
Palladium-catalyzed Br/D exchange of arenes: selective deuterium incorporation with versatile functional group tolerance and high efficiency†
Hong-Hai
Zhang
,
Peter V.
Bonnesen
and
Kunlun
Hong
*
Center for Nanophase Materials Sciences, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA. E-mail: hongkq@ornl.gov
First published on 13th July 2015
Abstract
A facile method for introducing one or more deuterium atoms onto an aromatic nucleus via Br/D exchange with high functional group tolerance and high incorporation efficiency is disclosed. Deuterium-labeled aryl chlorides and aryl borates which could be used as substrates in cross-coupling reactions to construct more complicated deuterium-labeled compounds can also be synthesized by this method.
Selectively–deuterated compounds have been increasingly attracting interest in such areas as drug discovery and development, in vitro and in vivo metabolic and pharmacokinetic probes, and advanced functional materials.1–3 Deuteration is also a powerful method in the study of structure and dynamics in soft matters through neutron scattering.3a,b The synthesis of selectively deuterium-labeled compounds often requires multiple steps sometimes under harsh conditions, such as strong acid or base, and high temperature and/or high pressure, particularly in H/D and Br/D exchange reactions.4,5 Accordingly, achieving high atom% deuterium incorporation in a manner that is tolerant to different functional groups has been a major challenge. Therefore, development of general methods to efficiently introduce deuterium at selected positions with versatile functional group tolerance is highly desirable.
We are interested in exploring the deuteron effect on the properties of materials through development of selectively deuterium-labeled compounds.6 Deuteration can be achieved either through reactions involving deuterium gas (D2), or with deuterium cation (D+) or deuterium anion (deuteride, D−). Although introduction of deuterium via D+ and D2 has been well developed,4,5 the deuterium anion D− was rarely employed in deuteration reactions other than the reduction of carbonyl groups.7 It is well known that in order to obtain highly efficient deuterium incorporation, a large excess of D2 or D+ is often required to overcome the influence of active hydrogen generated from the substrate, solvent, catalyst, or the reaction. In contrast, high atom% deuterium incorporation could be achieved in the reduction of carbonyl groups using equimolar amounts of D− as deuterium source.7 Based on the consideration that a deuterium anion reagent such as sodium formate-d is compatible with a wide-array of functional groups,8 we envisioned that such weak D− source could be a mild reagent to introduce deuterium with high functional group tolerance. Herein we report the results of our investigation into palladium catalyzed Br/D exchange reactions using deuterium anion as deuterium source to produce a wide variety of deuterium-labeled compounds.
Our study began with the Br/D exchange reaction with palladium acetate as the catalyst and sodium formate-d (D-COONa) as the deuterium source. We found the deuterium-labeled product formed with 20% conversion and 94% deuterium incorporation, which indicated most of the deuterium involved in the reaction was from DCOONa (entry 1 in Table 1). Based on the consideration that DCOONa is the only deuterium source in this reaction system and the reduced deuterium incorporation could be caused by the hydrogen from the catalyst, ligand or solvent, several palladium catalyst and phosphine ligand combinations were examined to determine the effect on conversion and deuterium incorporation (entries 1–6 in Table 1). We found the combination of tris(dibenzylideneacetone)dipalladium(0) (Pd2(dba)3)/t-Bu3P especially effective, with 100% conversion and 94% deuterium incorporation (entry 5). Since the deuterium-labeled compounds and related non-deuterium-labeled compounds are very difficult to separate, further improvement in the atom percent level of deuterium incorporation was still necessary. The similar level of deuterium incorporation obtained from different combinations of palladium catalyst and