
Brønsted base-catalyzed α-oxygenation of carbonyl compounds utilizing the [1,2]-phospha-Brook rearrangement†
Azusa
Kondoh
a and
Masahiro
Terada
*ab
aResearch and Analytical Center for Giant Molecules, Graduate School of Science, Tohoku University, Aoba-ku, Sendai 980-8578, Japan. E-mail: mterada@m.tohoku.ac.jp
bDepartment of Chemistry, Graduate School of Science, Tohoku University, Aoba-ku, Sendai 980-8578, Japan
First published on 29th April 2015
Abstract
A novel α-oxygenation reaction of carbonyl compounds was developed by utilizing the [1,2]-phospha-Brook rearrangement under Brønsted base catalysis. The reaction proceeds via the oxidation of enolates followed by the [1,2]-phospha-Brook rearrangement to afford α-oxygenated ester derivatives from easily accessible substrates having high functional group tolerance. In addition, the atom-economical tandem reaction of α-diethylphosphono ester derivatives with oxaziridines, which involves a carbon–carbon forming process in addition to α-oxygenation, was established under the low temperature conditions.
α-Oxygenated carbonyl compounds are useful building blocks in organic synthesis and common motifs in biologically active natural products as well as pharmaceuticals.1 Therefore, their synthesis is important in various fields of organic chemistry. One of the most well-known approaches for the synthesis of α-oxygenated carbonyl compounds is the enolate oxidation.2 Generally, this approach requires the use of a stoichiometric amount of a strong Brønsted base, such as LDA, or the preformation of a metal enolate, such as silyl enolate,2a tin enolate,3 titanium enolate,4 and boron enolate.5 On the other hand, direct catalytic oxygenation reactions that use Brønsted bases and metal complexes have been intensively investigated in recent years.6,7 While many catalytic reactions including asymmetric variants have been developed to date, the applicable substrates in most reactions are limited to compounds possessing relatively high acidity, such as 1,3-dicarbonyl compounds, which would facilitate the generation of an enolate. Very recently, the catalytic α-hydroxylation of less acidic carbonyl compounds was achieved by using a Brønsted base,8 transition metal complexes,9 and I2 or NBS.10 With these catalyst systems, less acidic substrates, such as simple ketones and esters, could be efficiently oxygenated under mild reaction conditions. Nevertheless, certain limitations remain regarding the substrates. For instance, some of those reactions provided only tertiary alcohols. Furthermore, the scope of applicable esters was extremely limited. Therefore, the development of an efficient method that would expand the scope of the catalytic synthesis of α-oxygenated carbonyl compounds is still being actively pursued. In this context, we designed a novel tandem reaction catalyzed by a Brønsted base with the aim of providing a practical method for the synthesis of α-oxygenated carbonyl compounds from easily accessible substrates. In our proposed system, α-hydroxylation is combined with the [1,2]-phospha-Brook rearrangement11,12 by using substrates bearing a diethylphosphono group α to a carbonyl group together with an oxaziridine2b as the oxidant. Our proposed reaction is shown in Scheme 1, path a.
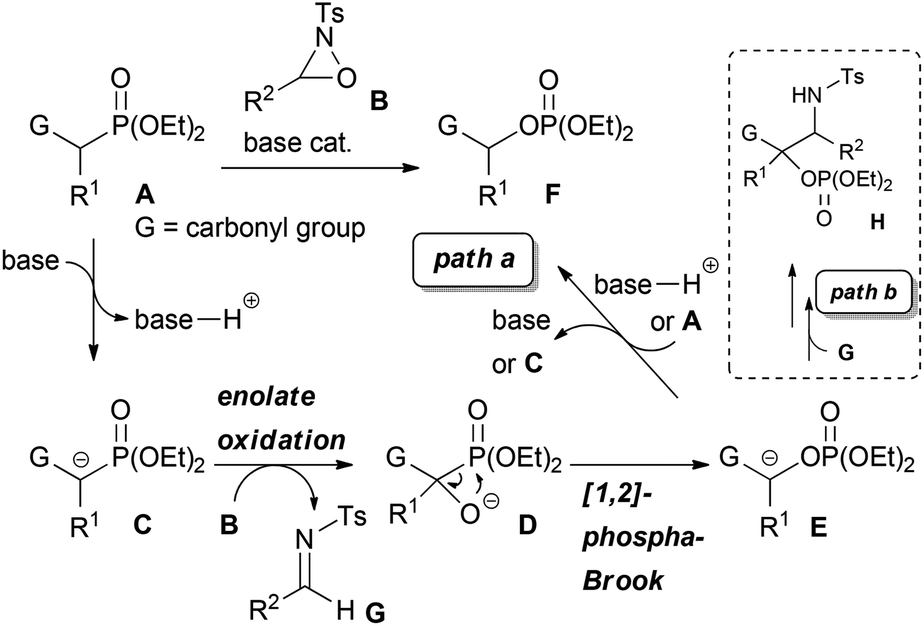 | ||
| Scheme 1 Proposed catalytic system. | ||
At first, deprotonation at the position α to the carbonyl group of substrate A, where the acidity of the proton is enhanced by the electron-withdrawing diethylphosphono group, by a Brønsted base occurs to generate enolate C. The following reaction of enolate C with oxaziridine B provides alkoxide D. Subsequently, the 1,2-rearrangement of the diethylphosphono group from carbon to oxygen, i.e. the [1,2]-phospha-Brook rearrangement, occurs to generate enolate E. Finally, protonation of enolate E by the conjugated acid of the Brønsted base or substrate A proceeds to afford product F along with the regeneration of the Brønsted base or enolate C. The key step of the catalytic reaction is the last step of the catalytic cycle, which involves the regeneration of the Brønsted base or the enolate of the substrate. In the conventional enolate oxidation of less acidic carbonyl compounds, this step is difficult because the acidity of the substrate is lower than that of the hydroxy proton of the product, and thus a stoichiometric amount of the Brønsted base is required. In contrast, the newly designed reaction system maintains the relatively high acidity of the substrate compared to that of the product because of the influence of the electron-withdrawing diethylphosphono group. As a consequence, protonation of enolate E occurs to regenerate the Brønsted base or enolate C, and the reaction can proceed catalytically. In the course of the reaction, a formal oxygen insertion into the carbon–phosphorus bond occurs under Brønsted base catalysis to yield α-oxygenated carbonyl compounds. We report herein the designed system, which is the tandem reaction of α-diethylphosphono ester derivatives with oxaziridines in the presence of a catalytic amount of the Brønsted base, providing α-oxygenated ester derivatives.
