
Stereoconvergent synthesis of 1-deoxynojirimycin isomers by using the 3 component 4 centred Ugi reaction†
Chandra S.
Azad
 and
Anil K.
Saxena
*
and
Anil K.
Saxena
*
Division of Medicinal and Process Chemistry CSIR-Central Drug Research Institute, Lucknow-226 031, U.P., India. E-mail: anilsak@gmail.com
First published on 16th February 2015
Abstract
A new reductive cyclization/Ugi multicomponent reaction sequence for the synthesis of 1-deoxyallonojirimycin and 1-deoxyaltronojirimycin has been developed. The method was successfully applied to the azido-hemiacetal derived from commercially available D-ribose. The very selective reagents were used for the synthesis of Ugi bis-amides which were sub sequentially hydrolysed to iminosugars.
Iminosugars are the carbohydrate mimics which have the nitrogen atom instead of endocyclic oxygen. This simple substitution raises numerous synthetic challenges and opens the approach to get novel molecules with significant biological properties. Iminosugars definitely form the most eye-catching class of carbohydrate mimics described so far.1 The real era of iminosugars was initiated by Paulsen in 1966 by the first synthesis of 1-deoxynojirimycin (1) (DNJ).2 In the same year, Inouye et al. identified its antibiotic properties by isolating it from bacteria (Streptomyces).3 Various iminosugars are currently in clinical trials and the first success was the drug Glyset (2) in 1996 for the treatment of complications associated with type II diabetes. Recently, iminosugar based Zavesca (3) was also developed as the first oral treatment for Gaucher disease (a severe lysosomal storage disorder) in 2003 and several other molecules are under clinical trials (Fig. 1).4 The importance of DNJ is clearly reflected as its two derivatives are drug molecules. Although iminosugars are highly active molecules nevertheless they did not gain much attention as compared to other therapeutically important scaffolds. The main reason is the challenging chemical synthesis, because of the polar nature of the polyhydroxylated structures and their stereochemical complexity.5 The D-and L-enantiomers of 1-DNJ were evaluated by Kato et al., where the L-DNJ isomer showed potential inhibition of α-glucosidase rice (IC50 4.3 μM) while the L-allo-DNJ showed the inhibition of rat epididymis (IC50 59 μM) and L-altro-DNJ showed inhibition of α-glucosidase (IC50 450 μM) as compared to their inactive D-isomers.6 The selectivity of L-iminosugars may contribute to its effective medicinal use, as the beginning of adverse effects by their D-enantiomers (due to their capacity to inhibit disaccharidases and/or their biosynthesis in intestinal brush border membrane) have often hindered in vivo clinical trials.7 These results