
Lewis acid catalyzed Nazarov type cyclization for the synthesis of a substituted indane framework: total synthesis of (±)-mutisianthol†
Dattatraya H.
Dethe
*,
Raghavender
Boda
and
Ganesh M.
Murhade
Department of Chemistry, Indian Institute of Technology Kanpur, Kanpur 208016, India. E-mail: ddethe@iitk.ac.in; Fax: +91-512-2597436; Tel: +91-512-2596537
First published on 11th February 2015
Abstract
A new synthetic route has been developed for the synthesis of a substituted indane derivative using Lewis acid catalyzed modified Nazarov type cyclization, which was further applied in the total synthesis of mutisianthol and epi-mutisianthol in a nine step longest linear sequence.
The indane motif is an important cyclic component present in many natural products and drug candidates possessing interesting biological activities (Fig. 1).1 It is also used as functional materials2 and ligands for transition metal complexes, which are widely utilized in catalytic reactions such as catalytic olefin polymerization.3
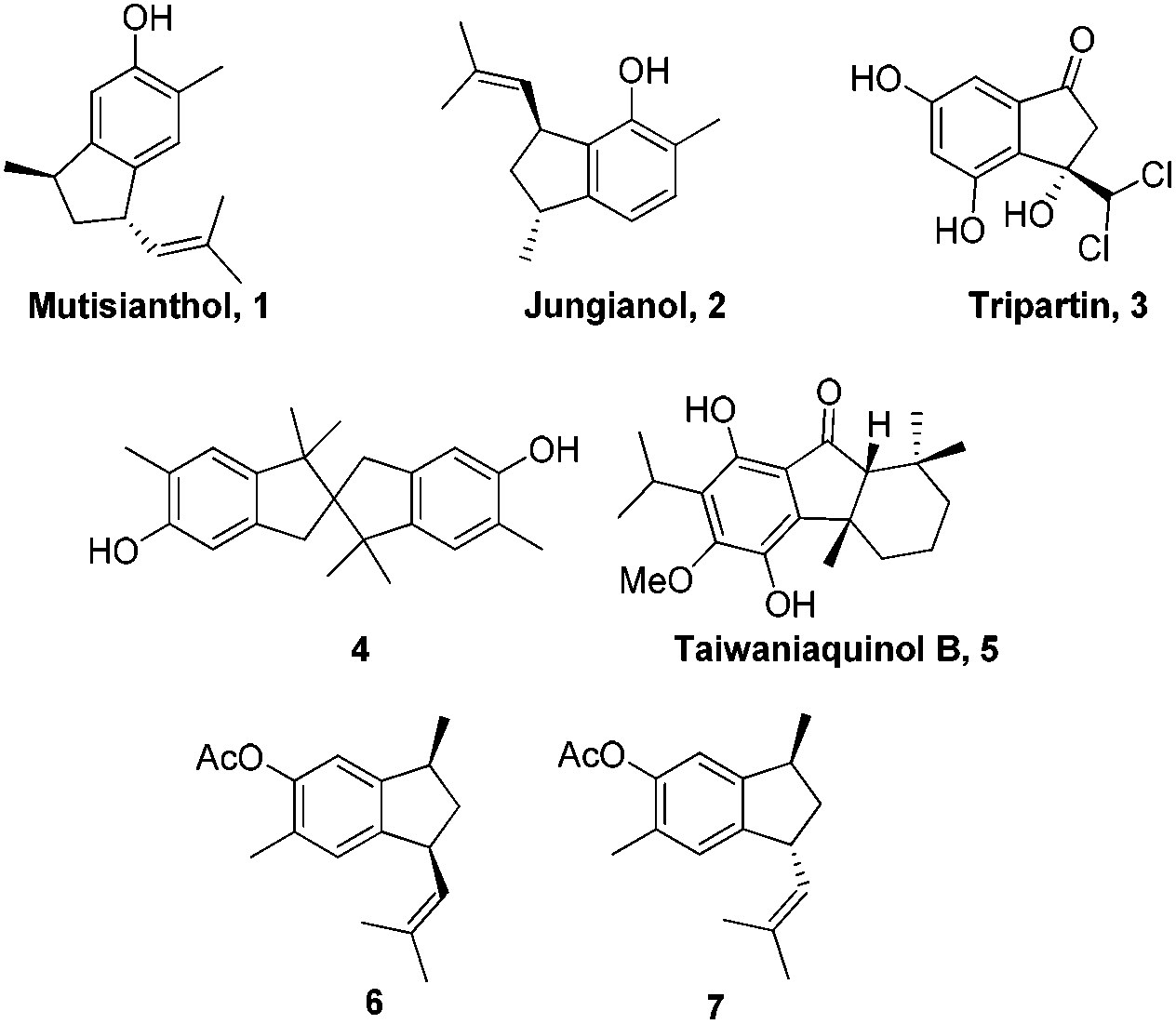 | ||
| Fig. 1 Biologically active indane natural products and their analogues. | ||
In 1979, Bohlmann et al. reported the isolation of a phenolic sesquiterpene mutisianthol 1 from the roots of Mutisia homoeantha.4 Interestingly, a diastereomer of mutisianthol 1, jungianol 2 was isolated from Jungia malvaefolia by the same group.5 Jungianol and mutisianthol are plausibly derived in nature from an α-curcumene type precursor by intramolecular ortho and para alkylation respectively. The structure and stereochemical assignment of these natural products were made on the basis of spectroscopy data. Initially the cis relationship was assigned for the two side chains on the five membered rings in mutisianthol 1 and jungianol 2 by the isolation group. Later Ho et al. revised the relative stereochemistry of two side chains to trans by synthesizing both cis and trans isomers 6 and 7 starting from the indanone derivative.6 Later Silva et al. also achieved the enantioselective synthesis of mutisianthol 1 using asymmetric hydrogenation and thalium mediated ring contraction of the tetraline derivative.7
Recently, we have reported the total synthesis of jungianol 2 using Lewis acid catalyzed Prins type cyclization.8 So far only two syntheses of each natural product have been reported in the literature. Herein we report a concise total synthesis of mutisianthol 1 using modified Nazarov type cyclization.9
It was envisaged that mutisianthol 1 could be prepared from ester 8 by functional group manipulations. Ester 8 in turn could be generated from compound 9 by the Friedel–Crafts type reaction in Michael fashion. Compound 9 could be prepared from 10 using the five step protocol (Scheme 1).
