
Target-oriented design and biosynthesis of thiostrepton-derived thiopeptide antibiotics with improved pharmaceutical properties†
Shoufeng
Wang‡
a,
Qingfei
Zheng‡
a,
Jianfeng
Wang
b,
Zhixiong
Zhao
a,
Qingye
Li
c,
Yunsong
Yu
b,
Renxiao
Wang
a and
Wen
Liu
*ac
aState Key Laboratory of Bioorganic and Natural Products Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: wliu@mail.sioc.ac.cn
bDepartment of Infectious Diseases, Sir Run Run Shaw Hospital, College of Medicine, Zhejiang University, Hangzhou, Zhejiang 310016, China
cHuzhou Center of Bio-Synthetic Innovation, 1366 Hongfeng Road, Huzhou 313000, China
First published on 5th December 2014
Abstract
Thiostrepton is a potent archetypal thiopeptide antibiotic. According to its mechanism known to target bacterial ribosome, we show here a rational design upon modeling of this molecule into the ribosome complex and an effective biosynthesis of new thiopeptide antibiotics through regioselective modifications. The resulting derivatives exhibit a series of anticipated and unanticipated pharmaceutical advantages, including improvement in activity against a number of drug-resistant pathogens and in water solubility that has largely affected the clinical use of thiostrepton.
Thiopeptide antibiotics,1 a class of sulfur-rich, highly modified peptides, share the macrocyclic loop1 and the extended tail, both of which constitute a family-specific core system featuring a nitrogen-containing domain central to multiple azoles and dehydroamino acids (Fig. 1A). In this natural product family, thiostrepton (TSR) is often referred to as the parent compound among the nearly 100 members for which structures are known. TSR is bi-cyclic and possesses loop2, the side ring system containing a quinaldic acid (QA) moiety, appended onto the characteristic core system (Fig. 1A). This molecule exhibits the potent activity against Gram-positive bacteria and particularly serves as an active drug component approved by the Pure Food and Drug Administration (FDA) for animal use. TSR has not been developed for human therapy, largely due to its poor aqueous solubility.1a The development of new analogs has extensively been carried out to overcome this physical drawback;2 however, the complex architecture of TSR poses a tremendous challenge to chemical synthesis and modification.