ligands also indicated that the palladium catalyst and ligands were not the reason for less than full deuterium incorporation (entries 1–6). Therefore, we next examined the effect of different solvents (entries 7–10) for the Pd2(dba)3/t-Bu3P combination. As shown in Table 1, dioxane and THF (entries 8 and 9) were inefficient with no conversion, probably due to the poor solubility of DCOONa. DMSO (entry 10) gave the best result with 100% conversion and >98% deuterium incorporation. Entry 11 shows the importance of the t-Bu3P ligand, as Pd2(dba)3 alone only provided 25% conversion. We next varied the reagent conditions for the deuterium source, and found that in situ generated DCOOK or DCOONa (entries 12 and 13) were also good deuterium reagents for this reaction. Other common deuterium sources such as CD3OD, D2O (entries 14–15) afforded no conversion. These results indicated that a strong polar aprotic solvent, such as DMSO, plays a key role in these reactions. A key finding was that the acid form DCOOD (entry 16) also afforded no conversion, highlighting the importance of the salt form (DCOOM) of the deuterated formate as the D-source for this Br/D exchange reaction (see mechanism discussion below).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | “Pd” catalyst | Temp (°C) | Solvent | “D-source” | Conv.a | D incorporationb |
| a Coversion was determined by GC/MS or 1H NMR. b The deuterium incorporation was determined by GC/MS or 1H NMR. c 2 equiv. K3PO4 were used. d 2 equiv. Na2CO3 were used. | ||||||
| 1 | Pd(OAc)2 | 80 | DMF | DCOONa | 20 | 94 |
| 2 | Pd(OAc)2 + XPhos | 80 | DMF | DCOONa | 38 | 94 |
| 3 | Pd(OAc)2 + S-Phos | 80 | DMF | DCOONa | 50 | 94 |
| 4 | Pd(OAc)2 + t-Bu3P | 80 | DMF | DCOONa | 50 | 94 |
| 5 | Pd2(dba)3 + t-Bu3P | 80 | DMF | DCOONa | 100 | 94 |
| 6 | Pd2(dba)3 + S-Phos | 80 | DMF | DCOONa | 80 | 93 |
| 7 | Pd2(dba)3 + t-Bu3P | 80 | DMA | DCOONa | 100 | 88 |
| 8 | Pd2(dba)3 + t-Bu3P | 80 | Dioxane | DCOONa | 0 | — |
| 9 | Pd2(dba)3 + t-Bu3P | 80 | THF | DCOONa | 0 | — |
| 10 | Pd2(dba)3 + t-Bu3P | 80 | DMSO | DCOONa | 100 | 98 |
| 11 | Pd2(dba)3 | 80 | DMSO | DCOONa | 25 | 98 |
| 12c | Pd2(dba)3 + t-Bu3P | 80 | DMSO | DCOOD | 100 | 98 |
| 13d | Pd2(dba)3 + t-Bu3P | 80 | DMSO | DCOOD | 100 | 98 |
| 14 | Pd2(dba)3 + t-Bu3P | 80 | DMSO | CD3OD | 0 | — |
| 15 | Pd2(dba)3 + t-Bu3P | 80 | DMSO | D2O | 0 | — |
| 16 | Pd2(dba)3 + t-Bu3P | 80 | DMSO | DCOOD | 0 | — |
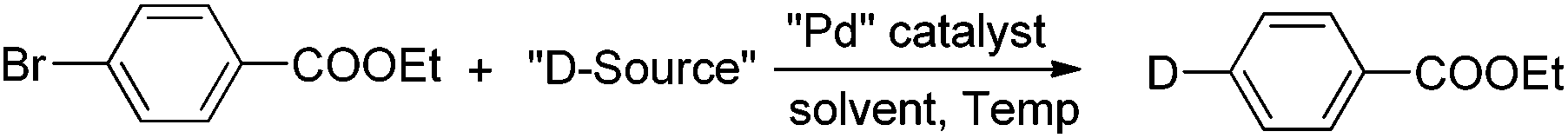
With the optimized condition of Pd2(dba)3/t-Bu3P with DCOOM in DMSO (entries 10, 12–13 in Table 1) in hand, several aryl bromide compounds with a variety of different functional groups were examined for this Br/D exchange reaction. As shown in Table 2, we found that aryl bromide compounds containing ester, ketone, nitrile, nitro, amine, aldehyde, alkene, or sulfonamide functional groups successfully afforded the deuterium-labeled products in good yields without degradation of the functional group. Several aryl bromide compounds containing active hydrogen (e.g., hydroxyl) were also suitable substrates, when 3 equivalents of DCOONa were used (2h, 2j, 2k). Usually, high atom% deuterium incorporation for these types of substrates could only be achieved by using a large excess of deuterium reagent or extremely strong base.9 The active hydrogen in the substrate did not affect the deuterium incorporation and the deuterium anion did not react with active hydrogen (such as 2h, 2j, 2k), which means DCOONa is a very mild deuterium source.4i,10 It is noteworthy to mention that functional groups such as alkene, aldehyde, and nitro which are sensitive to reducing conditions also showed high stability with the optimized reaction conditions. When other deuteration methods are employed,4,5 these types of deuterium-labeled compounds would require several protection/deprotection steps to prepare. Several heterocyclic bromide compounds were also examined and they also afforded the deuterium-labeled compounds in good yields. Under the optimized reaction conditions all the functionalized deuterium-labeled compounds were obtained with >98% deuterium incorporation using only 2–3 equivalents of sodium formate-d as deuterium reagent.
