
Synthesis of α-enamino esters via Cu(II)-promoted dehydrogenation of α-amino acid esters: application to the synthesis of polysubstituted pyrroles†
Nini
Zhou
,
Zhongle
Li
and
Zhixiang
Xie
*
State Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou, 730000, PR China. E-mail: xiezx@lzu.edu.cn
First published on 29th September 2015
Abstract
α-Enamino esters were efficiently prepared directly from readily available α-amino acid esters using a Cu(II)-promoted dehydrogenation with broad functional group tolerance and good to excellent yields. Moreover, the first application of α-enamino esters for the synthesis of polysubstituted pyrroles was achieved using an oxidative coupling with alkynes under Cu(II)-promoted conditions via intermolecular carbometalation of the alkyne in high yields.
Introduction
Enamino esters play an important role in organic chemistry because of their versatile reactivity which is composed of the nucleophilicity of an enamine and the electrophilicity of an α,β-unsaturated ester.1 Therefore, they have been widely used as nucleophiles and electrophiles in organic synthesis.2 In recent years, the preparation and application of β-enamino esters have both undergone substantial development. For the synthesis of β-enamino esters, based on the classical condensation reaction between keto-esters and amines,3 there have been general and efficient methods developed by Wang4 and Menéndez5 as well as several novel and efficient routes reported by Pellicciari,6 Muthusubramanian1a and Burgess.7 In organic synthetic chemistry, β-enamino esters are usually used as valuable reagents in the synthesis of various heterocycles, such as pyrroles,8 indoles,9 pyridines,10 quinolines,11 pyrimidineones,12 pyrazoles,13 amino-furanones14 and so on. Meanwhile, they also act as key intermediates in the total synthesis of natural products.15 However, in comparison with β-enamino esters, progress in the synthesis16 and application17 of α-enamino esters has rarely been reported. For the synthesis of α-enamino esters, a strategy which starts from α-amino acid esters is straightforward to visualize. In 1977, Öhler reported a methodology for the preparation of α-enamino esters from α-amino acid esters in two steps (Scheme 1a).16b Later, Lewin used this method for an investigation of the enamine/imine tautomerism in α,β-unsaturated amino acid esters.16e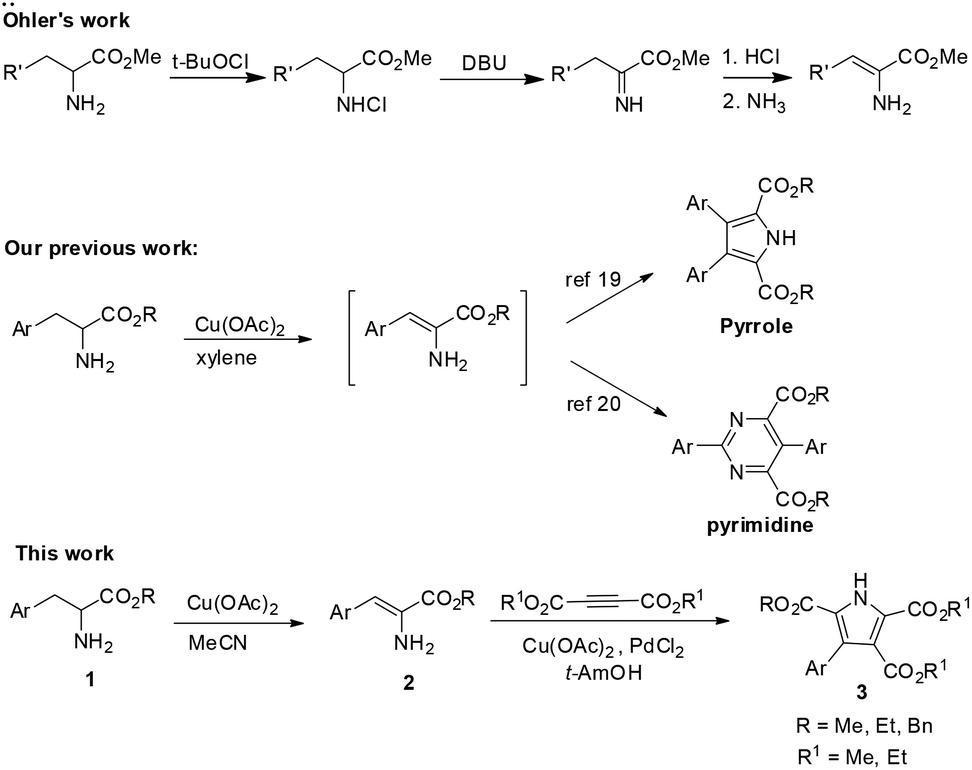 | ||
| Scheme 1 Synthesis and application of α-enamino esters from α-amino acid esters. | ||
α-Enamino esters which have been used as components of antibiotic peptides and biosynthetic intermediates should deserve more attention from chemists.18 Perhaps the β-positions of the α-enamino esters possess both nucleophilicity and electrophilicity, which would make α-enamino esters challenging substrates for metal-mediated coupling reactions. Therefore, it will be an important contribution to organic chemistry to investigate the synthesis and application of α-enamino esters. Very recently, we have reported that α-amino acid esters could be transformed to pyrroles19 and pyrimidines20 with promotion by Cu(OAc)2 (Scheme 1b). During our study, we found that α-enamino esters could be obtained as key intermediates for the construction of pyrroles and pyrimidines in the reaction process. However, a method which provides access to α-enamino esters directly from α-amino acid esters in one step has not been reported so far.16b In this article, we want to report an effective method for the synthesis of α-enamino esters from readily available α-amino acid esters through a Cu(II)-promoted oxidative dehydrogenation and the first application of α-enamino esters to the synthesis of polysubstituted pyrroles via oxidative coupling with alkynes (Scheme 1c).