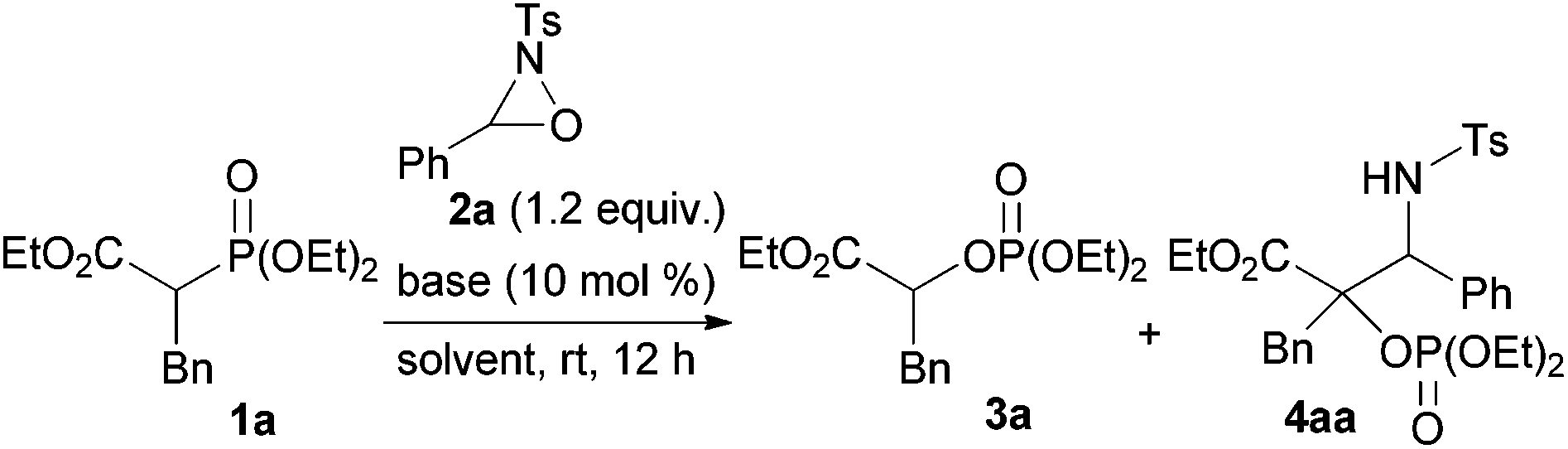
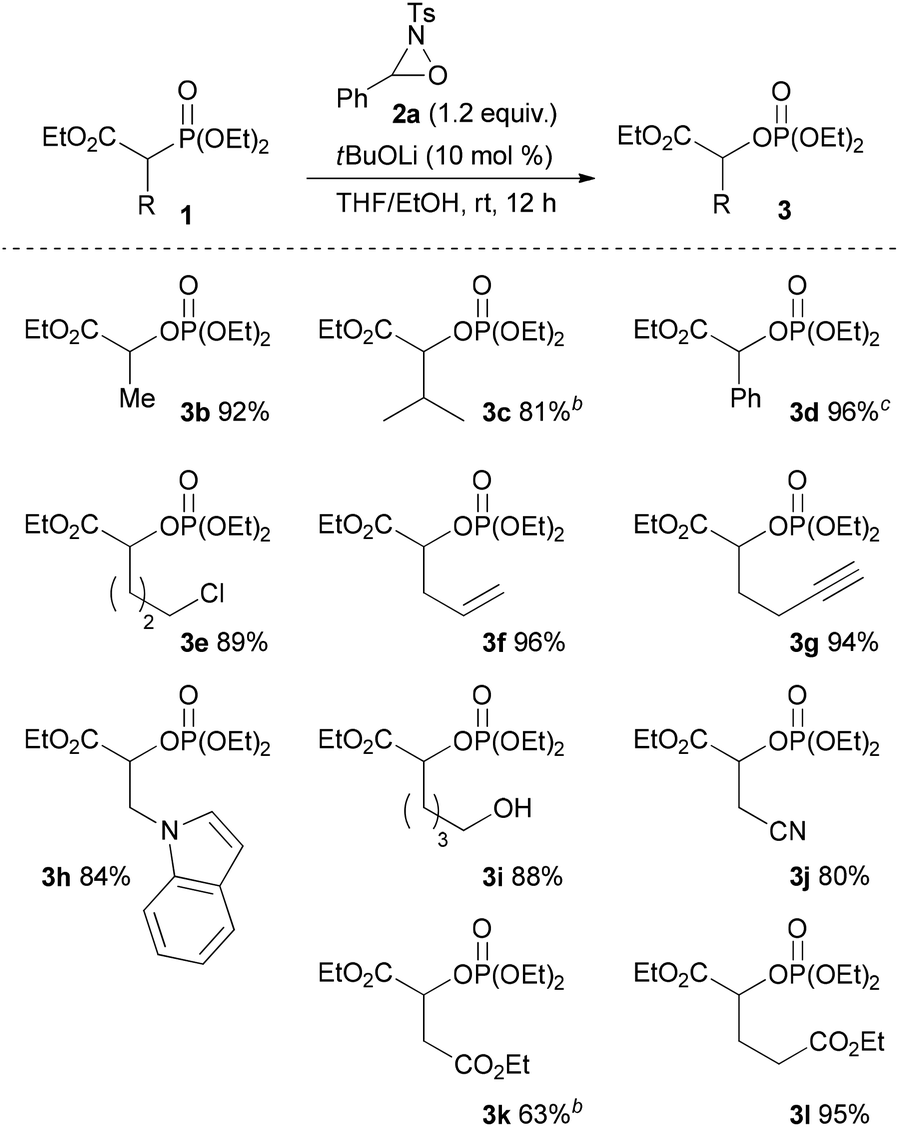
To ascertain the viability of the proposed tandem reaction system, we attempted the reaction of 1a as the primary substrate. An initial experiment was performed in which 1a was treated with oxaziridine 2a in the presence of 10 mol% tBuOK in THF. Gratifyingly, the desired product 3a was obtained in 60% yield along with the formation of 16% of 4aa as a diastereomixture (Table 1, entry 1). The formation of 4aa occurs through the addition of the enolate intermediate generated by the [1,2]-phospha-Brook rearrangement to tosyl imine derived from 2a in preference to the protonation, as shown in Scheme 1, path b. Screening for Brønsted bases revealed that 1a was quantitatively converted into a mixture of 3a and 4aa with an inorganic base, such as tBuONa or tBuOLi (entries 2 and 3), whereas inferior results were obtained with organic bases, such as TBD, P1-tBu, and P2-tBu (entries 4–6). Next, with tBuOLi as the Brønsted base, the solvent was screened for the selective formation of 3a (entries 7–13). As a result, 3a was exclusively formed in ethanol, albeit in moderate yield (entry 12). Finally, the use of a mixed solvent of THF and ethanol (4/1) was found to be optimal, and the desired product 3a was obtained in quantitative yield (entry 13).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Solvent | Yield of 3a (%)b | Yield of 4aa (%)b | dr of 4aac |
| a Reaction conditions: 1a (0.15 mmol), 2a (0.18 mmol), base (0.015 mmol), solvent (1.5 mL), rt, 12 h. b NMR yields. CHBr3 was used as the internal standard. The isolated yield is shown in the parenthesis. c Determined by 1H NMR analysis of the crude mixture. d A mixed solvent of THF (1.2 mL) and EtOH (0.30 mL) was used. | |||||