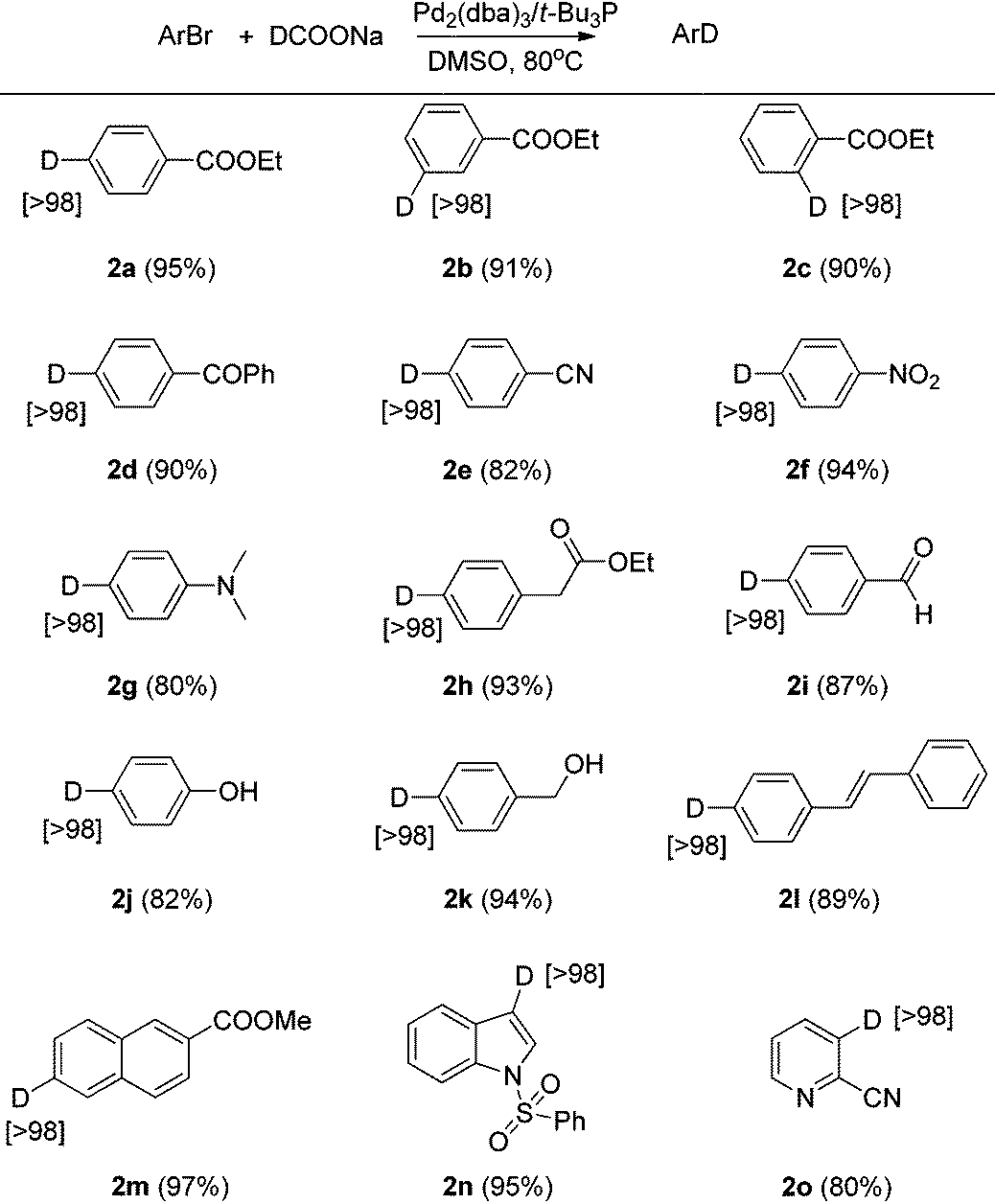
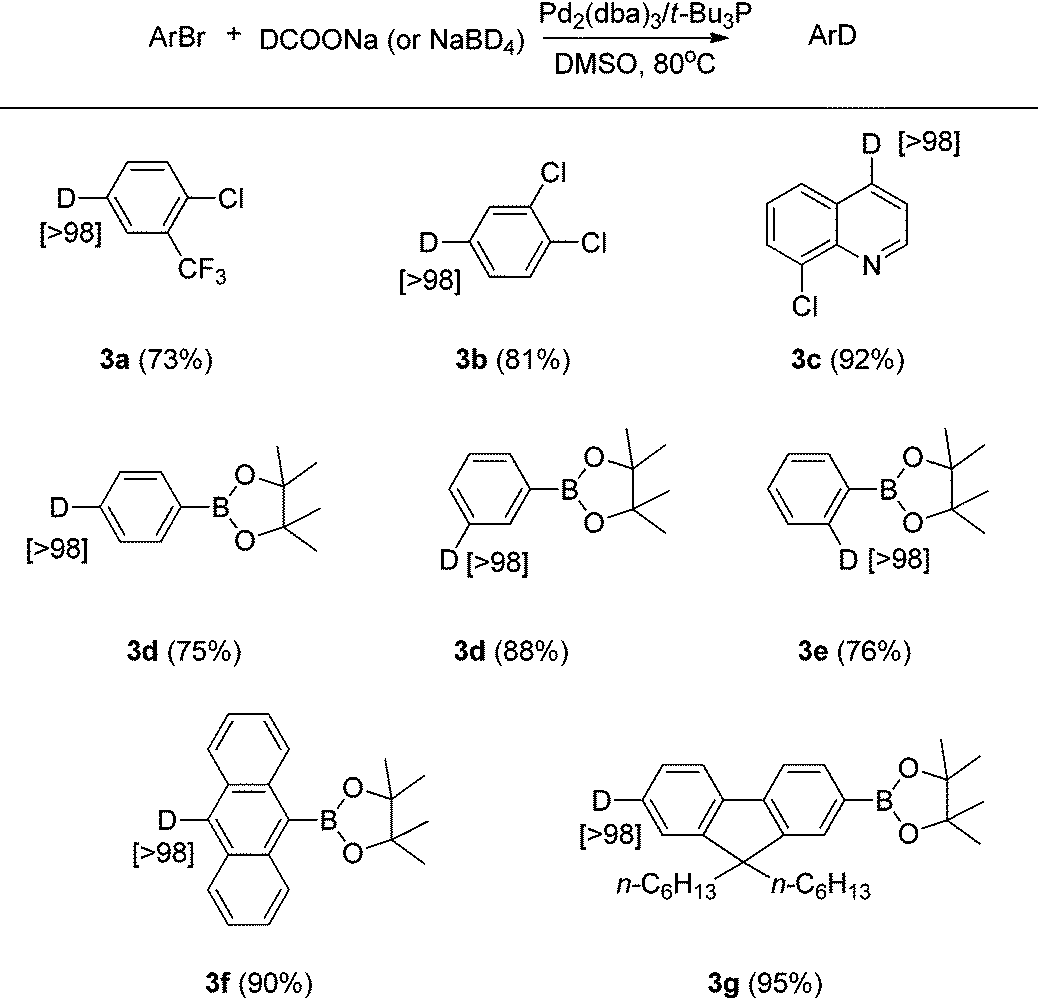
Aryl chlorides and boronic acid esters are very important building blocks since they are widely used in different cross-coupling reactions in synthetic organic chemistry to build more complicated compounds.11 To further explore the scope of this Br/D exchange reaction, a number of different aryl bromides containing chloride or boronic acid ester functionality were examined for Br/D exchange under the optimized reaction conditions. Though the combination of Pd2(dba)3/t-Bu3P has been well reported for the cross-coupling of aryl chlorides in THF,12 we found that this catalyst/ligand combination used under the optimized reaction conditions afforded deuterium-labeled aryl chlorides in good yield in DMSO, with no degradation of carbon-chloride bond being observed even after 16 hours (Table 3). However, when 3-bromobenzene boronic acid ester was employed as the substrate, the self-coupling dimer and trimer products were observed. Based on this observation, we considered that competition between the boronic acid ester and deuterium ion in the transmetalation step of the reaction could be the reason for the self-coupled products.11a,b To suppress this self-coupling reaction, 2 equivalents of the more reactive sodium borohydride-d4 were employed as the deuterium source. Several deuterium-labeled boronic acid esters were thus obtained in good yields and excellent deuterium incorporation using sodium borohydride-d4 as the D− source.