Results and discussion
In our previous work, we found that Cu(OAc)2 was the best metallic reagent for the transformation of α-amino acid esters to pyrroles19 and pyrimidines20via α-enamino ester intermediates. Therefore, in order to obtain the α-enamino esters as the product, we began our investigation with the reaction of 1a and Cu(OAc)2. Given the poor stability and reactivity of α-enamino esters, we first screened various solvents, such as toluene, xylene, MeCN, dioxane, THF, and DMF, and the results manifested that MeCN is the most suitable solvent for this transformation (Table 1, entries 1–6). When the reaction was conducted in the presence of 0.5 equivalents or 1.5 equivalents of Cu(OAc)2, the yield of 2a failed to improve (Table 1, entries 7 and 8), which illustrated that 1.0 equivalent of Cu(OAc)2 was favourable for the oxidative dehydrogenation of 1a. Further experiments showed that additives, such as NaOAc, KOAc, Na2CO3, NH3–SiO2 (silica gel basified with NH3), SiO2 and 4 Å MS, resulted in a decrease of the yield of 2a (Table 1, entries 9–14). Therefore, the screening of different conditions suggested that this transformation of 1a proceeded most efficiently in the presence of Cu(OAc)2 (1.0 equiv.) in refluxing MeCN under argon.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
|
||||
|---|---|---|---|---|
| Entry | Oxidant (equiv.) | Additive (equiv.) | Solvent | Yieldb % |
| a All reactions were carried out under the following conditions, unless otherwise noted: 0.3 mmol of 1a and a specified amount of Cu(OAc)2 in a refluxing solvent (7.0 mL) under argon. b Yield of isolated product. c An equivalent mass to that of 1a was used. | ||||
| 1 | Cu(OAc)2 1.0 | — | Toluene | 55 |
| 2 | Cu(OAc)2 1.0 | — | Xylene | 77 |
| 3 | Cu(OAc) 2 1.0 | — | MeCN | 78 |
| 4 | Cu(OAc)2 1.0 | — | Dioxane | 68 |
| 5 | Cu(OAc)2 1.0 | — | THF | 18 |
| 6 | Cu(OAc)2 1.0 | — | DMF | <5 |
| 7 | Cu(OAc)2 0.5 | — | MeCN | 50 |
| 8 | Cu(OAc)2 1.5 | — | MeCN | 74 |
| 9 | Cu(OAc)2 1.0 | NaOAc 1.0 | MeCN | 68 |
| 10 | Cu(OAc)2 1.0 | KOAc 1.0 | MeCN | 60 |
| 11 | Cu(OAc)2 1.0 | Na2CO3 1.0 | MeCN | 70 |
| 12 | Cu(OAc)2 1.0 | NH3–SiO2c | MeCN | 53 |
| 13 | Cu(OAc)2 1.0 | SiO2c | MeCN | 74 |
| 14 | Cu(OAc)2 1.0 | 4 Å MSc | MeCN | 56 |

With the optimized reaction conditions in hand, the substrate scope of this copper-promoted oxidative dehydrogenation was investigated (Table 2). The transformation was extended to a variety of aryl-substituted α-amino acid esters. When the ester groups of the α-amino acid esters ranged from CO2Me to CO2Et and CO2Bn, the desired α-enamino esters 2a–c were smoothly obtained in good to excellent yield. Both the electron-withdrawing and -donating para-substituents on the benzene ring were well tolerated under the reaction conditions to give the corresponding α-enamino esters 2c–j in good yield. 2k could be successfully produced in 73% yield with Me as a meta-substituent on the benzene ring while 2l was afforded in reduced yield because of the steric hindrance effects from Me as an ortho-substituent. When there were two or three OMe meta- and para-substituents on the benzene ring, the transformation also smoothly proceeded to construct the α-enamino esters 2m–o. Furthermore, the α-amino acid esters which were substituted with a naphthalene ring or heteroaromatic ring with large steric hindrance were also suitable for this reaction, to form 2p–q in moderate to good yield. It is notable that only the α-enamino esters (Z)-2a–q were obtained, and that (E)-2a–q were not detected. Moreover, using N-substituted (Ac, Tos, Bn) amino esters as substrates, the amino esters did not undergo dehydrogenation.
