
Palladium-catalyzed alkylation of unactivated C(sp3)–H bonds with primary alkyl iodides at room temperature: facile synthesis of β-alkyl α-amino acids†
B.
Wang
,
X.
Wu
,
R.
Jiao
,
S.-Y.
Zhang
,
W. A.
Nack
,
G.
He
and
G.
Chen
*
Department of Chemistry, The Pennsylvania State University, University Park, Pennsylvania 16802, USA. E-mail: guc11@psu.edu; Fax: +1-814-863-5319
First published on 23rd July 2015
Abstract
An efficient protocol for the synthesis of β-alkyl α-amino acids via palladium-catalyzed β-C(sp3)–H alkylation of aminoquinoline-coupled phthaloyl alanine is developed. The new TFA-promoted reaction conditions provide excellent C–H alkylation reactivity with alkyl iodides bearing moderately electron-withdrawing groups at room temperature. A range of β-alkyl α-amino acid products, including some difficult to access by other means, can be quickly prepared from readily available primary alkyl iodides and alanine precursors.
Over the past decade, the synthetic methodology for forming C–C bonds via palladium-catalyzed directing group-controlled functionalization of unactivated C(sp3)–H bonds has been quickly advanced.1 Despite the realization of various prototypical transformations, Pd-catalyzed C(sp3)–H functionalization chemistry requires further development to become truly synthetically useful.2 While C(sp3)–H arylation chemistry has steadily matured, the development of C(sp3)–H alkylation reactions is more challenging, paralleling the metal-catalyzed cross-coupling reactions of alkyl groups.3 Compared with aryl coupling partners, the reactivity of alkyl coupling partners is strongly influenced by their sterics and electronics. The competing side reactions of alkyl coupling partners also often cause problems.
Among the choices of alkyl coupling precursors, alkyl halides are particularly attractive due to their high accessibility and versatility.4–11 In 2010, Daugulis first reported that the primary β-C(sp3)–H bonds of 8-aminoquinoline (AQ)-coupled propanamide could be alkylated with isobutyl iodide in moderate yield under palladium catalysis (eqn (1), Scheme 1).6b In 2012, we discovered that picolinamide-coupled alkylamines can undergo Pd-catalyzed γ-C(sp3)–H alkylation with primary alkyl halides with high efficiency in the presence of Ag2CO3 and the dibenzylphosphate additive (BnO)2PO2H (eqn (2)).7b Soon after this discovery, these phosphate-promoted reaction conditions were successfully applied by the Shi group8 and us7c in the Pd-catalyzed methylene β-C(sp3)–H alkylation of AQ-coupled amino acid substrates with primary alkyl halides (eqn (3)). However, despite these improvements, there are considerable limitations of these Pd-catalyzed AQ-directed C(sp3)–H alkylation reactions, especially regarding the scope of alkyl halides. While simple primary alkyl halides bearing less interfering substituents such as α-haloacetates worked well in previous reaction systems, many other alkyl halides carrying more transformable functional groups such as α-iodomethyl ketones (see 10 in Scheme 2) gave significantly lower yield. Furthermore, alkylation of the β methyl C–H bonds of substrates e.g. AQ-coupled phthaloyl alanine 1 with more reactive alkyl iodides such as MeI7b,c and α-haloacetates often formed undesired multi-alkylated side products. Herein, we report the development of an improved protocol for Pd-catalyzed β-C(sp3)–H alkylation of aminoquinoline (AQ)-coupled phthaloyl alanine with a range of primary alkyl iodides bearing moderately electron withdrawing groups at room temperature. Monoselective methyl C–H alkylation of phthaloyl alanine with readily available alkyl iodides offers a convenient method for the stereoselective synthesis of β-alkyl α-amino acids bearing novel and transformable side chains.