highlight the significance of the synthesis and screening of iminosugars with unnatural L-configurations having little stereo chemical resemblance to the enzyme's natural substrates. Still, few attempts have been made towards the syntheses of such isomers of 1-DNJ, viz. L-1-deoxyallonojirimycin (L- allo-DNJ) and L-1-deoxyaltronojirimycin (L-altro-DNJ). In continuation of our interest in the synthesis of nitrogen containing heterocycles, we envisaged the syntheses of L-1-deoxyallonojirimycin (L-allo-DNJ) and L-1-deoxyaltronojirimycin (L-altro-DNJ) by a chiral pool strategy using D-ribose as the starting material and Ugi reaction as the core approach (Fig. 2). The main synthetic challenges for the L-allo-DNJ and L-altro-DNJ syntheses were the development of one pot cost effective reductive Ugi cyclization for the construction of a piperidine moiety, preservation of hydroxy groups with correct stereochemistry and mild hydrolysis of Ugi bis-amide.
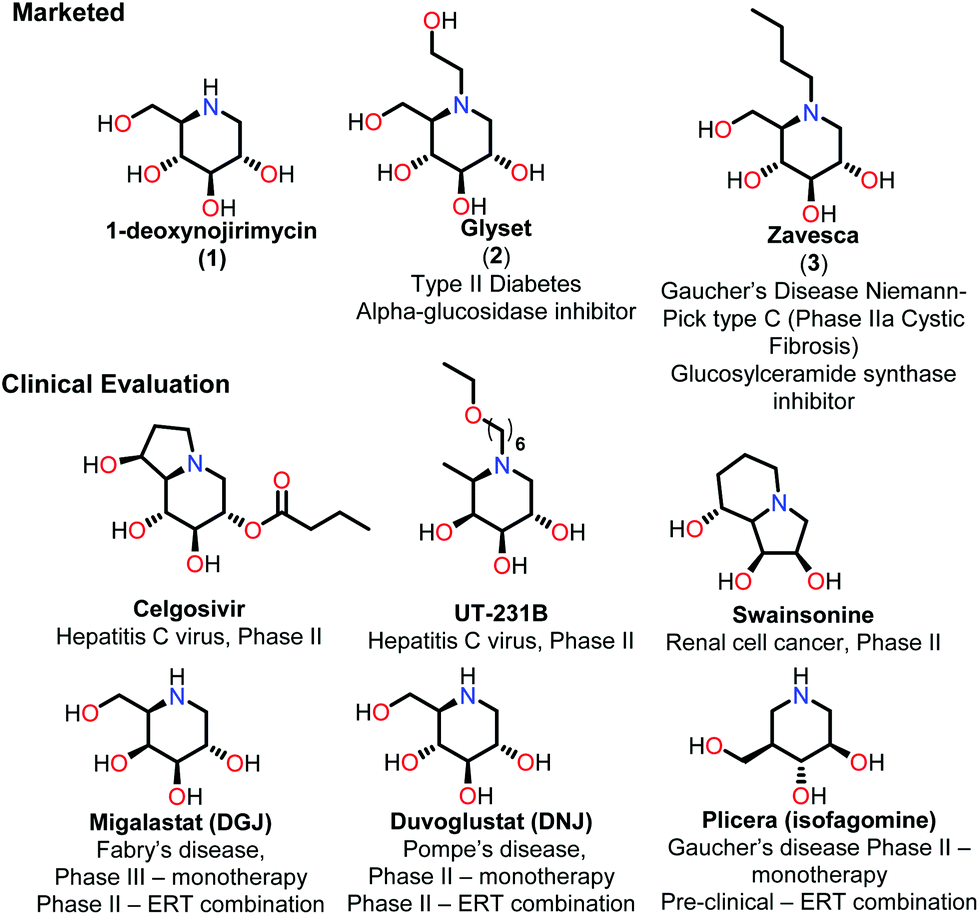 | ||
| Fig. 1 Marketed drug molecules and current status of other iminosugars. | ||
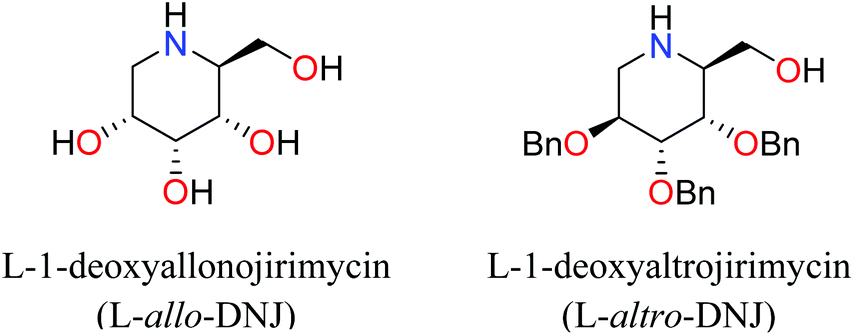 | ||
| Fig. 2 Structures of synthesised DNJ-isomers. | ||
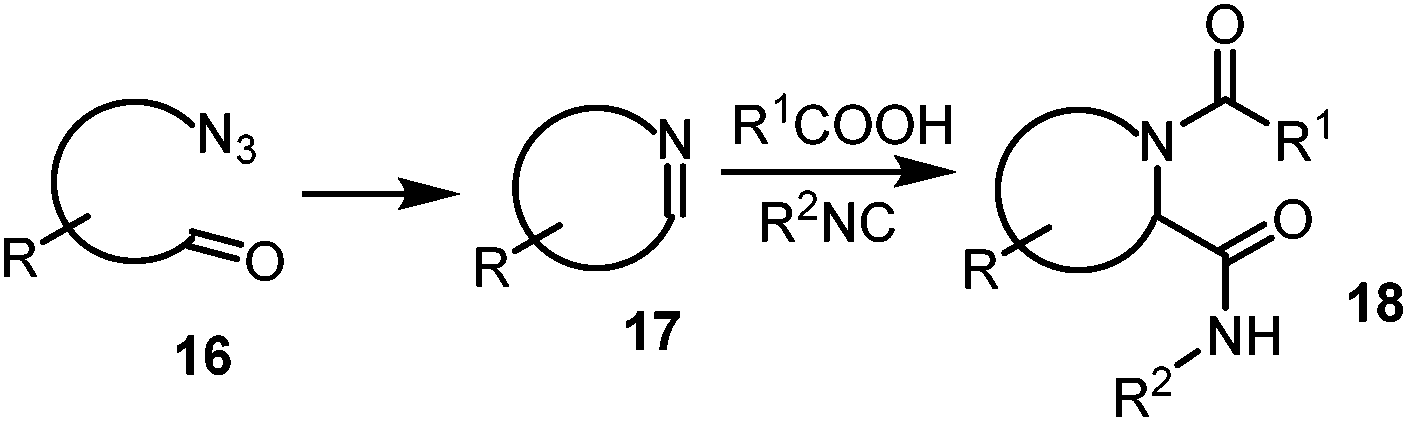
A careful examination of the DNJ structure 1 in Fig. 1 reveals that its six membered ring can be constructed by the Ugi reaction on the Schiffs base 5 through 4.85 can easily be constructed by the Staudinger–aza-Wittig cyclization of azido hemi-acetal 6. Finally DNJ 1 can be obtained by the hydrolysis of the bis-amide 4 (Scheme 1).
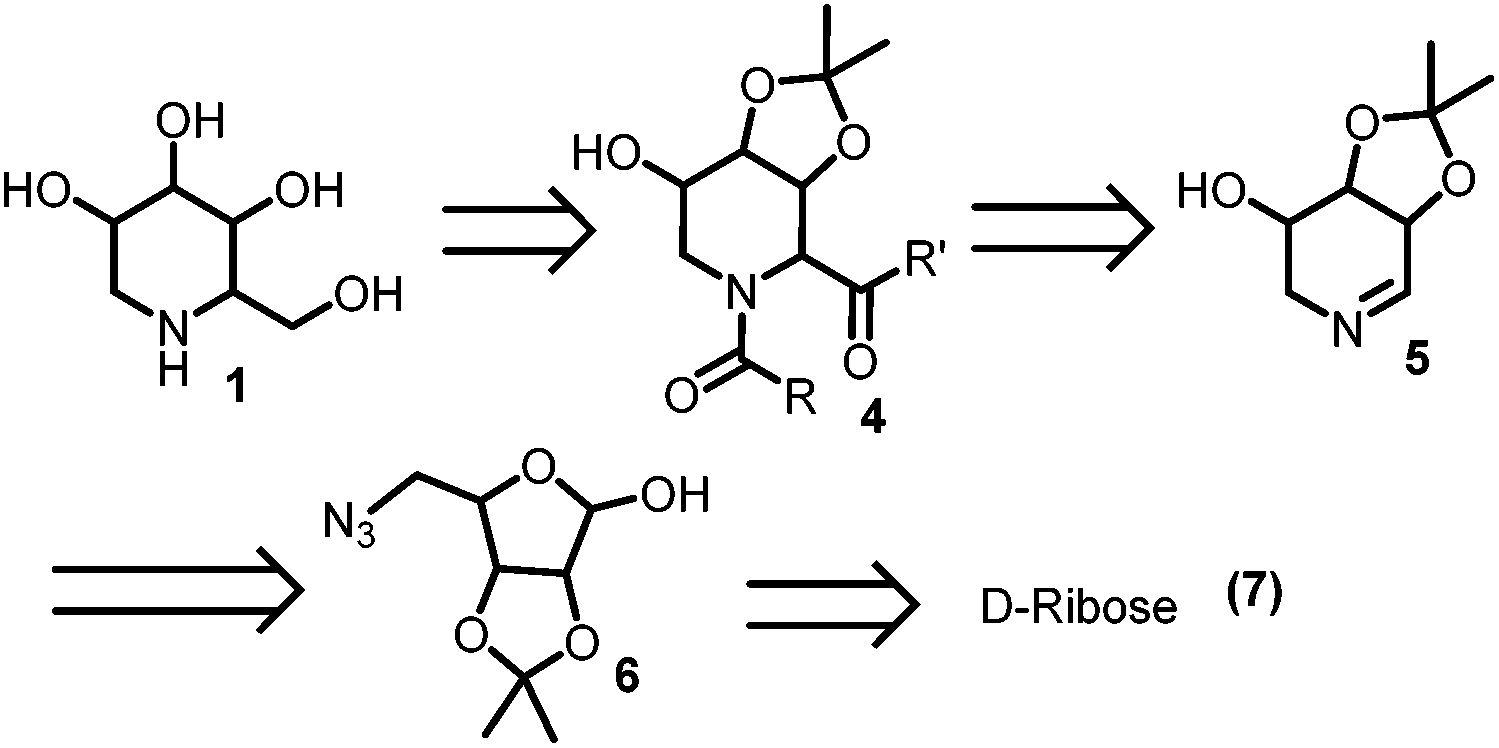 | ||
| Scheme 1 Retro-synthetic pathway of 1-DNJ's isomers. | ||
The generation of the bis-substituted imine 10, which is a key component in the Ugi-4CR reaction, could be accomplished by performing a tandem Staudinger–aza-Wittig reaction.9
Therefore the reaction of the azide 14 with trialkyl(aryl)phosphine would lead to the formation of the intermediate phosphazene 15 which, in sequence, may go through an aza-Wittig reaction with the aldehyde 8 to yield the imine 10 and the inert trialkyl(aryl)phosphine oxide (Scheme 2). The substrate having both an azide and an aldehyde, in addition to functional groups (R) may provide access to a substituted cyclic imine, thus opening a new dimension to the synthesis of cyclic dipeptides via reductive cyclization Ugi reaction (Scheme 3). Although this Staudinger–aza-Wittig reaction provides a good methodology nevertheless it has a major drawback of removal of side products e.g. phosphine oxide which subsequently lowers its yield.10 So to perform this reductive cyclization step a novel, mild and cost effective method has been developed for the synthesis of 1-DNJ's isomers and is reported in this manuscript.
