
Palladium catalyzed amide-oxazoline directed C–H acetoxylation of arenes†
Bin
Liu
,
Xuhu
Huang
,
Xin
Wang
,
Zemei
Ge
* and
Runtao
Li
*
Skate Key Laboratory of Natural and Biomimetic Drugs, Peking University, Beijing 100191, China. E-mail: lirt@bjmu.edu.cn
First published on 29th April 2015
Abstract
Herein we reported a Pd-catalyzed ortho-acetoxylation of arenes through a six-membered system using an amide-oxazoline as the directing group. A wide range of aryls and heteroaryls are tolerated. The approach provides general and straightforward access to phenol esters without the need for extra oxidants.
The direct acetoxylation of a C–H bond into a C–O bond has attracted considerable attention in recent years due to the industrially important roles of phenol or ester compounds.1 For example, both of them are key structural motifs in numerous drugs, carbohydrates and natural products. Transition-metal-catalyzed C–H bond activation is a new strategy for the step-economical synthesis. It can be used for the direct modification of many biologically important molecules without prefunctionalization.2 Pd-catalyzed pyridine-directed acetoxylation of the C–H bond was first demonstrated by Sanford and co-workers in 2004.3 Later, Yu et al.4 reported various Cu-mediated ortho-acetoxylations of aryl C–H bonds, which showed high regio-selectivity and efficiency (Scheme 1, eqn (1)). Thereafter, various directing groups (DGs) such as pyridine,5 amine,6 oxime7 and carbonyl8 have been used in the transition-metal-catalyzed C–H activation. Despite these advances, the directed acetoxylation has been rarely used for aryls. In addition, the directing groups are hard to be removed after the reaction. Recently, Zhang9 reported a Pd-catalyzed oxidative acetoxylation of the benzylic C(sp3)–H bond with PhI(OAc)2 as an oxidant (Scheme 1, eqn (2)). Meanwhile, Liang10 also demonstrated the Pd-catalyzed acetoxylations with amide-pyridine and Daugulis amide-quinoline11 as DGs. However, longer reaction times and higher reaction temperatures are required in these acetoxylations. Moreover, electron-deficient arenes and heteroarenes have not been demonstrated. Therefore, an efficient synthesis pathway under milder conditions is still a challenge.
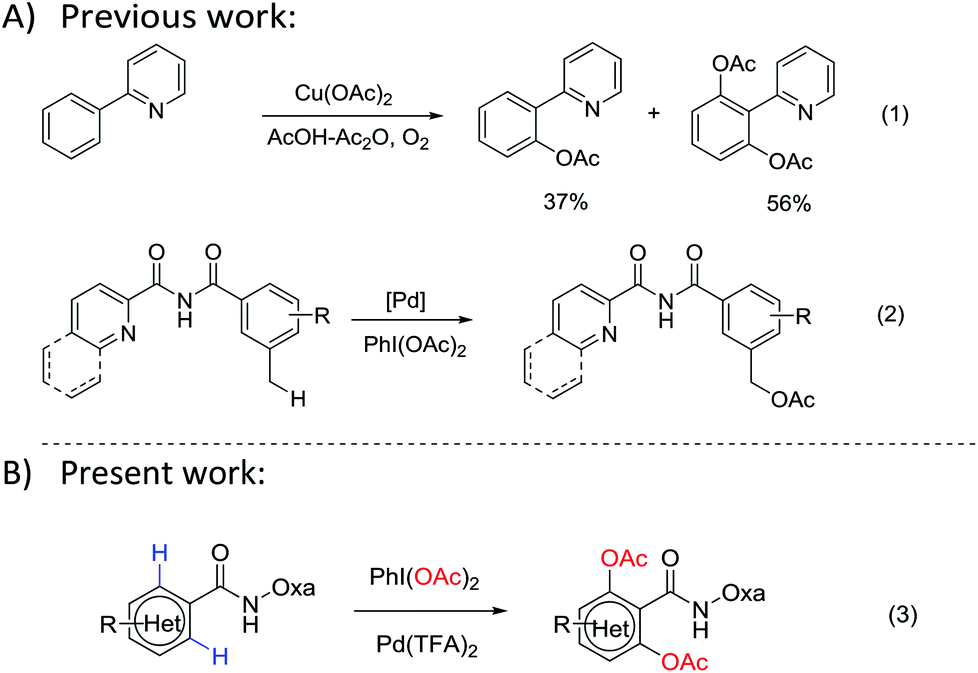 | ||
| Scheme 1 Transition-metal-catalyzed acetoxylation of C–H bonds. | ||
Very recently, Yu reported an amide-tethered oxazoline as a directing group (DG).12 It can form a six-membered bis-dentate complex with Cu(II) or Pd(II),13 which is different from the amide-quinoline DG. In the present work, we propose a direct ortho-acetoxylation of amide-tethered oxazoline through the Pd(TFA)2-catalyzed C–H activation in aromatic rings and heteroaryl amides with PhI(OAc)2 as both the oxidant and acetate source (Scheme 1, eqn (3)).