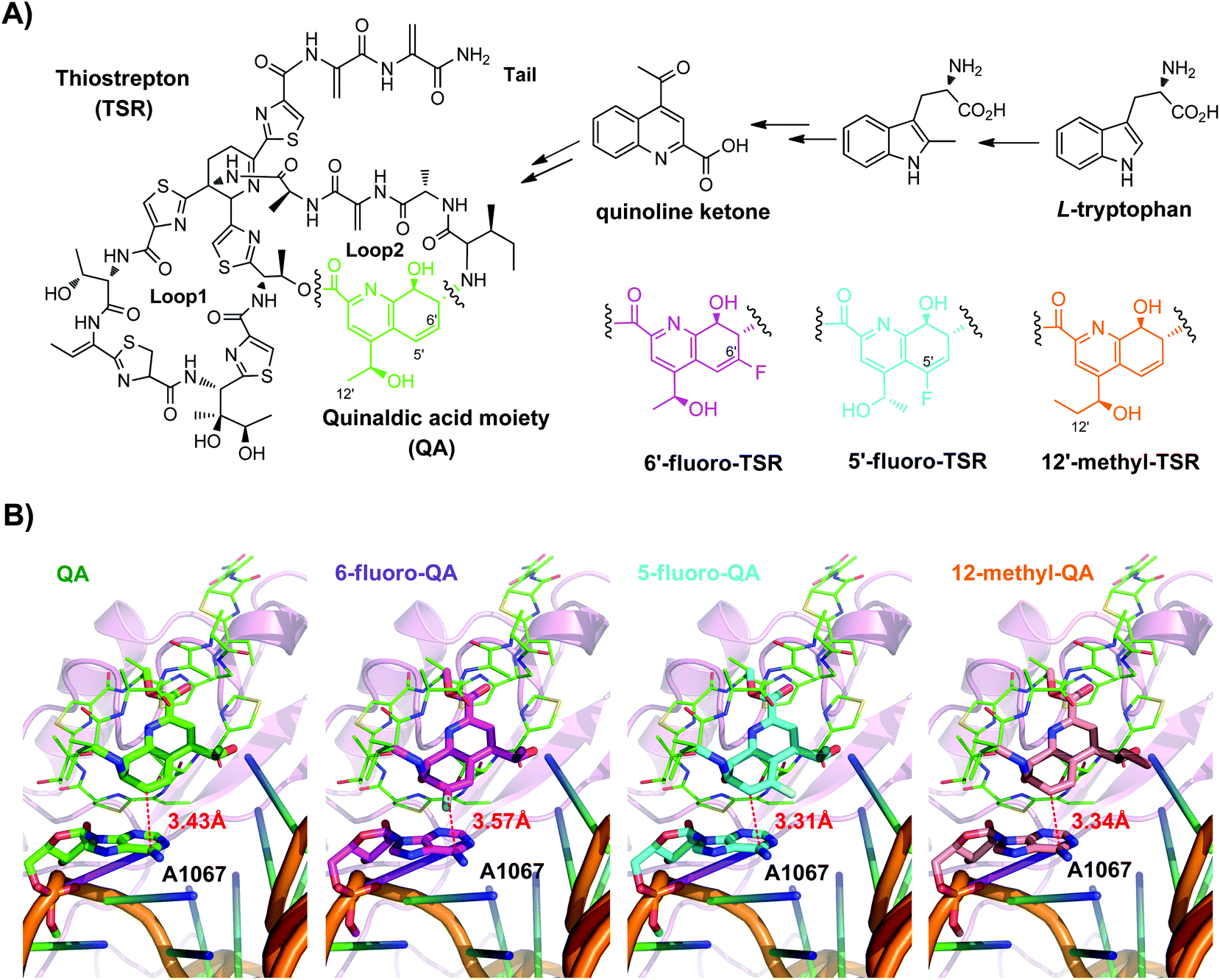 | ||
| Fig. 1 TSR, 6′-fluoro-TSR and newly designed derivatives and their interaction with the bacterial ribosome. (A) Chemical structures and the biosynthetic pathway for QA incorporation. The difference in QA modification is shown by color, with green representing the unmodified QA of TSR, purple representing the 6-fluoro-QA of 6′-fluoro-TSR, blue representing the 5-fluoro-QA of 5′-fluoro-TSR, and yellow representing the 12-methyl-QA of 12′-methyl-TSR. (B) 50S ribosomal subunit in complex with TSR or the derivative. The 23S rRNA (orange) and L11 protein (pink) are shown as cartoon models, and the TSR or its derivative is shown as a stick model. The dashed red line indicates the distance between QA and A1067 of the 23S rRNA. | ||
In the past several years, we and others have uncovered a common paradigm for thiopeptide biosynthesis that involves conserved posttranslational modifications of a ribosomally synthesized precursor peptide to construct the core system.3 Various approaches of genetic engineering have thus been developed upon sequence permutation of the precursor peptides,4 largely complementing the synthetic efforts in diverse modification of the peptidyl skeletons of thiopeptides. Consequently, structural modifications of the core system often lead to a significant decrease in antibacterial activity, which, in contrast, is essentially retained when changes occur on loop2 of the bi-cyclic members such as TSR. Unlike other chemotherapeutics targeting the bacterial ribosome,5 many thiopeptides are known to bind within a cleft located between the L11 protein and the 23S rRNA of the 50S large ribosomal subunit (Fig. 1B), thereby perturbing the translation factor binding and subsequent protein synthesis. Specifically, 60% and 26% of the surface buried by TSR on the ribosome are attributed to loop1 and the tail, respectively, and the remaining 14% is from loop2.6 Thus, modifications of the biologically more important core system (particularly loop1) are apparently liable to cause a decrease in binding affinity, and change of the biologically relevant but less dependent loop2 of bi-cyclic members could be an ideal strategy to modulate pharmaceutical properties of thiopeptides.