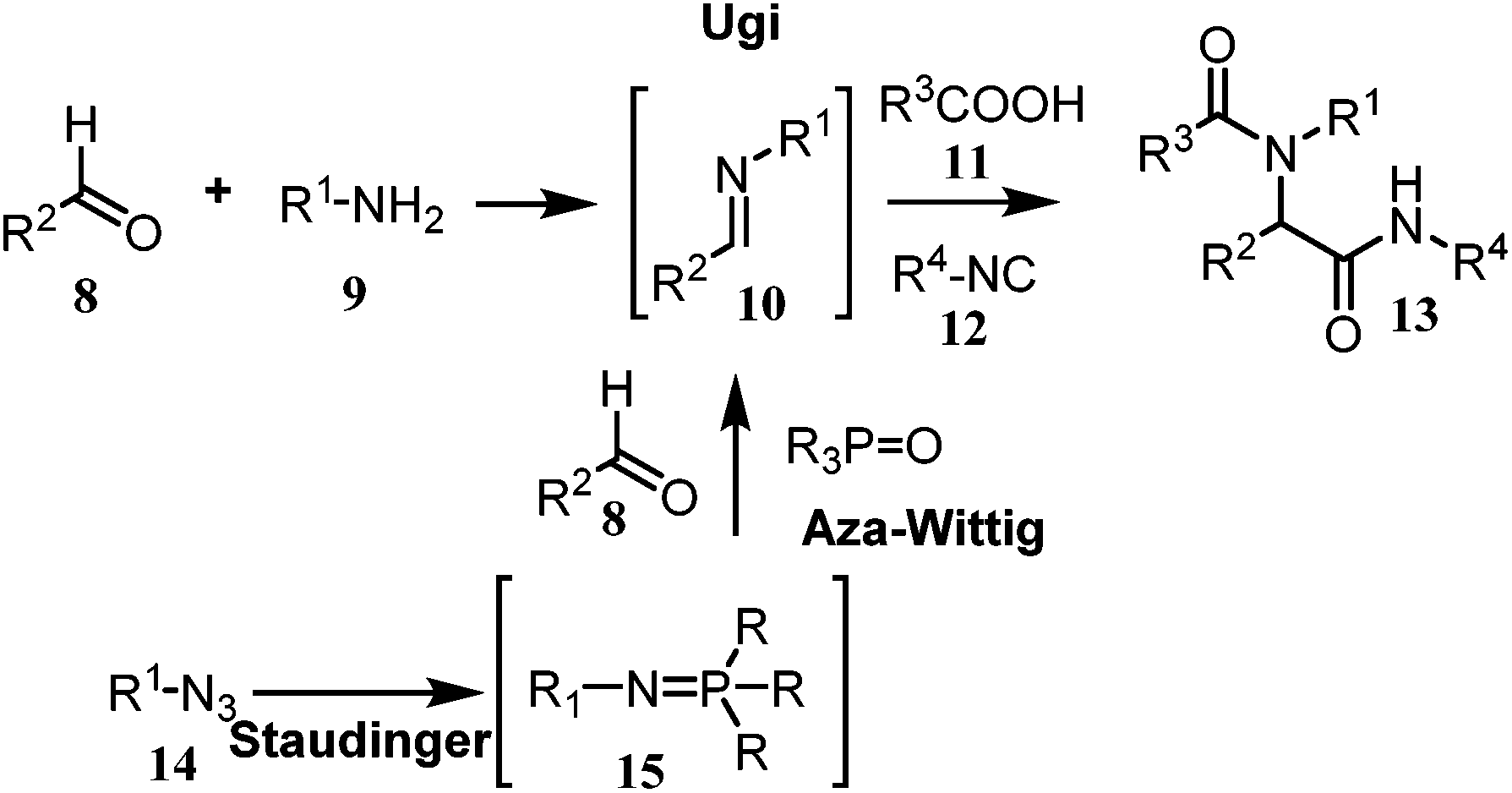 | ||
| Scheme 2 Schematic presentation of Staudinger–aza-Wittig reaction coupled with Ugi reaction. | ||
 | ||
| Scheme 3 Cyclic dipeptides via reductive cyclization Ugi reaction. | ||
The required azido hemi-acetal 24 for the synthesis of DNJ isomers was prepared by the commercially available D-ribose (19). Firstly C2–C3 cis-hydroxy of 19 was protected with the freshly distilled cyclohexanone using p-toluenesulphonic acid as the catalyst. The primary hydroxyl in 2,3-O-cyclohexylidene-D-ribose (20) was selectively tosylated (21) and the anomeric hydroxy was subsequently benzylated to give the fully protected ribofuranoside (22). 22 was treated with sodium azide in DMF at high temperature to give azide 23, which on successive anomeric debenzoylation using a catalytic amount of sodium methoxide in methanol afforded the target azido hemi-acetal 24 in an overall good yield (Scheme 4).11
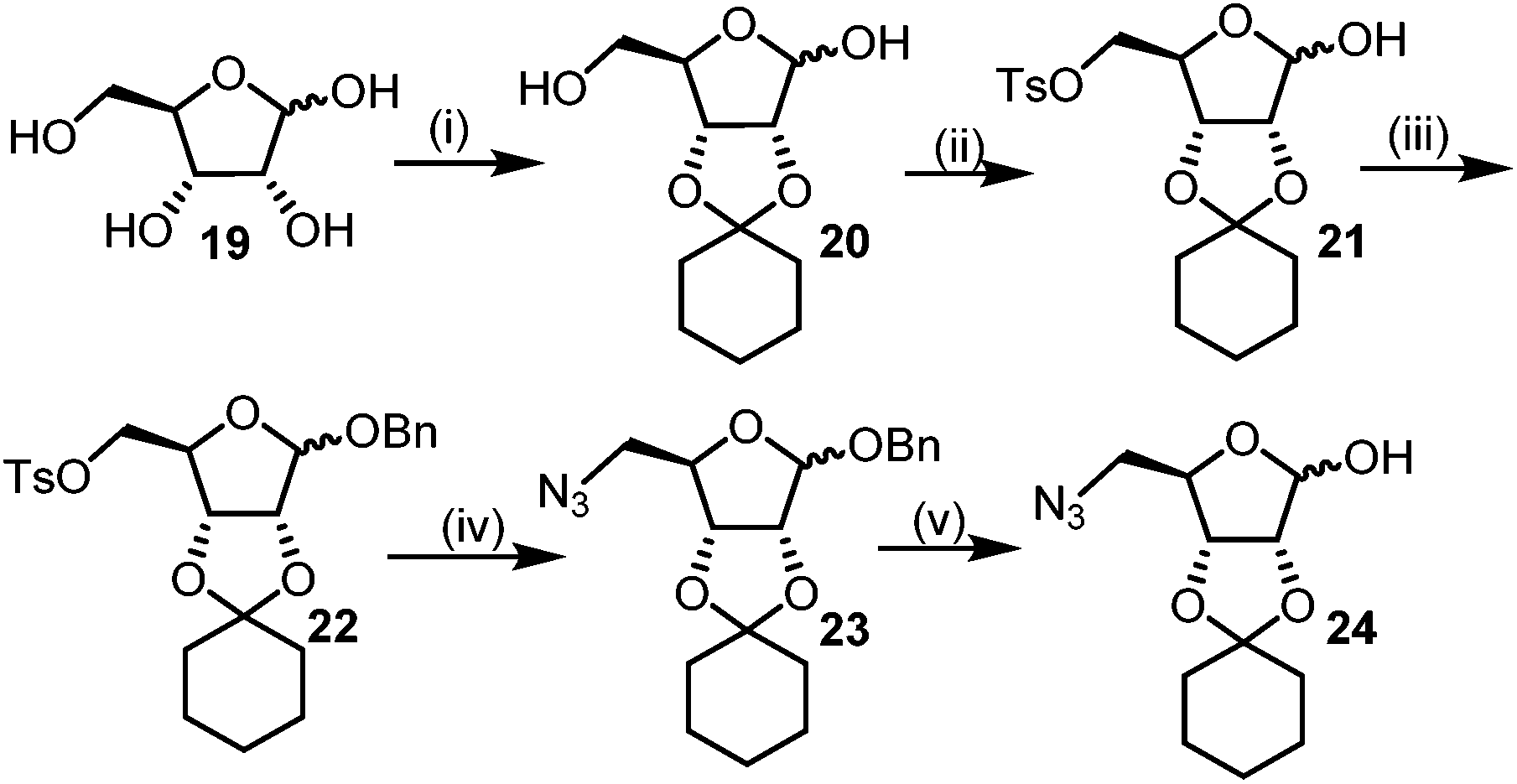 | ||
