
Solvent-controlled nucleophilic trifluoromethylthiolation of Morita–Baylis–Hillman carbonates: dual roles of DABCO in activating the Zard's trifluoromethylthiolation reagent and the MBH carbonates†
Hai-Bin
Yang
,
Xing
Fan
,
Yin
Wei
* and
Min
Shi
*
State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, P. R. China. E-mail: weiyin@mail.sioc.ac.cn; mshi@mail.sioc.ac.cn
First published on 20th July 2015
Abstract
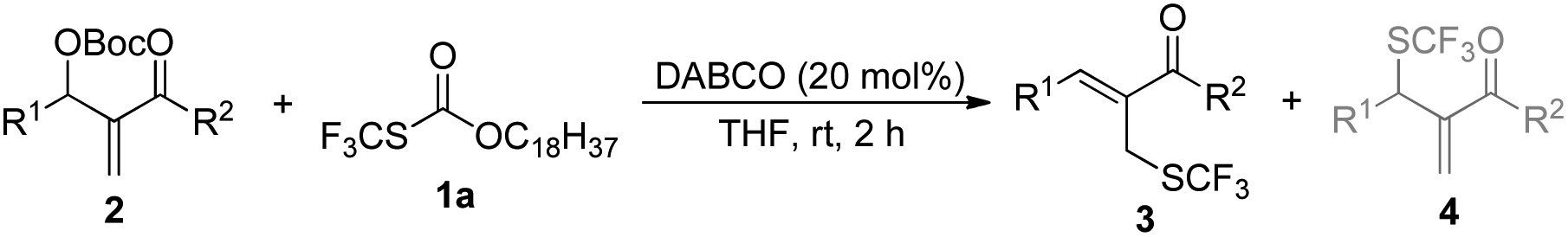
A novel amine-catalyzed nucleophilic trifluoromethylthiolation between Morita–Baylis–Hillman carbonates and O-octadecyl-S-trifluorothiolcarbonate has been developed. The regioselectivity of this reaction can be controlled by choosing different solvents, affording primary allylic SCF3 products in THF and secondary allylic SCF3 products in CHCl3 as major products. The mechanistic investigation indicated that DABCO plays a dual role in activating the Zard's trifluoromethylthiolation reagent and the MBH carbonates.
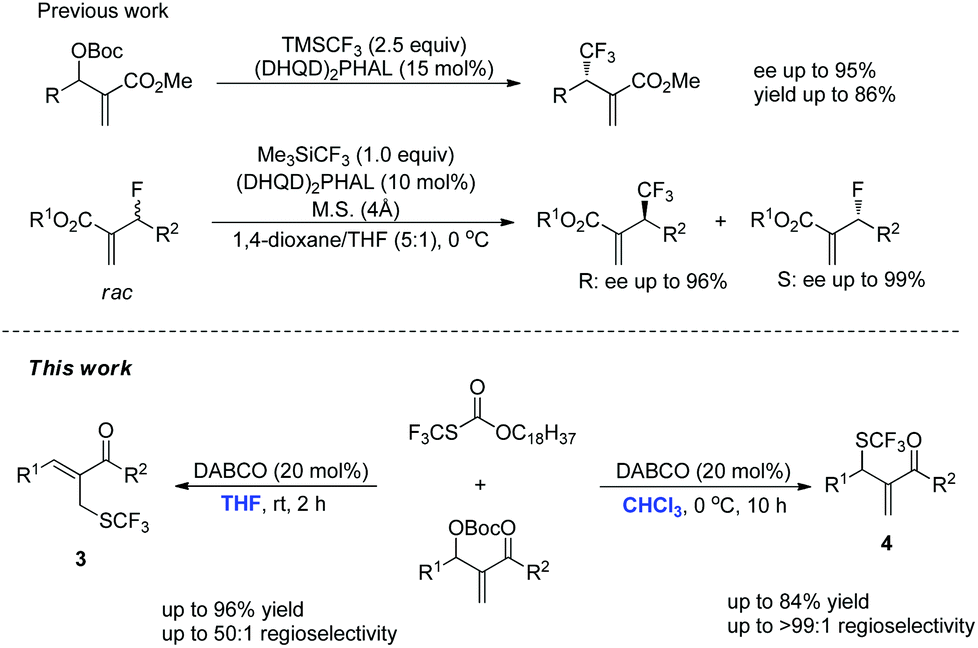
Fluorinated functional groups are key structural motifs found in various agrochemicals and pharmaceuticals.1 Approximately 30% of all agrochemicals and 20% of all pharmaceuticals on the market contain fluorine. Hence, the development of efficient methods for the selective introduction of fluorine into organic molecules has already become one of the hottest fields in modern chemistry.2 Among these substituents, the trifluoromethylthio group (CF3S–) plays an important part because of its high lipophilicity and strong electron-withdrawing effect. These characteristics have the similarity with those of trifluoromethyl (CF3–) and trifluoromethoxy (CF3O–) groups.3 Although impressive progress has been made in the formation of a C(sp2)–SCF3 bond in the past several years, the methods for the direct formation of C(sp3)–SCF3 bonds have been less developed. Thus far, successful examples include: (1) the trifluoromethylthiolations of β-ketoesters with electrophilic trifluoromethylthiolation reagents;4 (2) Lewis acid mediated difunctionalization of alkene with electrophilic trifluoromethylthiolation reagents;5 (3) the substitution reactions of aliphatic halides with nucleophilic trifluoromethylthiolation reagents;6 (4) copper-mediated trifluoromethylthiolations of α-diazo compounds with AgSCF3, etc.7
Lewis bases are widely used to catalyze asymmetric allylic alkylation using Morita–Baylis–Hillman (MBH) adducts as electrophiles, through an SN2′/SN2′ cascade and this synthetic method has emerged as a powerful strategy for the construction of multifunctional compounds.8 Recently, this synthetic method has been used to synthesize organofluorine compounds. For example, Shibata and Jiang have independently reported (DHQD)2PHAL-catalyzed asymmetric allylic trifluoromethylation of Morita–Baylis–Hillman carbonates using the Rupert–Prakash reagent (Scheme 1).9 Moreover, Shibata has also reported a (DHQD)2PHAL-catalyzed kinetic resolution of allyl fluorides to synthesize chiral allyl fluorides and trifluoromethylated compounds (Scheme 1).10 To the best of our knowledge, only one case of forming the C(sp3)–SCF3 bond using Lewis base catalyzed SN2′/SN2′ substitution has been reported during the preparation of this manuscript.11 With these precedents in mind and in connection with our ongoing efforts on developing novel reactions using nitrogen-containing Lewis bases as nucleophilic catalysts, we envisaged that an appropriate nucleophilic SCF3 reagent could be utilized in the direct introduction of a SCF3 unit in a catalytic manner. Herein, we report the solvent-controlled allylic trifluoromethylthiolation reaction of MBH adducts catalyzed by DABCO.