The X-ray crystal structure of the 50S ribosomal subunit in a complex with TSR showed that most loop2 moieties are solvent exposed, with the exception of the QA group.6 QA approaches A1067 of the 23S rRNA, one of the key nucleobases contributing to the mutation-induced bacterial resistance as well as to interactions with most thiopeptides.7 We have previously biosynthesized 6′-fluoro-TSR (Fig. 1A), a TSR derivative generated by fluorinating QA at C6′ (the carbon atom closest to A1067), and have shown that this functionalization has a positive effect on antibacterial activity.8 Quantum mechanics (QM)-molecular mechanics (MM) modeling (Fig. S1 and S2†) suggests that there is a changed action on A1067, as exemplified by the distance between the fluorinated C6′ of QA and A1067, which is accordingly shifted from 3.43 Å to 3.57 Å (Fig. 1B).
Focusing on the biologically relevant but tunable QA moiety, in this study, we have considered to introduce the pharmaceutically important fluorine and methyl groups onto TSR by taking into account the electronic and steric effects pertinent to drug design.9 This effort aims to improve the antibacterial potency by elaborately modulating the interaction between QA and A1067, on the premise that the overall binding nature of molecules to the bacterial ribosome is maintainable. Besides, according to the molecular modeling in silico (Table S4†) and previously reported results,2c large group modified QA moieties might crash into A1067 and destroy the binding affinity between TSR and the 50S ribosome. As a result, regioselective modifications of the QA group on such a complex molecule was achieved via biosynthesis by employing the QA-forming biochemistry and indeed produced a series of anticipated and unanticipated advantages of developing TSR-type thiopeptide antibiotics.