| 1 | tBuOK | THF | 60 | 16 | 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 :50 |
| 2 | tBuONa | THF | 73 | 24 | 59:41 |
| 3 | tBuOLi | THF | 49 | 50 | 64:36 |
| 4 | TBD | THF | 0 | 0 | |
| 5 | P1-tBu | THF | 0 | 0 | |
| 6 | P2-tBu | THF | 26 | 0 | |
| 7 | tBuOLi | Toluene | 45 | 55 | 64:36 |
| 8 | tBuOLi | 1,4-Dioxane | 58 | 42 | 61:39 |
| 9 | tBuOLi | CH3CN | 64 | 36 | 62:38 |
| 10 | tBuOLi | DMF | 30 | 41 | 73:27 |
| 11 | tBuOLi | DMSO | 0 | 0 | |
| 12 | tBuOLi | EtOH | 56 | 0 | |
| 13 | tBuOLi | THF/EtOHd | 99 (98) | 0 | |
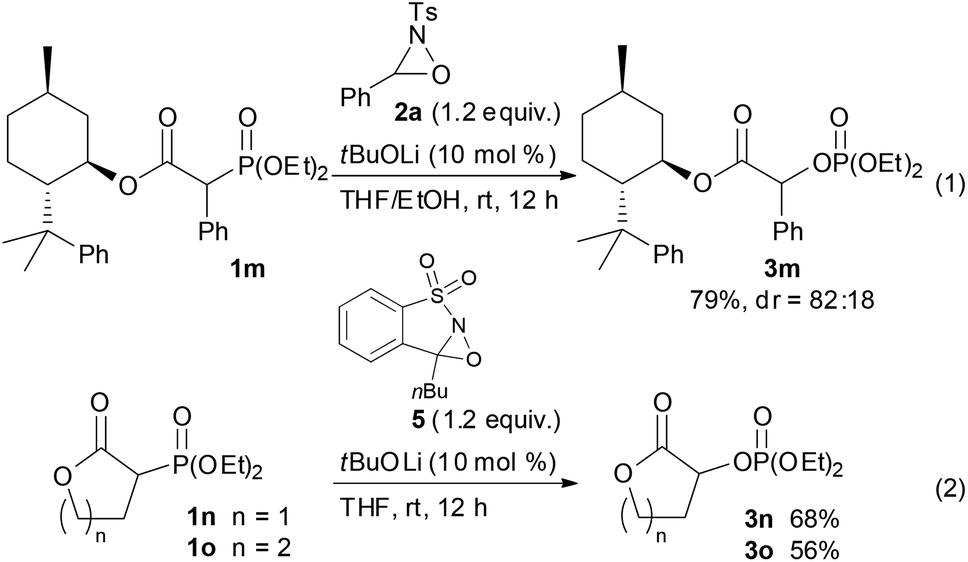
With the optimum conditions in hand, the scope of the substrates was investigated. At first, ethyl diethylphosphonoacetate derivatives possessing various substituents at the α position were subjected to the reaction conditions (Scheme 2). Not only alkyl-substituted substrates but also phenyl-substituted substrate 1d underwent the reaction to provide the corresponding product 3d in high yield. A variety of functional groups were compatible under the reaction conditions. In addition to a chloro group, the functional groups that were potentially oxidizable under oxidation conditions, such as alkene, alkyne, free hydroxy, and indole moieties, were intact under the reaction conditions to afford the corresponding products in high yields. Furthermore, it is noteworthy that the oxygenation of cyanoester 1j and diesters 1k and 1l selectively occurred at the position α to the diethylphosphono group to afford mono-oxygenated cyanoester 3j and diesters 3k