 | ||
| Scheme 1 Amine catalyzed allylic alkylation of MBH adducts for synthesizing fluorine-bearing compounds. | ||
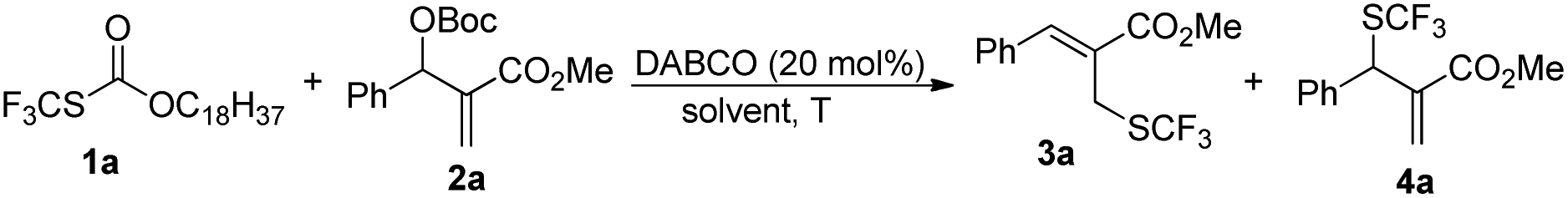
We began our studies on direct allylic trifluoromethylthiolation of MBH carbonates by utilizing O-octadecyl-S-trifluorothiolcarbonate (CF3SCO2C18H37) as the nucleophilic trifluoromethylthiolation reagent in the presence of DABCO.12 We chose O-octadecyl-S-trifluorothiolcarbonate as the SCF3 source based on two factors described as following: (1) O-octadecyl-S-trifluorothiolcarbonate is an efficient, cheap, air-stable, and easily available reagent; (2) O-octadecyl-S-trifluorothiolcarbonate can be activated by using amine to generate the trifluoromethylthiolate anion in situ. The results are summarized in Table 1. We found that primary allylic SCF3 product 3a was obtained in 40% yield as a major product along with concomitant formation of secondary allylic SCF3 product 4a in 14% yield when the reaction was carried out in CH3CN under the catalysis of DABCO (20 mol%) at room temperature for 2 h (Table 1, entry 1). Instead of DABCO, other commonly used Lewis bases such as DMAP, DBU, Et3N and PPh3 were also tested; however, no reaction occurred under the same reaction conditions. Further solvent screening indicated that (1) primary allylic SCF3 product 3a was obtained favourably in THF; (2) the reaction afforded secondary allylic SCF3 product 4a as the major product in a halohydrocarbon solvent. For example, using CHCl3 as the solvent afforded the corresponding secondary allylic SCF3 product 4a in 47% yield along with a ratio of 4a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3a = 23.0:1 (Table 1, entries 2–8). In THF, 3a was formed exclusively (Table 1, entries 9–11). When the reaction was carried out in THF, lowering the temperature to 0 °C did not improve the yield of product 3a (Table 1, entry 10). However, increasing the amount of O-octadecyl-S-trifluorothiolcarbonate to 2.0 equiv., the corresponding primary allylic SCF3 product 3a was obtained in 90% yield in THF (Table 1, entry 11). When CHCl3 was chosen as a solvent, lowering the temperature to 0 °C and increasing the amount of O-octadecyl-S-trifluorothiolcarbonate to 2.0 equiv., the corresponding secondary allylic SCF3 product 4a was obtained in 71% yield along with a ratio of 4a:3a = 26.0:1 (Table 1, entry 13).
:3a = 23.0:1 (Table 1, entries 2–8). In THF, 3a was formed exclusively (Table 1, entries 9–11). When the reaction was carried out in THF, lowering the temperature to 0 °C did not improve the yield of product 3a (Table 1, entry 10). However, increasing the amount of O-octadecyl-S-trifluorothiolcarbonate to 2.0 equiv., the corresponding primary allylic SCF3 product 3a was obtained in 90% yield in THF (Table 1, entry 11). When CHCl3 was chosen as a solvent, lowering the temperature to 0 °C and increasing the amount of O-octadecyl-S-trifluorothiolcarbonate to 2.0 equiv., the corresponding secondary allylic SCF3 product 4a was obtained in 71% yield along with a ratio of 4a:3a = 26.0:1 (Table 1, entry 13).