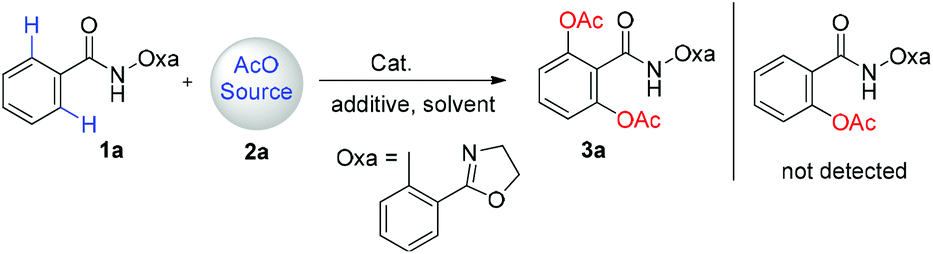
Amide (1a) was prepared from the acylation of benzoyl chloride and 2-(2-oxazolyl)aniline and used as the substrate. In our initial study, iodobenzene diacetate (2a) was used as the AcO source (Table 1). However, no desired product 3a was observed by treating 1a with 2a in the presence of Cu(OAc)2 (1 equiv.) in o-xylene at 100 °C (Table 1, entry 1). Product 3a was formed in 20% yield with Pd(OAc)2 as the catalyst under the same conditions (Table 1, entry 2). Its yield was slightly increased when the reaction temperature was increased to 130 °C (Table 1, entry 3). Other metal catalysts including Co(OAc)2, Mn(OAc)3·2H2O, Ni(OAc)2, Rh2(OAc)4, CuCl, InCl3 and Fe(acac)3 showed no catalytic activity for the reaction, indicating the unique catalytic ability of Pd in this transformation reaction (Table 1, entry 4). Therefore, a series of Pd salts including Pd(CH3CN)2Cl2, Pd(acac)2, Pd(PPh3)2Cl2 and Pd(TFA)2 were screened (Table 1, entries 5–8). All of them showed catalytic activity for the transformation reaction and Pd(TFA)2 gave the highest yield of 40% (Table 1, entry 8). With Pd(TFA)2 as the catalyst, various solvents were tested (Table 1, entries 9–12). The results indicate that no desired product can be formed in aprotic polar solvents, such as DMF and DMSO (Table 1, entries 9–10). A small amount of the product was observed with chlorobenzene as the reaction solvent (Table 1, entry 11). DCE is the best solvent for the reaction in which 3a was formed in 62% yield (Table 1, entry 12). Next, different AcO sources including Cu(OAc)2, AgOAc and AcOH/Ac2O were investigated (Table 1, entries 13–15). It is noteworthy that only a trace amount of product 3a was observed with AcOH/Ac2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) as the AcO source (Table 1, entry 15). Meanwhile, the yield of 3a was significantly increased to 73% with both PIDA and additive added (Table 1, entry 16). The yield of 3a decreased to 55% when the catalyst loading was decreased to 5 mol% (Table 1, entry 17). No desired product was obtained in the absence of the Pd catalyst (Table 1, entry 18).