and 3l. In the reaction of the diesters, the length of the alkyl chain between the two ethoxycarbonyl groups affected the reaction outcome; the reaction of the butanedioate derivative 1k was slow and 3k was obtained only in moderate yield. In contrast, the reaction of the pentanedioate derivative 1l proceeded smoothly to provide 3l in high yield. Next, a substrate possessing a chiral auxiliary was tested (eqn (1)). As a result, the corresponding α-oxygenated product 3m was obtained in good yield as an 82:18 mixture of diastereomers that were separable by column chromatography. The reaction of lactones 1n and 1o was then examined (eqn (2)). In those substrates, partial opening of the lactone ring by ethoxide was observed under the optimum reaction conditions, whereas ethanol was essential to suppress the formation of the overreaction product 4. Therefore, the reaction conditions were slightly changed. Specifically, oxaziridine 5 derived from a ketimine was employed instead of 2a derived from an aldimine in THF. Under the modified reaction conditions, the desired product was successfully obtained as a sole product. In order to expand the scope of the substrates, the α-oxygenation of amides and nitriles was investigated (Scheme 3). In both cases, the phenyl substituent at the α position was essential for occurrence of the reaction. The reaction of phenylacetamide derivative 6 required a higher catalyst loading to achieve acceptable yield. The reaction of phenylacetonitrile derivative 8 provided the corresponding product in quantitative yield by using 5 as the oxidant. In contrast, employment of alkyl-substituted acetamides and acetonitriles resulted in no reaction.