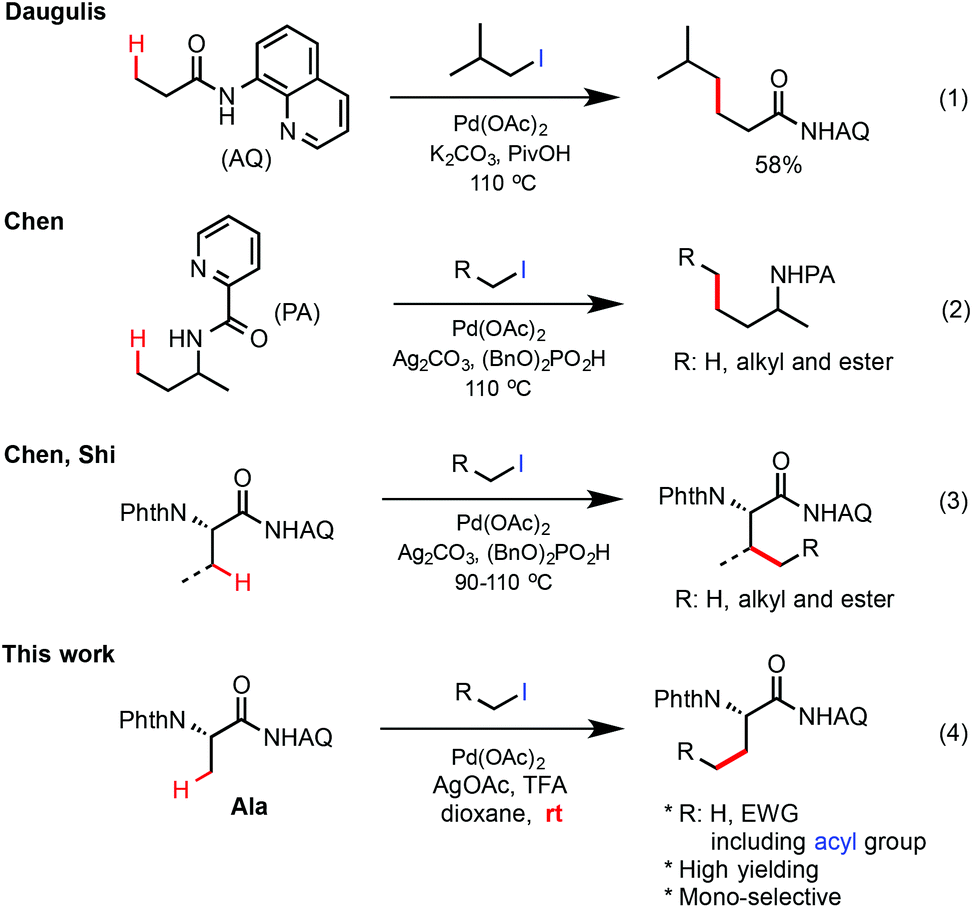 | ||
| Scheme 1 Pd-catalyzed C(sp3)–H alkylation with primary alkyl halides. | ||
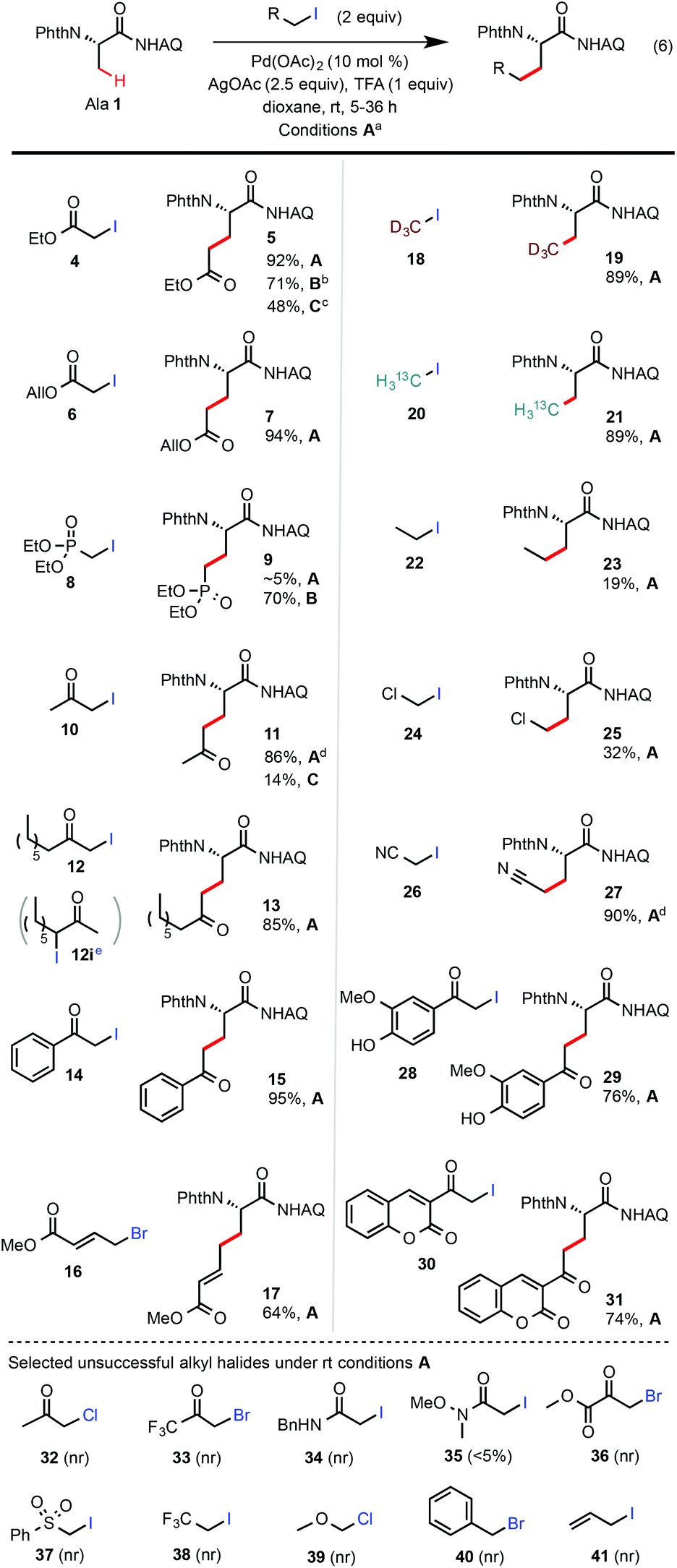 | ||
Scheme 2 Scope of alkyl iodides. (a) Isolated yields on a 0.2 mmol scale under general conditions A (see entry 14 of Table 1). (b) Conditions B: the same as A except at 70 °C. (c) Conditions C: Ag2CO3, (BnO)2PO2H, t-AmylOH, 110 °C, 24 h (see entry 1 of Table 1). (d) Isolated yields on a 3 mmol scale (1 gram of Ala 1). (e) A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.6 mixture of 12 and 12i prepared from the methyl ketone precursor was used as the starting material; no alkylation product with 12i was formed under the conditions A. :1.6 mixture of 12 and 12i prepared from the methyl ketone precursor was used as the starting material; no alkylation product with 12i was formed under the conditions A. | ||
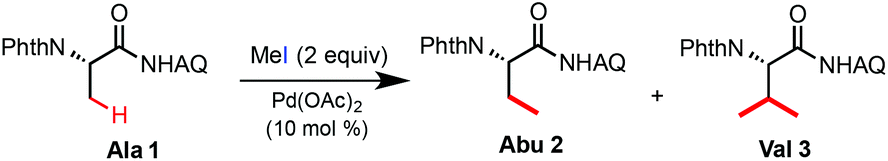
In a previous study, we discovered that AQ-coupled phthaloyl alanine 1 can readily undergo Pd-catalyzed C(sp3)–H alkylation with MeI at the β-methyl position in the presence of Ag2CO3 and (BnO)2PO2H at 110 °C (eqn (5)).7c However, the reaction of Ala 1 cannot be stopped at the mono-alkylation stage and gave β-dimethylated Val 3 as the major product (entry 1, Table 1). A considerable amount of Val 3 was formed with 1 equiv. of MeI at 110 °C or 2 equiv. of MeI at 60 °C (entries 2 and 3). Interestingly, the reaction can proceed in the MeI solvent at rt to give Abu 2 in low yield but high selectivity (entry 5). Recently, we discovered that the combination of the silver cation and trifluoroacetate (TFA) served as excellent promoters for Pd-catalyzed β-C(sp3)–H functionalization of Ala 1 with aryl iodides