|
|
|||||
|---|---|---|---|---|---|
| Entrya | Solvent | T (°C) | Yield of 3ab (%) | Yield of 4ab (%) |
3a:4a |
| a 2a (0.05 mmol) and 1a (0.05 mmol) were mixed in 0.5 mL of solvent. DABCO (20 mol%) was added and the mixture was stirred further for two hours. b The yields were determined using 1H NMR spectroscopy with 1,3,5-trimethoxybenzene as an internal standard. c The reaction time was prolonged to 10 hours. d 2.0 eq. 1a was used. | |||||
| 1 | CH3CN | Rt | 40 | 14 | 2.9:1 |
| 2 | Toluene | Rt | 40 | 31 | 1.3:1 |
| 3 | DMF | Rt | 5 | — | — |
| 4 | EtOAc | rt | 54 | — | — |
| 5 | n-Hexane | rt | 13 | 18 | 1:1.4 |
| 6 | CH2ClCH2Cl | rt | 13 | 39 | 1:3.0 |
| 7 | CH2Cl2 | rt | 2 | 40 | 1:20 |
| 8 | CHCl3 | rt | 2 | 47 | 1:23.0 |
| 9 | THF | rt | 74 | — | — |
| 10c | THF | 0 | 50 | — | — |
| 11d | THF | rt | 90 | — | — |
| 12c | CHCl3 | 0 | 2 | 50 | 1:25.0 |
| 13c,d | CHCl3 | 0 | 3 | 71 | 1:26.0 |
With the optimized reaction conditions in hand, we next investigated the generality of this allylic trifluoromethylthiolation for synthesizing primary allylic SCF3 products (Table 2). As for MBH carbonates 2b and 2c, the reaction afforded the desired products 3b and 3c with excellent yields and regioselectivities (Table 2, entries 1 and 2). However, when R1 was 4-BrC6H4 or 4-NO2C6H4 and the methyl ester group was changed to the t-butyl ester group, the corresponding products 3d and 3f were obtained in lower yields along with lower regioselectivities, presumably due to the steric hindrance (Table 2, entries 3 and 5). For MBH carbonates 2g and 2h having an electron-rich aromatic group, the reaction could not acquire good regioselectivities, perhaps due to the electronic effects (Table 2, entries 6 and 7). Using MBH carbonate 2i containing a 2-bromophenyl group, the corresponding product 3i was obtained in 90% yield and 25:1 regioselectivity (Table 2, entry 8). We were also pleased to find that MBH carbonates bearing the naphthalenyl group and the heteroaromatic group were also suitable for this reaction, affording the corresponding products 3j–3n in good yields and good regioselectivities (Table 2, entries 9–13). The structure of product 3l was confirmed by X-ray diffraction and its ORTEP drawing is shown in Fig. 1 (see the ESI† for the CIF data). Notably, the reaction afforded product 3o in 96% yield and 50.0:1 regioselectivity as for vinyl methyl ketone derived MBH carbonate 2o (Table 2, entry 14).
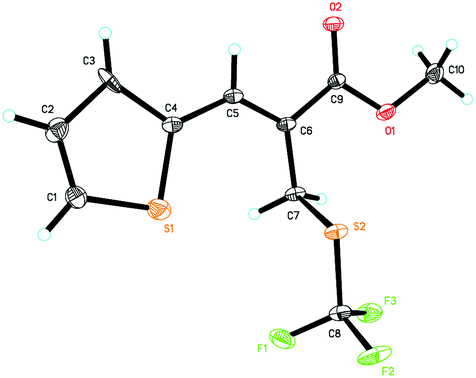 | ||
| Fig. 1 ORTEP drawing of 3l. | ||
|
|
|||
|---|---|---|---|
| Entrya | R1, R2 | Yield of 3b (%) |
3:4c |
|
a
2 (0.2 mmol) and 1 (0.4 mmol) were mixed in 2.0 mL of THF. DABCO (20 mol%) was added and the mixture was stirred further for two hours at rt.
b Isolated yields.
c The ratio of 3:4 was determined by 1H NMR spectroscopic data of the crude products.
d
Z:E = 16.3:1.
|
|||
| 1 | 2b, 4-ClC6H4, OMe | 3b, 93 | 40.0:1 |
| 2 | 2c, 4-CNC6H4, OMe | 3c, 91 | 42.0:1 |
| 3 | 2d, 4-BrC6H4, OtBu | 3d, 50 | 1.1:1 |
| 4 | 2e, 4-BrC6H4, OMe | 3e, 94 | 60.0:1 |
| 5d | 2f, 4-NO2C6H4, OtBu | 3f, 61 | 10.0:1 |
| 6 | 2g, 4-MeOC6H4, OEt | 3g, 47 | 1.4:1 |
| 7 | 2h, 3-MeC6H4, OMe | 3h, 80 | 5.0:1 |
| 8 | 2i, 2-BrC6H4, OMe | 3i, 90 | 25.0:1 |
| 9 | 2j, 1-naphthalenyl, OMe | 3j, 91 | 18.0:1 |
| 10 | 2k, 2-naphthalenyl, OMe | 3k, 89 | 15.0:1 |
| 11 | 2l, 2-thienyl, OMe | 3l, 94 | 32.0:1 |
| 12 | 2m, 2-furyl, OMe | 3m, 75 | 38.0:1 |
| 13 | 2n, 2-pyridyl, OMe | 3n, 87 | 49.0:1 |
| 14 | 2o, Ph, Me | 3o, 96 | 50.0:1 |
Under the optimized reaction conditions, we next investigated the generality of this allylic trifluoromethylthiolation for synthesizing secondary allylic SCF3 products and the results are summarized in Table 3. When R1 was 4-ClC6H4 or 4-NCC6H4 for MBH carbonate 2b or 2c, the reaction proceeded smoothly to give the corresponding trifluoromethylthiolated products 4b and 4c in 84% yield and 46% yield, respectively, but the regioselectivity for MBH carbonate 2c was not ideal, suggesting that the substituent on the phenyl ring plays a significant role in the reaction outcomes (Table 3, entries 1 and 2). The cyano substituents on the phenyl ring might have different electronic properties from the chlorine atom, causing a lower regioselectivity in the case of 2c. As for MBH carbonates 2d and 2f having the sterically hindered t-butyl ester group, the reaction had to be carried out at room temperature, affording the corresponding trifluoromethylthiolated products 3d and 3f in good yields and good regioselectivities (Table 3, entries 3 and 5). Comparing with MBH carbonates bearing an electron-deficient aromatic group, the reaction of 2g and 2h bearing an electron-rich aromatic ring with 1a could acquire better regioselectivities (Table 3, entries 6 and 7). In the case of MBH carbonate 2i, the trifluoromethylthiolated product 4i was obtained in 79% yield and 13.0:1 regioselectivity (Table 3, entry 8). With regard to MBH carbonates 2j and 2k bearing the naphthalenyl group, the reaction could afford the desired products 4j and 4k in good yields and regioselectivities (Table 3, entries 9 and 10). Using MBH carbonate 2l bearing a 2-thienyl aromatic ring as a substrate, the catalyst loading had to be increased to 40 mol% in order to consume the starting material completely, affording the corresponding product 4l in 60% yield and 5:1 regioselectivity (Table 3, entry 11). When R1 was a 2-furyl group for MBH carbonate, the reaction rate was sluggish at 0 °C, thus the reaction had to be conducted at room temperature, furnishing the corresponding product 4m in 26% yield along with 2.1:1 regioselectivity (Table 3, entry 12). As for MBH carbonate 2n, the corresponding product 4n was obtained in medium yield and excellent regioselectivity, presumably due to the electronic effect (Table 3, entry 13). In the case of vinyl methyl ketone derived MBH carbonate 2o, the reaction yielded 3o as the major product although chloroform was employed as a solvent, only affording the secondary allylic SCF3 product 4o in 37% yield (Table 3, entry 14).