The first designed TSR derivative is 5′-fluoro-TSR (Fig. 1A). Modeling of this molecule to the 50S large ribosomal subunit indicates that the distance between QA and A1067 is shortened to 3.31 Å (Fig. 1B), as a consequence of the potent electron-withdrawing effect arising from the adjacent fluoro-substitution. In contrast, C12′-methylation of QA, generating the second designed derivative 12′-methyl-TSR (Fig. 1A), results in a steric or hydrophobic effect, pushing the QA moiety toward A1067 with a distance of 3.34 Å (Fig. 1B). Because these two naturally unavailable TSR antibiotics are structurally complex and difficult to be prepared by using current chemical synthesis approaches,10 we produced them in a biosynthetic method.8
The formation of the QA moiety of loop2 in the TSR biosynthetic pathway involves methyl transfer onto and rearrangement of the indole of L-tryptophan (Fig. 1A and S3†), which is independent of the precursor peptide, to produce a quinoline ketone as the key intermediate.8 For QA modification, we synthesized the 5-fluorinated and 12-methylated ester analogs of this intermediate (Scheme 1 and ESI†). The exogenous feeding of the analogs into a non-TSR-producing strain,8 which lacks the methyl transfer step for endogenous quinoline ketone generation, resulted in the robust biosynthesis of 5′-fluoro-TSR (12–15 mg L−1) and 12′-methyl-TSR (20–25 mg L−1), respectively, with 11–31% yields of that achieved for TSR (80–110 mg L−1) produced in the wild-type strain (Fig. 2A).
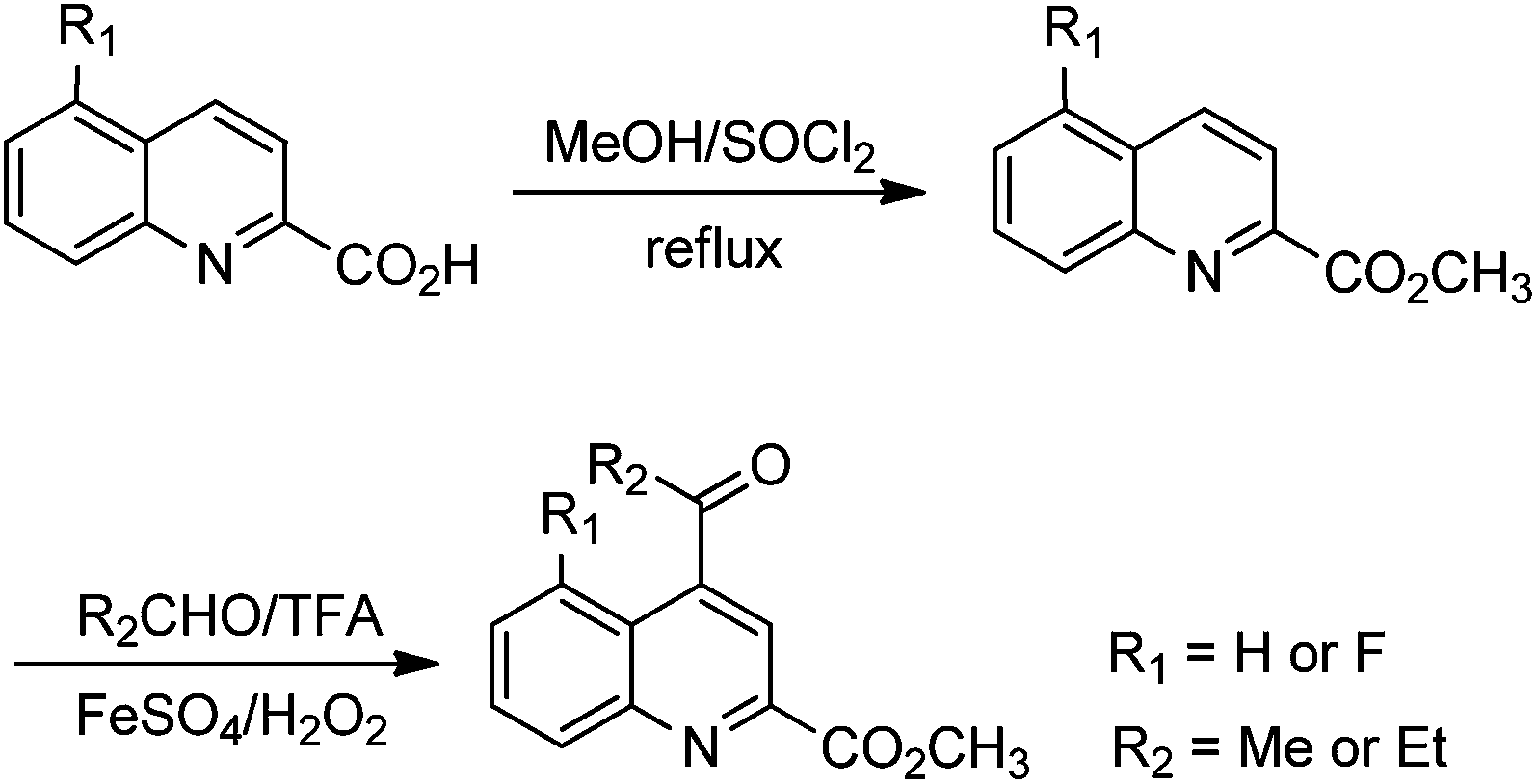 | ||
| Scheme 1 Chemical synthesis of the analogs of the quinolone ketone intermediate. | ||
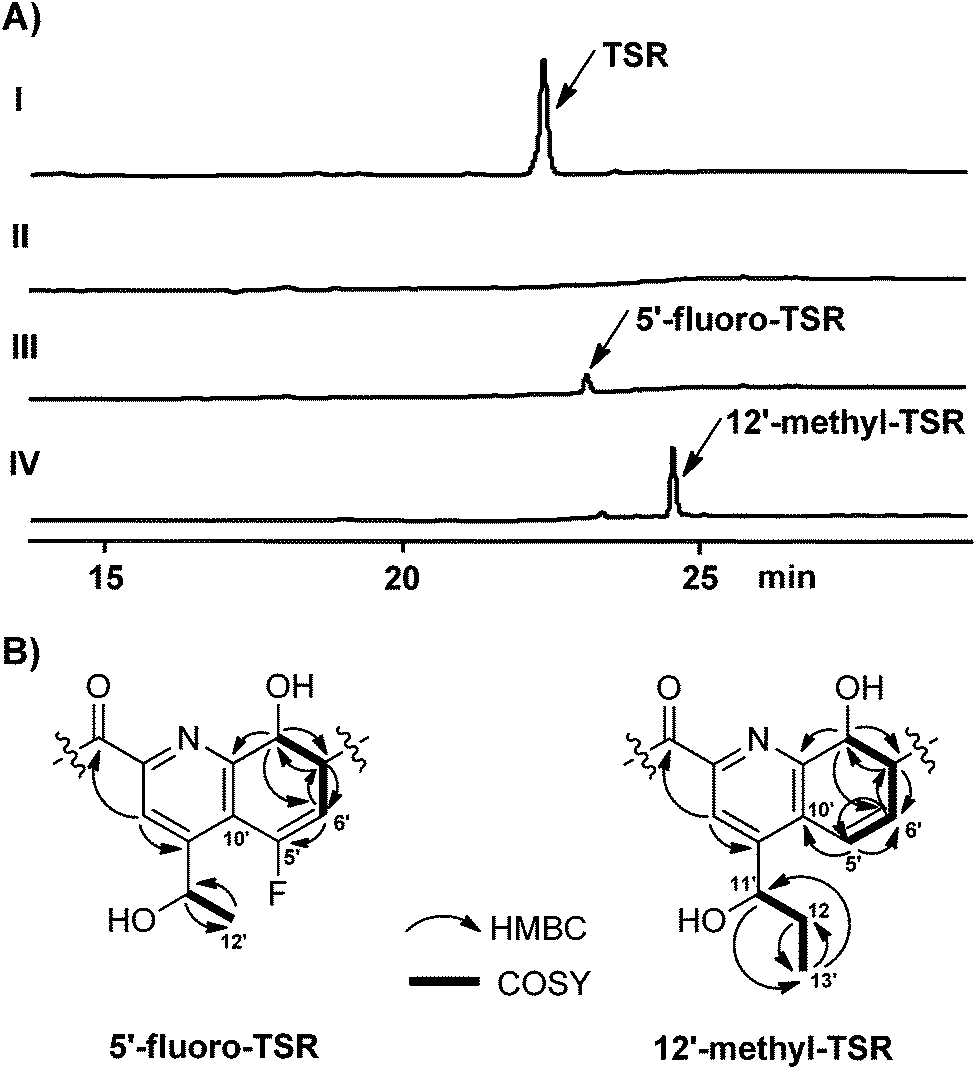 | ||