:2) as the AcO source (Table 1, entry 15). Meanwhile, the yield of 3a was significantly increased to 73% with both PIDA and additive added (Table 1, entry 16). The yield of 3a decreased to 55% when the catalyst loading was decreased to 5 mol% (Table 1, entry 17). No desired product was obtained in the absence of the Pd catalyst (Table 1, entry 18).
|
|
||||
|---|---|---|---|---|
| Entry | Cat. | Solvent | AcO source | Yield 3a (%) |
|
a Reaction conditions: 1a (0.2 mmol), 2a (0.5 mmol), catalyst (0.02 mmol), additive (0.2 mmol) in solvent (3 mL) was stirred at 130 °C for 10 h in a sealed tube.
b The temperature used was 100 °C.
c Other metals: Co(OAc)2, Mn(OAc)3·2H2O, Ni(OAc)2, Rh2(OAc)4, CuCl, InCl3, Fe(acac)3 respectively.
d Different AcO sources (2.5 eq.) were added.
e AcOH/Ac2O = 1:2 (1 eq.) or PhCOOH (1 eq.) was added as the additive.
f 5% of the catalyst was employed.
|
||||
| 1 | Cu(OAc)2 | o-Xylene | PIDA | N.R. |
| 2b | Pd(OAc)2 | o-Xylene | PIDA | 20 |
| 3 | Pd(OAc)2 | o-Xylene | PIDA | 29 |
| 4c | Other metals | o-Xylene | PIDA | N.R. |
| 5 | Pd(CH3CN)2Cl2 | o-Xylene | PIDA | 28 |
| 6 | Pd(acac)2 | o-Xylene | PIDA | 18 |
| 7 | Pd(PPh3)2Cl2 | o-Xylene | PIDA | 25 |
| 8 | Pd(TFA)2 | o-Xylene | PIDA | 40 |
| 9 | Pd(TFA)2 | DMF | PIDA | N.R. |
| 10 | Pd(TFA)2 | DMSO | PIDA | N.R. |
| 11 | Pd(TFA)2 | Chlorobenzene | PIDA | 25 |
| 12 | Pd(TFA)2 | DCE | PIDA | 62 |
| 13d | Pd(TFA)2 | DCE | Cu(OAc)2 | N.R. |
| 14d | Pd(TFA)2 | DCE | AgOAc | N.R. |
| 15d | Pd(TFA)2 | DCE | AcOH/Ac2O | Trace |
| 16e | Pd(TFA)2 | DCE | PIDA + additive | 73 |
| 17f | Pd(TFA)2 | DCE | PIDA + additive | 55 |
| 18 | — | DCE | PIDA + additive | N.R. |