|
|
|||
|---|---|---|---|
| Entrya | R1, R2 | Yield of 4b (%) |
4:3c |
|
a
2 (0.2 mmol) and 1 (0.2 mmol) were mixed in 2.0 mL of CHCl3. DABCO (20 mol%) was added and the mixture was stirred further for ten hours at 0 °C.
b Isolated yields.
c The ratio of 3:4 was determined by 1H NMR spectroscopic data of the crude products.
d Carried out at rt.
e 40 mol% of DABCO was added.
|
|||
| 1 | 2b, 4-ClC6H4, OMe | 4b, 84 | 12.0:1 |
| 2 | 2c, 4-CNC6H4, OMe | 4c, 46 | 1.6:1 |
| 3d | 2d, 4-BrC6H4, OtBu | 4d, 76 | 13.0:1 |
| 4 | 2e, 4-BrC6H4, OMe | 4e, 50 | 20.0:1 |
| 5d | 2f, 4-NO2C6H4, OtBu | 4f, 83 | 31.0:1 |
| 6e | 2g, 4-MeOC6H4, OEt | 4g, 63 | 50.1:1 |
| 7 | 2h, 3-MeC6H4, OMe | 4h, 84 | 34.0:1 |
| 8 | 2i, 2-BrC6H4, OMe | 4i, 79 | 13.0:1 |
| 9 | 2j, 1-naphthalenyl, OMe | 4j, 81 | 39.0:1 |
| 10 | 2k, 2-naphthalenyl, OMe | 4k, 77 | 15.0:1 |
| 11e | 2l, 2-thienyl, OMe | 4l, 60 | 5.0:1 |
| 12d | 2m, 2-furyl, OMe | 4m, 26 | 2.1:1 |
| 13 | 2n, 2-pyridyl, OMe | 4n, 51 | >99:1 |
| 14 | 2o, Ph, Me | 4o, 37 | 0.6:1 |
After investigating the substrate scope, we next focused on the exploration of the reaction mechanism. As reported by Deng and co-workers, tertiary amine catalyzed cyanation of ketones involved a quaternary ammonium salt intermediate generated in situ from cyanoformate and the tertiary amine.13 Due to the similar electronic properties between O-octadecyl-S-trifluorothiolcarbonate and cyanoformate, we supposed that this allylic trifluoromethylthiolation underwent a quaternary ammonium salt intermediate as well. To verify the existence of an ammonium salt intermediate, we treated 1a (1.0 equiv.), 2a (1.0 equiv.) with DABCO (1.0 equiv.) in CDCl3 and monitored the reaction proceeding by 19F NMR spectroscopy. As shown in Fig. 2, after a reaction time of 15 min, a signal at δ = −16.515 was observed. We hypothesized that this signal corresponded to the ammonium salt species generated in situ from 1a and DABCO because it