and vinyl iodides at rt.12 While the mechanism of the TFA-promoting effect is still unclear, we were delighted to find that the combined use of Ag2CO3 or AgOAc and TFA significantly improved the alkylation reactivity of Ala 1 with MeI at rt (entries 6–11). Importantly, the mono-alkylated product Abu 2 was obtained with excellent selectivity. Carboxylic acids such as AcOH provided little improvement in terms of reactivity or selectivity (entry 12). Finally, the reaction of Ala 1 with 2 equiv. of MeI, 2.5 equiv. of AgOAc and 1 equiv. of TFA in dioxane at rt for 24 h gave Abu 2 in 89% isolated yield at a 1 gram scale (entry 14).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Reagents (equiv.) | Solvents | t (°C)/time (h) | Yielda (%) | |
| 2 | 3 | ||||
| a Yields are based on 1H-NMR analysis on a 0.2 mmol scale under an ambient atmosphere. b 3 equiv. of MeI was used. c 1 equiv. of MeI was used. d Isolated yield on a 3 mmol scale (1 gram of Ala 1). e Product 2 was obtained in 96% ee based on chiral HPLC analysis. | |||||
| 1 | Ag2CO3 (2) (BnO)2PO2H (0.2)b | t-AmylOH | 110/24 | 5 | 92 |
| 2 | Ag2CO3 (2) (BnO)2PO2H (0.2)c | t-AmylOH | 110/24 | 42 | 15 |
| 3 | Ag2CO3 (2) (BnO)2PO2H (0.2) | t-AmylOH | 60/24 | 51 | 20 |
| 4 | Ag2CO3 (2) | t-AmylOH | 110/24 | 7 | 41 |
| 5 | Ag2CO3 (2) | MeI | rt/24 | 9 | <2 |
| 6 | Ag2CO3 (2) | Dioxane | rt/24 | <2 | <2 |
| 7 | AgOAc (2.5) | Dioxane | rt/24 | <2 | <2 |
| 8 | AgTFA (2) | Dioxane | rt/24 | 10 | <2 |
| 9 | Ag2CO3 (2), TFA (1) | Dioxane | rt/24 | 89 | 5 |
| 10 | Ag2CO3 (2), TFA (2) | Dioxane | rt/24 | 69 | 3 |
| 11 | Ag2CO3 (2), TFA (0.5) | Dioxane | rt/24 | 86 | 9 |
| 12 | Ag2CO3 (2), AcOH (1) | Dioxane | rt/24 | 8 | 7 |
| 13 | Ag2CO3 (2), TfOH (1) | Dioxane | rt/24 | 51 | 10 |