| Fig. 2 Production of new TSR derivatives and their structural elucidation. (A) HPLC analysis of the fermentation cultures of the wild type TSR-producing Streptomyces laurentii strain (I) and the mutant TSR non-producing strain,8 in which generation of the endogenous quinoline ketone intermediate was blocked, in the absence (II) or the presence of the exogenous C5-fluorinated (III) and C12-methylated (IV) intermediate analogs, respectively. (B) Selected HMBC and COSY correlations for the QA moieties of 5′-fluoro-TSR and 12′-methyl-TSR. | ||
Consequently, both the new compounds were purified and structurally compared with the parent compound, TSR. Extensive spectral analysis (Fig. 2B, S4, S5 and S6 and ESI†) revealed that they are extremely similar to TSR and that the only difference observed was in the substitution of the QA moiety. For 5′-fluoro-TSR, there was a 19F NMR signal (δF −72.97), corresponding to the disappearance of the 1H NMR signal of H-5′ (δH 6.73) found for TSR, and the 13C NMR signals of C-5′, C-6′ and C-10′ of the QA moiety were accordingly shifted. For 12′-methyl-TSR, the 13C NMR signal of C-13′ (δc 11.0) and the 1H NMR signal of H3-13′ (δH 1.10) appeared; and the relevant COSY correlation between C-13′ and C-12′ and the HMBC correlations of H3-13′ to C-12′ and C-11′, H2–12′ to C-13′ and H-11′ to C-13′ further supported that this molecule has a methyl substitution at C-12′, in comparison with TSR.
The newly obtained TSR derivatives, 5′-fluoro-TSR and 12′-methyl-TSR, were subjected to a wide variety of in vitro quantitative bioassays for comparative analysis with TSR and the previously obtained 6′-fluoro-TSR (Table 1). Consistent with previous findings,1a TSR and all of its derivatives exhibited substantially more potency than the chemotherapeutic control drugs. The sensitive strains include a number of Gram-positive clinical pathogens, such as methicillin-resistant Staphylococcus aureus (MRSA), penicillin-resistant Streptococcus pneumoniae (PRSP), and vancomycin-resistant Enterococcus faecium (VRE), as well as several Gram-negative colistin-resistant isolates, have not been reported in previous studies.