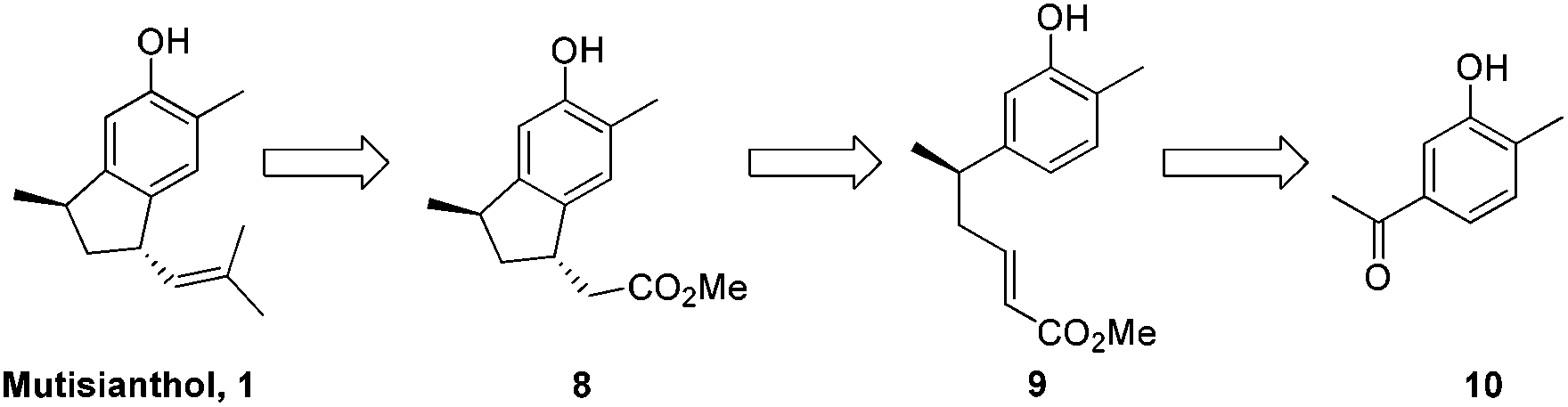 | ||
| Scheme 1 Retrosynthetic analysis of mutisianthol 1. | ||
To quickly check our strategy, we carried out the model studies starting from 3-methoxy acetophenone 11. To begin with, the Wittig–Horner reaction of 3-methoxy acetophenone 11 afforded α,β-unsaturated ester 12 in 80% yield. The sequential reduction of the double bond and the ester moiety generated alcohol 14via13. Oxidation of alcohol 14 using IBX in EtOAc under reflux conditions generated aldehyde 15 in 94% yield. The Wittig reaction of aldehyde 15 using Ph3PCHCO2Me afforded the α,β-unsaturated ester 16 in 93% yield (Scheme 2).
