
Polyethylene glycol-400/H3PO2: an eco-friendly reductive system for the synthesis of selanylesters†
G.
Perin
*,
M. B.
Silveira
,
A. M.
Barcellos
,
R. G.
Jacob
and
D.
Alves
*
LASOL, Universidade Federal de Pelotas, Pelotas, Brazil. E-mail: gelson_perin@ufpel.edu.br; diego.alves@ufpel.edu.br; Fax: +55 5332757533; Tel: +55 5332757533
First published on 21st September 2015
Abstract
An alternative green method was described for the synthesis of selanylesters by reactions of acyl chlorides with arylselenols, generated in situ by reaction of diaryl diselenides with hypophosphorous acid (H3PO2) using polyethylene glycol-400 (PEG-400) as the solvent. These reactions proceeded efficiently at room temperature under N2 using a range of acyl chlorides and diaryl diselenides, both containing electron-withdrawing and electron-donating groups, affording the corresponding selanylesters in moderate to excellent yields. Additionally, PEG-400 can be recovered and directly reused for further reactions.
Introduction
Interest in compounds containing sulfur, selenium and tellurium atoms has been continuously increasing due to their synthetic applications in the field of cross-coupling reactions,1 asymmetric synthesis,2 materials science,3 synthesis of natural products4 and their biological properties.5 More specifically, chalcogenylesters are versatile intermediates in several organic transformations, for example in synthetic chemistry as precursors of acyl anions and radicals. They have been used in the synthesis of more complex compounds6 and new natural products.7 In addition, these compounds demonstrated a wide spectrum of pharmacological activities.8Selanylesters were prepared from convenient methodologies that involve the condensation of nucleophilic selenium species with carbonyl compounds, such as acyl halides,9 anhydrides10 and aldehydes11 or alternative procedures with aryl iodides12 and alkynes.13 In general, the majority of these described methods for the synthesis of selanylesters requires long reaction times,9g high temperatures11b and the use of volatile solvents.9k However, there are some alternative methods using green solvents and conditions for the synthesis of selanylesters, such as the use of ionic liquids,9c–f water9l or microwave irradiation9h in solvent-free medium.11c
In the context of alternative solvents, polyethylene glycol-400 (PEG-400) has been considered as a green solvent because it has negligible vapor pressure, biodegradability, biocompatibility and low flammability, and is non-toxic and non-volatile. PEG-400 is known to be an inexpensive compound which serves as a suitable medium for environmentally friendly and safe chemical reactions.14 This green solvent is used in the widest range of organic transformations by multicomponent reactions,15 Michael additions,16 cycloaddition reactions17 and other carbon–carbon bond formation classic reactions.18 In the last few years, PEG has been described as the reaction medium for the addition of chalcogenolate anions to alkynes19 or electron-deficient alkenes,20 oxidation of disulfides21 and several procedures involving organochalcogen compounds.22
On the other hand, the most used synthetic protocol for the incorporation of selanyl units into organic compounds generally requires the preparation of nucleophilic selenium species, which can be generated via reductive cleavage of Se–Se bonds. Generally these species were generated in situ employing several reducing agents, such as NaBH4,23 PPh3,24 copper salts,25 SmI2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26 and other metals.27 In addition, in the literature the use of hypophosphorous acid (H3PO2) for this purpose was described and organylselenols could be in situ prepared using this reducing reagent.28 This acid is stable in air, it has no metal in its composition and could be used efficiently in aqueous solution. While the organylselenol is generated in situ under a nitrogen atmosphere, the bad smell of this selenating agent does not become an inconvenience. Our research group published three methodologies29 using H3PO2 for in situ generation of the organylselenols from diorganyl diselenides, with application in the synthesis of unsymmetrical diaryl selenides,29a organylselanyl pyridines29b and benzoselenazoles or benzoselenazolines.29c
26 and other metals.27 In addition, in the literature the use of hypophosphorous acid (H3PO2) for this purpose was described and organylselenols could be in situ prepared using this reducing reagent.28 This acid is stable in air, it has no metal in its composition and could be used efficiently in aqueous solution. While the organylselenol is generated in situ under a nitrogen atmosphere, the bad smell of this selenating agent does not become an inconvenience. Our research group published three methodologies29 using H3PO2 for in situ generation of the organylselenols from diorganyl diselenides, with application in the synthesis of unsymmetrical diaryl selenides,29a organylselanyl pyridines29b and benzoselenazoles or benzoselenazolines.29c
In this sense and due to our interest in green protocols correlated with organochalcogen chemistry, we report an alternative method for the preparation of selanylesters under mild reaction conditions without the use of a base or transition metal. This method involves reactions of acyl chlorides with arylselenols, generated in situ by the reaction of diaryl diselenides with H3PO2 using PEG-400 as the solvent (Scheme 1).