| Reaction conditions: amino acid ester 1 (0.3 mmol) and Cu(OAc)2 (1.0 equiv.) in refluxing MeCN (7 mL) under argon. Yields of the isolated products are given. |
|---|
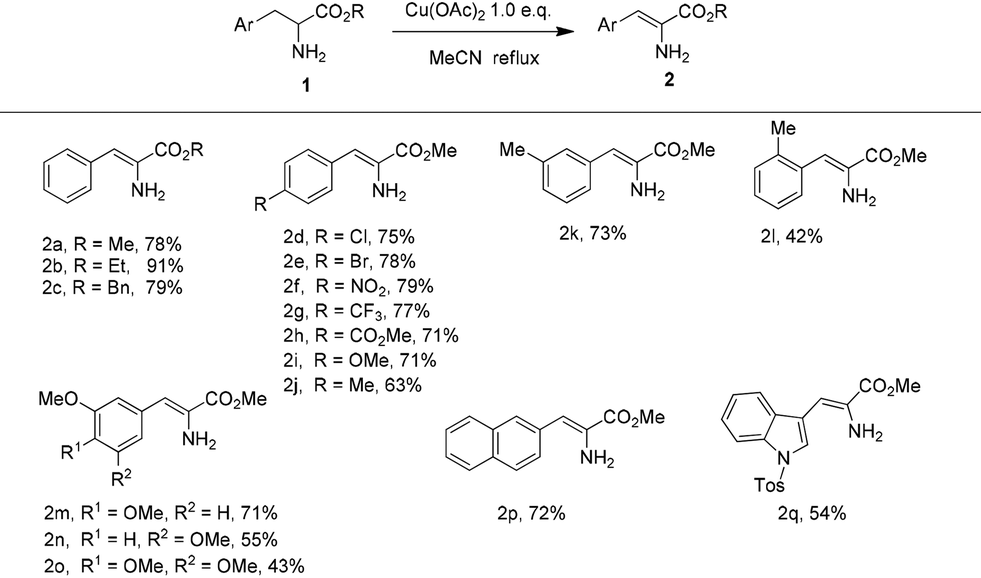
|
With the achievement of the preparation of the α-enamino esters, our investigation on the application of them started with the reaction of 2a and dimethyl but-2-ynedioate. According to Guan's report,21 phosphine ligands had shown positive effects in the reaction of enamides and alkynes. We were pleased to obtain the desired pyrrole 3a in 27% yield in the presence of 1.0 equiv. Cu(OAc)2, 5 mol% Pd(OAc)2, and 10 mol% phosphine ligand (Xantphos) in xylene at 120 °C (Table 3, entry 1). In order to improve the yield of 3a, firstly, solvents were screened and t-AmOH was found to be the most suitable solvent for the reaction (Table 3, entries 2 and 3). When the ligand was changed from Xantphos (dimethylbisdiphenyl-phosphinoxanthene) to dppp (1,3-bis(diphenylphosphino) propane) or dppf (1,1′-bis(diphenylphosphino)ferrocene), the yield of 3a was reduced by some degree (Table 3, entries 4 and 5). However, the yield of 3a was further improved to 50% when the reaction was conducted in the absence of ligand (Table 3, entry 6). Then we turned to the screening of palladium salts without using a ligand (Table 3, entries 7–11), such as Pd(PPh3)2Cl2, Pd(PPh3)4, Pd(dppf)2Cl2, PdCl2, and Pd(TFA)2, and the results showed that PdCl2 was more suitable for this reaction. The corresponding pyrrole 3a was obtained in 64% yield (Table 3, entry 10). In order to further improve the yield of 3a, we increased the amount of Cu(OAc)2 to 1.2 equivalents and obtained pyrrole 3a in 77% yield (Table 3, entry 12). However, further increasing the amount of Cu(OAc)2 to 1.5 equivalents decreased the yield of 3a (Table 3, entry 13). Increasing the amount of dimethyl but-2-ynedioate to 1.5 equivalents, the yield of 3a was further improved to 88% (Table 3, entry 14). Using t-BuOH as the solvent, with the remaining reaction conditions as for entry 14, the pyrrole 3a was obtained in 53% yield (Table 3, entry 15). After the careful examination of the different conditions, it was comforting to achieve the optimum conditions: 1.5 equivalents of dimethyl but-2-ynedioate, 1.2 equivalents of Cu(OAc)2, and 5 mol% PdCl2 in t-AmOH.
a
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Ligand | Solvent | Yieldb (%) |
| a All reactions were carried out under the following conditions, unless otherwise noted: 0.25 mmol of 1a, 1.1 equivalents of dimethyl but-2-ynedioate and a specified amount of Cu(OAc)2 in a solvent (2.0 mL) at 120 °C for 8 h. b Yield of the isolated product. c 1.0 equivalents of Cu(OAc)2 was used. d 1.2 equivalents of Cu(OAc)2 was used. e 1.5 equivalents of Cu(OAc)2 was used. f 1.5 equivalents of dimethyl but-2-ynedioate was used. | ||||
| 1c | Pd(OAc)2 5 mol% | Xantphos 10 mol% | Xylene | 27 |
| 2c | Pd(OAc)2 5 mol% | Xantphos 10 mol% | t-AmOH | 47 |
| 3c | Pd(OAc)2 5 mol% | Xantphos 10 mol% | MeCN | 9 |
| 4c | Pd(OAc)2 5 mol% | dppp 10 mol% | t-AmOH | 42 |
| 5c | Pd(OAc)2 5 mol% | dppf 10 mol% | t-AmOH | 35 |
| 6c | Pd(OAc)2 5 mol% | — | t-AmOH | 50 |
| 7c | Pd(PPh3)2Cl2 5 mol% | — | t-AmOH | 45 |
| 8c | Pd(PPh3)4 5 mol% | — | t-AmOH | 39 |
| 9c | Pd(dppf)2Cl2 5 mol% | — | t-AmOH | 59 |
| 10c | PdCl2 5 mol% | — | t-AmOH | 64 |
| 11c | Pd(TFA)2 5 mol% | — | t-AmOH | 41 |
| 12d | PdCl2 5 mol% | — | t-AmOH | 77 |
| 13e | PdCl2 5 mol% | — | t-AmOH | 55 |
| 14 , | PdCl 2 5 mol% | — | t-AmOH | 88 |
| 15d,f | PdCl2 5 mol% | — | t-BuOH | 53 |
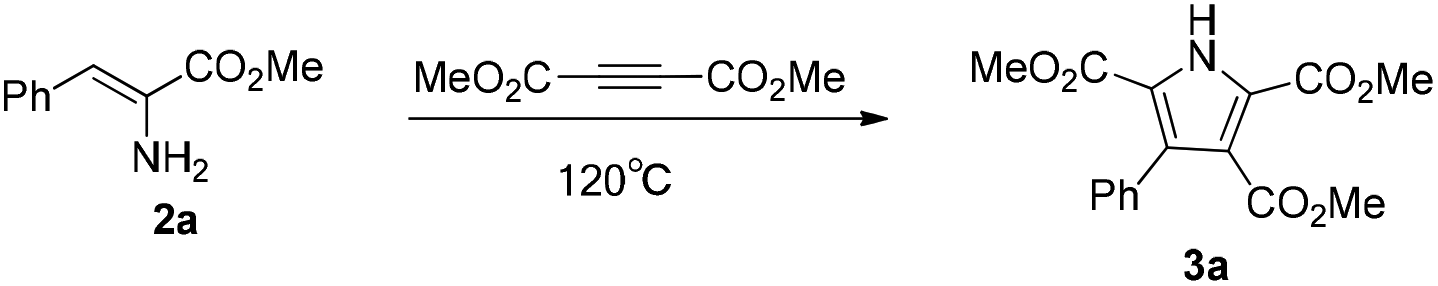