| Scheme 4 Synthesis of required azido hemi-acetal. Reagents: (i) cyclohexanone, pTSA (ii) TsCl, py (iii) BzCl, py (iv) NaN3, DMF, (v) NaOMe (cat.), MeOH. | ||
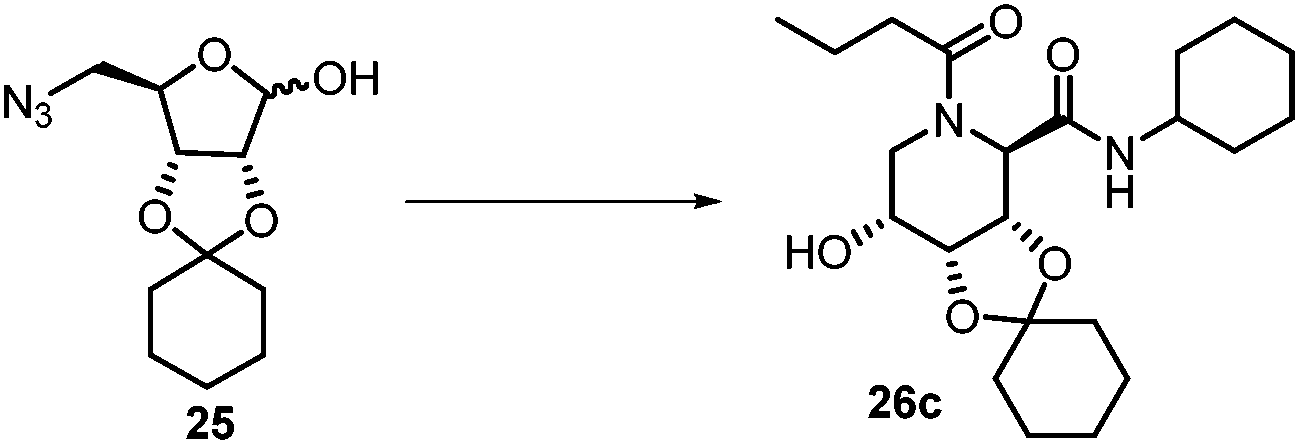
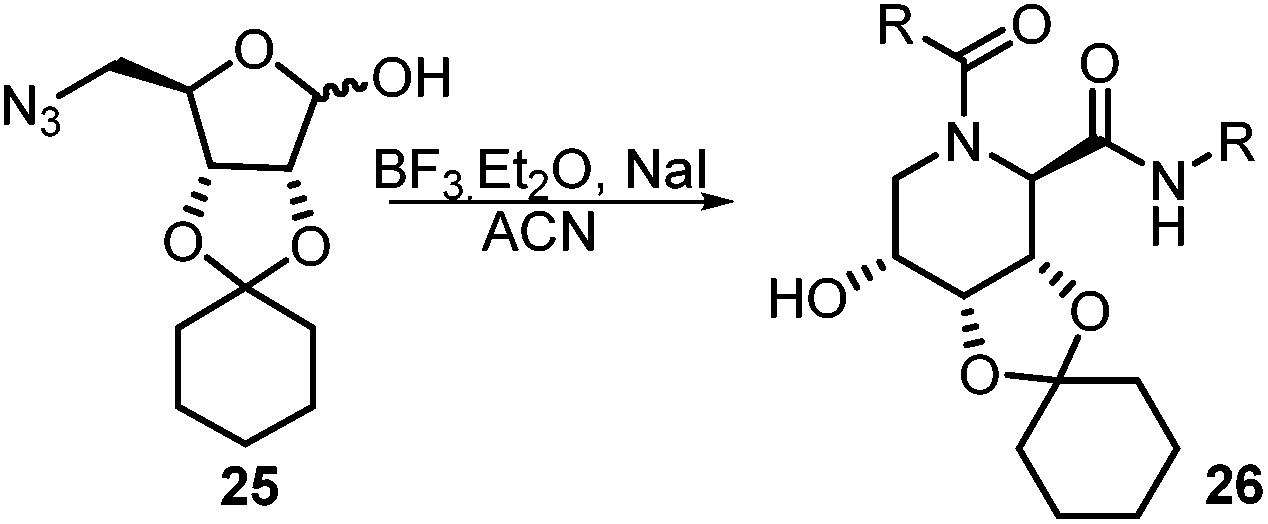
After getting the required 24 we searched the literature for a cost effective reductive cyclization method and we found a new methodology for the reduction of azide followed by cyclization reported by Kamal et al., in which azide was reduced with FeSO4·7H2O/NH3 in DCM.12 It has been thought that if azide is reduced in situ then it may probably react with the aldehyde generated from hemi-acetal under mild acidic conditions to form an imine which further undergoes Ugi reaction to give bisamide. To extend the scope of the reaction a model reaction having butanoic acid and cyclohexylisocyanide as Ugi reactants has been performed at room temperature with different reductive cyclization methodologies. The results of this study are summarized in Table 1.