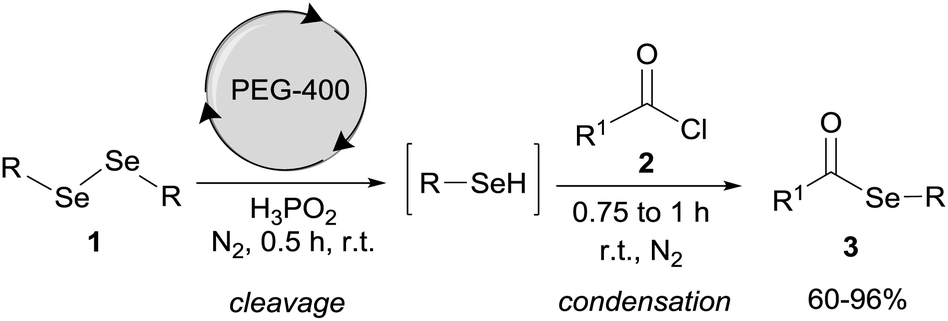 | ||
| Scheme 1 General scheme of the reaction. | ||
Results and discussion
In our preliminary experiments, we chose diphenyl diselenide 1a, H3PO2 (50 wt% in H2O) and benzoyl chloride 2a as model substrates to establish the best conditions for the selanylesterification reaction and some experiments, including stoichiometry and solvent tests, were performed to synthesize selanylester 3a (Table 1). Thus, a mixture of 0.50 mmol of diphenyl diselenide 1a and 0.10 mL of H3PO2 in THF (3.0 mL) was stirred at room temperature under a N2 atmosphere for 1.0 h to afford in situ the benzeneselenol 1a′. The diphenyl diselenide cleavage was accompanied by the change in the reaction solution color, from yellow to colorless. After this, benzoyl chloride 2a (1.0 mmol) was added in the reaction vessel and the reaction remained at room temperature for an additional 1.0 h. Under these reaction conditions the desired product 3a was obtained in 49% yield (Table 1, entry 1). To improve this result, we performed reactions by increasing the amount of diphenyl diselenide 1a or benzoyl chloride 2a, and fortunately, a good result was found when we used 0.60 mmol of reagent 1a (Table 1, entry 3).|
|
|||||
|---|---|---|---|---|---|
| # | 1a (mmol) | H3PO2 (mL) | Solvent | Time (h) cleavage/cond. | Yieldb (%) |
| a Reactions are performed using benzoyl chloride 2a (1.0 mmol) and solvent (3.0 mL) at room temperature under a N2 atmosphere. b Yields are given for isolated product 3a. c Reaction performed using 1.20 mmol of benzoyl chloride 2a. d The condensation step was performed at 0 °C. e The cleavage step was performed at 90 °C. | |||||
| 1 | 0.50 | 0.10 | THF | 1.0/1.0 | 49 |
| 2 | 0.55 | 0.10 | THF | 1.0/1.0 | 55 |
| 3 | 0.60 | 0.10 | THF | 1.0/1.0 | 62 |
| 4c | 0.50 | 0.10 | THF | 1.0/1.0 | 35 |
| 5d | 0.60 | 0.10 | THF | 1.0/1.0 | 52 |
| 6 | 0.60 | 0.05 | THF | 3.0/4.0 | 43 |
| 7 | 0.60 | 0.30 | THF | 0.7/1.0 | 65 |
| 8 | 0.60 | 0.50 | THF | 0.5/1.0 | 71 |
| 9 | 0.60 | 1.00 | THF | 0.5/1.0 | 73 |
| 10e | 0.60 | 0.50 | Toluene | — | — |
| 11 | 0.60 | 0.50 | MeCN | — | — |
| 12 | 0.60 | 0.50 | CH2Cl2 | — | — |
| 13 | 0.60 | 0.50 | DMF | 0.1/0.25 | 46 |
| 14 | 0.60 | 0.50 | EtOH | 1.5/0.7 | 68 |
| 15 | 0.60 | 0.50 | H2O | 1.5/3.0 | — |
| 16e | 0.60 | 0.50 | Glycerol | 1.0/5.0 | 7 |
| 17 | 0.60 | 0.50 | PEG-400 | 0.5/0.75 | 88 |
In another experiment using this amount of diphenyl diselenide 1a, when the reaction mixture was cooled to 0 °C in the condensation step, a decrease in the yield of product 3a was observed (Table 1, entry 5). We also analyzed the amount of H3PO2 in this reaction (Table 1, entries 5–9) and a good yield was achieved when we used 0.5 mL of this acid (Table 1, entry 8). Regarding the influence of the solvent on the reaction, a range of solvents were tested and in reactions using toluene, MeCN and CH2Cl2 the benzeneselenol 1a′ formation was not observed (Table 1, entries 10–12). When the reactions were carried out using DMF, EtOH, H2O or glycerol as solvents, the desired product 3a was obtained in lower yields compared to the reaction performed in THF (Table 1, entry 8 vs. 13–16). To our satisfaction, when we carried out the reaction using PEG-400 as the solvent, the corresponding product 3a was obtained in high yield (Table 1, entry 17). Thus, analyzing the results shown in Table 1, we established the best reaction conditions as those in the previous reaction of diphenyl diselenide 1a (0.6 mmol) with 50 wt% H3PO2 in H2O (0.5 mL) in PEG-400 (3.0 mL) at room temperature under N2 for 0.5 h (for the in situ formation of benzeneselenol 1a′), followed by the dropwise addition of benzoyl chloride 2a (1.0 mmol) and stirring for an additional 0.75 h at room temperature.30
One of the most attractive features of green solvents is that they frequently can be recycled. Thus, after reaction optimization, a study regarding the recovery and reuse of PEG-400 was performed. Subsequent to the formation of product 3a (monitored by TLC), the reaction mixture was diluted and extracted with a mixture of hexane/ethyl acetate 90:10 (3 × 15 mL). The upper phase was dried and the solvent evaporated. The inferior, PEG-400 phase was dried under vacuum and directly reused in the next cycle. However, a decrease in the yield of compound 3a was observed (63% yield). In view of this result, more experiments were performed with the recovered system, to which were added different amounts of H3PO2 (0.1, 0.3 and 0.5 mL) after each cycle. According to our studies, the obtained results indicate the necessity of the addition of 0.5 mL of H3PO2 in all runs, according to Fig. 1.31 It was observed that a moderate level of efficiency was maintained even after being reused three times. After these runs, the efficiency of recovered PEG-400 was reduced and the obtained yields of compound 3a were decreased.