Following the establishment of the optimized reaction conditions for this oxidative coupling, a series of α-enamino esters and two alkynes were investigated to extend the substrate scope (Table 4). This transformation presented good functional group tolerance and proved to be an efficient methodology for the preparation of polysubstituted NH pyrroles, which suggests that α-enamino esters could be versatile synthons for various heterocycles. As the ester groups of the α-enamino esters ranged from CO2Me to CO2Et and CO2Bn, the transformation smoothly proceeded to construct the corresponding pyrroles 3a–f in good to high yield. α-Enamino esters with the electron-neutral group Cl or electron-withdrawing group NO2 on the aryl ring were well tolerated in the reaction to give desired pyrroles 3g–j in moderate to good yield. In addition, 3,4-dimethoxylphenyl-α-enamino ester 2m was also suitable for this reaction, forming 3k in 65% yield and 3l in 58% yield. Furthermore, the α-enamino esters substituted with heteroaromatic rings capable of large steric hindrance, such as N-tosyl indole, could also be effectively transformed to the desired pyrroles 3m–n in good yield, which suggests that this oxidative coupling between α-enamino esters and dimethyl but-2-ynedioate is insensitive to steric hindrance from the α-enamino esters. However the reaction might be slightly sensitive to steric hindrance from the alkynes, because the yield of the corresponding pyrrole decreased (see 3h, 3j, 3l, and 3n) when diethyl but-2-ynedioate was subjected to the reaction instead of dimethyl but-2-ynedioate.
| Reaction conditions: α-enamino ester 2 (0.25 mmol), alkyne (1.5 equiv.), Cu(OAc)2 (1.2 equiv.) and PdCl2 (5 mol%) in t-AmOH (2.0 mL) at 120 °C for 8 h. Yields of the isolated products are given. |
|---|
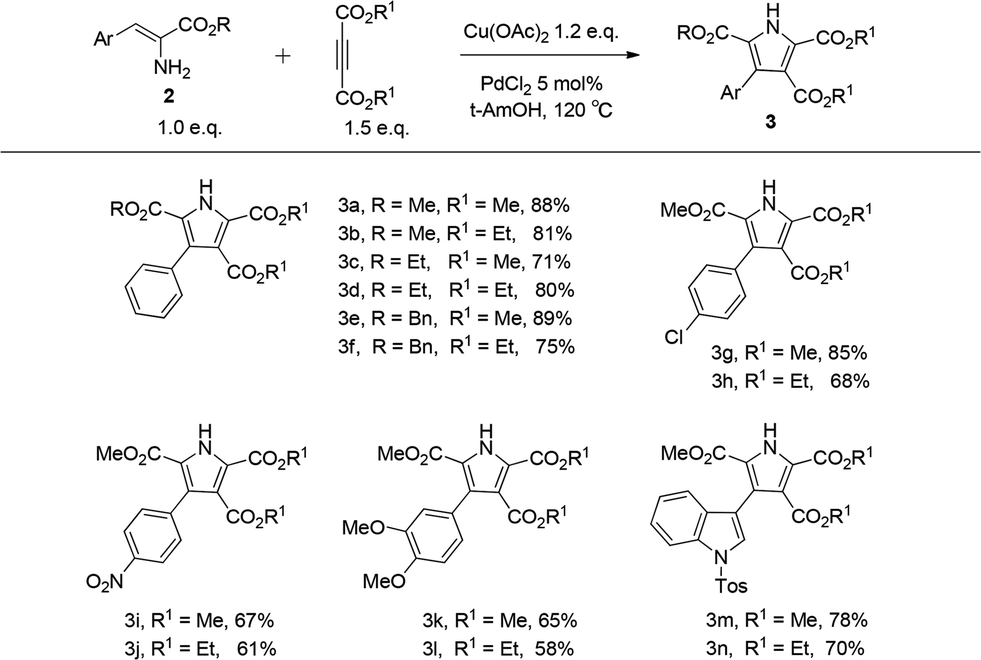
|
In order to gain some insight into the mechanism of the copper-promoted oxidative coupling between α-enamino esters and alkynes, a series of control experiments were conducted on the basis of the standard conditions (Scheme 2). Without Cu(OAc)2 and PdCl2, the polysubstituted pyrrole 3a was not obtained from the reaction (Scheme 2, eqn (1), control experiment A). In the absence of PdCl2, the yield of 3a was reduced to 66% (Scheme 2, eqn (1), control experiment B). The transformation could not proceed with PdCl2 (1.0 equiv.) alone (Scheme 2, eqn (1), control experiment C). These results show that this oxidative coupling was promoted by Cu(OAc)2 and that PdCl2 played an important role in this transformation to improve the yield of 3a. The yield of 3a was reduced to 60% when the reaction was performed in degassed solvent under argon (Scheme 2, eqn (1), control experiment D), which illustrates that the oxygen in the air acts as a co-oxidant in this reaction. When 2a (0.25 mmol) was reacted with dimethyl but-2-ynedioate (1.5 equiv.) in t-AmOH (2.0 mL) without Cu(OAc)2 and PdCl2, 4a and 4b were obtained at 95 °C in 20% and 7% yield, respectively, and a complex product mixture was obtained for a longer reaction time or at higher reaction temperatures (Scheme 2, eqn (2)). When 4a was subjected to the optimized reaction conditions, 3a was isolated in only 10% yield and much of 4a had decomposed under the standard conditions (Scheme 2, eqn (3)). This fact suggests that the coupling reaction could not be mainly initiated by nucleophilic addition of the amine group to the electron-deficient alkyne. However, 3a was obtained in 73% yield from 4b under the optimized reaction conditions, which suggests that the reaction could be initiated by Micheal addition of the α-enamino ester to the alkyne (Scheme 2, eqn (4)). Moreover, using the standard reaction conditions at a lower temperature, we failed to obtain 4b in the early stage of the process when the reaction was slowed down, which suggests that there might be an active form of 4b in the mechanism. Notably, using the standard conditions but reducing the reaction temperature resulted in a relatively low yield of 3a and a longer reaction time, which indicated that Micheal addition without Cu(II) promotion might lead to the low yield of the pyrrole and that the higher temperature might be helpful for Cu(II) promotion of the Micheal addition. Therefore, we speculated that, under the standard conditions, Micheal addition of an α-enamino ester activated by Cu(II) to the alkyne occurs to give 4b in an active form in the process.