A promising result was obtained when the reaction was performed with BF3·Et2O in acetonitrile solvent. After getting the results with BF3·Et2O/NaI we screened the solvents viz. DCM, DCE, CHCl3, DMF and DMSO in which ACN was found to be the best among all the solvents. This type of novel reductive cyclization method has never been performed coupled with Ugi reaction. To explain the applicability of the reaction, reductive cyclization/Ugi reaction is performed with a variety of acids and isocyanides (Table 2).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Acid | Isocyanide | Product | Time (h) | Yield |
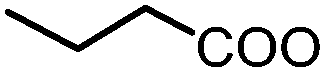
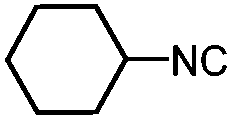
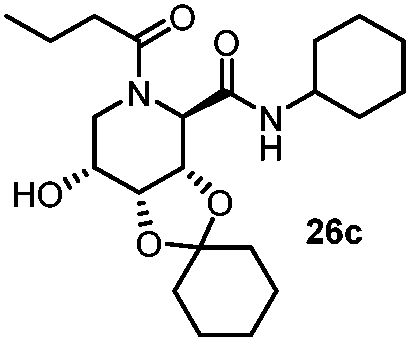
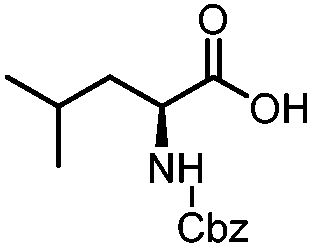
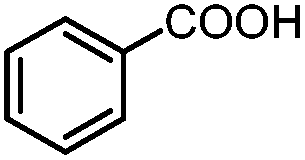
| 1 | Benzoic acid |
|
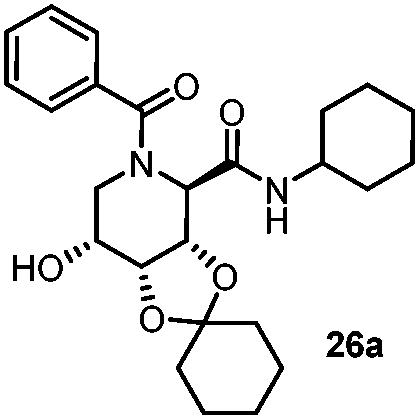
|
3 | 43 |
| 2 |
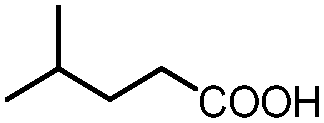
|
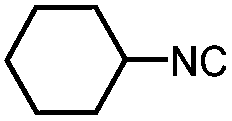
|
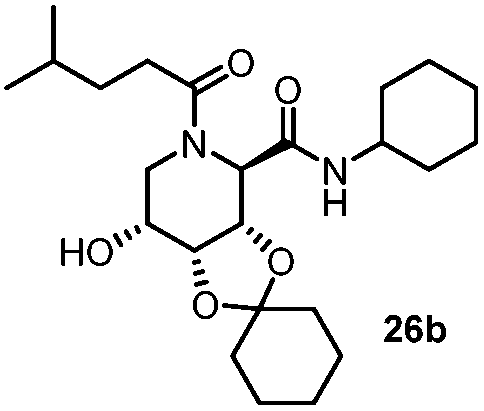
|
4.5 | 66 |
| 3 |
|
|
|
2 | 56 |
| 4 |
|
|
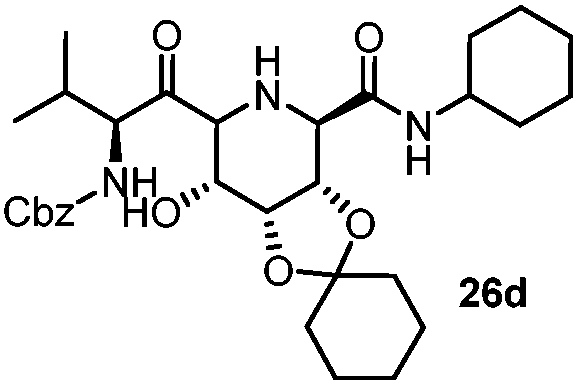
|
3 | 55 |
| 5 |
|
|
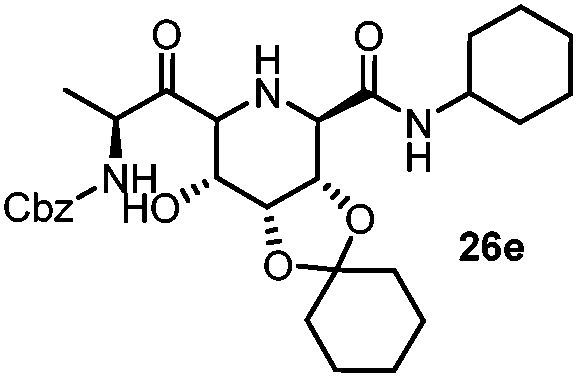
|
3.5 | 52 |
| 6 |
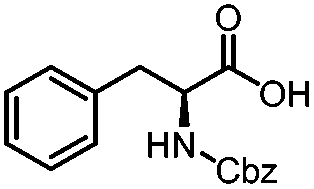
|
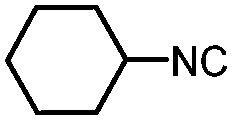
|
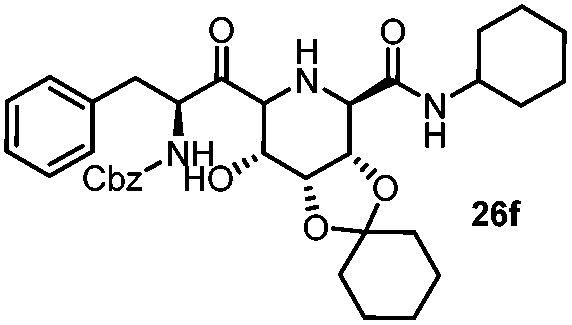
|
4 | 50 |
| 7 |
|
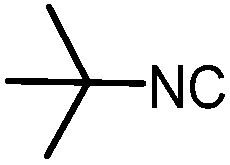
|
|
3 | 47 |
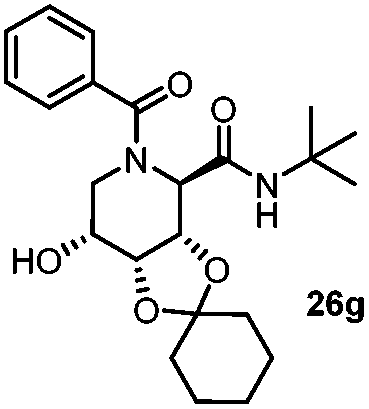
| 8 |
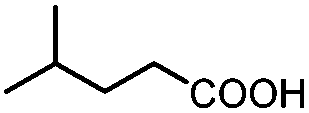
|
|
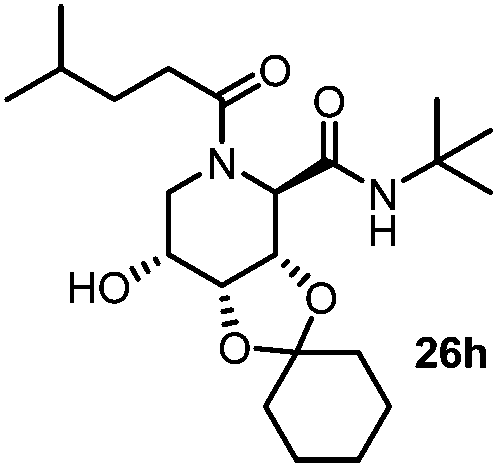
|
3.5 | 44 |
| 9 |
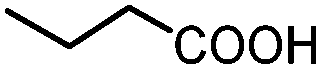