 | ||
| Scheme 2 Substrate scope.a aReaction conditions: 1 (0.10 mmol), 2a (0.12 mmol), base (0.010 mmol), solvent (1.0 mL), rt, 12 h. b20 mol% of tBuOLi was used. cTHF was used as the solvent. | ||
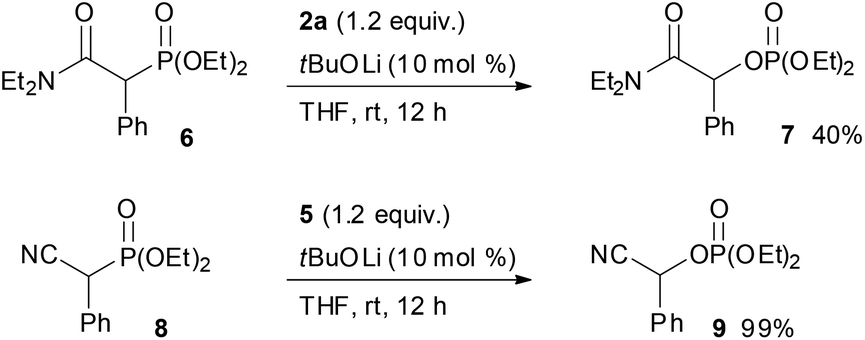 | ||
| Scheme 3 Reaction of amides and nitriles. | ||
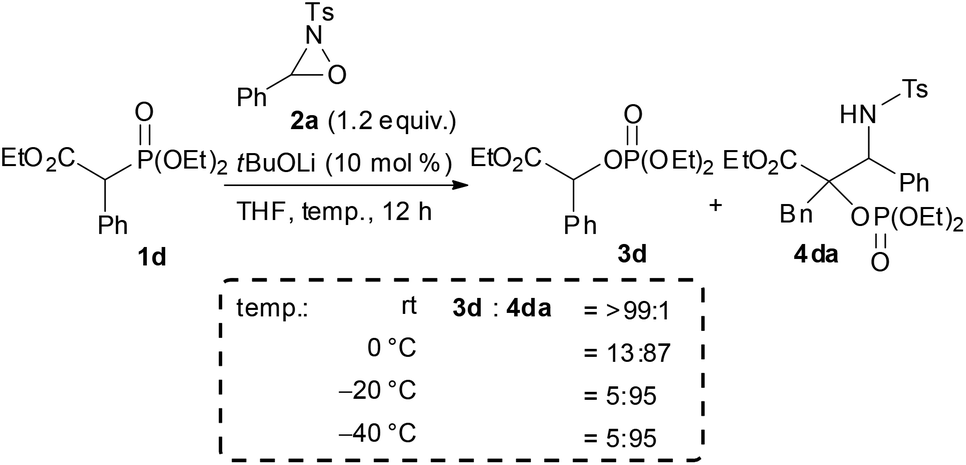
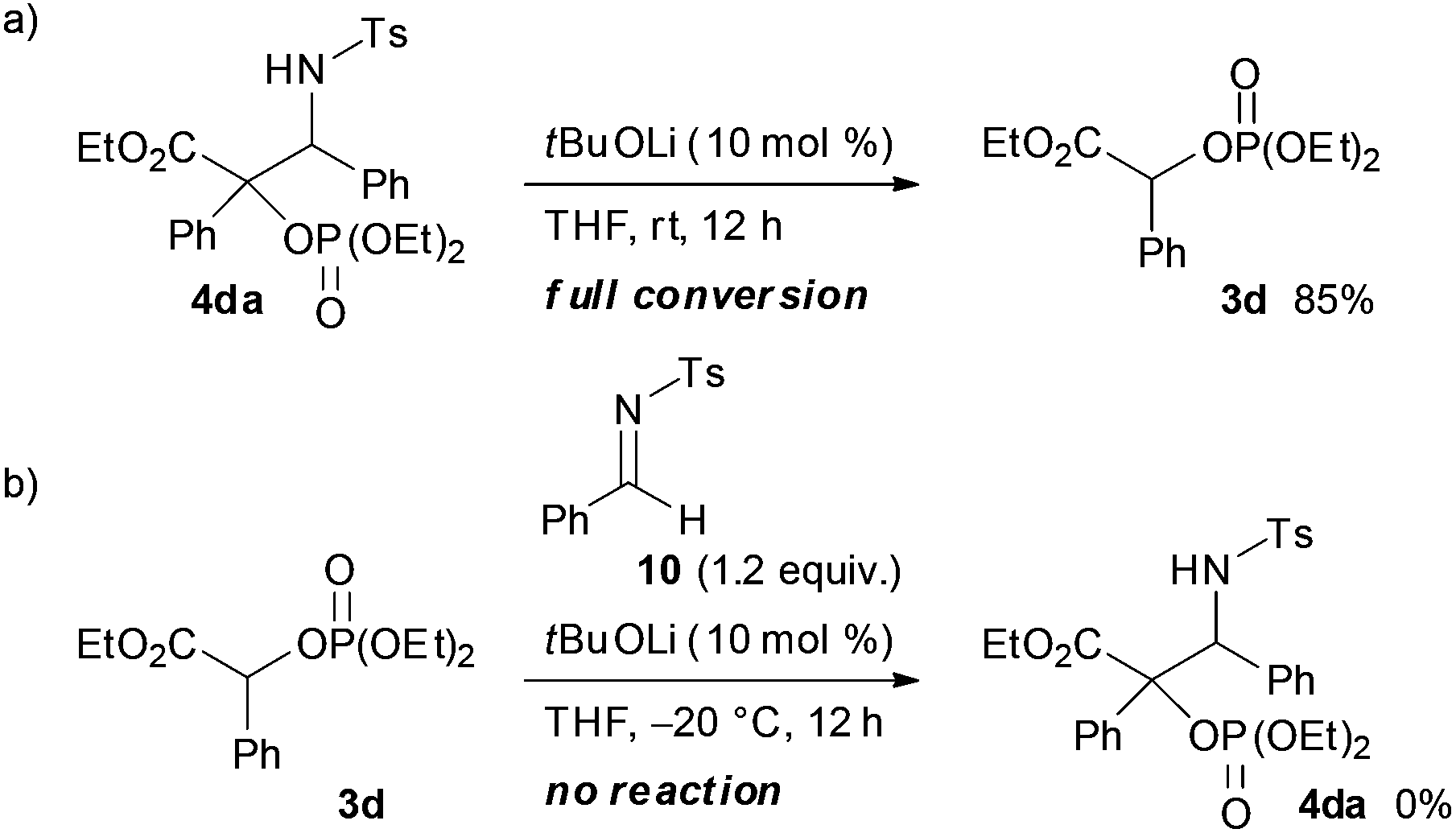
In the course of the investigation of this reaction, an interesting result was obtained (Scheme 4). Whereas the reaction of 1d with 2a in THF at room temperature afforded 3d as the sole product, 4da was formed as the major product (4da:3d = 87:13) when the reaction was performed at 0 °C. Further reducing the reaction temperature to −20 °C enhanced the selective formation of 4da (4da:3d = 95:5). In order to clarify the origin of this result, a control experiment was conducted (Scheme 5a). Treatment of 4da with a catalytic amount of tBuOLi in THF at room temperature resulted in the full conversion of 4da into 3d. This result clearly indicated that the retro-Mannich type reaction of 4da, which could be suppressed at low temperature, would occur at room temperature and thus 4da was not detected when the reaction was performed at that temperature. In addition, treatment of 3d with tosyl imine 10 in the presence of a catalytic amount of tBuOLi in THF at −20 °C resulted in no reaction (Scheme 5b). This result suggested that the formation of 4da occurred directly through the addition of the enolate generated by the [1,2]-phospha-Brook rearrangement (Scheme 1, path b), not by the deprotonation of 3d, in preference to the protonation providing 3d.