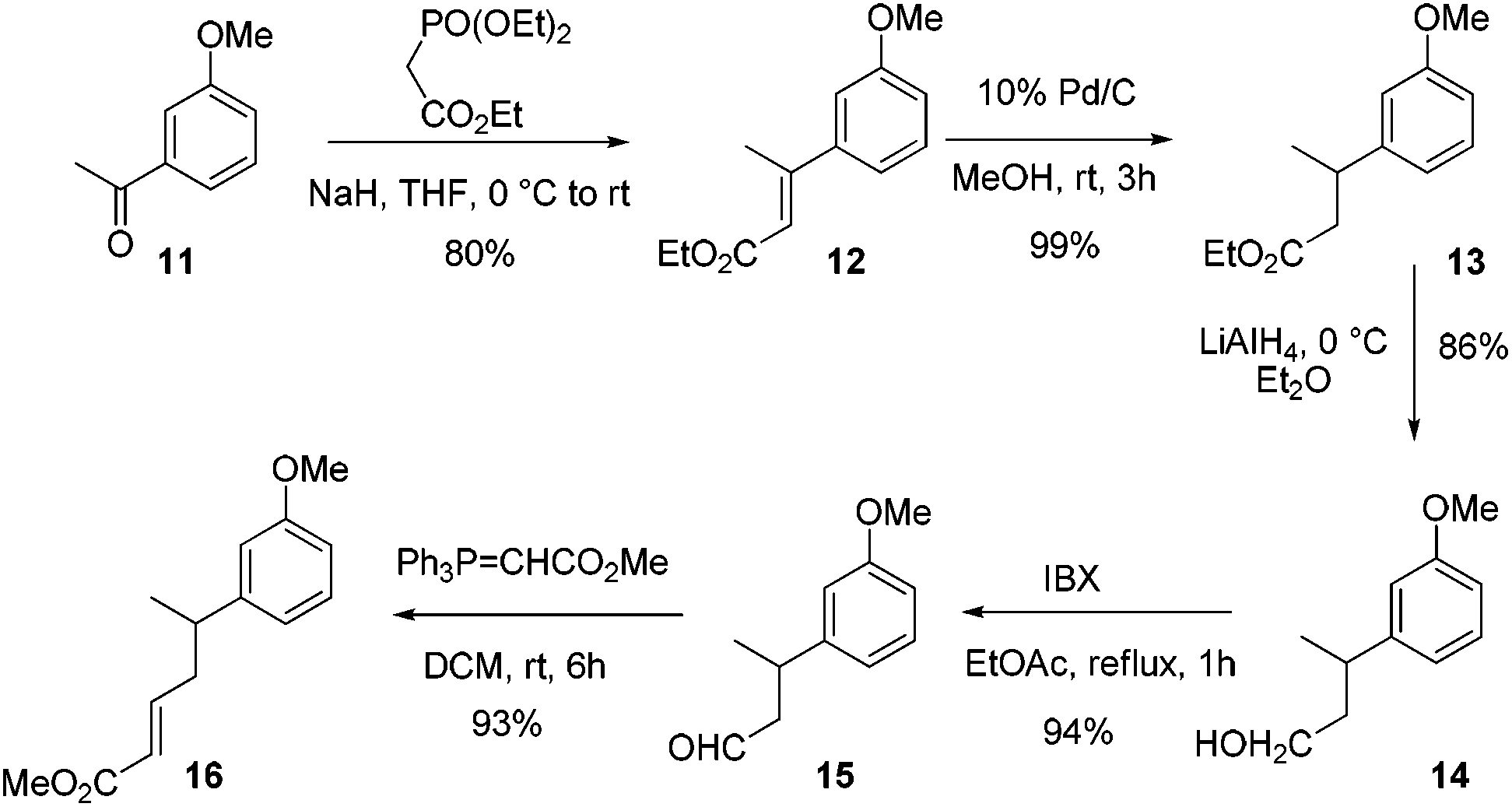 | ||
| Scheme 2 Preparation of α,β-unsaturated ester 16. | ||
Having the key precursor 16 in hand, we next turned our attention towards the key Friedel–Crafts reaction in Michael fashion. To our disappointment treatment of compound 16 with different Lewis acids such as BF3·OEt2, SnCl4, TiCl4, Cu(OTf)2, BiCl3 under various reaction conditions failed to generate the desired product and in most cases we recovered the starting material.
At this stage we also became interested to check the outcome of the reaction of diene ester 19 with Lewis acid. It was contemplated that diene ester 19 on treatment with Lewis acid might generate a Nazarov type cationic intermediate 19a which on cyclization could afford the indane derivative 20.9 Compound 19 was prepared from the earlier intermediate 12 by using alane reduction, oxidation and the Wittig reaction sequence (Scheme 3).
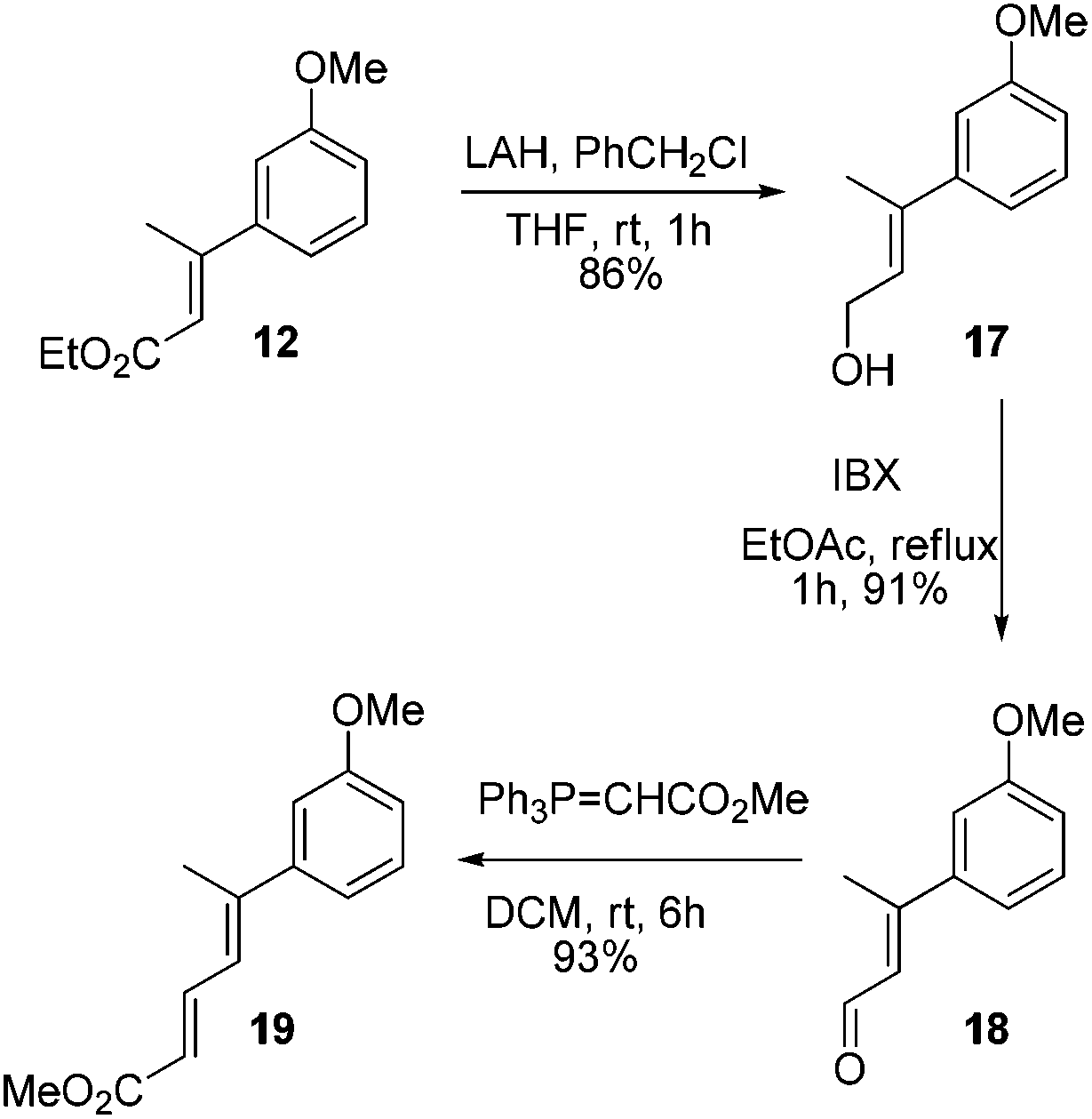 | ||
| Scheme 3 Synthesis of diene ester 19. | ||
To our delight treatment of diene ester 19 with BF3·OEt2 afforded the indane derivative 20 in 24% yield by Nazarov type cyclization followed by double isomerisation of the olefin to a more stable position between the ester group and the phenyl ring. After screening different Lewis acids, it was found that TiCl4 in DCM was the best reagent for this cyclization, generating the indane derivative 20 in 60% yield (Scheme 4, Table 1). The structure and stereochemistry of the double bond of compound 20 was unambiguously established by single crystal X-ray analysis.10