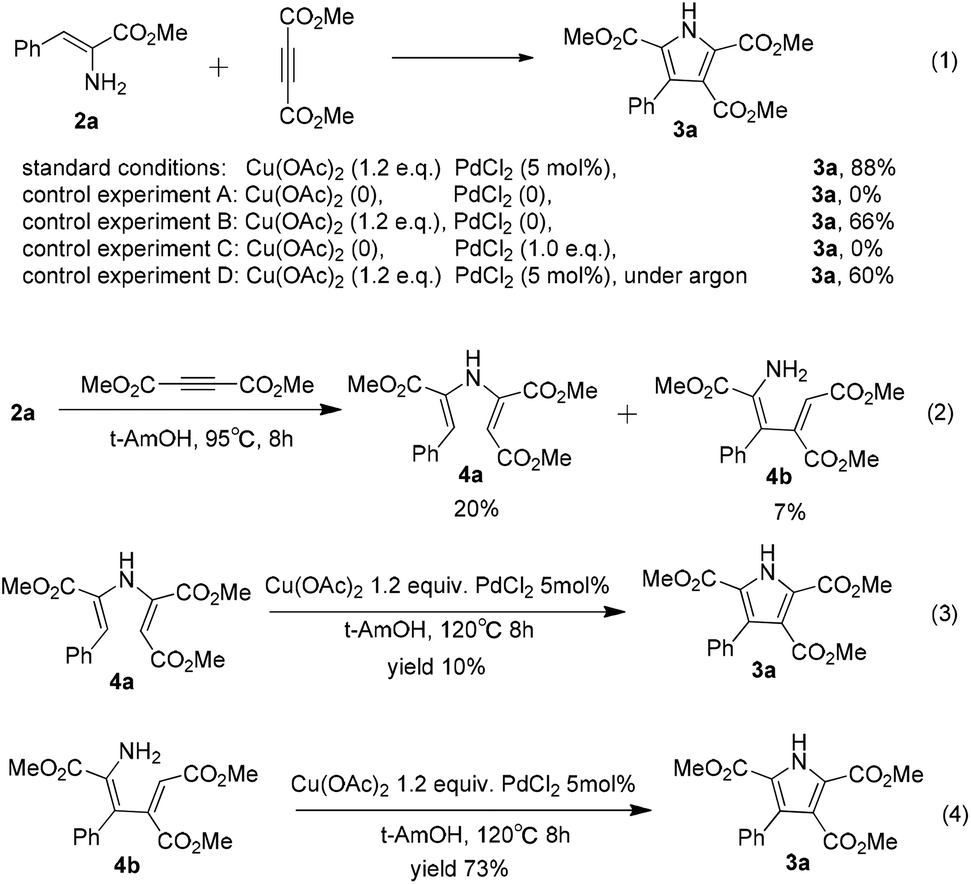 | ||
| Scheme 2 Exploration of the mechanism. Reaction conditions for the control experiments: α-enamino ester 2a (0.25 mmol), dimethyl but-2-ynedioate (1.5 equiv.), and a specified amount of Cu(OAc)2 and PdCl2 in t-AmOH (2.0 mL) at 120 °C for 8 h. Yields of the isolated products are given. | ||
On the basis of the above control experiment results, a plausible mechanism for the oxidative coupling between α-enamino esters and electro-deficient alkynes is proposed in Scheme 3. Reaction of α-enamino ester 2a with Cu(OAc)2 forms 5 and 6.8c Then, with an electron-deficient alkyne, the intermolecular carbocupration of the alkyne generates organocopper intermediates 7 and 8.11,22 The intramolecular deprotonation–cyclization of 8 generates a six-membered cupracycle 9. The formation of 9 followed by C–N bond-forming reductive elimination would give the pyrrole 3a.23 In the presence of Pd(II), 9 would transform to a six-membered palladacycle 10 through metal exchange. Finally, the reductive elimination of 10 affords pyrrole 3a and a Pd(0) species which could be reoxidized by Cu(OAc)2 or oxygen from the air to regenerate Pd(II). The metal exchange between copper and palladium likely promotes the reductive elimination of the six-membered metallacycle to give 3a.