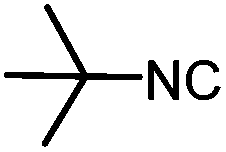
|
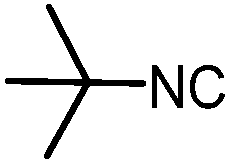
|
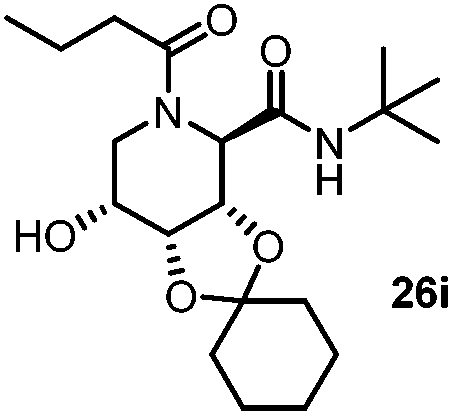
|
3 | 52 |
| 10 |
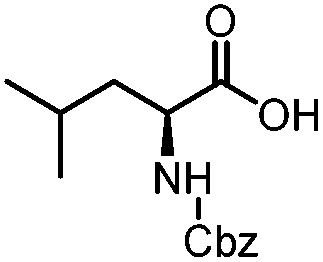
|
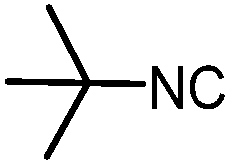
|
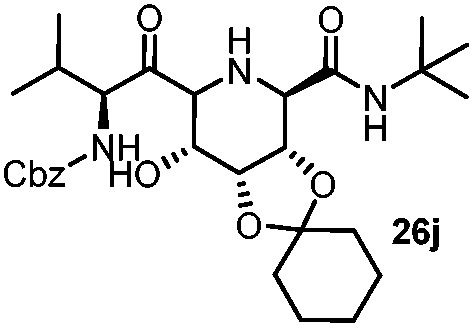
|
4 | 60 |
| 11 |
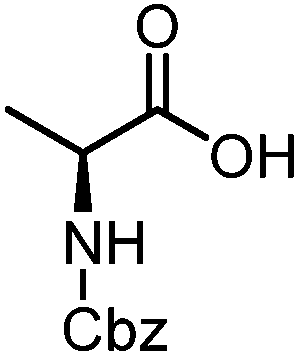
|
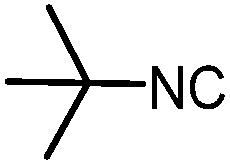
|
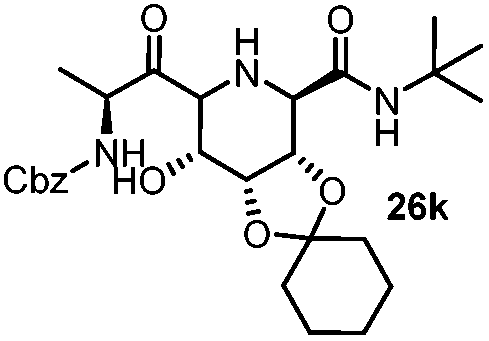
|
4 | 48 |
| 12 |
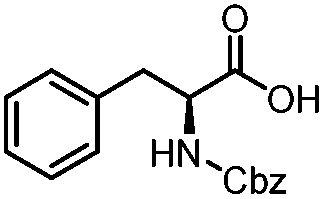
|
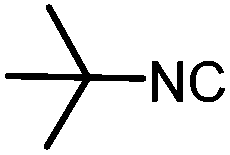
|
|
3.5 | 46 |
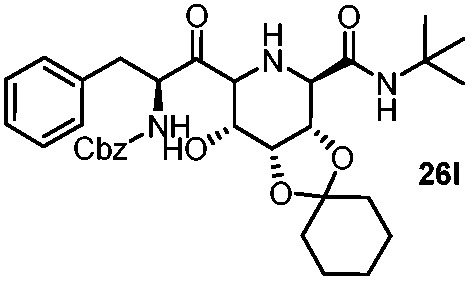
This Lewis acid mediated reductive cyclization followed by Ugi has been reported for the first time in this manuscript. After getting bisamides 26 several methods were applied for the amide bond hydrolysis but no satisfactory result was obtained. After visualizing the ease of hydrolysis of the amide bond with an appropriate substrate, pent-4-enoic acid and benzyl isocyanide were chosen as Ugi reagents. After reductive cyclization followed by Ugi the bis-amide 27 was formed. The cyclohexyl protection of the cis hydroxy group of 27 was cleaved under mild conditions by using hydrogen fluoride-pyridine.22 The resulting triol was subsequently benzylated to give 28, which was hydrolysed by iodine in the THF–H2O system to give amide 29.23 The other left amide bond was selectively reduced to aldehyde 30 by the treatment with triflic anhydride, 2-fluoro-pyridine and triethylsilylhydride in DCM.2430 was subsequently hydrogenated in Pd/C in MeOH to give L-1-deoxyallonojirimycin (31) via debenzylation coupled with aldehyde reduction (Scheme 5).