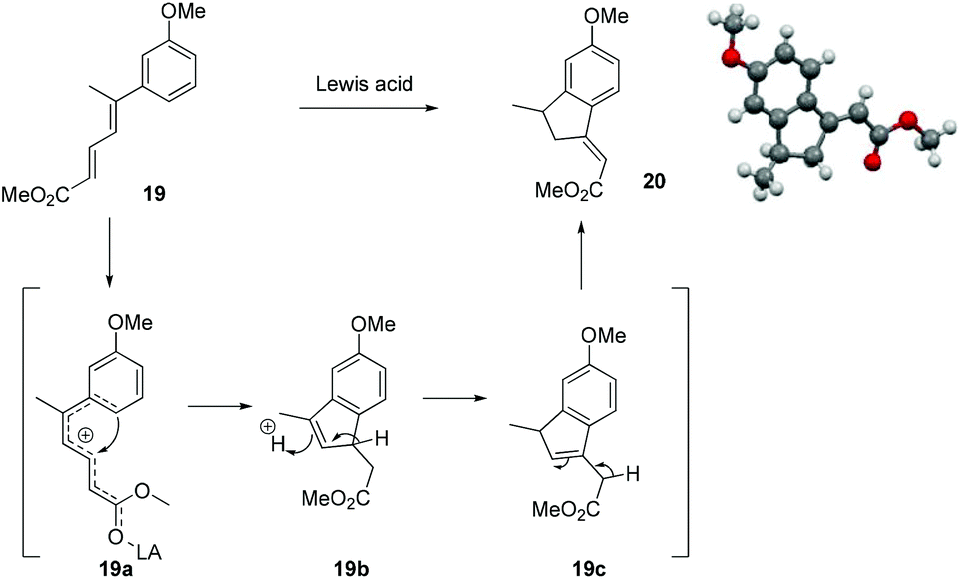 | ||
| Scheme 4 Modified Nazarov type cyclization. | ||
| Entry | Catalyst | Time | Solvent | Yield (%) |
|---|---|---|---|---|
| a Reaction conditions: 19 (0.2 mmol), catalyst (0.2 mmol), solvent (3 ml). | ||||
| 1 | p-TSA | 24 h | DCM | NR |
| 2 | Bi(OTf)3 | 24 h | DCM | NR |
| 3 | BiCl3 | 24 h | DCM | NR |
| 4 | InBr3 | 24 h | DCM | NR |
| 5 | TFA | 24 h | DCM | NR |
| 6 | AlCl3 | 24 h | DCM | Trace |
| 7 | BF3·OEt2 | 10 h | DCM | 24 |
| 8 | FeCl3 | 2 h | DCM | 45 |
| 9 | TfOH | 2 h | DCM | 54 |
| 10 | TiCl4 | 2 h | DCM | 60 |
After establishing the reaction conditions, we next targeted the total synthesis of mutisianthol 1. The suitably substituted diene ester 25 was prepared from the known compound 22.11 The Vilsmeier–Haack reaction of compound 23 using POCl3/DMF generated α,β-unsaturated aldehydes 24a,b as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 mixture of cis and trans isomers,12 which was carefully separated by silica gel column chromatography. An independent Wittig reaction of aldehydes 24a,b using Ph3PCHCO2Me afforded the α,β-unsaturated esters 25a,b. Both cis and trans isomers on independent treatment with TiCl4 in DCM afforded the indane derivative 26 in 62% yield. Also the mixture of isomers on treatment with TiCl4 in DCM at RT generated compound 26 in 62% yield (Scheme 5). The above observation shows that generation of a mixture of double bond isomers in the Vilsmeier–Haack reaction is inconsequential for the Nazarov type cyclization.
