martin, F. M. Miloserdov, M. A. Novikov, J. A. Panetier and L. V. Romasho, Organometallics, 2012, 31, 1315 CrossRef CAS.
- (a) C. M. Blackburn, D. A. England and F. Kolkmann, J. Chem. Soc., Chem. Commun., 1981, 930 RSC; (b) G. M. Blackburn, D. E. Kent and F. Kolkmann, J. Chem. Soc., Perkin Trans. 1, 1984, 1119 RSC; (c) T. Kitazume and T. Kamazaki, Experimental Methods in Organic Fluorine Chemistry, Gordon and Breach Science, Tokyo, 1998 Search PubMed.
- For related radical inhibition experiments studies, see: (a) X.-T. Huang and Q.-Y. Chen, J. Org. Chem., 2001, 66, 4651 CrossRef CAS PubMed; (b) R. N. Loy and M. S. Sanford, Org. Lett., 2011, 13, 2548 CrossRef CAS PubMed.
- J. E. Baldwin, Chem. Rev., 2003, 103, 1197 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, and analytical data for all new compounds. See DOI: 10.1039/c4qo00246f |
| This journal is © the Partner Organisations 2015 |