| a Conditions for deuterium-labeled aryl chloride (3a–3c): aryl bromide (1 mmol), Pd2dba3 (2 mol%), t-Bu3P (6 mol%), DCOONa (2 mmol), DMSO (1 mL), 80 °C. b Conditions for deuterium-labeled aryl boronic acid ester (3d–3g): aryl bromide (1 mmol), Pd2dba3 (2 mol%), t-Bu3P (6 mol%), NaBD4 (2 mmol), DMSO (2 mL), 80 °C. c Isolated yield is shown in parentheses. d Atom% D incorporation was determined by 1H NMR spectroscopic analysis (shown in square brackets.) |
|---|
|
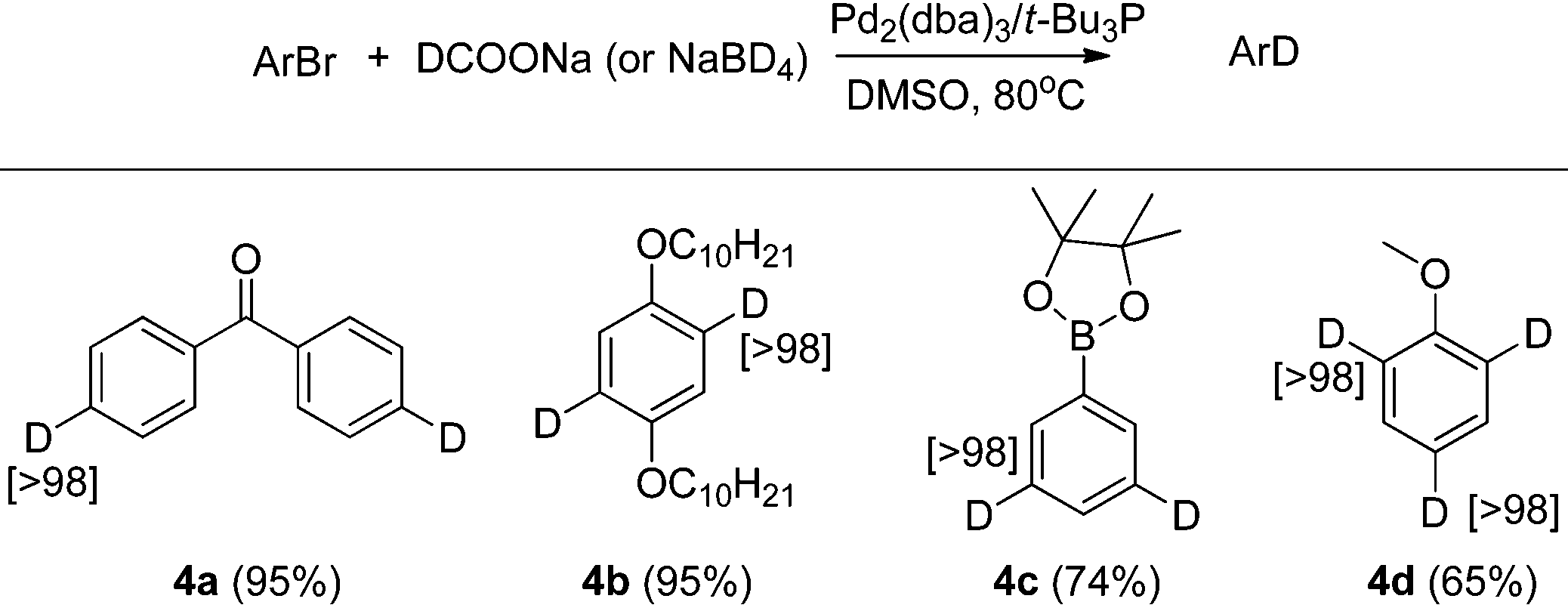
After testing the scope of the functional group compatibility, we turned our attention to deuteration of di- and tri-bromoarenes. A number of functionalized arenes with two bromides and one with three bromides were also examined for Br/D exchange. As shown in Table 4, all of these substrates afforded the deuterated products in good yields and with excellent deuterium incorporation. Compound 4b is an important building block for preparing conjugated polymers that are widely employed as optical and electronic materials.13
| a Conditions for 4a, 4b, 4d: aryl bromide (1 mmol), Pd2dba3 (2 mol%), t-Bu3P (6 mol%), DCOONa (2 equivalent to bromide), DMSO (2 mL), 80 °C. b Conditions for 4c: aryl bromide (1 mmol), Pd2dba3 (2 mol%), t-Bu3P (6 mol%), NaBD4 (4 mmol), DMSO (4 mL), 80 °C. c Isolated yield is shown in parentheses. d Atom% D incorporation was determined by 1H NMR spectroscopic analysis (shown in square brackets.) |