:5 mixture of cis and trans isomers,12 which was carefully separated by silica gel column chromatography. An independent Wittig reaction of aldehydes 24a,b using Ph3PCHCO2Me afforded the α,β-unsaturated esters 25a,b. Both cis and trans isomers on independent treatment with TiCl4 in DCM afforded the indane derivative 26 in 62% yield. Also the mixture of isomers on treatment with TiCl4 in DCM at RT generated compound 26 in 62% yield (Scheme 5). The above observation shows that generation of a mixture of double bond isomers in the Vilsmeier–Haack reaction is inconsequential for the Nazarov type cyclization.
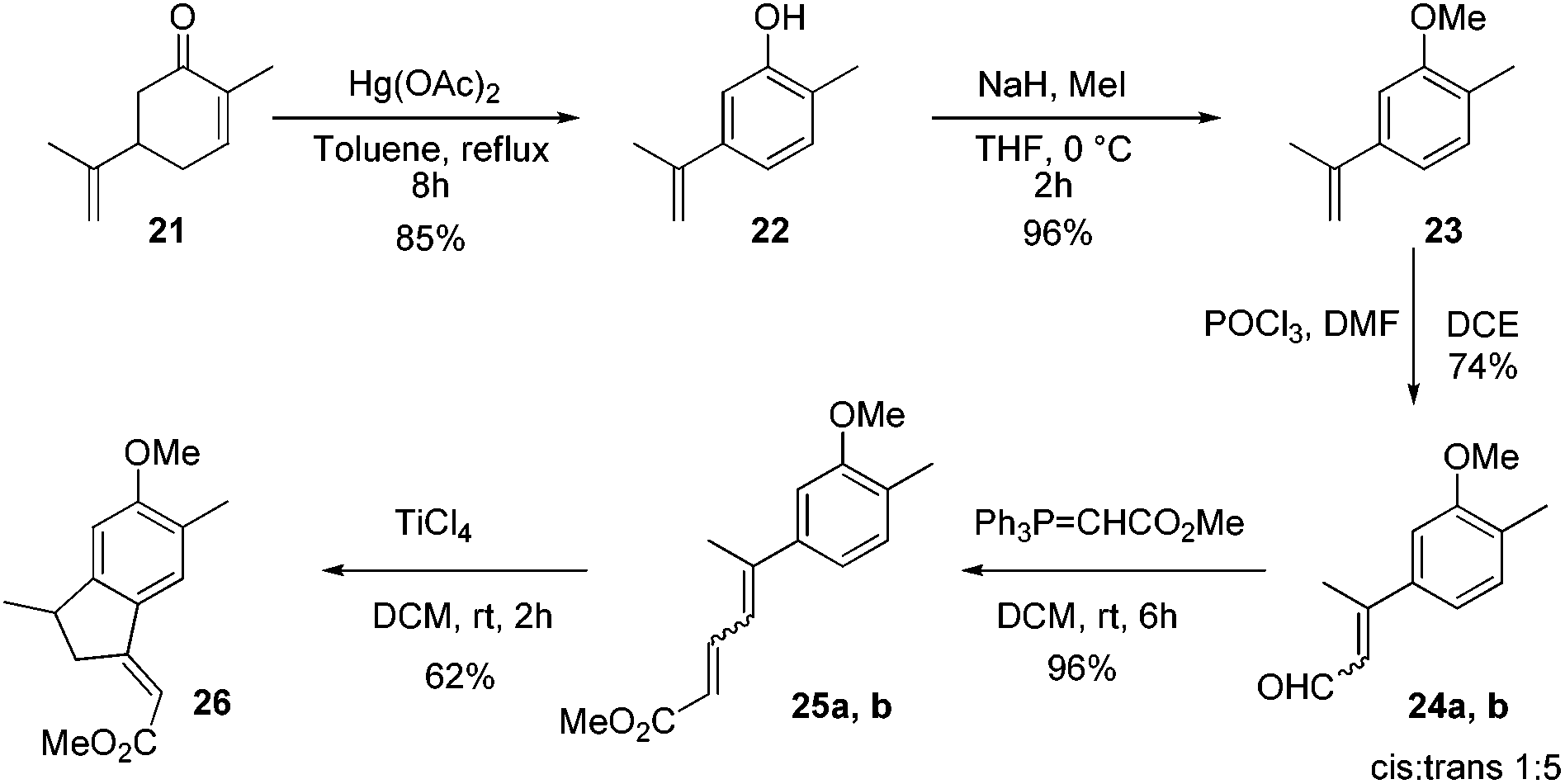 | ||
| Scheme 5 Preparation of α,β-unsaturated ester 26. | ||
After having the key indane derivative 26 in hand, our next target was stereoselective reduction of the double bond to generate the trans isomer with respect to methyl group and functional group manipulations of the ester group for the generation of an isobutene side