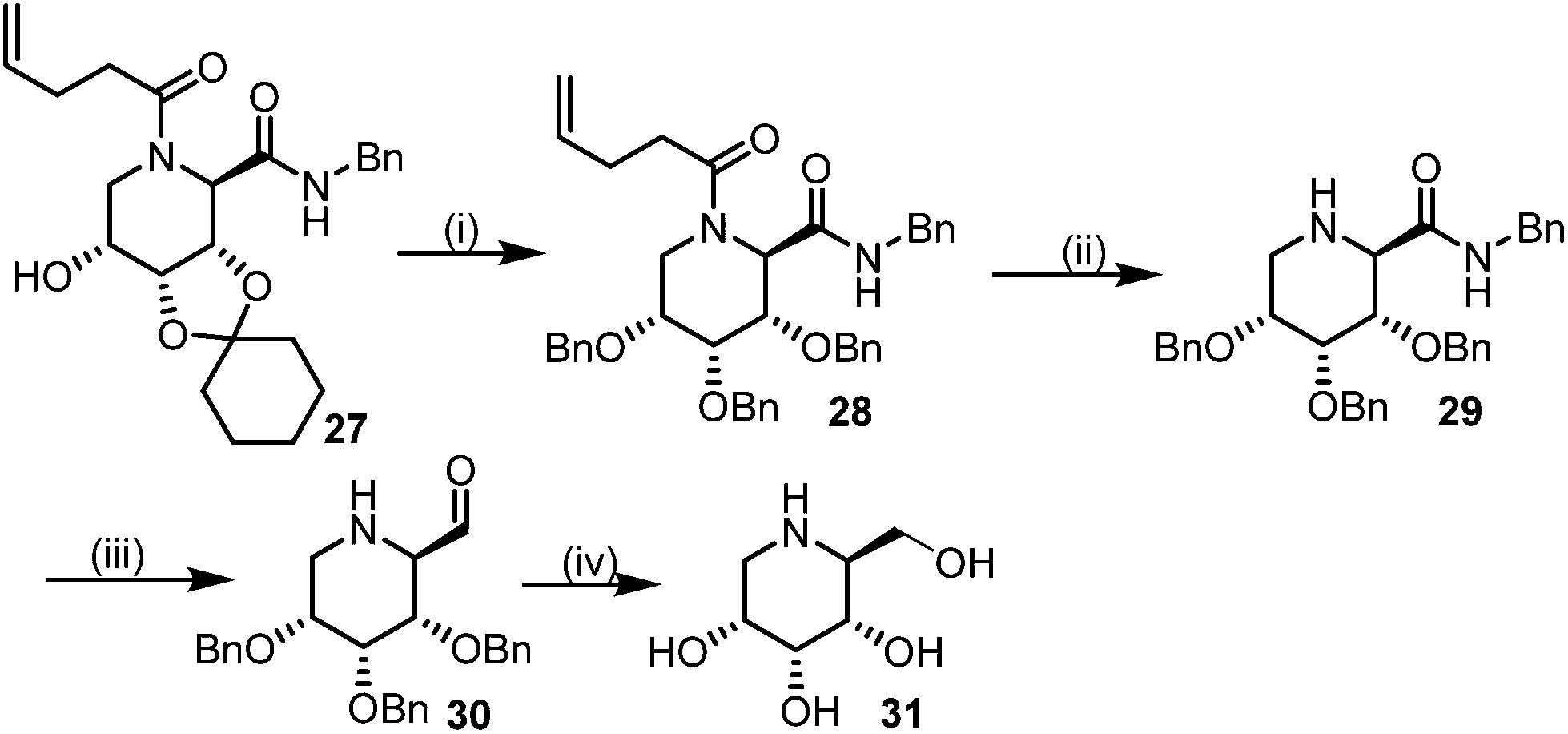 | ||
| Scheme 5 Synthetic route for (L-allo-DNJ). Reagents: (i) (a) HF(py), CHCl3 (b) BnBr, NaI, DMF (ii) I2, THF–H2O (iii) Tf2O, 2-FPyr, Et3SiH, DCM (iv) H2, Pd/C, MeOH. | ||
The other isomer L-1-deoxyaltronojirimycin (36) can easily be obtained by the inversion of configuration of the hydroxy of bis-amide 27 by Mitsunobu reaction. The free hydroxy group was converted into picolinic ester which was subsequently hydrolysed under neutral conditions to give bis-amide (32). After flipping the stereocenter the same synthetic methodologies have been applied for achieving (36) (Scheme 6).
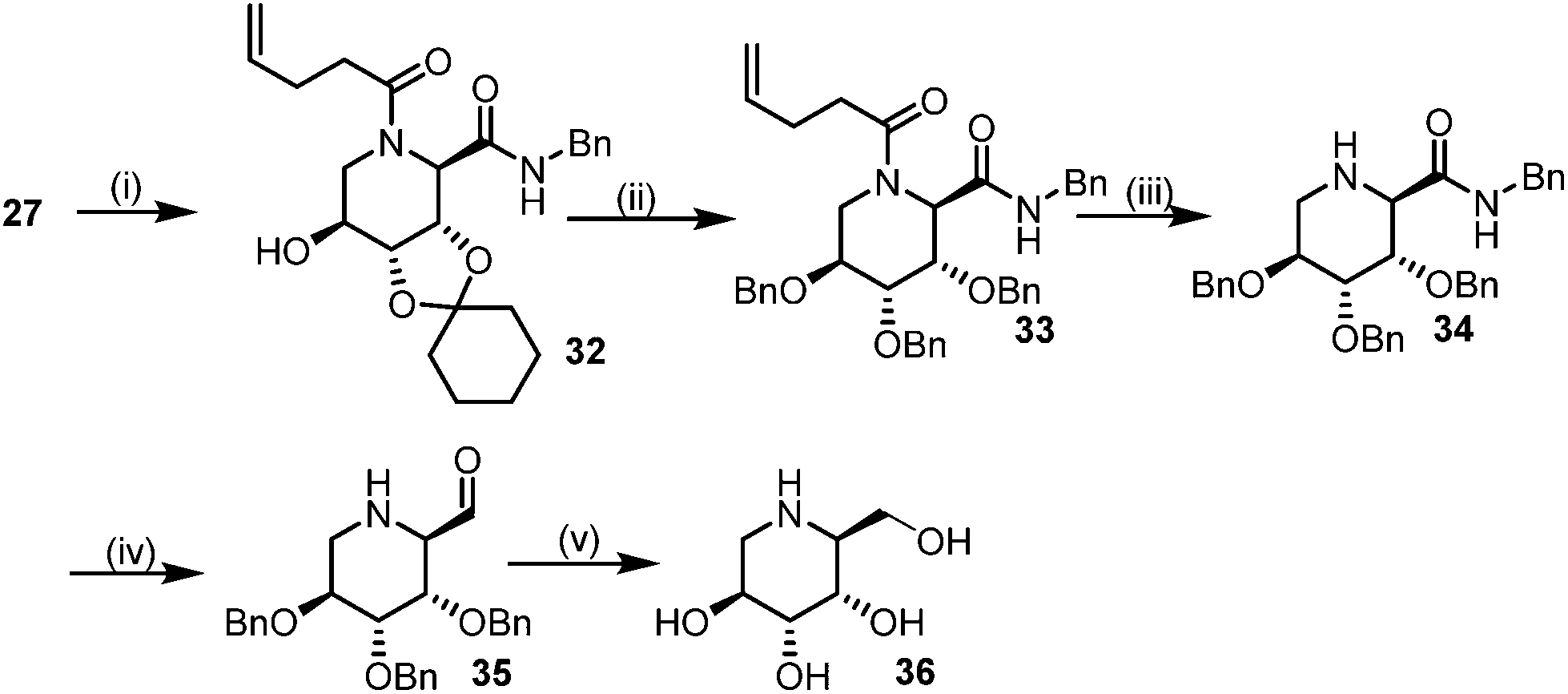 | ||
| Scheme 6 Synthetic route for (L-altro-DNJ). Reagents: (i) (a) PPh3, DIAD, picolinic acid, THF (b) Cu(OAc)2, MeOH (ii) (a) HF (py), CHCl3 (b) BnBr, NaI, DMF (iii) I2, THF–H2O (iv) Tf2O-FPyr, Et3SiH, DCM (v) H2, Pd/C, MeOH. | ||
Conclusions
In conclusion, we have synthesized L-1-deoxyallonojirimycin and L-1-deoxyaltronojirimycin by developing a cost effective mild and three component four centred Ugi reaction coupled with novel reductive cyclization as a key step from commercially available D-ribose.Acknowledgements
C.S.A. is thankful to CSIR, New Delhi for the award of SRF and acknowledges A.S. Kushwaha and Mr Zahid Ali for their technical support. The authors also acknowledge SAIF-CDRI for providing spectral and analytical data. The CDRI communication number allotted to this manuscript is 8919.Notes and references
- (a) N. Asano, Curr. Top. Med. Chem., 2003, 3, 471–484 CrossRef CAS PubMed; (b) A. K. Saxena and C. S. Azad, Curr. Res. Inf. Pharm. Sci., 2014, 15, 2–8 Search PubMed.