 | ||
| Scheme 4 Temperature effect in the reaction of 1d with 2a. | ||
 | ||
| Scheme 5 Control experiments. | ||
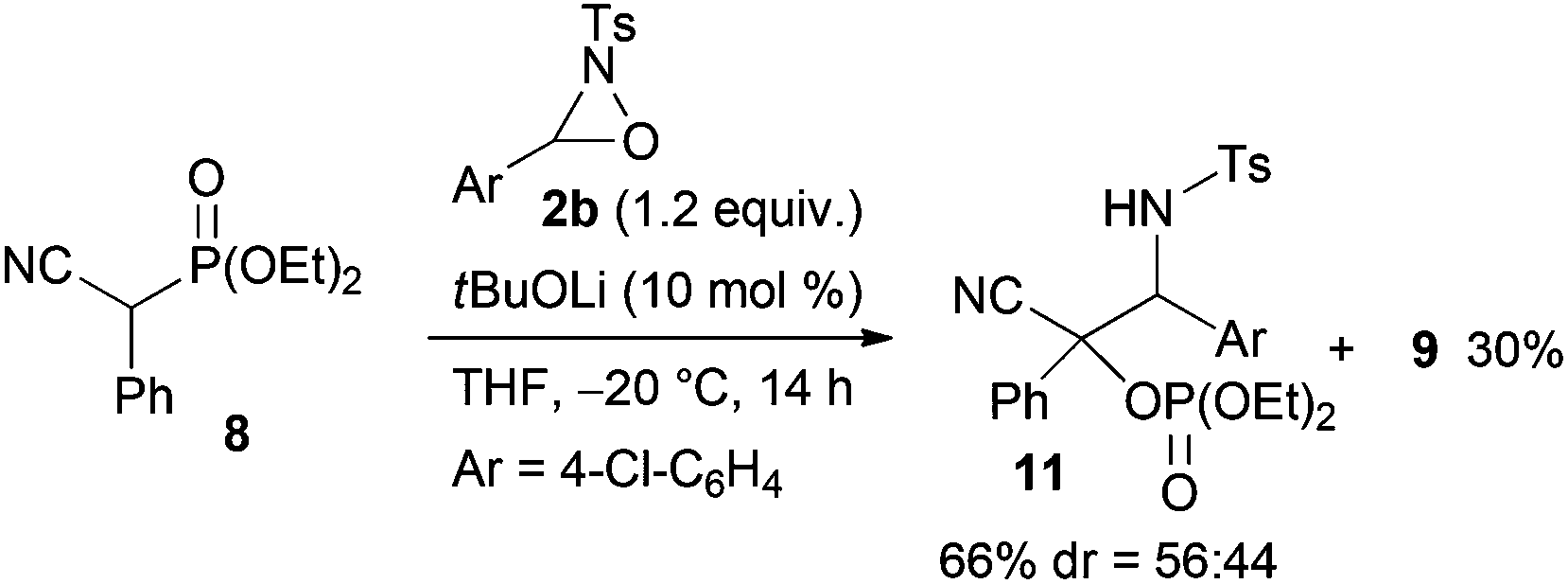
Next, the scope of the reaction under low temperature conditions, that is the atom-economical tandem reaction involving carbon–carbon bond formation in addition to α-oxygenation, was investigated (Table 2). Interestingly, the ester moiety of the substrate was found to influence both the reactivity and the diastereoselectivity significantly. Whereas the reaction of ethyl ester 1d with 2a provided 4da in 82% yield with 77:23 dr, the corresponding benzyl ester 1p afforded 4pa in 92% yield with 85:15 dr (entries 1 and 2).13 Oxaziridines 2 having a functional group on the aryl moiety, such as a methoxy, chloro, or nitro group, participated in the reaction, and the corresponding products were obtained in high yields although the diastereoselectivity was markedly dependent on the nature of the oxaziridines (entries 3–6). The reaction of 1a, which has an alkyl group at the α position, provided the desired products 4 in moderate yields along with the formation of significant amounts of protonated products 3 (entries 7 and 8). Lactone 1o and nitrile 8 also gave the desired products in moderate to good yields (entry 9 and eqn (3)).