|---|
|
The proposed reaction mechanism is shown in Scheme 1. Based on the observation that only the deuterium anion as deuterium source could afford high conversion in this Br/D exchange reaction we believe deuteride is essential to replace the bromide in ArPd(II)LBr complex. This is also the reason for high atom% deuterium incorporation, since active (cationic) hydrogen present in the substrate did not impact the Br/D replacement step.
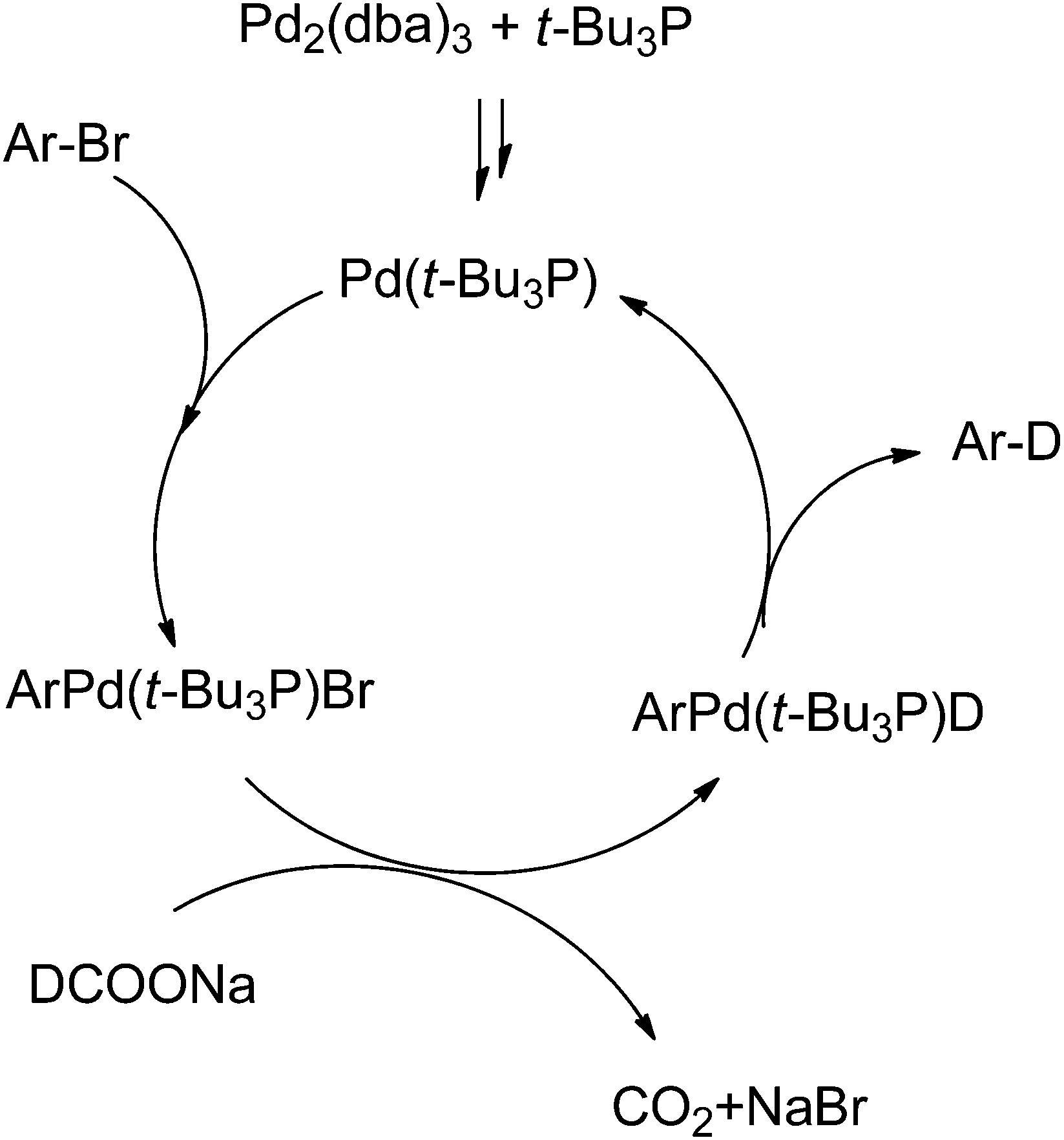 | ||
| Scheme 1 Mechanism for palladium catalyzed Br/D exchange. | ||
The parallel reactions with sodium formate-h and sodium formate-d were performed to examine the kinetic isotope effect. As shown in Scheme 2, comparable levels of conversion were achieved in both cases after reaction times of 10 and 30 minutes. The results indicated that formation and breaking of a carbon–hydrogen/carbon–deuterium bond is not involved in the rate-limiting step. Therefore, the oxidative addition step could be the rate-limiting step of this Br/D exchange reaction.
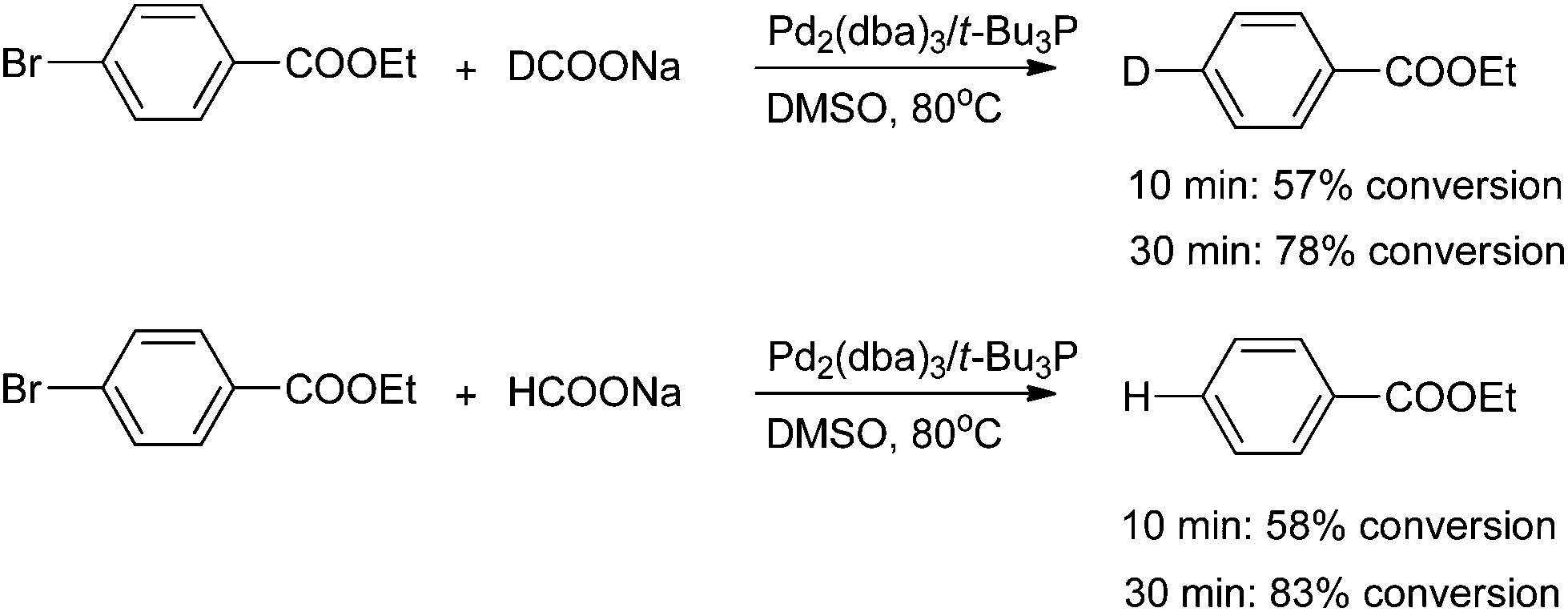 | ||
| Scheme 2 Kinetic isotope effect study. | ||