chain. Hydrogenation of the double bond using 10% Pd/C exclusively generated the cis isomer 27 in 94% yield. Stereochemistry of the compound 27 was further confirmed by converting it into epi-mutisianthol 30. Thus, treatment of ester 27 with excess of MeMgI generated the tertiary alcohol 28 which under p-TSA conditions afforded the dehydrated product 29. Deprotection of methyl ether under basic conditions afforded epi-mutisianthol 30 in 77% yield (Scheme 6).7 For the stereo selective reduction of the double bond to generate the trans isomer with respect to the methyl group, we have tried different reduction conditions, in which, refluxing with Cr(CO)6 under dioxane and Mg–ZnCl2 conditions failed to generate the desired product and in both cases the starting material was recovered. Compound 26 with Zn–KOH in refluxing EtOH and also with NaCNBH3 in TFA furnished a complex reaction mixture. Surprisingly, the Birch reduction protocol furnished exclusively the cis isomer. The reaction of unsaturated ester 26 with magnesium and NH4Cl in 1:1 mixture of methanol and THF yielded 1:1 inseparable mixture of cis and trans esters 31a,b in 51% yield. The addition of excess methyl Grignard reagent to esters 31a,b generated the mixture of tertiary alcohols 32a,b in 90% yield, which on dehydration using p-TSA in refluxing benzene afforded compounds 33a,b in 86% yield. Deprotection of methyl ether under basic conditions using ethane thiol and sodium hydride in DMF generated the 1:1 mixture of mutisianthol 1 and epi-mutisianthol 30 in 77% combined yield (Scheme 7).7
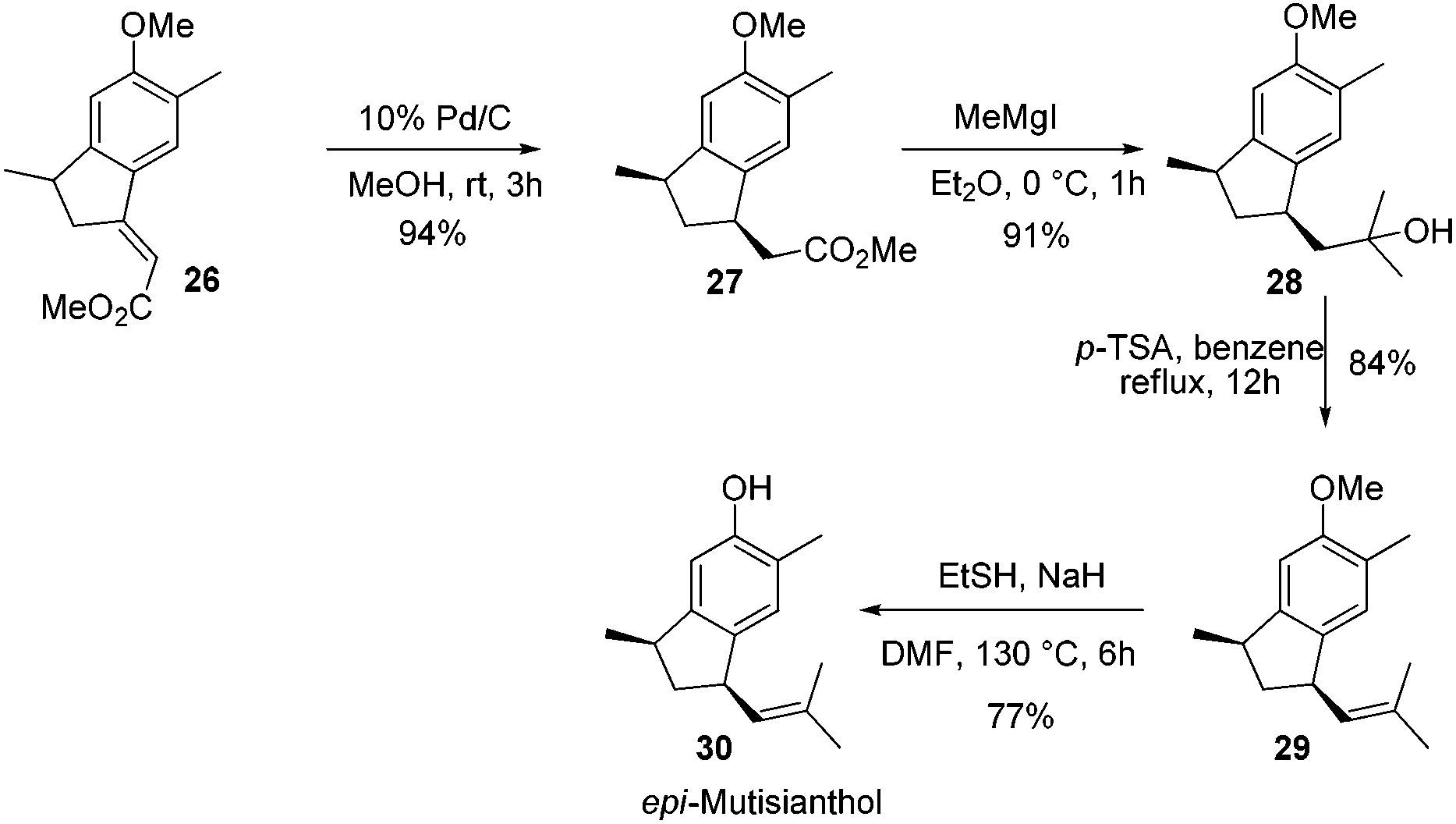 | ||
| Scheme 6 Synthesis of epi-mutisianthol. | ||
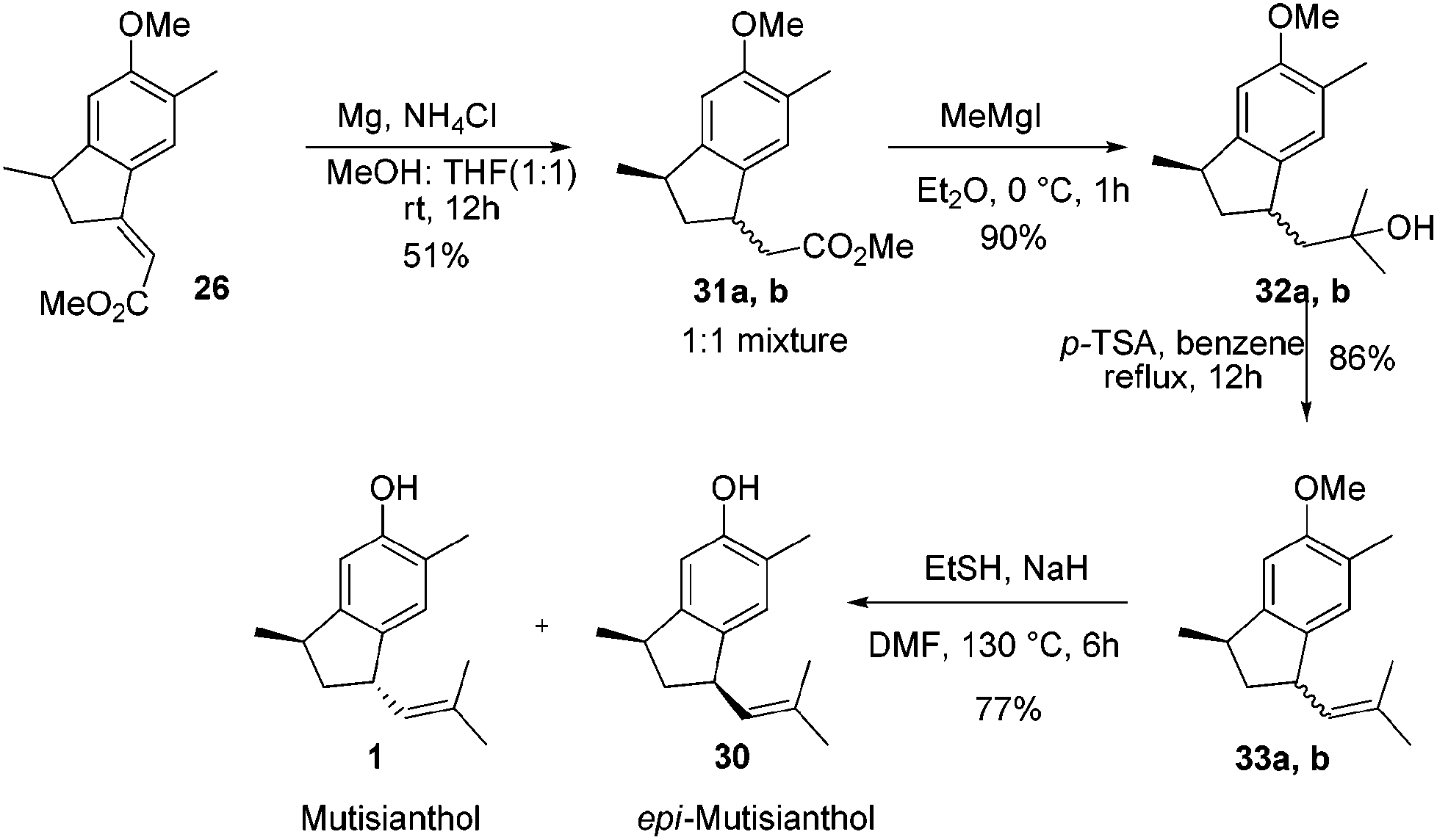 | ||
| Scheme 7 Synthesis of mutisianthol and epi-mutisianthol. | ||
Separation of mutisianthol 1 and epi-mutisianthol 30 was done by careful silica gel column chromatography. The spectral data of compound 1 (1H, 13C, IR, HRMS) were identical to the natural mutisianthol.