 | (3) |
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | 1 | R1 | R2 | 2 | Ar | 4 | Yieldb (%) | drc |
4:3c |
| a Reaction conditions: 1 (0.10 mmol), 2 (0.12 mmol), tBuOLi (0.010 mmol), THF (1.0 mL), −20 °C, 14 h. b Determined by 1H NMR measurements after column chromatography. See the ESI for details. c Determined by 1H NMR analysis of the crude mixture. | |||||||||
| 1 | 1d | Et | Ph | 2a | Ph | 4da | 82 | 77:23 |
95:5 |
| 2 | 1p | Bn | Ph | 2a | Ph | 4pa | 92 | 85:15 |
95:5 |
| 3 | 1p | Bn | Ph | 2b | 4-Cl-C6H4 | 4pb | 95 | 78:22 |
97:3 |
| 4 | 1p | Bn | Ph | 2c | 2-Cl-C6H4 | 4pc | 95 | 81:19 |
97:3 |
| 5 | 1p | Bn | Ph | 2d | 4-NO2-C6H4 | 4pd | 86 | 62:38 |
>99:1 |
| 6 | 1p | Bn | Ph | 2e | 3-MeO-C6H4 | 4pe | 89 | 86:14 |
95:5 |
| 7 | 1a | Et | Bn | 2a | Ph | 4aa | 57 | 67:33 |
64:36 |
| 8 | 1a | Et | Bn | 2b | 4-Cl-C6H4 | 4ab | 59 | 63:37 |
70:30 |
| 9 | 1o | –(CH2)3– | 2b | 4-Cl-C6H4 | 4ob | 79 | 75:25 |
92:8 |
|
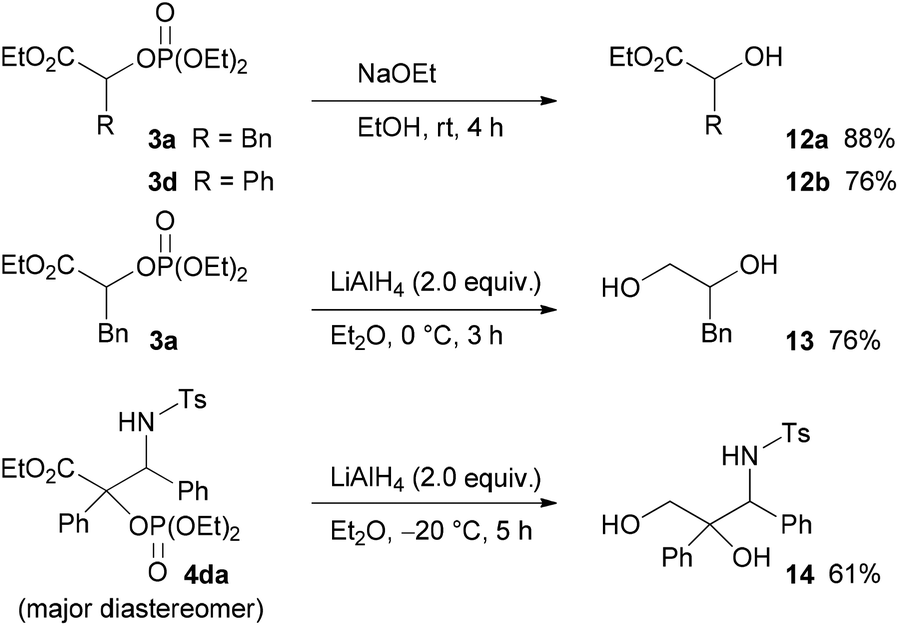
Finally, the diethoxyphosphoryloxy group of the product, which can be regarded as a protected hydroxy group, was converted into a hydroxy group (Scheme 6). For example, treatment of 3a and 3d with sodium ethoxide in ethanol provided the corresponding hydroxy esters 12a and 12b in good yields. In addition, the reduction of 3a and 4da with LiAlH4 afforded the corresponding diol 13 and 3-amino-1,2-diol 14 in good yields, respectively.
 | ||
| Scheme 6 Conversion of the diethoxyphosphoryloxy group into the hydroxy group. | ||
Conclusions
In conclusion, a novel α-oxygenation reaction of carbonyl compounds was developed by utilizing the [1,2]-phospha-Brook rearrangement. This operationally simple reaction under Brønsted base catalysis can furnish α-oxygenated ester derivatives from easily accessible substrates having high functional group tolerance. In addition, the atom-economical tandem reaction of α-diethylphosphono ester derivatives with oxaziridines, which involves carbon–carbon formation in addition to α-oxygenation, was also established under the low temperature conditions. Further investigation of this methodology, including an enantioselective reaction, is in progress.Acknowledgements
This research was partially supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Advanced Molecular Transformations by Organocatalysts” from MEXT (Japan) and a Grant-in-Aid for Scientific Research from the JSPS.Notes and references
- For selected reviews, see: (a) J. Christoffers, A. Baro and T. Werner, Adv. Synth. Catal., 2004, 346, 143–151 CrossRef CAS PubMed; (b) R. L. Webb, N. Schiering, R. Sedrani and J. Maibaum, J. Med. Chem., 2010, 53, 7490–7520 CrossRef CAS PubMed; (c) A. Armstrong and T. J. Blench, Tetrahedron, 2002, 58, 9321–9349 CrossRef CAS; (d) G. G. I. Kingston, Chem. Commun., 2001, 867–880 RSC; (e) Y.-Z. Shu, J. Nat. Prod., 1998, 61, 1053–1071 CrossRef CAS PubMed; (f) A. Nadin and K. C. Nicolaou, Angew. Chem., Int. Ed. Engl., 1996, 35, 1622–1656 CrossRef CAS PubMed.
- For reviews, see: (a) B.-C. Chen, P. Zhou, F. A. Davis and E. Ciganek, Org. React., 2003, 62, 1–356 CAS; (b) F. A. Davis and B.-C. Chen, Chem. Rev., 1992, 92, 919–934 CrossRef CAS.