| 14 | AgOAc (2.5), TFA (1) | Dioxane | rt/24 | 91(89)d,e | 5 |
Next, we proceeded to evaluate the scope of primary alkyl iodides amenable to β-C–H alkylation of Ala 1 under the general rt reaction conditions A (Scheme 2). α-Iodoacetates bearing various ester appendants gave the corresponding glutamate products in excellent yields (>90%) and monoselectivity. The allyl group of Glu 7 was completely conserved. In comparison, the reaction of Ala 1 with ethyl iodoacetate 4 under our previous (BnO)2PO2H-promoted conditions at 110 °C (conditions C) gave Glu 5 in <50% yield along with other unidentified side products. In contrast to α-iodoacetates, α-iodoacetamide 34 shows little reactivity probably due to its lower electrophilicity. In addition to α-iodoacetates, we were very pleased to find that reactions of α-iodomethyl ketones, e.g.10, 12, 14, 28, and 30, proceeded cleanly to give the corresponding δ-ketocarboxamide products in good to excellent yields. It is worth noting that the reactions of these α-iodomethyl ketones under our previous conditions C at 110 °C gave only a small amount of the desired products (see 11) due to the competing esterification side reactions caused by their high electrophilicity. It should also be noted that secondary alkyl iodide 12i, a regioisomer of 12, was completely unreactive under the rt conditions A.13 Compared with MeI, the reactions of Ala 1 with sterically bulkier alkyl iodides such as EtI gave much lower yield at rt (see 23). Iodoacetonitrile 26 worked very well to give the nitrile product 27 in excellent yield. Overall, sterically unhindered primary alkyl iodides bearing moderately electron-withdrawing substituents performed well under the standard rt conditions.14