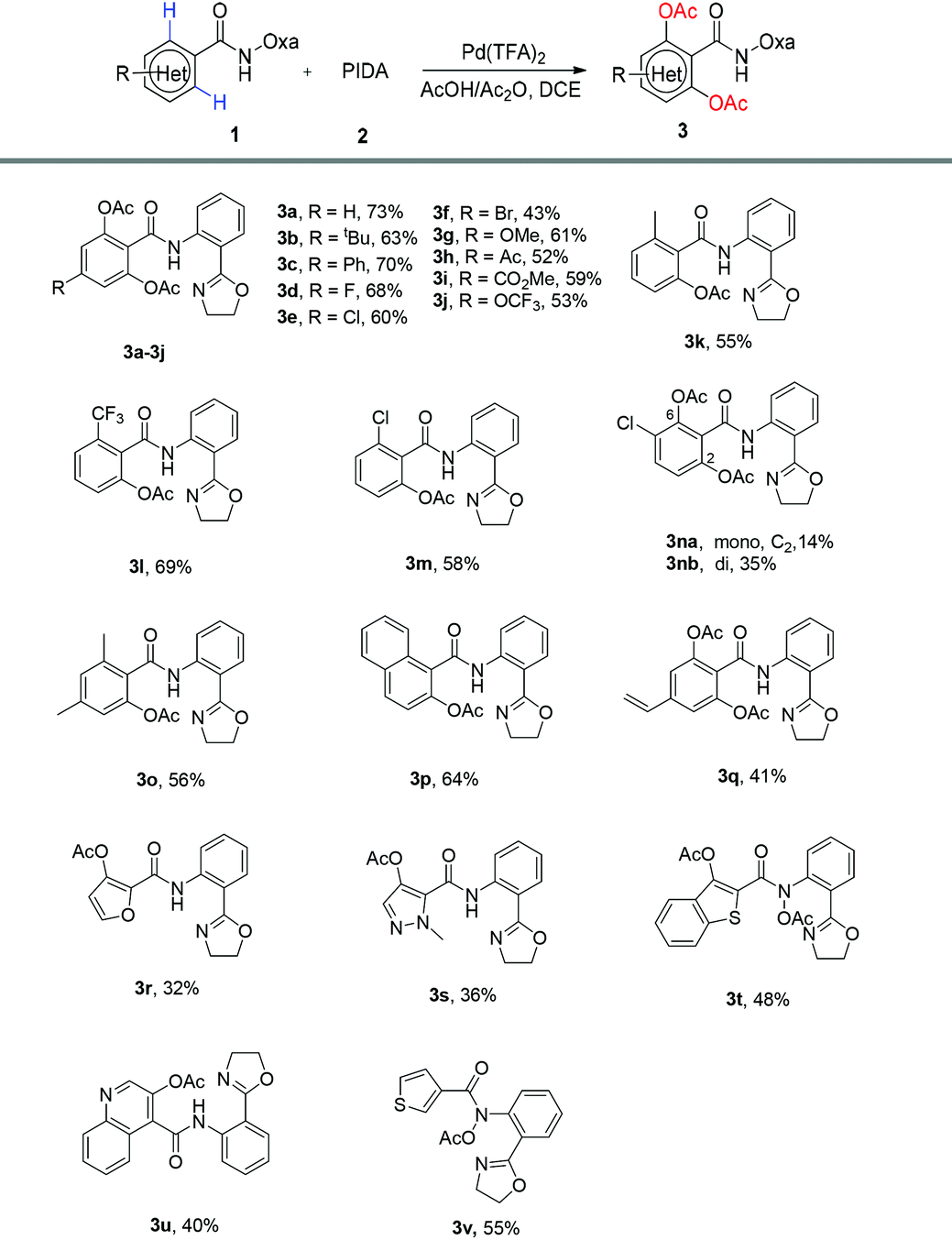
Next, various benzamide substrates were tested under the optimized reaction conditions (Table 1, entry 16). As shown in Table 2, the transformations of most functionalized benzamides were able to produce the desired products 3 under the optimized reaction conditions. Both electron-donating and electron-withdrawing groups on the phenyl ring are compatible with the transformation reaction. The transformation reaction can be applied to compound 1 with a variety of substituents including alkyl, aryl, halo (F, Cl, Br), methoxy, acetyl, ester and trifluoromethoxy to afford the desired products (3a–3j) in moderate to good yields (43%–73%). Remarkably, the reaction with ortho-substituted 1 as a substrate afforded mono-products 3k–3m and 3o–3p in similar yields. For the meta-substituted substrate 1n, the double ortho-acetoxylation product (3nb) was major and only a small amount of the mono ortho-acetoxylation product (3na) was separated. The vinylated arene can still be transformed into 3q in 41% yield. It is worth noting that heterocyclic substrates, including furan, pyrazole, benzothiophene and quinoline, were also transformed into 3r–3u in 32%–55% yields, respectively. To the best of our knowledge the ortho-acetoxylation of these heterocycles with other directing groups has not yet been reported. Interestingly, substrate 1t with benzothiophene produced not only the expected acetoxylation, but also N-acetoxylation, affording the product 3t in a moderate yield, and the substrate 1v with thiophene gave only the N-acetoxylation product 3v. Further work is needed to understand these two transformations.