- (a) N. Momiyama and H. Yamamoto, J. Am. Chem. Soc., 2003, 125, 6038–6039 CrossRef CAS PubMed; (b) N. Momiyama and H. Yamamoto, J. Am. Chem. Soc., 2004, 126, 5360–5361 CrossRef CAS PubMed.
- (a) P. J. Mabe and A. Zakarian, Org. Lett., 2014, 16, 516–519 CrossRef CAS PubMed; (b) A. Gómez-Palomino, M. Pellicena, J. M. Romo, R. Solá, P. Romea, F. Urpí and M. Font-Bardia, Chem. – Eur. J., 2014, 20, 10153–10159 CrossRef PubMed.
- (a) M. Pouliot, P. Renaud, K. Schenk, A. Studer and T. Vogler, Angew. Chem., Int. Ed., 2009, 48, 6037–6040 CrossRef CAS PubMed; (b) Y. Li, M. Pouliot, T. Vogler, P. Renaud and A. Studer, Org. Lett., 2012, 14, 4474–4477 CrossRef CAS PubMed.
- For reviews, see: (a) A. Russo, C. De Fiscp and A. Lattanzi, RSC Adv., 2012, 2, 385–397 RSC; (b) G. Guillena and D. J. Ramón, Tetrahedron: Asymmetry, 2006, 17, 1465–1492 CrossRef CAS PubMed; (c) A. M. R. Smith and K. K. Hii, Chem. Rev., 2011, 111, 1637–1656 CrossRef CAS PubMed.
- For selected examples of Brønsted base-catalyzed reactions, see: (a) T. Watanabe and T. Ishikawa, Tetrahedron Lett., 1999, 40, 7795–7798 CrossRef CAS; (b) M. R. Acocella, O. G. Mancheño, B. Bella and K. A. Jørgensen, J. Org. Chem., 2004, 69, 8165–8167 CrossRef CAS PubMed; (c) L. Zou, B. Wang, H. Mu, H. Zhang, Y. Song and J. Qu, Org. Lett., 2013, 15, 3106–3109 CrossRef CAS PubMed; (d) M. Odagi, K. Furukori, T. Watanabe and K. Nagasawa, Chem. – Eur. J., 2013, 19, 16740–16745 CrossRef CAS PubMed; (e) Z. Li, M. Lian, F. Yang, Q. Meng and Z. Gao, Eur. J. Org. Chem., 2014, 3491–3495 CrossRef CAS PubMed , and references therein.
- Y.-F. Liang and N. Jiao, Angew. Chem., Int. Ed., 2014, 53, 548–552 CrossRef CAS PubMed.
- (a) G. J. Chuang, W. Wang, E. Lee and T. Ritter, J. Am. Chem. Soc., 2011, 133, 1760–1762 CrossRef CAS PubMed; (b) O. A. Hamed, A. El-Qisairi, H. Qaseer, E. M. Hamed, P. M. Henry and D. P. Becker, Tetrahedron Lett., 2012, 53, 2699–2701 CrossRef CAS PubMed; (c) T. Arai, T. Sato, H. Noguchi, H. Kanoh, K. Kaneko and A. Yanagisawa, Chem. Lett., 2006, 35, 1094–1095 CrossRef CAS; (d) A. K. El-Qisairi and H. A. Qaseer, J. Organomet. Chem., 2002, 659, 50–55 CrossRef CAS.
- Y.-F. Liang, K. Wu, S. Song, X. Li, X. Huang and N. Jiao, Org. Lett., 2015, 17, 876–879 CrossRef CAS PubMed.
- For selected examples, see: (a) C. C. Bausch and J. S. Johnson, Adv. Synth. Catal., 2005, 347, 1207–1211 CrossRef CAS PubMed; (b) A. S. Demir, Ö. Reis, A. Ç. İğdir, İ. Esiringü and S. Eymur, J. Org. Chem., 2005, 70, 10584–10587 CrossRef CAS PubMed; (c) A. S. Demir, B. Reis, Ö. Reis, S. Eymür, M. Göllü, S. Tural and G. Saglam, J. Org. Chem., 2007, 72, 7439–7442 CrossRef CAS PubMed; (d) A. S. Demir, I. Esiringü, M. Göllü and Ö. Reis, J. Org. Chem., 2009, 74, 2197–2199 CrossRef CAS PubMed; (e) L. El Kaïm, L. Gaultier, L. Grimaud and A. Dos Santos, Synlett, 2005, 2335–2336 CrossRef PubMed; (f) M. Hayashi and S. Nakamura, Angew. Chem., Int. Ed., 2011, 50, 2249–2252 CrossRef CAS PubMed; (g) M. T. Corbett, D. Uraguchi, T. Ooi and J. S. Johnson, Angew. Chem., Int. Ed., 2012, 51, 4685–4689 CrossRef CAS PubMed.
- (a) A. Kondoh and M. Terada, Org. Lett., 2013, 15, 4568–4571 CrossRef CAS PubMed; (b) A. Kondoh, T. Aoki and M. Terada, Org. Lett., 2014, 16, 3528–3531 CrossRef CAS PubMed.
- The structure of the major diastereomer of 4pa was confirmed by single-crystal X-ray diffraction analysis (CCDC no. 1046578). See the ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data. CCDC 1046578. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00108k |
| This journal is © the Partner Organisations 2015 |