 | (9) |
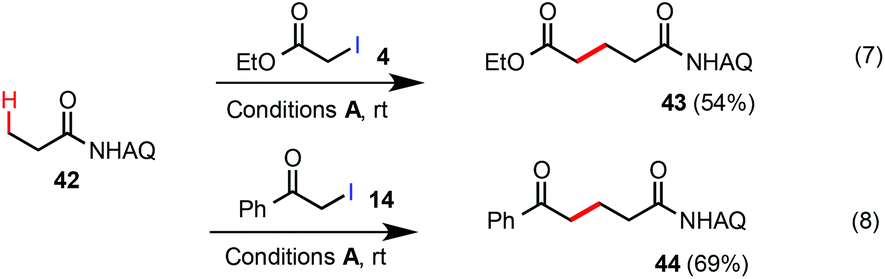
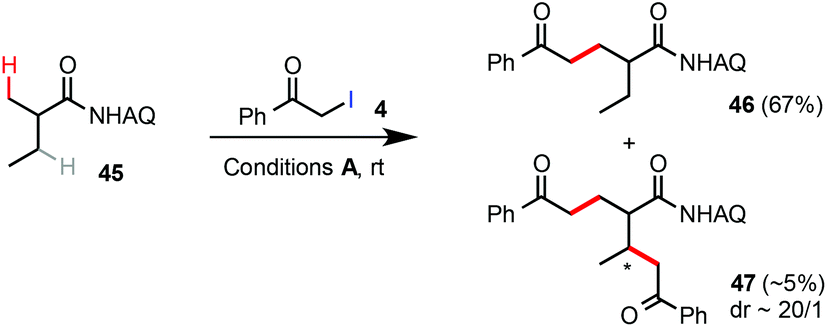
Unsubstituted propanamide 42 was less reactive than Ala 1 under the standard rt C–H alkylation conditions. Alkylation of 42 with α-iodoacetate 4 gave the product 43 in 54% yield along with 40% unreacted 42 (eqn (7)). Alkylation of 42 with α-iodomethylphenylketone 14 proceeded in higher conversion and gave compound 44 in 69% yield (eqn (8)). As shown in eqn (9), compound 45 was selectively alkylated at the β-methyl position to give 46 in 67% yield along with a trace amount of the dialkylated side product 47. While the substrate scope and reactivity still need further improvement, these Pd-catalyzed β-C(sp3)–H alkylation reactions of easily accessible carboxamides with α-iodoacetates or ketones at rt could offer an attractive strategy to construct 5-ketoacid and 1,5-diacid scaffolds, privileged structural motifs in organic synthesis, in an unprecedented fashion.15
As shown in Scheme 3A, compound 11 bearing a ketone group underwent efficient addition reaction with benzyloxyamine to form the oxime product 48 in excellent yield at rt.16 The nitrile group of compound 27 can be cleanly hydrolyzed with conc. H2SO4 to give the glutamine product 49 at rt. The AQ group of compounds 9, 21 and 27 can be readily removed to give the corresponding β-alkyl α-amino acids bearing phosphonate,1713C-labeled methyl,18 and nitrile groups respectively under mild conditions using our previously reported Boc activation and amide cleavage procedure (Scheme 3B).12a
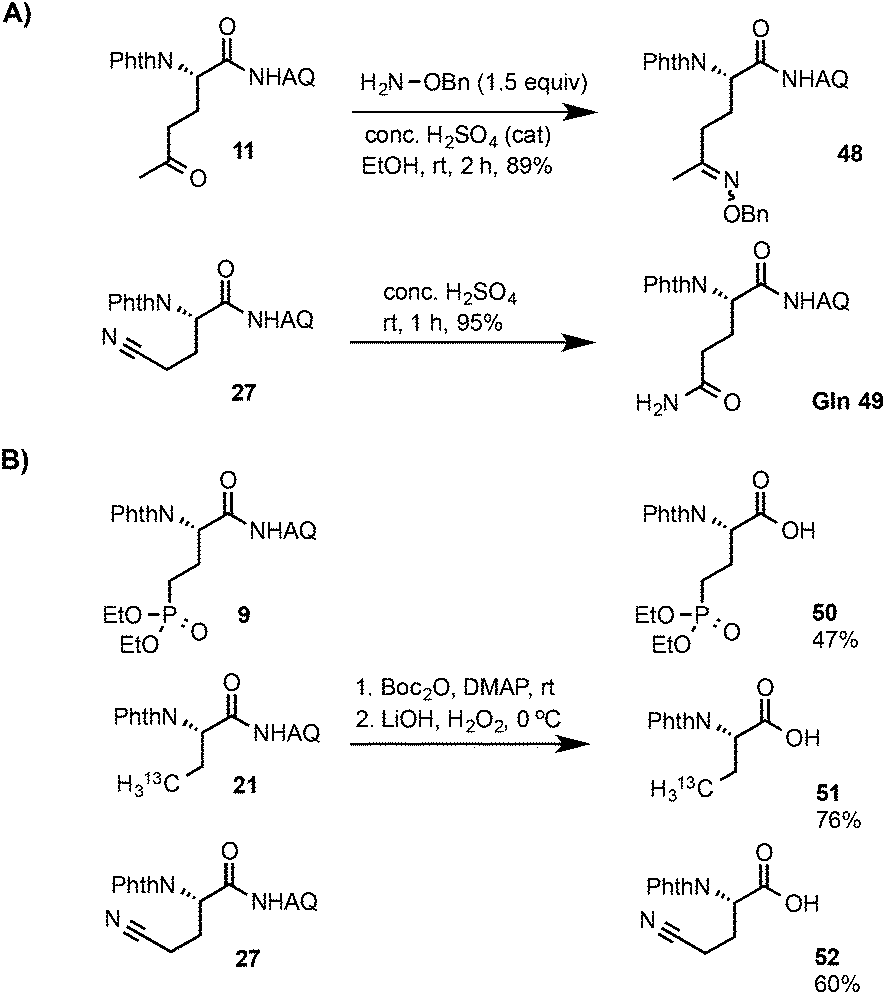 | ||
| Scheme 3 Further transformations. | ||