was close to the signal of [Me4N][SCF3] in the 19F NMR spectrum (δ = −6.49, CD3CN).14 Treating 2a with 1b in THF/CH3CN in the absence of DABCO, no reaction occurred, suggesting that the newly generated SCF3 anion could not nucleophilically attack 2a without the assistance of DABCO (Scheme 2). These experiments indicated that DABCO played a dual role in activating the Zard's trifluoromethylthiolation reagent and the MBH carbonates. When THF was chosen as a solvent, the trifluoromethylthiolated product 3a was obtained in 82% yield under the catalysis of quinuclidine, which might suggest that the mechanism of activating the Zard's trifluoromethylthiolation reagent and the MBH carbonate by the two nitrogen atoms of one DABCO molecule was unlikely (Scheme 2).
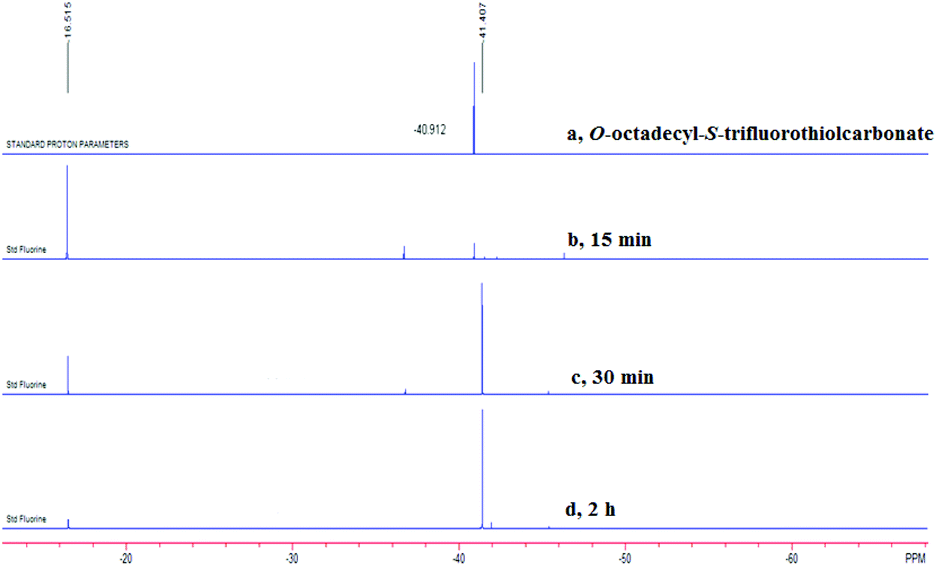 | ||
| Fig. 2 19F NMR spectroscopic tracing experiment (376 MHz, CDCl3, CFCl3): (a) the 19F NMR spectrum of O-octadecyl-S-trifluorothiolcarbonate (δ = −40.912); (b) the reaction was carried out for 15 min; (c) the reaction was carried out for 30 min; (d) the reaction was carried out for 2 h. | ||
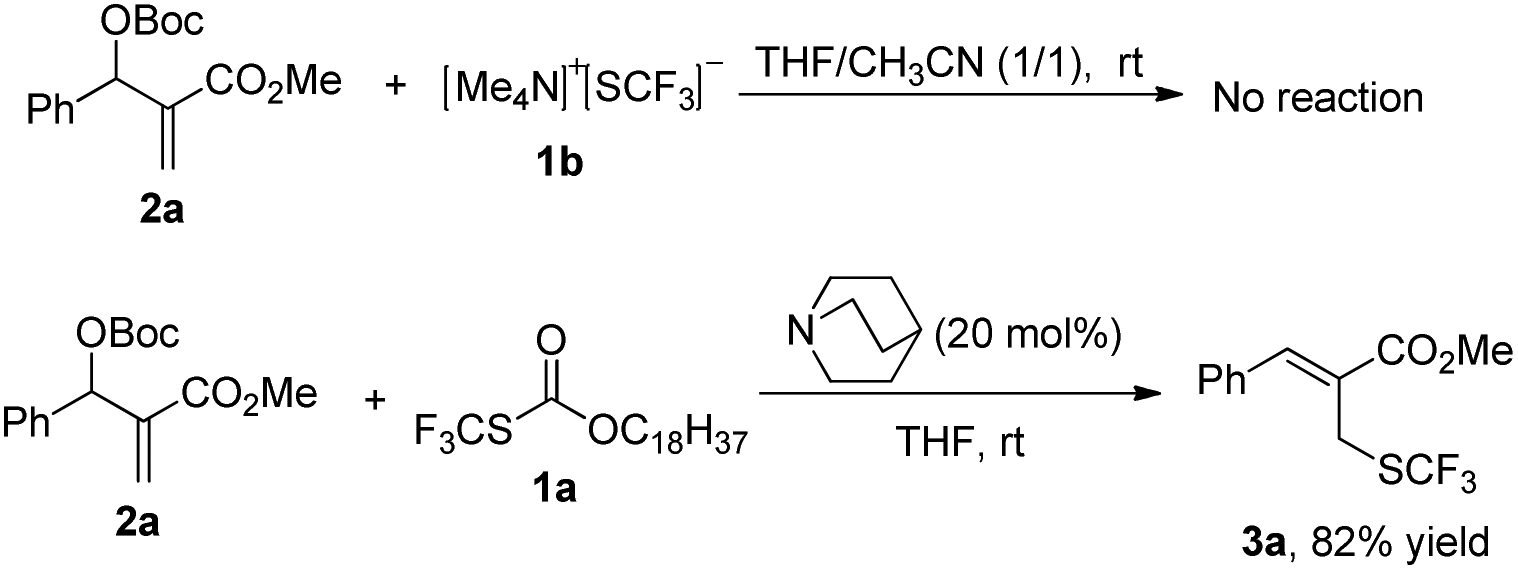 | ||
| Scheme 2 Control experiments to probe the reaction mechanisms. | ||
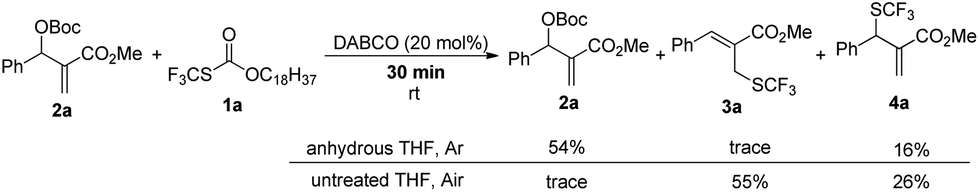
When the reaction was conducted in THF and CHCl3, carbonate 5a was isolated in 35% and 70% yields, respectively. Treating 1a, 2a and 6a in CHCl3 under the catalysis of DABCO, we could recover 6a completely by silica gel column chromatography, suggesting that 6a could not be transformed into 5a (Scheme 3). During further exploration of the reaction mechanism, we found that water played a crucial role in the reaction. When the reaction was carried out in anhydrous THF under an Ar atmosphere for 30 min, 4a was obtained in 16% yield along with 54% of 2a recovered after the reaction was quenched by hydrochloric acid. However, treating 1a with 2a in untreated THF under an