Conclusions
In conclusion, we have developed a Lewis acid mediated Nazarov type cyclization for the construction of a substituted indane, which was further applied in the concise total synthesis of natural product mutisianthol 1 in 9 steps with 5.5% overall yield from carvone and epi-mutisianthol 30 in 9 steps with 19.9% overall yield.Acknowledgements
BR and GM thank CSIR, New Delhi, for the award of research fellowship. Financial support from IIT Kanpur is gratefully acknowledged. We thank Prosenjit Daw from IIT Kanpur for his help.Notes and references
- (a) R. J. Capon, J. K. MacLeod and P. J. Scammells, Tetrahedron, 1986, 42, 6545 CrossRef CAS; (b) T. L. Ho, K. Y. Lee and C. K. Chen, J. Org. Chem., 1997, 62, 3365 CrossRef CAS PubMed; (c) M. F. Gross, S. Beaudoin, G. Mc Naughton-Smith, G. S. Amato, N. A. Castle, C. Huang, A. Zou and W. Yu, Bioorg. Med. Chem. Lett., 2007, 17, 2849 CrossRef CAS PubMed; (d) S. A. Snyder and Z. G. Brill, Org. Lett., 2011, 13, 5524 CrossRef CAS PubMed; (e) K. P. Bogeso, A. V. Christensen, J. Hyttel and T. Liljefors, J. Med. Chem., 1985, 28, 1817 CrossRef CAS; (f) K. P. Bogeso, J. Arnt, K. Frederiksen, H. O. Hansen, J. Hyttel and H. Pedersen, J. Med. Chem., 1995, 38, 4380 CrossRef CAS; (g) H. Sugimoto, Pure Appl. Chem., 1999, 71, 2031 CrossRef CAS; (h) J. Guillon, P. Dallemagne, J.-M. Leger, J. Sopkova, P. R. Bovy, C. Jarry and S. Rault, Bioorg. Med. Chem., 2002, 10, 1043 CrossRef CAS; (i) V. Molteni, D. Rhodes, K. Rubins, M. Hansen, F. D. Bushman and J. S. Siegel, J. Med. Chem., 2000, 43, 2031 CrossRef CAS PubMed; (j) S.-H. Kim, S. H. Kwon, S.-H. Park, J. K. Lee, H.-S. Bang, S.-J. Nam, H. C. Kwon, J. Shin and D.-C. Oh, Org. Lett., 2013, 15, 1834 CrossRef CAS PubMed.
- (a) A. C. Grimsdale and K. Mullen, Angew. Chem., Int. Ed., 2005, 44, 5592 CrossRef CAS PubMed; (b) X. Zhu, H. Tsuji, K. Nakabayashi, S.-I. Ohkoshi and E. Nakamura, J. Am. Chem. Soc., 2011, 133, 16342 CrossRef CAS PubMed.
- (a) H. G. Alt and A. Koppl, Chem. Rev., 2000, 100, 1205 CrossRef CAS PubMed; (b) S. Ren, E. Igarashi, K. Nakajima, K.-I. Kanno and T. Takahashi, J. Am. Chem. Soc., 2009, 131, 7492 CrossRef CAS PubMed.
- F. Bohlmann, C. Zdero and N. Le Van, Phytochemistry, 1979, 18, 99 CrossRef CAS.
- F. Bohlmann and C. Zdero, Phytochemistry, 1977, 16, 239 CrossRef CAS.
- T.-L. Ho, K.-Y. Lee and C.-K. Chen, J. Org. Chem., 1997, 62, 3365 CrossRef CAS PubMed.
- (a) G. G. Bianco, H. M. C. Ferraz, A. M. Costa, L. V. Costa-Lotufo, C. Pessoa, M. O. de Moraes, M. G. Schrems, A. Pfaltz and L. F. Silva Jr., J. Org. Chem., 2009, 74, 2561 CrossRef CAS PubMed; (b) H. M. C. Ferraz, A. M. Aguilar and L. F. Silva Jr., Tetrahedron, 2003, 59, 5817 CrossRef CAS.
- D. H. Dethe and G. Murhade, Org. Lett., 2013, 15, 429 CrossRef CAS PubMed.
- (a) C. J. Rieder, K. J. Winberg and F. G. West, J. Am. Chem. Soc., 2009, 131, 7504 CrossRef CAS PubMed; (b) M. J. Riveira and M. P. Mischne, J. Org. Chem., 2014, 79, 8244 CrossRef CAS PubMed; (c) G. R. Elia, R. F. Childs and G. S. Shaw, Can. J. Chem., 1992, 70, 2065 CrossRef CAS.
- CCDC 1035966 (20) contains the supplementary crystallographic data for this paper.
- W. Treibs, Justus Liebigs Ann. Chem., 1953, 59, 581 Search PubMed.
- C. Schmidle and P. G. Barnett, J. Am. Chem. Soc., 1956, 78, 3209 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1035966. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00005j |
| This journal is © the Partner Organisations 2015 |