To further understand the catalytic cycle of the reaction, a 1:1 mixture of 1a and [d5]-1a was used as a substrate and transformed under the optimized reaction conditions (Scheme 2(a)). No kinetic isotopic effects (KIEs ≈ 1.0) were observed, suggesting that C–H activation of arenes might not be a rate-limiting step. Furthermore, control experiments were designed (Scheme 2(b)), in which H/D exchange was clearly observed in the reaction of [d5]-1a. Meanwhile, with the excess introduction of D2O, the deuterated product 1a–b was obtained (52% D incorporation). These observations suggest that the C–H activation step is reversible.
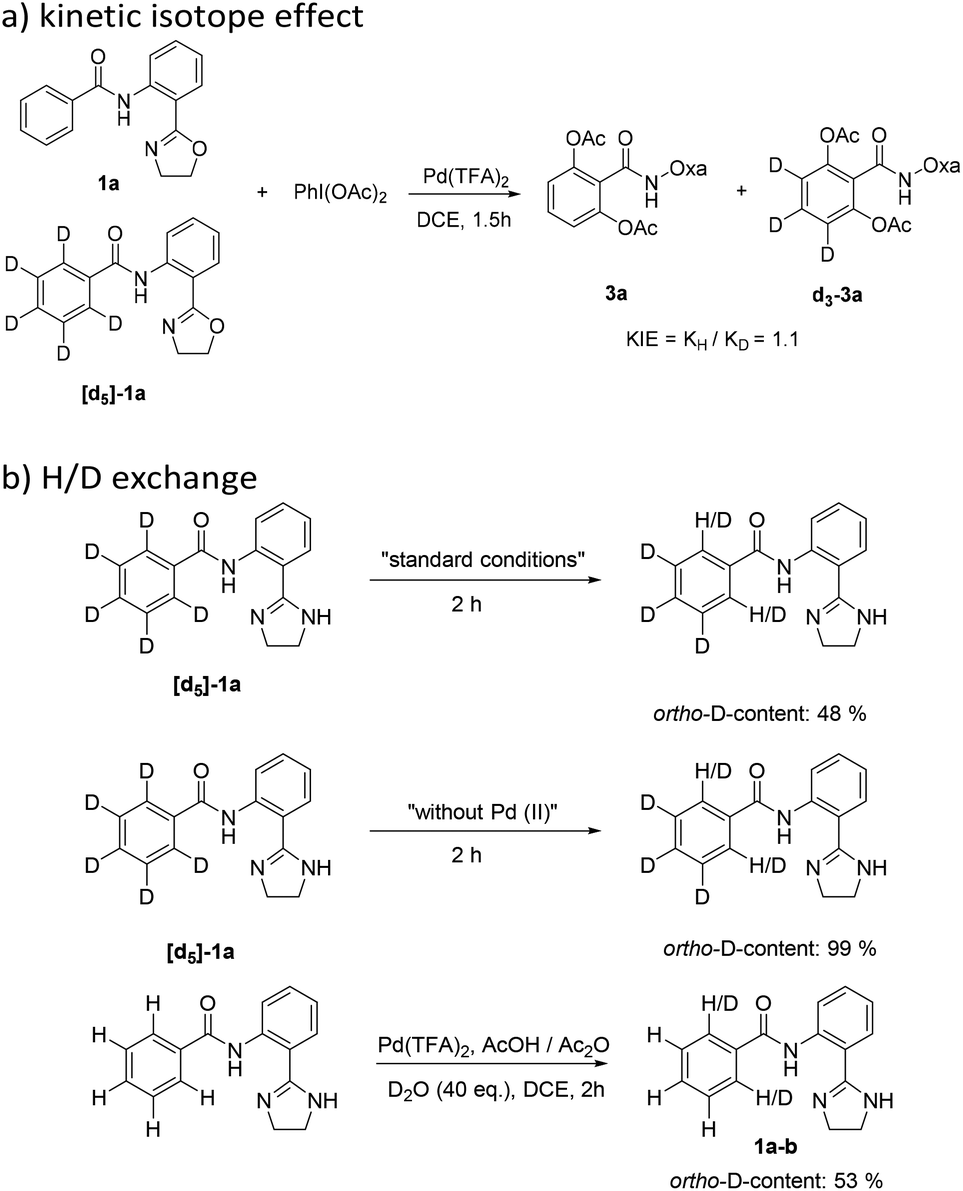 | ||
| Scheme 2 Determination of the kinetic isotope effect and H/D exchange. | ||
Although details about the mechanism remain to be ascertained, based on the previous reports13,14 and above results, a plausible mechanism for the Pd-catalyzed transformation is proposed (Scheme 3). The catalytic cycle starts with an initial C–H bond activation of 1a (1a′), followed by the oxidation of palladium (II–IV) complex A–B, which undergoes reductive elimination to afford product 1a′ followed by another catalytic cycle to produce 3a.