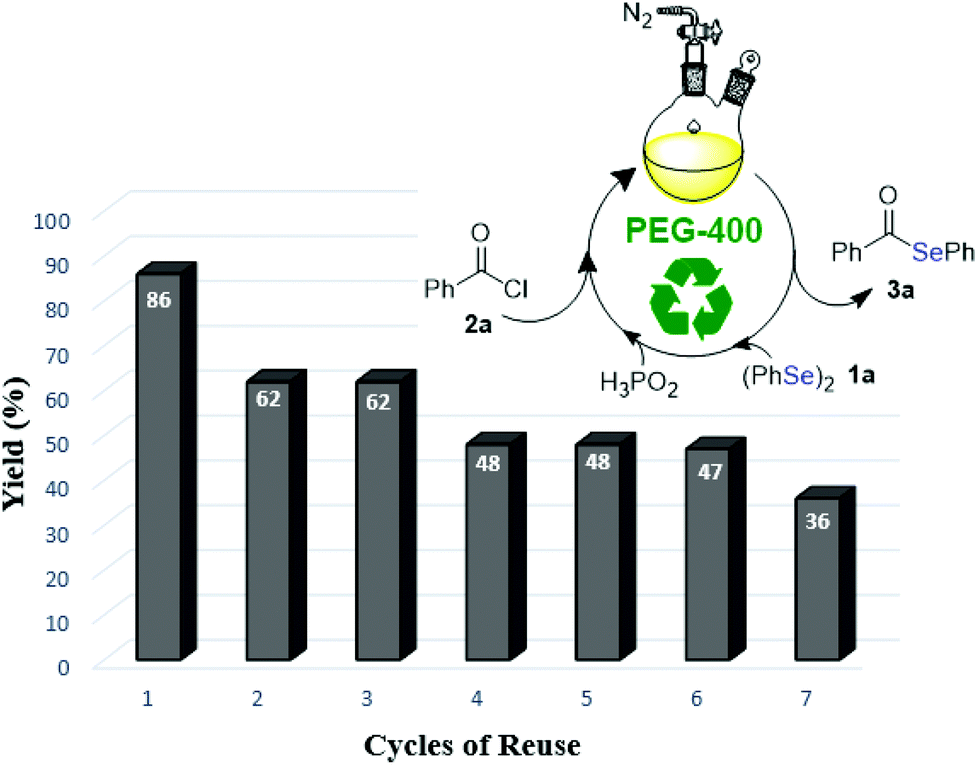 | ||
| Fig. 1 Reuse of solvent PEG-400. | ||
In addition, under optimized reaction conditions, the scope and limitations of this methodology were evaluated by reactions of a range of diorganyl diselenides 1a–f with a variety of acyl chlorides 2a–g (Table 2). The results shown in Table 2 reveal that our protocol worked well for a range of substrates employed. In a general way, the reactions are not sensitive to the electronic effect of the aromatic ring in the benzoyl chloride. According to the results, benzoyl chlorides containing electron-donating (Me), electron-withdrawing (Cl, Br) and electron-neutral groups at the aromatic ring gave good yields of desired selanylesters (Table 2, entries 1–5). The reaction performed with furan-2-carbonyl chloride 2f furnished the respective product 3f in good yield (81%) (Table 2, entry 6). Unfortunately, when the reaction was carried out with pivaloyl chloride 2g, only traces of the corresponding product 3g was obtained, even after 12.0 h at 60 °C (Table 2, entry 7).