The alkylation reaction of Ala 1 likely begins with AQ-directed C–H palladation, forming a 5-membered palladacycle intermediate (see 54, Scheme 4). A primary KIE (kH/D ∼ 2.5 based on parallel experiments) was observed for the C–H alkylation of Ala 1 with α-iodoacetate 4 at rt, suggesting C–H palladation of 1 as the rate-limiting step.19
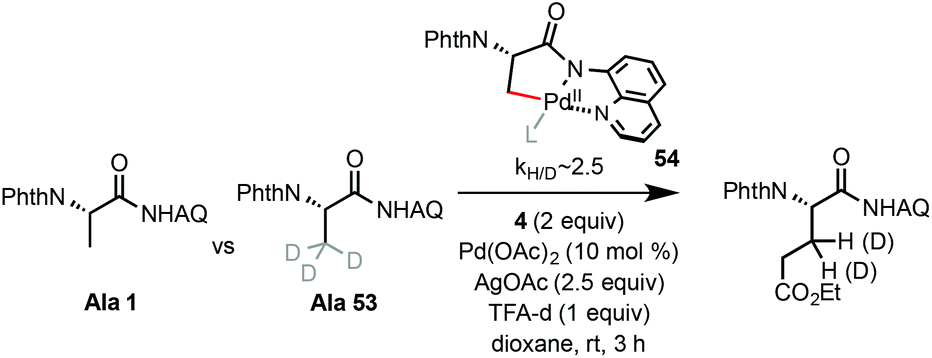 | ||
| Scheme 4 KIE experiments. | ||
In summary, we have developed an improved protocol for the synthesis of β-alkyl α-amino acids via palladium-catalyzed β-C(sp3)–H alkylation of aminoquinoline-coupled phthaloyl alanine. Compared with previous methods, TFA-promoted conditions provide excellent reactivity with alkyl iodides bearing moderately electron-withdrawing groups at room temperature. Critically, mild reaction conditions prevent competing esterification reactions of alkyl iodide electrophiles and di-alkylation, providing higher coupling yield and monoselectivity. A range of β-alkyl α-amino acid products, including some difficult to access using the previous conditions or other methods, can be quickly prepared from readily available primary alkyl iodides and alanine precursors.
We gratefully thank the Pennsylvania State University and NSF (CAREER CHE-1055795) for financial support of this work.
Notes and references
- For selected reviews on Pd-catalyzed C(sp3)–H functionalization: (a) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed; (b) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (c) R. Jazzar, J. Hitce, A. Renaudat, J. Sofack-Kreutzer and O. Baudoin, Chem. – Eur. J., 2010, 16, 2654 CrossRef CAS PubMed.
- For selected reviews on C(sp3)–H for synthesis of complex molecules. (a) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS PubMed; (b) H. M. Davies and J. R. Manning, Nature, 2008, 451, 417 CrossRef CAS PubMed; (c) W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976 RSC; (d) L. McMurray, F. O'Hara and M. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (e) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed.