| TSR | 6′-F-TSR | 5′-F-TSR | 12′-Me-TSR | Vancomycin | Colistin | |
|---|---|---|---|---|---|---|
| Bacillus subtilis SIPI-DJ100a | 0.016 | 0.004 | 0.002 | 0.008 | 0.125 | — |
| Staphylococcus aureus SIPI-DJ1002a | 0.032 | 0.008 | 0.004 | 0.016 | 1.0 | — |
| Staphylococcus aureus ATCC25923a | 0.064 | 0.016 | 0.008 | 0.064 | 1.0 | — |
| Streptococcus pneumoniae PRSP1063a | 0.001 | <0.000125 | <0.000125 | <0.000125 | 0.25 | — |
| Streptococcus pneumoniae PRSP2831a | 0.001 | <0.000125 | <0.000125 | 0.001 | 0.25 | — |
| Streptococcus pneumoniae PRSP224588a | 0.008 | 0.002 | 0.001 | 0.004 | 0.25 | — |
| Staphylococcus aureus MRSA-s1a | 0.032 | 0.008 | 0.004 | 0.032 | 0.5 | — |
| Staphylococcus aureus MRSA-SAU3a | 0.064 | 0.008 | 0.008 | 0.032 | 0.5 | — |
| Staphylococcus aureus MRSA-SAU5a | 0.064 | 0.008 | 0.008 | 0.032 | 1.0 | — |
| Enterococcus faecium VRE3a | 0.032 | 0.008 | 0.004 | 0.032 | >256 | — |
| Enterococcus faecium VRE73a | 0.064 | 0.016 | 0.008 | 0.064 | >256 | — |
| Enterococcus faecium VRE83a | 0.064 | 0.016 | 0.008 | 0.064 | >256 | — |
| Acinetobacter baumannii Azj06-200b | 0.008 | 0.004 | 0.002 | 0.004 | — | >256 |
| Acinetobacter junii A1322b | 4.0 | 2.0 | 1.0 | 4.0 | — | 12 |
| Chryseobacterium meningosepticum A2757b | 4.0 | 2.0 | 2.0 | 4.0 | — | 12 |
Remarkably, all three derivatives displayed increased activity compared with TSR, in the following potency order: 5′-fluoro-TSR (2- to 8-fold) > 6′-fluoro-TSR (2- to 4-fold) > 12′-methyl-TSR (1- to 2-fold) ≥ TSR, thus confirming the rationale for QA modification to develop TSR-based antibiotics. In addition, measurement of the water solubility of these compounds revealed the following order: 12′-methyl-TSR (5.9 ± 1.2 μg mL−1) > 6′-fluoro-TSR (3.5 ± 0.9 μg mL−1) > 5′-fluoro-TSR (2.5 ± 0.9 μg mL−1) ≈ TSR (2.4 ± 0.8 μg mL−1), indicating an unanticipated advantage of QA modification, i.e., improvement of the solubility compared to TSR.
In conclusion, according to the known mechanism by acting on bacterial ribosome, we have designed two TSR derivatives, 5′-fluoro-TSR and 12′-methyl-TSR, and provided a biosynthetic approach to efficiently accomplish their preparation that challenges the current ways of chemical synthesis. As anticipated, these newly obtained TSRs along with previously obtained 6′-fluoro-TSR displayed increased antibacterial activity, demonstrating the rationality for drug design by regioselective modification of the biologically tunable QA moiety. Excitingly, the derivative 12′-methyl-TSR had an unanticipated advantage, showing approximately 1.5-fold improvement of water solubility in comparison with the parent compound. These findings are significant and indicate the possibility to lower the therapeutic dose of TSRs and mitigate the major physical disadvantage affecting their clinical use. Given the finding that their activity against a wide range of bacterial pathogens was much higher than those of the clinically used control chemotherapeutics, including vancomycin, and the fact that TSR has proven to be safe in animal therapy, these TSR derivatives may serve as the promising leads to develop new potent and low cytotoxic antibiotics.
Acknowledgements
We thank Prof. Heinz G. Floss, University of Washington, for providing the TSR-producing strain S. laurentii ATCC 31255. This work was supported in part by grants from NSFC (31430005 and 91413101), STCSM (13XD1404500 and 14JC1407700), “973 program” (2012CB721100), and MST (2012AA02A706) of China. This research was also funded in part by the Youth Innovation Foundation Fellowship, Chinese Academy of Sciences (to Dr Shoufeng Wang).Notes and references
- (a) M. C. Bagley, J. W. Dale, E. A. Merritt and X. Xiong, Chem. Rev., 2005, 105, 685 CrossRef CAS PubMed; (b) J. Li, X. Qu, X. He, L. Duan, G. Wu, D. Bi, Z. Deng, W. Liu and H. Y. Ou, PLoS One, 2012, 7, e45878 CAS.