ambient atmosphere afforded 3a in 55% yield along with 4a in 26% yield (Scheme 4). Moreover, the control experiment shown in Scheme 5 indicated that 4a can be transformed into 3a under the catalysis of DABCO in THF, suggesting that the formation of 4a was a kinetically controlled process and the formation of 3a was a thermodynamic-controlled process (Scheme 5).15 A similar phenomenon has been also observed by Cahard and co-workers.11
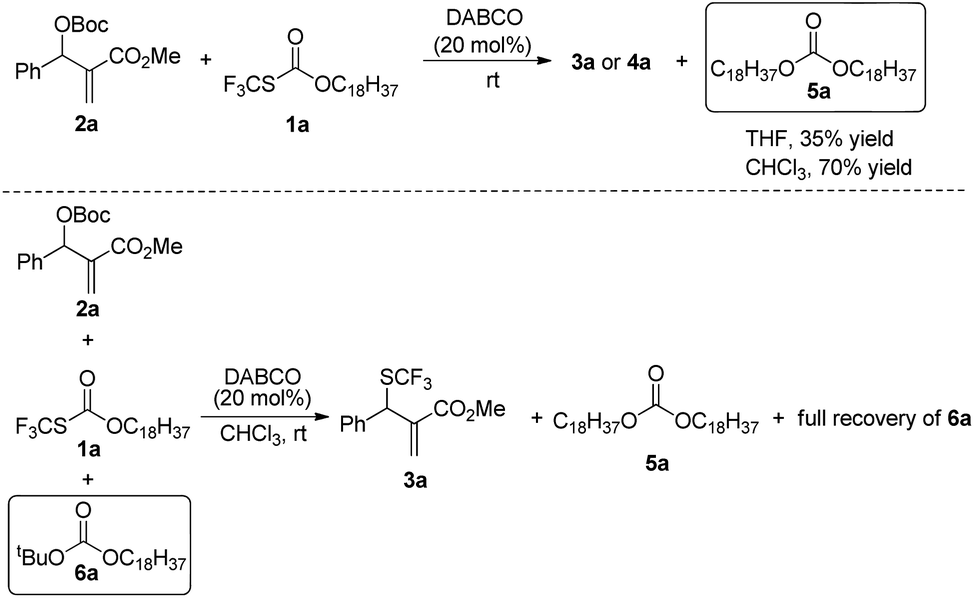 | ||
| Scheme 3 The isolation of carbonate 5a. | ||
 | ||
| Scheme 4 The water effect in the allylic trifluoromethylthiolation reaction. | ||
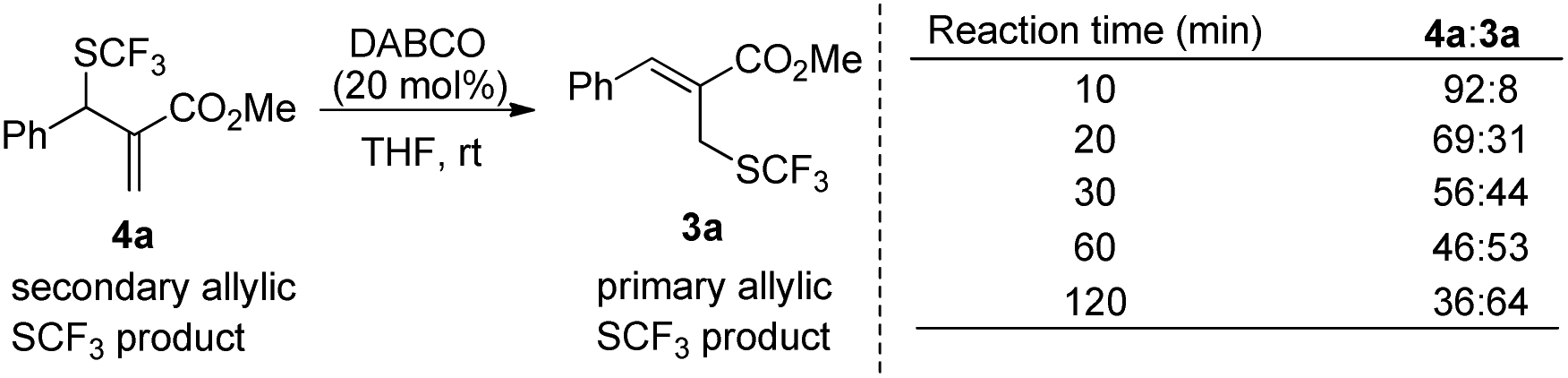 | ||
| Scheme 5 The transformations of 4a into 3a in THF. | ||
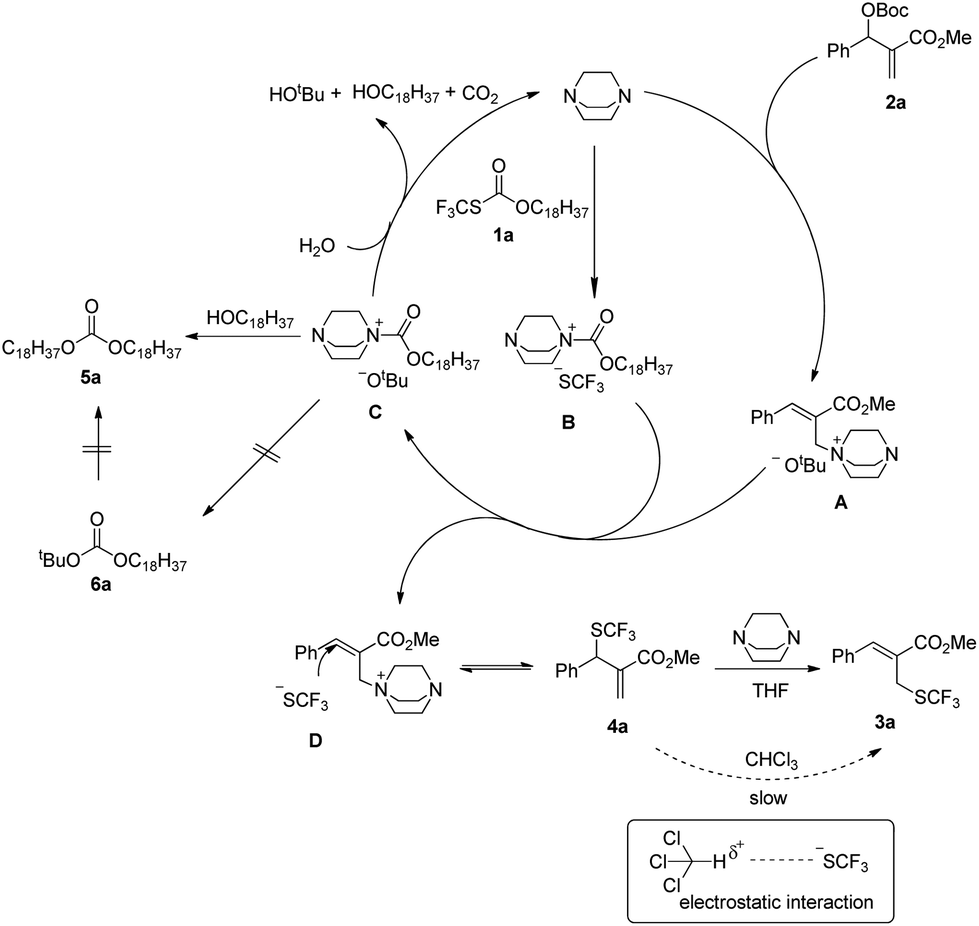
A plausible mechanism has been proposed in Scheme 6 on the basis of the above investigations. The solvent-controlled allylic trifluoromethylthiolation reactions are initiated by the formation of ammonium salt intermediates A and B derived from the nucleophilic addition of DABCO to 2a and 1a, respectively. The exchange between the SCF3 anion and the t-butoxyl anion delivers ammonium salt intermediates C and D. An intermolecular SN2′ reaction converts allylammonium ion D to the kinetic product 4a. Under the catalysis of DABCO, the transformation of 4a to 3a can take place readily in THF. However, when the reaction was conducted in CHCl3, the transformation of 4a to 3a can hardly take place because the electrostatic interaction between the SCF3 anion and CHCl3 weakens the nucleophilicity of the SCF3 anion. The nucleophilic attack of H2O to ammonium salt intermediate C yields tert-butanol, N-octadecanol, carbon dioxide and regenerates DABCO for the catalytic cycle. Carbonate 5a is generated via the nucleophilic addition of N-octadecanol to ammonium salt intermediate C, but carbonate 6a cannot be formed because of the weak nucleophilicity of the t-butoxyl anion.