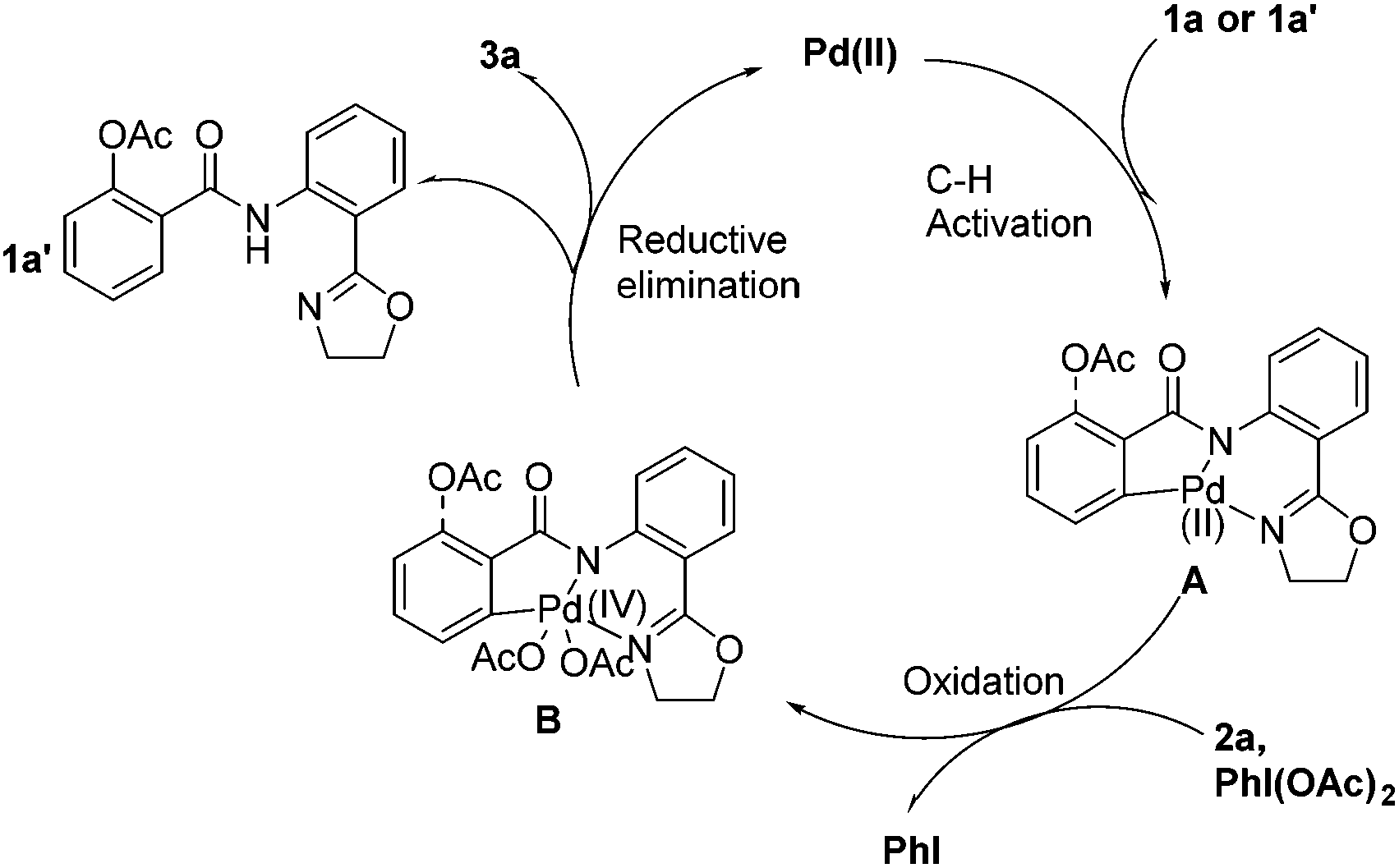 | ||
| Scheme 3 Proposed mechanism. | ||
Conclusions
In summary, we have developed a Pd(II) catalyzed ortho-acetoxylation of arenes including heterocycles, with PhI(OAc)2 as both the AcO source and oxidant. This synthesis pathway provides a new strategy for the ortho-acetoxylation of aryl compounds with high regio-selectivity. Further investigations and applications of this reaction are currently underway in our laboratory.Financial support from the National Science Foundation of China (no. 21172011) is acknowledged.