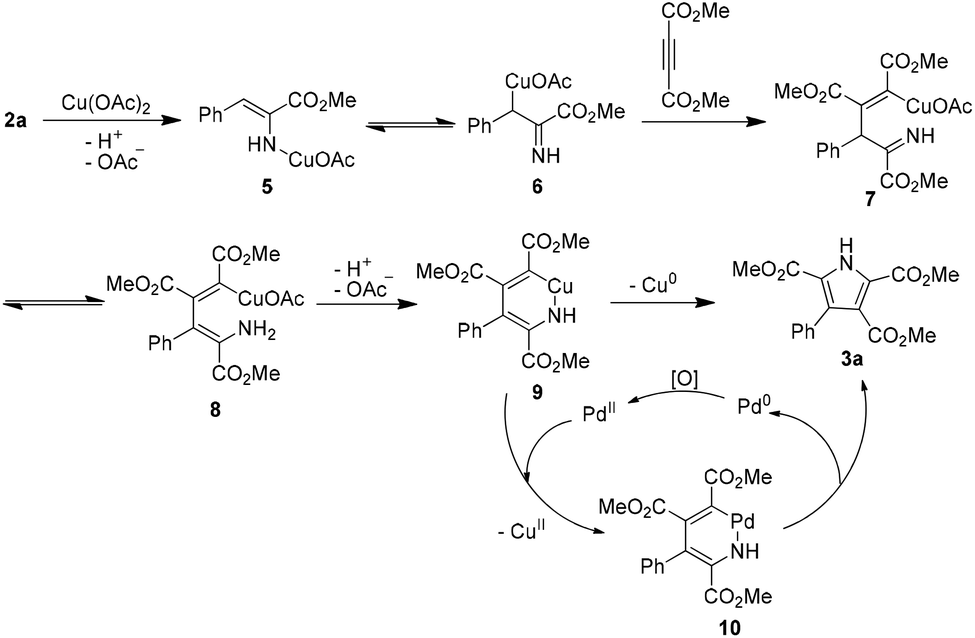 | ||
| Scheme 3 A plausible mechanism for the oxidative coupling between α-enamino esters and alkynes. | ||
Conclusions
In summary, a novel and efficient methodology for the synthesis of α-enamino esters from readily available α-amino acid esters has been successfully developed, which tolerates a wide range of functional groups and provides a favorable means for the research of α-enamino esters. Assisted by this methodology, α-enamino esters have been firstly used as valuable synthons for the construction of polysubstituted NH pyrroles with broad substrate scope and in good yield. α-Enamino esters have been successfully prepared and applied, and will be valuable reagents in organic chemistry as attractive alternatives to β-enamino esters. Further studies on α-enamino esters are ongoing in our group.Experimental
General experimental methods
1H NMR spectra were recorded using 400 MHz spectrometers, and 13C NMR spectra were recorded using a 100 MHz spectrometer. Chemical shifts (in ppm) are referenced to tetramethylsilane (δ = 0 ppm) in CDCl3 as an internal standard. The 13C NMR spectra were obtained using the same NMR spectrometers and were calibrated with CDCl3 (δ = 77.00 ppm). The following abbreviations are used for the multiplicities: s, singlet; d, doublet; t, triplet; q, quartet, m, multiplet, brs, broad singlet, for the proton spectra. Coupling constants (J) are reported in hertz (Hz). High-resolution mass spectra (HRMS) were obtained on a 4G mass spectrometer using electrospray ionization (ESI) analyzed by quadrupole time-of-flight (QTof). The IR spectra were recorded using a FT-IR spectrometer, and only the major peaks are reported in cm−1. Melting points were determined on X-4 microscopic apparatus and are uncorrected. Unless otherwise noted, the materials, including solvents, obtained from commercial suppliers were used without further purification. Flash column chromatography was performed using silica gel (200–300 mesh) with the solvents distilled prior to use.Typical procedure for synthesis of the α-enamino esters
In a 25 mL round-bottomed flask, α-amino acid ester 1 (0.3 mmol) and Cu(OAc)2 (0.3 mmol, 55 mg) were stirred in refluxing CH3CN (7 mL) under argon. After completion of the reaction (detected using TLC), the reaction mixture was cooled to room temperature, and the solvent evaporated under reduced pressure. Finally the resulting residue was purified using flash column chromatography to give the pure α-enamino ester.Typical procedure for synthesis of the polysubstituted pyrroles
In a 10 mL reaction tube, α-enamino ester 2 (0.25 mmol), an alkyne (0.38 mmol), Cu(OAc)2 (0.3 mmol, 55 mg) and PdCl2 (0.013 mmol, 2 mg) were stirred in t-AmOH (2 mL) at 120 °C for 8 h. After completion of the reaction, the reaction mixture was cooled to room temperature, and the solvent evaporated under reduced pressure. Finally the resulting residue was purified using flash column chromatography to give the pure pyrrole.Synthesis of dimethyl 2-(((E)-3-methoxy-3-oxo-1-phenylprop-1-en-2-yl)amino)maleate (4a)30 and trimethyl 4-amino-3-phenylbuta-1,3-diene-1,2,4-tricarboxylate (4b)
In a 10 mL reaction tube, α-enamino ester 2a (45 mg, 0.25 mmol) and an alkyne (54 mg, 0.38 mmol) were stirred in t-AmOH (2 mL) at 95 °C for 8 h. After completion of the reaction, the reaction mixture was cooled to room temperature, and the solvent evaporated under reduced pressure. Finally the resulting residue was purified using flash column chromatography on silica gel (petroleum ether/ethyl acetate = 10/1 to 4/1, v/v) to give 4a, 16 mg (20%), as a yellow oil and 4b, 6 mg (7%), as a yellow oil.4a: 1H NMR (400 MHz, CDCl3) δ ppm 9.65 (brs, 1H), 7.60 (d, J = 7.2 Hz, 2H), 7.38 (t, J = 7.2 Hz, 2H), 7.33–7.29 (m, 1H), 7.10 (s, 1H), 5.60 (s, 1H), 3.79 (s, 3H), 3.74 (s, 3H), 3.71 (s, 3H); 13C NMR (100 MHz, CDCl3) δ ppm 170.0, 165.8, 163.7, 147.5, 133.6, 129.6 (2C), 129.0, 128.8 (2C), 128.0, 125.6, 92.5, 52.6, 52.4, 51.2; HRMS (ESI) m/z calcd for C16H17NO6Na (M + Na)+ 342.0948, found 342.0942; IR (neat) 3253, 3024, 3001, 2953, 1735, 1720, 1672, 1634, 1611, 1477, 1437, 1368, 1279, 1227, 1192, 1160, 1133, 1030, 1003, 980, 938, 923, 889, 832, 817, 782, 770, 732, 694, 603, 496, 442 cm−1.