In summary, we have demonstrated that the commercially available deuterium-labeled formate salt can serve as a mild deuterium reagent in a palladium-catalyzed Br/D exchange reaction. When a boronic acid ester functional group is present, sodium borohydride-d4 was used to suppress the self-coupling side reaction. Our method provides introduction of deuterium at selected positions of arenes in high atom% deuterium incorporation with high tolerance to a wide range of functional groups. This will facilitate the synthesis of more complicated deuterium-labeled compounds through further modification of the functional groups. We believe this method will be a powerful tool to provide deuterium-labeled compounds for various applications. Further extension to substrates other than aryl bromides in this palladium-catalyzed Br/D exchange reaction is now under investigation.
Acknowledgements
This research was conducted at the Center for Nanophase Materials Sciences, which is a DOE Office of Science User Facility.Notes and references
-
(a) T. Giagou and M. P. Meyer, Chem. – Eur. J., 2010, 16, 10616–10628 CrossRef CAS PubMed
; (b) P. Krumbiegel, Isotopes Environ. Health Stud., 2011, 47, 1–17 CrossRef CAS PubMed
- For deuterium in drug discovery:
(a) S. L. Harbeson and R. D. Tung, Annu. Rep. Med. Chem., 2011, 46, 403–417 CAS
- For deuterium in material discovery:
(a) Y. B. Melnichenko and G. D. Wignall, J. Appl. Phys., 2007, 102, 021101 CrossRef PubMed