|
|
|||
|---|---|---|---|
| # | Product 3 | Time (h) | Yieldb (%) |
| a Reactions were performed in the presence of diorganyl diselenides 1a–f (0.6 mmol), acyl chlorides 2a–g (1.0 mmol), 50 wt% H3PO2 (0.5 mL) and PEG-400 (3.0 mL) at room temperature under a N2 atmosphere. b Yields are given for isolated products. c The condensation step was performed at 60 °C. | |||
| 1 |
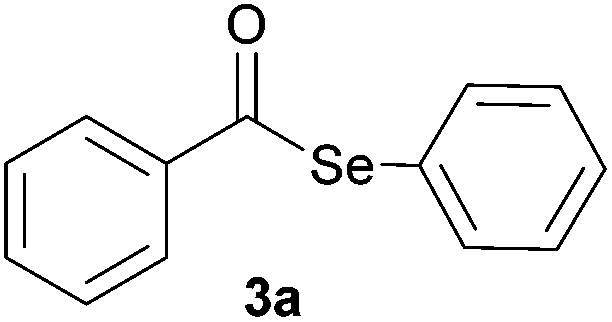
|
0.75 | 88 |
| 2 |
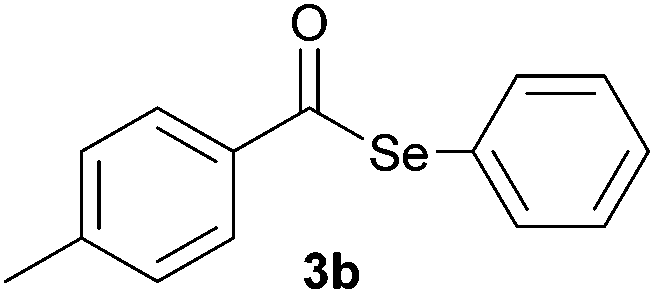
|
0.75 | 74 |
| 3 |
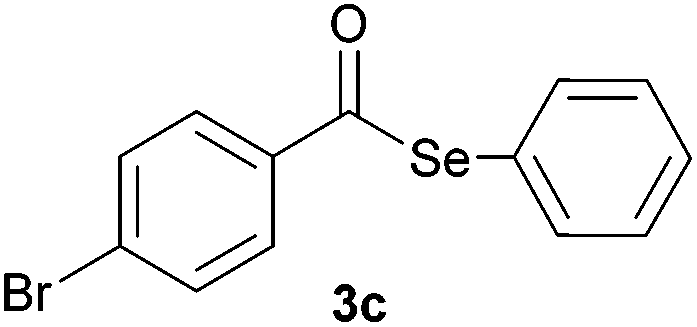
|
0.75 | 81 |
| 4 |
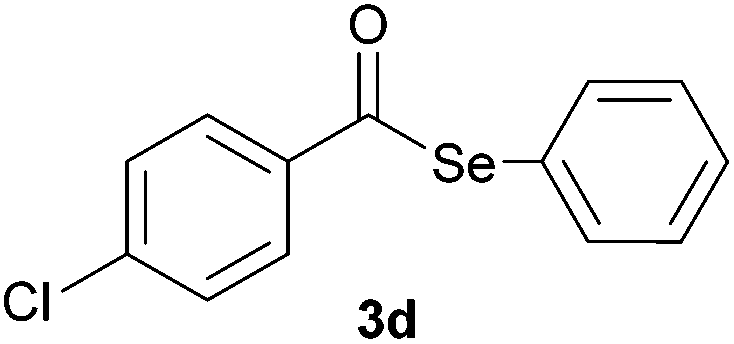
|
0.75 | 75 |
| 5 |
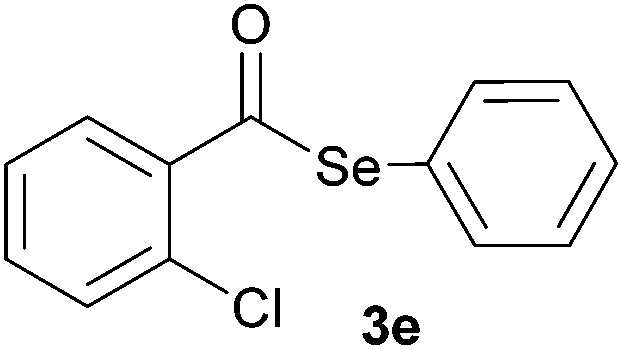
|
0.75 | 71 |
| 6 |
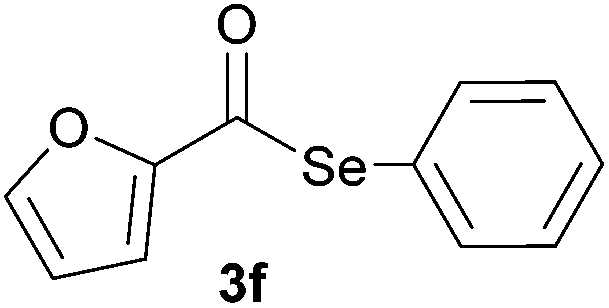
|
1.0 | 81 |
| 7c |
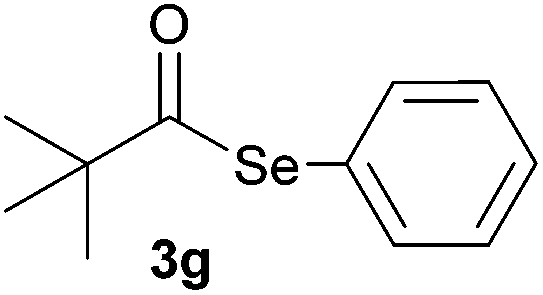
|
12.0 | Traces |
| 8 |
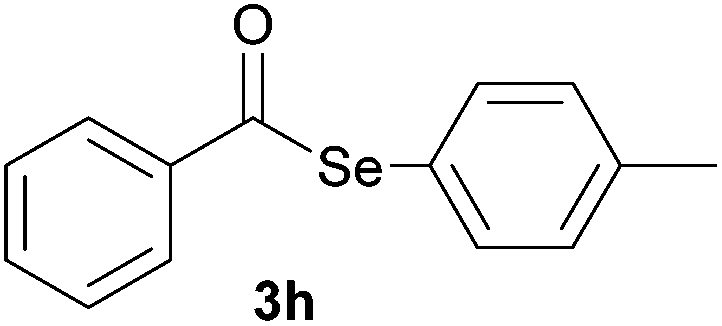
|
1.0 | 60 |
| 9 |
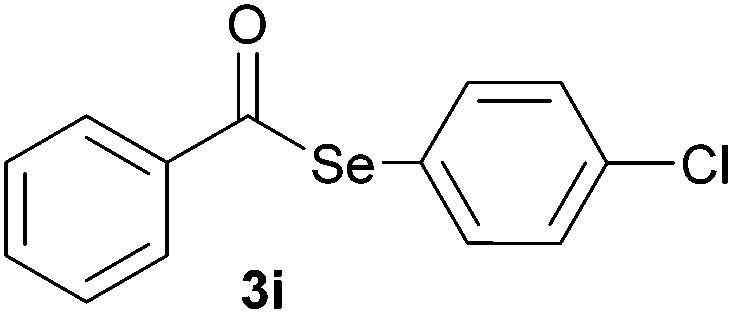
|
1.0 | 91 |
| 10 |

|
1.0 | 93 |
| 11 |
|
0.75 | 96 |
| 12c |
|
15.0 | Traces |
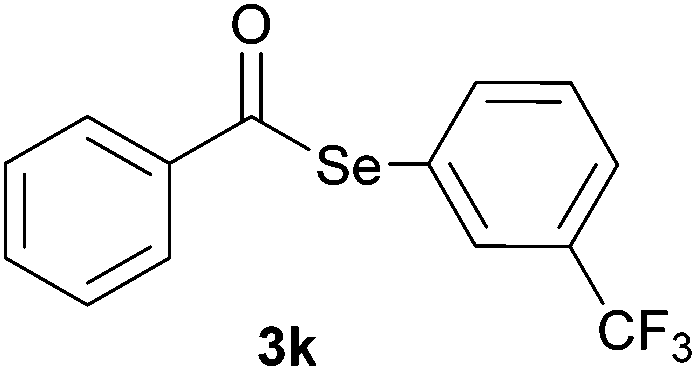
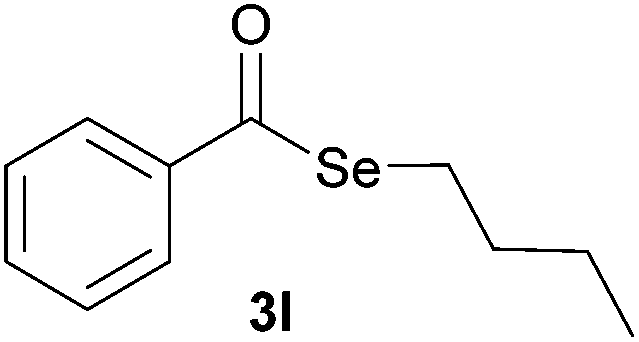
Later, the possibility of performing these reactions with other diorganyl diselenides 1b–f was also investigated. Different diaryl diselenides containing electron-donating and electron-withdrawing groups at the aromatic ring were efficiently reacted with benzoyl chloride 2a, affording the respective selanylesters 3h–k in acceptable yields (Table 2, entries 8–11). As shown in Table 2, the electronic effects on the aryl moiety of the diaryl diselenide seemed to have an influence on the product yield. For example, diaryl diselenide 1b, with an electron-donating group at the aromatic ring gave a lower result than those with electron-withdrawing groups (Table 2, entry 8 vs. 9–11). We also examined the reaction of the benzoyl chloride 2a with dibutyl diselenide 1f and only traces of desired product 3l was obtained, even after 15.0 h at 60 °C (Table 2, entry 12).
Conclusions
In conclusion, we developed an alternative and green method for the synthesis of selanylesters using PEG-400 as the solvent. This method involves reactions of acyl chlorides with arylselenols, generated in situ by reactions of diaryl diselenides with H3PO2. These reactions proceeded efficiently at room temperature under N2 using a range of acyl chlorides and diaryl diselenides, both containing electron-withdrawing and electron-donating groups, affording the corresponding selanylesters in moderate to excellent yields. Additionally, PEG-400 can be easily recovered and directly reused for further reactions without significant loss of efficiency.Acknowledgements
We thank the CNPq, CAPES, FAPERGS and FINEP for the financial support. CNPq is also acknowledged for the fellowship for G.P., R.G.J. and D.A.Notes and references
- I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596 CrossRef CAS PubMed.