4b: 1H NMR (400 MHz, CDCl3) δ ppm 7.69 (s, 1H), 7.37–7.35 (m, 2H), 7.33–7.28 (m, 3H), 3.79 (s, 3H), 3.70 (s, 3H), 3.52 (s, 3H), 1.59 (brs, 1H), 1.25 (s, 1H); 13C NMR (100 MHz, CDCl3) δ ppm 169.6, 168.8, 164.0, 146.4, 139.9, 135.6, 129.7 (2C), 128.8, 128.5 (2C), 127.6, 96.7, 52.9, 52.2, 51.5; HRMS (ESI) m/z calcd for C16H17NO6Na (M + Na)+ 342.0948, found 342.0956; IR (neat) 3450, 3325, 3025, 2952, 2952, 2927, 2851, 2371, 2255, 1718, 1674, 1605, 1543, 1527, 1493, 1435, 1375, 1252, 1101, 1029, 913, 864, 775, 734, 696, 649, 495 cm−1.
Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (grant no. 21272104), the Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT: IRT1138), the Research Fund for the Doctoral Program of Higher Education of China (no. 20110211110009), the Program for New Century Excellent Talents in University (NCET-12-0247 and lzujbky-2012-56), and the Fundamental Research Funds for the Central Universities (no. lzujbky-2013-ct02).Notes and references
- (a) T. Ponpandian and S. Muthusubramanian, Tetrahedron, 2013, 69, 527 CrossRef CAS PubMed; (b) H. J. Jeon, D. J. Jung, J. H. Kim, Y. Kim, J. Bouffard and S. Lee, J. Org. Chem., 2014, 79, 9865 CrossRef CAS PubMed.
- (a) J. V. Greenhill, Chem. Soc. Rev., 1977, 6, 277 RSC; (b) P. Leu and J. V. Greenhill, Adv. Heterocycl. Chem., 1996, 67, 207 CrossRef; (c) B. Stanovnik and J. Svete, Chem. Rev., 2004, 104, 2433 CrossRef CAS PubMed; (d) Y. Cheng, Z. T. Huang and M. X. Wang, Curr. Org. Chem., 2004, 8, 325 CrossRef CAS.
- (a) H. M. C. Ferraz, E. O. Oliveira, M. E. Payret-Arrua and C. A. Brandt, J. Org. Chem., 1995, 60, 7357 CrossRef CAS; (b) V. A. Soloshonok and V. Kukhar, Tetrahedron, 1996, 52, 6953 CrossRef CAS.
- Z.-H. Zhang, L. Yin and Y.-M. Wang, Adv. Synth. Catal., 2006, 348, 184 CrossRef CAS PubMed.
- V. Sridharan, C. Avendaño and J. C. Menéndez, Synlett, 2007, 881 CAS.
- A. Gioiello, F. Venturoni, B. Natalini and R. Pellicciari, J. Org. Chem., 2009, 74, 3520 CrossRef CAS PubMed.
- D. Xin and K. Burgess, Org. Lett., 2014, 16, 2108 CrossRef CAS PubMed.
- (a) L. Meng, K. Wu, C. Liu and A. Lei, Chem. Commun., 2013, 49, 5853 RSC; (b) J. Ke, C. He, H. Liu, M. Li and A. Lei, Chem. Commun., 2013, 49, 7549 RSC; (c) K. K. Toh, Y.-F. Wang, E. P. J. Ng and S. Chiba, J. Am. Chem. Soc., 2011, 133, 13942 CrossRef CAS PubMed; (d) M. Zhao, F. Wang and X. Li, Org. Lett., 2012, 14, 1412 CrossRef CAS PubMed.
- (a) Z. Jia, T. Nagano, X. Li and A. S. C. Chan, Eur. J. Org. Chem., 2013, 858 CrossRef CAS PubMed; (b) J. J. Neumann, S. Rakshit, T. Droege, S. Wuertz and F. Glorius, Chem. – Eur. J., 2011, 17, 7298 CrossRef CAS PubMed; (c) H. H. Nguyen and M. J. Kurth, Org. Lett., 2013, 15, 362 CrossRef CAS PubMed; (d) S. Kramer, K. Dooleweerdt, A. T. Lindhardt, M. Rottlander and T. Skrydstrup, Org. Lett., 2009, 11, 4208 CrossRef CAS PubMed.
- (a) S. Yamamoto, K. Okamoto, M. Murakoso, Y. Kuninobu and K. Takai, Org. Lett., 2012, 14, 3182 CrossRef CAS PubMed; (b) N. R. Gade, V. Devendram, M. Pal and J. Iqbal, Chem. Commun., 2013, 49, 7926 RSC; (c) A. Noole, M. Borissova, M. Lopp and T. Kanger, J. Org. Chem., 2011, 76, 1538 CrossRef CAS PubMed.
- K. K. Toh, S. Sanjaya, S. Sahnoun, S. Y. Chong and S. Chiba, Org. Lett., 2012, 14, 2290 CrossRef CAS PubMed.