- Selected reference for arene H/D exchange:
(a) J. Atzrodt, V. Derdau, T. Fey and J. Zimmermann, Angew. Chem., Int. Ed., 2007, 46, 7744–7765 CrossRef CAS PubMed
- Selected arene Br/D exchange:
(a) Y. Miura, H. Oka, E. Yamano and M. Morita, J. Org. Chem., 1997, 62, 1188–1190 CrossRef CAS
-
(a) J. Yang, K. Hong and P. V. Bonnesen, J. Labelled Compd. Radiopharm., 2012, 55, 463–466 CrossRef CAS PubMed
-
(a) H. V. Thulasiram, R. M. Phan, S. B. Rivera and C. D. Poulter, J. Org. Chem., 2006, 71, 1739–1741 CrossRef CAS PubMed
- S. Korsager, R. H. Taaning, A. T. Lindhardt and T. Skrydstrup, J. Org. Chem., 2013, 78, 6112–6120 CrossRef CAS PubMed
-
(a) T. Furuyama, M. Yonehara, S. Arimoto, M. Kobayashi, Y. Matsumoto and M. Uchiyama, Chem. – Eur. J., 2008, 114, 10348–10356 CrossRef PubMed
-
(a) S. R. Klei, J. T. Golden, T. D. Tilley and R. G. Bergman, J. Am. Chem. Soc., 2002, 124, 2092–2093 CrossRef CAS PubMed
-
(a) A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176–4211 CrossRef CAS
-
(a) A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 1998, 37, 3387–3388 CrossRef CAS
-
(a) Y. J. Cheng, S. H. Yang and C. S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details of Br/D exchange, identification of products. See DOI: 10.1039/c5qo00181a |
| This journal is © the Partner Organisations 2015 |