 | ||
| Scheme 6 A proposed mechanism. | ||
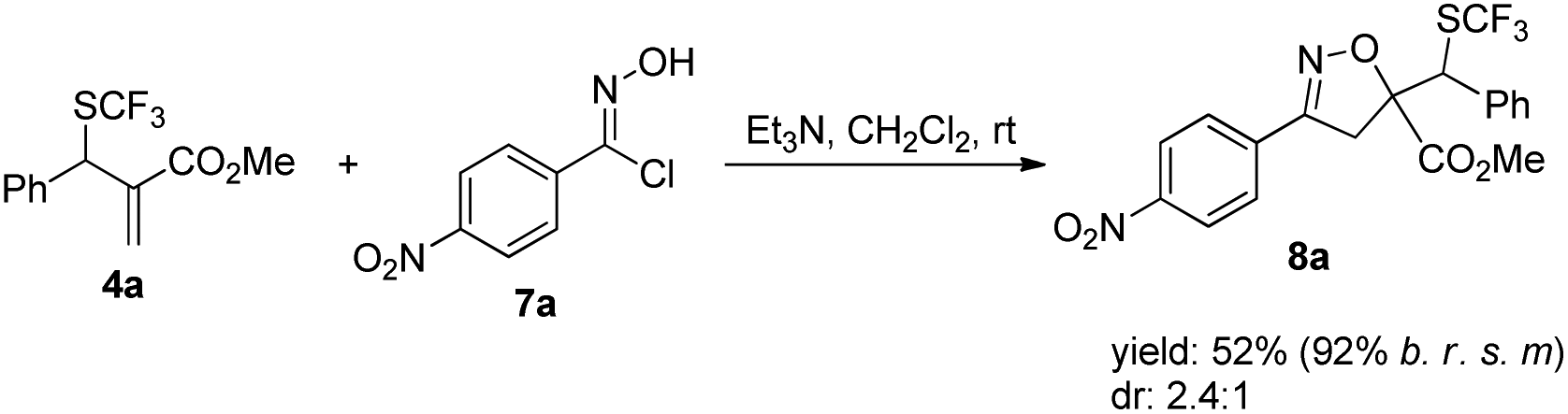
Furthermore, product 4a could be smoothly transformed into isoxazole 8a incorporating a SCF3 group as diastereoisomeric mixtures via a 1,3-dipolar cycloaddition (Scheme 7).
 | ||
| Scheme 7 The transformations of 4a into 8a. | ||
In summary, we have developed a novel amine-catalyzed nucleophilic trifluoromethylthiolation between Morita–Baylis–Hillman carbonates and O-octadecyl-S-trifluorothiolcarbonates under mild conditions. The regioselectivity of this reaction can be controlled by choosing different solvents, affording primary allylic SCF3 products in THF and secondary allylic SCF3 products in CHCl3 as major products. The mechanistic investigation showed that DABCO plays a dual role in activating the Zard's trifluoromethylthiolation reagent and the MBH carbonates. Various cinchona alkaloid derived catalysts did not catalyze this reaction. Therefore, at the present stage, we did not achieve any success in the asymmetric version of this reaction. Nevertheless, the asymmetric variant of this reaction will be further explored in our laboratory.
Acknowledgements
We thank the National Basic Research Program of China (973)-2015CB856603 and the National Natural Science Foundation of China (20472096, 21372241, 21361140350, 20672127, 21421091, 21302203, 21102166, 21372250 and 20732008).Notes and references
- (a) T. Yamazaki, T. Taguchi and I. Ojima, Fluorine in Medicinal Chemistry and Chemical Biology, ed. I. Ojima, Wiley-Black-well, Chichester, 2009 Search PubMed; (b) P. Jeschke, ChemBioChem, 2004, 5, 570 CrossRef CAS PubMed; (c) K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (d) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (e) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed; (f) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed.
- For reviews, see: (a) C. Alonso, E. M. de Marigorta, G. Rubiales and F. Palacios, Chem. Rev., 2015, 115, 1847 CrossRef CAS PubMed; (b) X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826 CrossRef CAS PubMed; (c) X.-H. Xu, K. Matsuzaki and N. Shibata, Chem. Rev., 2015, 115, 731 CrossRef CAS PubMed; (d) X. Shao, C. Xu, L. Lu and Q. Shen, Acc. Chem. Res., 2015, 48, 1227 CrossRef CAS PubMed; (e) C. Ni, M. Hu and J. Hu, Chem. Rev., 2015, 115, 765 CrossRef CAS PubMed; (f) L. Chu and F.-L. Qing, Acc. Chem. Res., 2014, 47, 1513 CrossRef CAS PubMed.
- (a) A. Leo, C. Hansch and D. Elkins, Chem. Rev., 1971, 71, 525 CrossRef CAS; (b) C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS; (c) S. Manteau, S. Pazenok, J. P. Vors and F. R. Leroux, J. Fluorine Chem., 2010, 131, 140 CrossRef PubMed; (d) F. Leroux, P. Jeschke and M. Schlosser, Chem. Rev., 2005, 105, 827 CrossRef CAS PubMed.
- (a) T. Bootwicha, X. Liu, R. Pluta, I. Atodiresei and M. Rueping, Angew. Chem., Int. Ed., 2013, 52, 12856 CrossRef CAS PubMed; (b) X. Wang, T. Yang, X. Cheng and Q. Shen, Angew. Chem., Int. Ed., 2013, 52, 12860 CrossRef CAS PubMed; (c) M. Rueping, X. Liu, T. Bootwicha, R. Pluta and C. Merkens, Chem. Commun., 2014, 50, 2508 RSC; (d) X.-L. Zhu, J.-H. Xu, D.-J. Cheng, L.-J. Zhao, X.-Y. Liu and B. Tan, Org. Lett., 2014, 16, 2192 CrossRef CAS PubMed.