- (a) T. Wirth, Angew. Chem., Int. Ed., 2000, 39, 3740 CrossRef CAS; (b) A. L. Braga, D. S. Lüdtke and F. Vargas, Curr. Org. Chem., 2006, 10, 1921 CrossRef CAS; (c) C. Santi, S. Santoro and B. Battistelli, Curr. Org. Chem., 2010, 14, 442 CrossRef; (d) E. E. Alberto, V. Nascimento and A. L. Braga, J. Braz. Chem. Soc., 2010, 21, 2032 CrossRef CAS; (e) D. M. Freudendahl, S. A. Shahzad and T. Wirth, Eur. J. Org. Chem., 2009, 1649 CrossRef CAS PubMed.
- (a) D. B. Rasale, I. Maity and A. K. Das, Chem. Commun., 2014, 50, 11397 RSC; (b) B. L. Ahuja, V. Raykar, R. Joshi, S. Tiwari, S. Talreja and G. Choudhary, Physica B, 2015, 465, 21 CrossRef CAS PubMed.
- (a) R. D. Mazery, M. Pullez, F. Lopez, S. R. Harutyunyan, A. J. Minnaard and B. L. Feringa, J. Am. Chem. Soc., 2005, 127, 9966 CrossRef PubMed; (b) B. M. Trost, J. Waser and A. Meyer, J. Am. Chem. Soc., 2007, 129, 14556 CrossRef CAS PubMed; (c) W. J. Wever, J. W. Bogart, J. A. Baccile, A. N. Chan, F. C. Schroeder and A. A. Bowers, J. Am. Chem. Soc., 2015, 137, 3494 CrossRef CAS PubMed.
- (a) C. Jacob, G. I. Giles, N. M. Giles and H. Sies, Angew. Chem., Int. Ed., 2003, 42, 4742 CrossRef CAS PubMed; (b) C. W. Nogueira, G. Zeni and J. B. T. Rocha, Chem. Rev., 2004, 104(5), 6255 CrossRef CAS PubMed; (c) B. K. Sarma and G. Mugesh, Org. Biomol. Chem., 2008, 6, 965 RSC; (d) M. Doering, L. A. Ba, N. Lilienthal, C. Nicco, C. Scherer, M. Abbas, A. A. P. Zada, R. Coriat, T. Burkholz, L. Wessjohann, M. Diederich, F. Batteux, M. Herling and C. Jacob, J. Med. Chem., 2010, 53, 6954 CrossRef CAS PubMed; (e) M. Ninomiyaa, D. R. Garudb and M. Koketsua, Coord. Chem. Rev., 2011, 255, 2968 CrossRef PubMed; (f) M. Ibrahim, W. Hassan, A. M. Deobald, A. L. Braga and J. B. T. Rocha, Biol. Trace Elem. Res., 2012, 149, 399 CrossRef CAS PubMed; (g) W. G. Salgueiro, M. C. D. F. Xavier, L. F. B. Duarte, D. F. Câmara, D. A. Fagundez, A. T. G. Soares, G. Perin, D. Alves and D. S. Avila, Eur. J. Med. Chem., 2014, 75, 448 CrossRef CAS PubMed; (h) F. N. Victoria, R. Anversa, F. Penteado, M. Castro, E. J. Lenardão and L. Savegnago, Eur. J. Pharmacol., 2014, 742, 131 CrossRef CAS PubMed; (i) L. Orian and S. Toppo, Free Radicals Biol. Med., 2014, 66, 65 CrossRef CAS PubMed; (j) J. C. Goldbeck, F. N. Victoria, A. M. Motta, L. Savegnago, R. G. Jacob, G. Perin, E. J. Lenardão and W. P. Silva, LWT - Food Sci. Technol., 2014, 59, 813 CrossRef CAS PubMed; (k) L. C. C. Gonçalves, F. N. Victória, D. B. Lima, P. M. Y. Borba, G. Perin, L. Savegnago and E. J. Lenardão, Tetrahedron Lett., 2014, 55, 5275 CrossRef PubMed; (l) F. Donato, N. F. Pavin, A. T. R. Goes, L. C. Souza, L. C. Soares, O. E. D. Rodrigues, C. R. Jesse and L. Savegnago, Pharm. Biol., 2015, 53, 395 CrossRef CAS PubMed.
- (a) T. M. Coleman, N. Li and F. Huang, Tetrahedron Lett., 2005, 46, 4307 CrossRef CAS PubMed; (b) G. Pattenden, D. A. Stoker and J. M. Winne, Tetrahedron, 2009, 65, 5767 CrossRef CAS PubMed.
- (a) M. Inoue, S. Yamashita, Y. Ishihara and M. Hirama, Org. Lett., 2006, 8, 5805 CrossRef CAS PubMed; (b) B. T. Horst, B. L. Feringa and A. J. Minnaard, Org. Lett., 2007, 9, 3013 CrossRef PubMed.