- (a) C. L. Myers, P. C. Hang, G. Ng, J. Yuen and J. F. Honek, Bioorg. Med. Chem., 2010, 18, 4231 CrossRef CAS PubMed; (b) M. N. Aminake, S. Schoof, L. Sologub, M. Leubner, M. Kirschner, H. D. Arndt and G. Pradel, Antimicrob. Agents Chemother., 2011, 55, 1338 CrossRef CAS PubMed; (c) N. S. Hegde, D. A. Sanders, R. Rodriguez and S. Balasubramanian, Nat. Chem., 2011, 3, 725 CrossRef CAS PubMed.
- (a) H. D. Arndt, S. Schoof and J. Y. Lu, Angew. Chem., Int. Ed., 2009, 48, 6770 CrossRef CAS PubMed; (b) C. T. Walsh, M. G. Acker and A. A. Bowers, J. Biol. Chem., 2010, 285, 27525 CrossRef CAS PubMed; (c) C. Li and W. L. Kelly, Nat. Prod. Rep., 2010, 27, 153 RSC; (d) S. Wang, S. Zhou and W. Liu, Curr. Opin. Chem. Biol., 2013, 17, 626 CrossRef CAS PubMed; (e) S. J. Malcolmson, T. S. Young, J. G. Ruby, P. Skewes-Cox and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8483 CrossRef CAS PubMed; (f) S. Hayashi, T. Ozaki, S. Asamizu, H. Ikeda, S. Omura, N. Oku, Y. Igarashi, H. Tomoda and H. Onaka, Chem. Biol., 2014, 21, 679 CrossRef CAS PubMed.
- (a) F. Zhang and W. L. Kelly, Methods Enzymol., 2012, 516, 3 CAS; (b) T. S. Young, P. C. Dorrestein and C. T. Walsh, Chem. Biol., 2012, 19, 1600 CrossRef CAS PubMed; (c) W. Y. Liu, M. Ma, Y. J. Xue, N. Liu, S. Z. Wang and Y. J. Chen, ChemBioChem, 2013, 14, 573 CrossRef CAS PubMed; (d) A. Tocchetti, S. Maffioli, M. Iorio, S. Alt, E. Mazzei, C. Brunati, M. Sosio and S. Donadio, Chem. Biol., 2013, 20, 1067 CrossRef CAS PubMed; (e) Q. Zhang and W. Liu, Nat. Prod. Rep., 2013, 30, 218 RSC; (f) H. Guo, J. Wang, Y. M. Li, Y. Yu, Q. F. Zheng, J. Q. Wu and W. Liu, Chem. Sci., 2014, 5, 240 RSC.
- D. N. Wilsson, Nat. Rev. Microbiol., 2014, 12, 35 CrossRef PubMed.
- J. M. Harms, D. N. Wilson, F. Schluenzen, S. R. Connell, T. Stachelhau, Z. Zaborowska, C. M. T. Spahn and P. Fucini, Mol. Cell, 2008, 30, 26 CrossRef CAS PubMed.
- S. Baumann, S. Schoof, M. Bolten, C. Haering, M. Takagi, K. Shin-ya and H. D. Arndt, J. Am. Chem. Soc., 2010, 132, 6973 CrossRef CAS PubMed.
- L. Duan, S. Wang, R. Liao and W. Liu, Chem. Biol., 2012, 19, 443 CrossRef CAS PubMed.
- (a) E. J. Barreiro, A. E. Kummerle and C. A. Fraga, Chem. Rev., 2011, 111, 5215 CrossRef CAS PubMed; (b) J. Wang, C. D. Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed.
- K. C. Nicolaou, B. S. Safina, M. Zak, S. H. Lee, M. Nevalainen, M. Bella, A. A. Estrada, C. Funke and S. J. Bulat, J. Am. Chem. Soc., 2005, 127, 11159 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4qo00288a |
| ‡ These authors equally contributed to this work. |
| This journal is © the Partner Organisations 2015 |