- For selected reviews on metal-catalyzed C–H alkylations: (a) L. Ackermann, Chem. Commun., 2010, 46, 4866 RSC; (b) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814 CrossRef CAS PubMed.
- For selected reviews on metal-catalyzed cross coupling of alkyl halides: (a) A. C. Frisch and M. Beller, Angew. Chem., Int. Ed., 2005, 44, 674 CrossRef CAS PubMed; (b) A. Rudolph and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 2656 CrossRef CAS PubMed; (c) M. R. Netherton and G. C. Fu, Top. Organomet. Chem., 2005, 14, 85 CAS.
- For selected examples of Pd-catalyzed C–H alkylations with alkyl halides: (a) S. J. Tremont and H. U. Rahmen, J. Am. Chem. Soc., 1984, 106, 5759 CrossRef CAS; (b) M. Catellani, F. Frignani and A. Rangoni, Angew. Chem., Int. Ed. Engl., 1997, 36, 119 CrossRef CAS PubMed; (c) C. Bressy, D. Alberico and M. Lautens, J. Am. Chem. Soc., 2005, 127, 13148 CrossRef CAS PubMed; (d) Y.-H. Zhang, B.-F. Shi and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 609 Search PubMed; (e) D. Lapointe and K. Fagnou, Org. Lett., 2009, 11, 4160 CrossRef CAS PubMed; (f) L. Jiao and T. Bach, J. Am. Chem. Soc., 2011, 133, 12990 CrossRef CAS PubMed.
- (a) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS PubMed; (b) D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965 CrossRef CAS PubMed; (c) L. D. Tran and O. Daugulis, Angew. Chem., Int. Ed., 2012, 51, 5188 CrossRef CAS PubMed; (d) E. T. Nadres, G. I. F. Santos, D. Shabashov and O. Daugulis, J. Org. Chem., 2013, 78, 9689 CrossRef CAS PubMed.