Notes and references
- (a) E. P. Fuerst, C. J. Arntzen, K. Pfister and D. Penner, Weed Sci., 1986, 34, 344 CAS; (b) J. H. P. Tyman, Synthetic and Natural Phenols, Elsevier, New York, 1996 Search PubMed; (c) J. A. May, H. Ratan, J. R. Glenn;, W. Losche, P. Spangenberg and S. Heptinstall, Platelets, 1998, 9, 27 Search PubMed; (d) M. G. Charest, C. D. Lerner, J. D. Brubaker, D. R. Siegel and A. G. Myers, Science, 2007, 318, 783 CrossRef PubMed.
- For selected reviews on transition-metal-catalyzed C–H activation, see: (a) G. Parkin, Acc. Chem. Res., 2009, 42, 315 CrossRef CAS PubMed; (b) C. J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS PubMed; (c) O. Daugulis, Top. Curr. Chem., 2010, 292, 57 CrossRef CAS; (d) F. Bellina and R. Rossi, Chem. Rev., 2010, 110, 1182 Search PubMed; (e) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (f) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814 CrossRef CAS PubMed; (g) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed; (h) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053 CrossRef CAS PubMed.
- A. R. Dick, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 2300 CrossRef CAS PubMed.
- X. Chen, X.-S. Hao, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 6790 CrossRef CAS PubMed.
- Y.-P. Yan, P. Feng, Q.-Z. Zheng, Y.-F. Liang, J.-F. Lu, Y.-X. Cui and N. Jiao, Angew. Chem., Int. Ed., 2013, 52, 1 CrossRef PubMed.
- (a) G.-W. Wang, T.-T. Yuan and X.-L. Wu, J. Org. Chem., 2008, 73, 4717 CrossRef CAS PubMed; (b) B. V. S. Reddy, L. R. Reddy and E. J. Corey, Org. Lett., 2006, 8, 3391 CrossRef CAS PubMed; (c) F.-J. Chen, S. Zhao, F. Hu, Q. Zhang, S.-Q. Zhang and B.-F. Shi, Chem. Sci., 2013, 4, 4187 RSC.
- (a) L. V. Desai, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 9543 Search PubMed; (b) S. R. Neufeldt and M. S. Sanford, Org. Lett., 2010, 12, 532 CrossRef CAS PubMed; (c) P. Lennartz, G. Raabe and C. Bolm, Adv. Synth. Catal., 2012, 354, 3237 CrossRef CAS PubMed; (d) Z. Ren, F.-Y. Mo and G.-B. Dong, J. Am. Chem. Soc., 2012, 134, 16991 CrossRef CAS PubMed.
- M.-N. Wang, Y. Yang, Z.-L. Fan, Z. Cheng, W.-L. Zhu and A. Zhang, Chem. Commun., 2015, 51, 3219 RSC.