- A. Ferry, T. Billard, B. R. Langlois and E. Bacqué, Angew. Chem., Int. Ed., 2009, 48, 8551 CrossRef CAS PubMed.
- (a) S. Munavalli, G. W. Wagner, B. Hashemi, D. K. Rohrbaugh and H. D. Durst, Synth. Commun., 1997, 27, 2847 CrossRef CAS PubMed; (b) W. Tyrra, D. Naumann, B. Hoge and Y. L. Yagupolskii, J. Fluorine Chem., 2003, 119, 101 CrossRef CAS; (c) D. Kong, Z. Jiang, S. Xin, Z. Bai, Y. Yuan and Z. Weng, Tetrahedron, 2013, 69, 6046 CrossRef CAS PubMed; (d) Q. Lin, L. Chen, Y. Huang, M. Rong, Y. Yuan and Z. Weng, Org. Biomol. Chem., 2014, 12, 5500 RSC; (e) C. Chen, X.-H. Xu, B. Yang and F.-L. Qing, Org. Lett., 2014, 16, 3372 CrossRef CAS PubMed.
- (a) M. Hu, J. Rong, W. Miao, C. Ni, Y. Han and J. Hu, Org. Lett., 2014, 16, 2030 CrossRef CAS PubMed; (b) X. Wang, Y. Zhou, G. Ji, G. Wu, M. Li, Y. Zhang and J. Wang, Eur. J. Org. Chem., 2014, 3093 CrossRef CAS PubMed; (c) Q. Lefebvre, E. Fava, P. Nikolaienko and M. Rueping, Chem. Commun., 2014, 50, 6617 RSC.
- For reviews, see: (a) T.-Y. Liu, M. Xie and Y.-C. Chen, Chem. Soc. Rev., 2012, 41, 4101 RSC; (b) Y. Wei and M. Shi, Chem. Rev., 2013, 113, 6659 CrossRef CAS PubMed. For selected examples, see: (c) C.-W. Cho, J.-R. Kong and M. Krische, J. Org. Lett., 2004, 6, 1337 CrossRef CAS PubMed; (d) D. J. V. C. van Steenis, T. Marcelli, M. Lutz, A. L. Spek, J. H. van Maarseveen and H. Hiemstra, Adv. Synth. Catal., 2007, 349, 281 CrossRef CAS PubMed; (e) Y.-Q. Jiang, Y.-L. Shi and M. Shi, J. Am. Chem. Soc., 2008, 130, 7202 CrossRef CAS PubMed; (f) H.-L. Cui, X. Feng, J. Peng, J. Lei, K. Jiang and Y.-C. Chen, Angew. Chem., Int. Ed., 2009, 48, 5737 CrossRef CAS PubMed; (g) W. Sun, L. Hong, C. Liu and R. Wang, Org. Lett., 2010, 12, 3914 CrossRef CAS PubMed; (h) T. Furukawa, J. Kawazoe, W. Zhang, T. Nishimine, E. Tokunaga, T. Matsumoto, M. Shiro and N. Shibata, Angew. Chem., Int. Ed., 2011, 50, 9684 CrossRef CAS PubMed; (i) F. Zhong, J. Luo, G.-Y. Chen, X. Dou and Y. Lu, J. Am. Chem. Soc., 2012, 134, 10222 CrossRef CAS PubMed; (j) H.-B. Mao, A.-J. Lin, Y. Shi, Z.-J. Mao, X.-B. Zhu, W.-P. Li, H.-W. Hu, Y.-X. Cheng and C.-J. Zhu, Angew. Chem., Int. Ed., 2013, 52, 6288 CrossRef CAS PubMed.
- (a) T. Furukawa, T. Nishimine, E. Tokunaga, K. Hasegawa, M. Shiro and N. Shibata, Org. Lett., 2011, 13, 3972 CrossRef CAS PubMed; (b) Y. Li, F. Liang, Q. Li, Y.-C. Xu, Q.-R. Wang and L. Jiang, Org. Lett., 2011, 13, 6082 CrossRef CAS PubMed.
- T. Nishimine, K. Fukushi, N. Shibata, H. Taira, E. Tokunaga, A. Yamano, M. Shiro and N. Shibata, Angew. Chem., Int. Ed., 2014, 53, 517 CrossRef CAS PubMed.
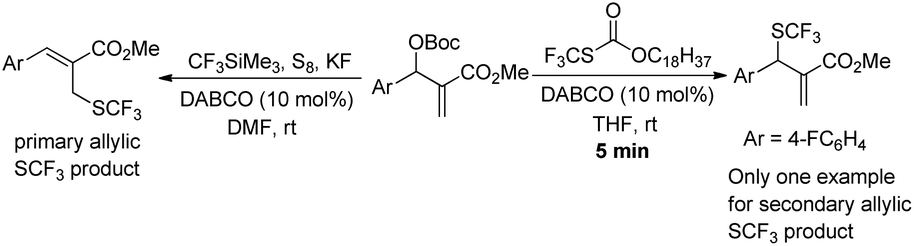
- During the preparation of this manuscript, Cahard reported a similar nucleophilic trifluoromethylthiolation of Morita–Baylis–Hillman carbonates: the primary allylic SCF3 derivatives were synthesized by means of the metal-free combination of CF3SiMe3/S8/KF/DMF and the kinetic secondary allylic SCF3 derivatives were obtained by using the Zard's reagent, however, only one example of nucleophilic trifluoromethylthiolation using the Zard's reagent was disclosed and the authors did not investigate the mechanism in detail. X. Dai and D. Cahard, Synlett, 2015, 40 CAS .
. - S.-G. Li and S. Z. Zard, Org. Lett., 2013, 15, 5898 CrossRef CAS PubMed.
- S.-K. Tian and L. Deng, J. Am. Chem. Soc., 2001, 123, 6195 CrossRef CAS.
- W. Tyrra, D. Naumann, B. Hoge and Y. L. Yagupolskii, J. Fluorine Chem., 2003, 119, 101 CrossRef CAS.
- K.-Y. Ye, X. Zhang, L.-X. Dai and S.-L. You, J. Org. Chem., 2014, 79, 12106 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data of new compounds. CCDC 1048652. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00198f |
| This journal is © the Partner Organisations 2015 |