- (a) Y. Zhao and G. Chen, Org. Lett., 2011, 13, 4850 CrossRef CAS PubMed; (b) S.-Y. Zhang, G. He, W. A. Nack, Y.-S. Zhao, Q. Li and G. Chen, J. Am. Chem. Soc., 2013, 135, 2124 CrossRef CAS PubMed; (c) S.-Y. Zhang, Q. Li, G. He, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2013, 135, 12135 CrossRef CAS PubMed; (d) G. He, S.-Y. Zhang, W. A. Nack, Q. Li and G. Chen, Angew. Chem., Int. Ed., 2013, 52, 11124 CrossRef CAS PubMed; (e) S.-Y. Zhang, Q. Li, G. He, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2015, 137, 531 CrossRef CAS PubMed.
- (a) K. Chen, F. Hu, S.-Q. Zhang and B.-F. Shi, Chem. Sci., 2013, 4, 3906 RSC; (b) K. Chen and B.-F. Shi, Angew. Chem., Int. Ed., 2014, 53, 11950 CrossRef CAS PubMed.
- For reports on AQ-directed Fe-catalyzed C–H alkylation with alkyl halides: (a) S. Asako, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2013, 135, 17755 CrossRef CAS PubMed; (b) L. Ilies, T. Matsubara, S. Ichikawa, S. Asako and E. Nakamura, J. Am. Chem. Soc., 2014, 136, 13126 CrossRef CAS PubMed; (c) E. R. Fruchey, B. M. Monks and S. P. Cook, J. Am. Chem. Soc., 2014, 136, 13130 CrossRef CAS PubMed.
- For reports on AQ-directed Ni-catalyzed C–H alkylation with alkyl halides: (a) Y. Aihara and N. Chatani, J. Am. Chem. Soc., 2013, 135, 5308 CrossRef CAS PubMed; (b) X. Wu, Y. Zhao and H. Ge, J. Am. Chem. Soc., 2014, 136, 1789 CrossRef CAS PubMed; (c) W. Song, S. Lackner and L. Ackermann, Angew. Chem., Int. Ed., 2014, 53, 2477 CrossRef CAS PubMed.
- G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed.
- (a) B. Wang, W. A. Nack, G. He, S.-Y. Zhang and G. Chen, Chem. Sci., 2014, 5, 3592 Search PubMed; (b) B. Wang, C. Lu, S.-Y. Zhang, G. He, W. A. Nack and G. Chen, Org. Lett., 2014, 16, 6260 CrossRef CAS PubMed.
- I2-mediated iodination of the corresponding methylketone precursor provided an inseparable mixture of 12 and 12i, see the ESI† for details. No secondary alkyl halides have been successfully applied in a reported Pd-catalyzed C(sp3)–H alkylation system.
- Selected unsuccessful alkyl halides in this rt reaction system were listed at the bottom of Scheme 2.
- Michael addition of soft enolate precursors to α,β-unsaturated carbonyl compounds has been commonly used to construct 1,5-dicarbonyl motifs.
- L. K. Mahal, K. J. Yarema and C. R. Bertozzi, Science, 1997, 276, 1125 CrossRef CAS . αAAs bearing a ketone functional group are valuable tools for orthogonal labelling of peptides in chemical biology.
- Removal of the Phth group of 50 would give L-2-amino-4-phosphonobutyric acid (L-AP4), a highly potent group III mGluR agonist. C. Selvam, C. Goudet, N. Oueslati, J.-P. Pin and F. Acher, J. Med. Chem., 2007, 50, 4656 CrossRef CAS PubMed.
- For a related preparation of a Val product bearing two γ-13C-labeled methyl groups, see ref. 7c.
- In comparison, a small KIE (1.2) was observed for Pd-catalyzed TFA-promoted C–H arylation of Ala 1 with aryl iodide at rt, see ref. 12a.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00112a |
| This journal is © the Partner Organisations 2015 |