- (a) M. Baca, T. W. Muir, M. SchniSlzer and S. B. H. Kent, J. Am. Chem. Soc., 1995, 117, 1881 CrossRef CAS; (b) E. D. Alvarez, D. Plano, M. Font, A. Calvo, C. Prior, C. Jacob, J. A. Palop and C. Sanmartin, Eur. J. Med. Chem., 2014, 73, 153 CrossRef PubMed.
- (a) C. C. Silveira, A. L. Braga and E. L. Larghi, Organometallics, 1999, 18, 5183 CrossRef CAS; (b) K. Ren, M. Wang, P. Liu and L. Wang, Synthesis, 2010, 1078 CAS; (c) D. Singh, S. Narayanaperumal, K. Gul, M. Godoi, O. E. D. Rodrigues and A. L. Braga, Green Chem., 2010, 12, 957 RSC; (d) G. Tabarelli, E. E. Alberto, A. N. Deobald, G. Marin, O. E. D. Rodrigues, L. Dornelles and A. L. Braga, Tetrahedron Lett., 2010, 51, 5728 CrossRef CAS PubMed; (e) K. Gul, S. Narayanaperumal, L. Dornelles, O. E. D. Rodrigues and A. L. Braga, Tetrahedron Lett., 2011, 52, 3592 CrossRef CAS PubMed; (f) S. Narayanaperumal, E. E. Alberto, K. Gul, C. Y. Kawasoko, L. Dornelles, O. E. D. Rodrigues and A. L. Braga, Tetrahedron, 2011, 67, 4723 CrossRef CAS PubMed; (g) G. Marin, A. L. Braga, A. S. Rosa, F. Z. Galetto, R. A. Burrow, H. Gallardo and M. W. Paixão, Tetrahedron, 2009, 65, 4614 CrossRef CAS PubMed; (h) M. Godoi, E. W. Ricardo, G. V. Botteselle, F. Z. Galetto, J. B. Azeredo and A. L. Braga, Green Chem., 2012, 14, 456 RSC; (i) O. A. Wallner and K. J. Szabó, J. Org. Chem., 2005, 70, 9215 CrossRef CAS PubMed; (j) D. Taher and J. F. Corrigan, Organometallics, 2011, 30, 5943 CrossRef CAS; (k) A. Capperucci, A. Degl'Innocenti and C. Tiberi, Synlett, 2011, 2248 CrossRef CAS; (l) C. Santi, B. Battistelli, L. Testaferri and M. Tiecco, Green Chem., 2012, 14, 1277 RSC.
- W. Dan, H. Deng, J. Chen, M. Liu, J. Ding and H. Wu, Tetrahedron, 2010, 66, 7384 CrossRef CAS PubMed.
- (a) T. Inoue, T. Takeda, N. Kambe, A. Ogawa, I. Ryu and N. Sonoda, J. Org. Chem., 1994, 59, 5824 CrossRef CAS; (b) C. Hea, X. Qiana and P. Sun, Org. Biomol. Chem., 2014, 12, 6072–6075 RSC; (c) J. C. Liou, S. S. Badsara, Y. Huang and C. Lee, RSC Adv., 2014, 4, 41237 RSC.
- Y. Nishiyama, K. Tokunaga, H. Kawamatsu and N. Sonoda, Tetrahedron Lett., 2002, 43, 1507 CrossRef CAS.
- (a) A. L. Braga, T. L. C. Martins, C. C. Silveira and O. E. D. Rodrigues, Tetrahedron Lett., 2001, 57, 3297 CrossRef CAS; (b) M. Tiecco, L. Testaferri, A. Temperini, L. Bagnoli, F. Marini, C. Santi and R. Terlizzi, Eur. J. Org. Chem., 2004, 3447 CrossRef CAS PubMed.
- (a) F. M. Kerton and R. Marriott, RSC Green Chemistry Book Series - Alternative Solvents for Green Chemistry, RSCPublishing, Cambridge, UK, 2nd edn, 2013 Search PubMed; (b) C. Reichardt and T. Welton, Solvents and Solvent Effects in Organic Chemistry, WILEY-VCH, Weinheim, 4th edn, 2011 Search PubMed.
- Y. Gu, Green Chem., 2012, 14, 2091 RSC.
- K. S. Feu, A. F. Torre, S. Silva, M. A. F. Moraes Jr., A. G. Corrêa and M. W. Paixão, Green Chem., 2014, 16, 3169 RSC.
- I. Billault, F. Pessel, A. Petit, R. Turgis and M.-C. Scherrmann, New J. Chem., 2015, 39, 1986 RSC.
- M. Vafaeezadeh and M. M. Hashemi, J. Mol. Liq., 2015, 207, 73 CrossRef CAS PubMed.
- R. G. Lara, P. C. Rosa, L. K. Soares, M. S. Silva, R. G. Jacob and G. Perin, Tetrahedron, 2012, 68, 10414 CrossRef CAS PubMed.