- (a) H. Paulsen, Angew. Chem., Int. Ed. Engl., 1966, 5, 495–510 CrossRef CAS; (b) H. Paulsen and K. Todt, Chem. Ber., 1967, 100, 3385–3396 CrossRef CAS.
- S. Inouye, T. Tsuruoka and T. Nida, J. Antibiot., 1966, 19, 288 CAS.
- (a) P. Compain and O. R. Martin, Iminosugars: from synthesis to therapeutic applications, John Wiley & Sons, 2007 Search PubMed; (b) T. D. Butters, R. A. Dwek and F. M. Platt, Curr. Top. Med. Chem., 2003, 3, 561–574 CrossRef CAS PubMed; (c) R. J. Nash, A. Kato, C.-Y. Yu and G. W. Fleet, Future Med. Chem., 2011, 3, 1513–1521 CrossRef CAS PubMed; (d) G. Horne and F. X. Wilson, Prog. Med. Chem., 2011, 50, 135 CAS.
- G. Horne, F. X. Wilson, J. Tinsley, D. H. Williams and R. Storer, Drug Discovery Today, 2011, 16, 107–118 CrossRef CAS PubMed.
- A. Kato, N. Kato, E. Kano, I. Adachi, K. Ikeda, L. Yu, T. Okamoto, Y. Banba, H. Ouchi, H. Takahata and N. Asano, J. Med. Chem., 2005, 48, 2036–2044 CrossRef CAS PubMed.
- D. D'Alonzo, A. Guaragna and G. Palumbo, Curr. Med. Chem., 2009, 16, 473–505 CrossRef.
- I. Ugi, Angew. Chem., Int. Ed. Engl., 1962, 1, 8–21 CrossRef.
- (a) D. C. Kim, K. H. Yoo, D. J. Kim, B. Y. Chung and S. W. Park, Tetrahedron Lett., 1999, 40, 4825–4828 CrossRef CAS; (b) T. M. Chapman, S. Courtney, P. Hay and B. G. Davis, Chem. – Eur. J., 2003, 9, 3397–3414 CrossRef CAS PubMed; (c) M. A. Maughan, I. G. Davies, T. D. Claridge, S. Courtney, P. Hay and B. G. Davis, Angew. Chem., Int. Ed., 2003, 42, 3788–3792 CrossRef CAS PubMed; (d) B. G. Davis, M. A. Maughan, T. M. Chapman, R. Villard and S. Courtney, Org. Lett., 2002, 4, 103–106 CrossRef CAS PubMed; (e) D. H. Valentine Jr. and J. H. Hillhouse, Synthesis, 2003, 0317–0334 Search PubMed.
- M. S. Timmer, M. D. Risseeuw, M. Verdoes, D. V. Filippov, J. R. Plaisier, G. A. van der Marel, H. S. Overkleeft and J. H. van Boom, Tetrahedron: Asymmetry, 2005, 16, 177–185 CrossRef CAS.
- Y. Ichikawa, Y. Igarashi, M. Ichikawa and Y. Suhara, J. Am. Chem. Soc., 1998, 120, 3007–3018 CrossRef CAS.
- A. Kamal, E. Laxman and M. Arifuddin, Tetrahedron Lett., 2000, 41, 7743–7746 CrossRef CAS.
- A. Kamal, B. Reddy and B. Reddy, Tetrahedron Lett., 1996, 37, 6803–6806 CrossRef CAS.
- A. Kamal, Y. Damayanthi, B. N. Reddy, B. Lakminarayana and B. P. Reddy, Chem. Commun., 1997, 1015–1016 RSC.
- A. Kamal, E. Laxman, N. Laxman and N. Venugopal Rao, Bioorg. Med. Chem. Lett., 2000, 10, 2311–2313 CrossRef CAS PubMed.
- A. Kamal, A. H. Babu, A. V. Ramana, K. V. Ramana, E. V. Bharathi and M. S. Kumar, Bioorg. Med. Chem. Lett., 2005, 15, 2621–2623 CrossRef CAS PubMed.
- A. Kamal, P. Reddy and D. R. Reddy, Tetrahedron Lett., 2002, 43, 6629–6631 CrossRef CAS.
- A. Kamal, K. Srinivasa Reddy, B. Rajendra Prasad, A. Hari Babu and A. V. Ramana, Tetrahedron Lett., 2004, 45, 6517–6521 CrossRef CAS.
- A. Kamal, N. Shankaraiah, K. L. Reddy and V. Devaiah, Tetrahedron Lett., 2006, 47, 4253–4257 CrossRef CAS.
- A. Kamal, S. Prabhakar, N. Shankaraiah, N. Markandeya, P. Venkat Reddy, V. Srinivasulu and M. Sathish, Tetrahedron Lett., 2013, 54, 4435–4441 CrossRef CAS.
- A. Kamal, N. Shankaraiah, N. Markandeya and C. S. Reddy, Synlett, 2008, 1297–1300 CrossRef CAS.
- Y. Watanabe, Y. Kiyosawa, A. Tatsukawa and M. Hayashi, Tetrahedron Lett., 2001, 42, 4641–4643 CrossRef CAS.
- R. Madsen, C. Roberts and B. Fraser-Reid, J. Org. Chem., 1995, 60, 7920–7926 CrossRef CAS.
- G. Pelletier, W. S. Bechara and A. B. Charette, J. Am. Chem. Soc., 2010, 132, 12817–12819 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Preparation of substrates, characterization data, 1H, 13C NMR, CHN analysis and ESI. See DOI: 10.1039/c5qo00019j |
| This journal is © the Partner Organisations 2015 |