- Y. S. Chun, Z. Xuan, J. H. Kim and S. Lee, Org. Lett., 2013, 15, 3162 CrossRef CAS PubMed.
- M. Suri, T. Jousseaume, J. J. Neumann and F. Glorius, Green Chem., 2012, 14, 2193 RSC.
- V. Toum, C. Kadouri-Puchot, L. Hamon, G. Lhommet, V. Mouriès-Mansuy, C. Vanucci-Bacqué and L. Dechoux, Synthesis, 2011, 2781 CAS.
- (a) S. R. Hussaini and M. G. Moloney, Org. Biomol. Chem., 2006, 4, 2600 RSC; (b) J. P. Michael, C. B. Koning, G. D. Hosken and T. V. Stanbury, Tetrahedron, 2001, 57, 9635 CrossRef CAS.
- (a) C. C. Shin, M. Masaki and M. Ohta, J. Org. Chem., 1967, 32, 1860 CrossRef CAS; (b) U. Schmidt and E. Öhler, Angew. Chem., Int. Ed. Engl., 1977, 16, 327 CrossRef CAS PubMed; (c) C. Shia, Y. Yonezawa, K. Unoki and J. Yoshimura, Bull. Chem. Soc. Jpn., 1979, 52, 1657 CrossRef; (d) T. Moriga, K. Matsumoto and M. Miyoshi, Synthesis, 1981, 915 CrossRef; (e) S.-P. Lu and A. H. Lewin, Tetrahedron, 1998, 54, 15097 CrossRef CAS.
- (a) L. Marinus, United States Patent US, 4798619A, 1989 Search PubMed; (b) A. S. Moskalenko, A. V. Prosyanik, N. Yu. Kol'tsov, E. Yu. Nesterova, T. D. Novobranets and R. G. Kostyanovskii, Izv. Akad. Nauk SSSR, Ser. Khim., 1992, 1, 167 Search PubMed; (c) Y. Zheng, X. Li, C. Ren, D. Zhang-Negrerie, Y. Du and K. Zhao, J. Org. Chem., 2012, 77, 10353 CrossRef CAS PubMed.
- (a) C. H. Poisrl and U. Schmidr, Chem. Ber., 1975, 108, 2547 CrossRef PubMed; (b) B. W. Bycroft, Nature, 1969, 224, 595 CrossRef CAS PubMed.
- N. Zhou, T. Xie, L. Liu and Z. Xie, J. Org. Chem., 2014, 79, 6061 CrossRef CAS PubMed.
- N. Zhou, T. Xie, Z. Li and Z. Xie, Chem. – Eur. J., 2014, 20, 17311 CrossRef CAS PubMed.
- M.-N. Zhao, Z.-H. Ren, Y.-Y. Wang and Z.-H. Guan, Org. Lett., 2014, 16, 608 CrossRef CAS PubMed.
- S. Peng, L. Wang, J. Huang, S. Sun, H. Guo and J. Wang, Adv. Synth. Catal., 2013, 355, 2550 CrossRef CAS PubMed.
- (a) K. K. Toh, A. Biswas, Y.-F. Wang, Y. Y. Tan and S. Chiba, J. Am. Chem. Soc., 2014, 136, 6011 CrossRef CAS PubMed; (b) K. Hirano and M. Miura, Top. Catal., 2014, 57, 878 CrossRef CAS; (c) Z. Chen, Q. Yan, Z. Liu, Y. Xu and Y. Zhang, Angew. Chem., Int. Ed., 2013, 52, 13324 CrossRef CAS PubMed; (d) K. Hirano and M. Miura, Chem. Commun., 2012, 48, 10704 RSC; (e) M. M. Guru, M. A. Ali and T. Punniyamurthy, Org. Lett., 2011, 13, 1194 CrossRef CAS PubMed; (f) G. Brasche and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 1932 CrossRef CAS PubMed.
- (a) Y. Hirose, E. Ohta, Y. Kawai and S. Ohta, J. Nat. Prod., 2013, 76, 554 CrossRef CAS PubMed; (b) J. Smodis, R. Zupet, A. Petric, B. Stanovnik and M. Tisler, Heterocycles, 1990, 30, 393 CrossRef CAS; (c) S. C. Shim, Y. K. Yeo, K. N. Choi and H. K. Lee, TaehanHwahakhoe Chi, 1988, 32, 156 CAS; (d) D. Knittel, Monatsh. Chem., 1984, 115, 1335 CrossRef CAS; (e) H. Poisel, Chem. Ber., 1977, 110, 942 CrossRef CAS PubMed.
- (a) M. R. Maddani, S. K. Moorthy and K. R. Prabhu, Tetrahedron, 2010, 66, 329 CrossRef CAS PubMed; (b) V. Kvita, H. Sauter and G. Rihs, Helv. Chim. Acta, 1989, 72, 457 CrossRef CAS PubMed; (c) C.-G. Shin, Y. Yonezawa and J. Yoshimura, Chem. Lett., 1976, 10, 1095 CrossRef.
- C. G. Shin, Y. Yonezawa and T. Yamada, Chem. Pharm. Bull., 1984, 32, 3934 CrossRef CAS.
- D. Knittel, Monatsh. Chem., 1985, 116, 1133 CrossRef CAS.
- D. Knittel, Monatsh. Chem., 1986, 117, 491 CrossRef CAS.
- CAS registry number: 1026541-77-4.
- C. Balsamini, A. Bedini, R. Galarini and G. Spadoni, Tetrahedron, 1994, 50, 12375 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and 13C NMR spectra of the products 2a–2q, 3a–3m, and 4a–4b. See DOI: 10.1039/c5qo00182j |
| This journal is © the Partner Organisations 2015 |