- L. Ju, J.-Z. Yao, Z.-H. Wu, Z.-X. Liu and Y.-H. Zhang, J. Org. Chem., 2013, 78, 10821 CrossRef CAS PubMed.
- F.-R. Gou, X.-C. Wang, P.-F. Huo, H.-P. Bi, Z.-H. Guan and Y.-M. Liang, Org. Lett., 2009, 11, 5726 CrossRef CAS PubMed.
- For selected examples on Daugulis amide-quinoline as the DG, see: (a) L. D. Tran, I. Popov and O. Daugulis, J. Am. Chem. Soc., 2012, 134, 18237 CrossRef CAS PubMed; (b) L. D. Tran, J. Roane and O. Daugulis, Angew. Chem., Int. Ed., 2013, 52, 1 CrossRef PubMed; (c) T. Truong, K. Klimovica and O. Daugulis, J. Am. Chem. Soc., 2013, 135, 9342 CrossRef CAS PubMed; (d) Y. Aihara and N. Chatani, J. Am. Chem. Soc., 2013, 135, 5308 CrossRef CAS PubMed; (e) A. M. Suess, M. Z. Ertem, C. J. Cramer and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 9797 CrossRef CAS PubMed; (f) M. Nishino, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2013, 52, 4457 CrossRef CAS PubMed; (g) W.-F. Song, S. Lackner and L. Ackermann, Angew. Chem., Int. Ed., 2014, 53, 1 CrossRef PubMed; (h) M.-L. Li, J.-X. Dong, X.-L. Huang, K.-Z. Li, Q. Wu, F.-J. Song and J.-S. You, Chem. Commun., 2014, 50, 3944 RSC; (i) J.-X. Dong, F. Wang and J.-S. You, Org. Lett., 2014, 16, 2884 CrossRef CAS PubMed; (j) S. Wang, R. Guo, G. Wang, S.-Y. Chen and X.-Q. Yu, Chem. Commun., 2014, 16, 12718 RSC; (k) L. Ilies, T. Matsubara, S. Ichikawa, S. Asako and E. Nakamura, J. Am. Chem. Soc., 2014, 136, 13126 CrossRef CAS PubMed; (l) E. R. Fruchey, B. M. Monks and S. P. Cook, J. Am. Chem. Soc., 2014, 136, 13130 CrossRef CAS PubMed; (m) K. Shibata and N. Chatani, Org. Lett., 2014, 16, 5148 CrossRef CAS PubMed.
- For selected examples on amide-oxazoline as the DG, see: (a) M. Shang, S.-Z. Sun, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 3354 CrossRef CAS PubMed; (b) M. Shang, H.-L. Wang, S.-Z. Sun, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 11590 CrossRef CAS PubMed; (c) M. Shang, S.-Z. Sun, H.-L. Wang, B. N. Laforteza, H.-X. Dai and J.-Q. Yu, Angew. Chem., Int. Ed., 2014, 53, 10439 CrossRef CAS PubMed; (d) M. Shang, S.-Z. Sun, H.-X. Dai and J.-Q. Yu, Org. Lett., 2014, 16, 5666 CrossRef CAS PubMed; (e) H.-L. Wang, M. Shang, S.-Z. Sun, Z.-L. Zhou, B. N. Laforteza and J.-Q. Yu, Org. Lett., 2015, 17, 1228 CrossRef CAS PubMed.
- R. Giri, N. Maugel, B. M. Foxman and J.-Q. Yu, Organometallics, 2008, 27, 1667 CrossRef CAS.
- (a) L. Ackermann, Chem. Rev., 2011, 11, 1315 CrossRef PubMed; (b) D. C. Powers and T. Ritter, Acc. Chem. Res., 2012, 45, 840 CrossRef CAS PubMed; (c) Y.-F. Yang, G.-J. Cheng, P. Liu, D. Leow, T.-Y. Sun, P. Chen, X. Zhang, J.-Q. Yu, Y.-D. Wu and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 344 CrossRef CAS PubMed; (d) A. K. Cook and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 3109 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00104h |
| This journal is © the Partner Organisations 2015 |