- (a) G. Perin, E. L. Borges, P. C. Rosa, P. N. Carvalho and E. J. Lenardão, Tetrahedron Lett., 2013, 54, 1718 CrossRef CAS PubMed; (b) G. Perin, E. L. Borges, T. J. Peglow and E. J. Lenardão, Tetrahedron Lett., 2014, 55, 5652 CrossRef CAS PubMed.
- M.-T. Cai, G.-S. Lv, J.-X. Chen, W.-X. Gao, J.-C. Ding and H.-Y. Wu, Chem. Lett., 2010, 39, 368 CrossRef CAS.
- (a) K. H. V. Reddy, G. Satish, K. Ramesh, K. Karnakar and Y. V. D. Nageswar, Chem. Lett., 2012, 41, 585 CrossRef CAS; (b) G. Perin, E. L. Borges and D. Alves, Tetrahedron Lett., 2012, 53, 2066 CrossRef CAS PubMed; (c) D. Kundu, N. Mukherjee and B. C. Ranu, RSC Adv., 2013, 3, 117 RSC.
- (a) H. C. Braga, A. D. Wouters, F. B. Zerillo and D. S. Lüdtke, Carbohydr. Res., 2010, 345, 2328 CrossRef CAS PubMed; (b) E. J. Lenardão, L. C. C. Gonçalves, S. R. Mendes, M. T. Saraiva, D. Alves, R. G. Jacob and G. Perin, J. Braz. Chem. Soc., 2010, 21, 2093 CrossRef.
- S. Banerjee, L. Adak and B. C. Ranu, Tetrahedron Lett., 2012, 53, 2149 CrossRef CAS PubMed.
- (a) N. Taniguchi, J. Org. Chem., 2007, 72, 1241 CrossRef CAS PubMed; (b) A. L. Braga, T. Barcellos, M. W. Paixão, A. M. Deobald, M. Godoi, H. A. Stefani, R. Cella and A. Sharma, Organometallics, 2008, 27, 4009 CrossRef CAS; (c) V. G. Ricordi, C. S. Freitas, G. Perin, E. J. Lenardão, R. G. Jacob, L. Savegnago and D. Alves, Green Chem., 2012, 14, 1030 RSC.
- Y. W. Tu, L. J. Zhou, X. Lv and X. X. Wang, Indian J. Chem., 2014, 53B, 435 CAS.
- (a) J. Dowsland, F. McKerlie and D. J. Procter, Tetrahedron Lett., 2000, 41, 4923 CrossRef CAS; (b) W. Munbunjong, E. H. Lee, P. Ngernmaneerat, S. J. Kim, G. Singh, W. Chavasiri and D. O. Jang, Tetrahedron, 2009, 65, 2467 CrossRef CAS PubMed.
- W. H. H. Günther, J. Org. Chem., 1966, 31, 1202 CrossRef.
- (a) R. A. Balaguez, V. G. Ricordi, C. S. Freitas, G. Perin, R. F. Schumacher and D. Alves, Tetrahedron Lett., 2014, 55, 1057 CrossRef CAS PubMed; (b) S. Thurow, R. Webber, G. Perin, E. J. Lenardão and D. Alves, Tetrahedron Lett., 2013, 54, 3215 CrossRef CAS PubMed; (c) R. A. Balaguez, R. Krüger, C. S. Radatz, R. S. Rampon, E. J. Lenardão, P. H. Schneider and D. Alves, Tetrahedron Lett., 2015, 56, 2735 CrossRef CAS PubMed.
- General procedure for the synthesis of selanylesters 3a–l: To a 10.0 mL round-bottomed flask containing a solution of diorganyl diselenide 1a–f (0.6 mmol) in PEG-400 (3.0 mL) under N2 atmosphere, was added H3PO2 50 wt% in H2O (0.5 mL). The resulting solution was stirred for 0.5 hour at room temperature, when its color changes from yellow to colorless. After this, the corresponding acyl chlorides 2a–g (1.0 mmol) were added and the mixture was stirred at room temperature for the time indicated in Table 2. The reactions were monitored by TLC until total disappearance of the starting materials. After that, the reaction mixture was mixed with water (50.0 mL), extracted with ethyl acetate (3 × 15.0 mL), dried over MgSO4, and concentrated under vacuum. The residue was purified by column chromatography on silica gel using hexane as the eluent.
- General procedure for the recycling of PEG-400: The aforementioned procedure was used with diphenyl diselenide 1a (0.60 mmol), H3PO2 50 wt% in H2O (0.5 mL), benzoyl chloride 2a (1.0 mmol) and PEG-400 (3.0 mL). After the reaction was complete, the reaction mixture was washed with a mixture of hexane/ethyl acetate (90:10) (3 × 15.0 mL) and the upper organic phases were separated from PEG-400. The product was isolated according to the procedure above. The resulting PEG-400 phase was dried under vacuum and reused for further reactions without previous purification. For the best performance of recycling experiments, it was necessary for the addition of 0.5 mL of H3PO2 in each successive run.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00256g |
| This journal is © the Partner Organisations 2015 |