
Synthesis of aromatic β-keto esters via a carbonylative Suzuki–Miyaura coupling reaction of α-iodo esters with arylboronic acids†
Shuhei
Sumino
,
Takahito
Ui
and
Ilhyong
Ryu
*
Department of Chemistry, Graduate School of Science, Osaka Prefecture University, Sakai, Osaka 599-8531, Japan. E-mail: ryu@c.s.osakafu-u.ac.jp
First published on 14th July 2015
Abstract
Aromatic β-keto esters were synthesized via a carbonylative cross-coupling reaction of alkyl iodides and arylboronic acids in the presence of a catalytic amount of Pd catalyst. A cooperative radical and Pd-catalyzed mechanism is proposed.
Carbonylation reactions with carbon monoxide are one of the most useful tools for the synthesis of carbonyl compounds.1 Thus far, our group has focused on the potential of the Pd/light-induced system for the carbonylation of alkyl iodides,2 and we previously reported that arylboronic acids can serve as a coupling reagent to modify the Pd/light-induced system to produce alkyl aryl ketones.3
β-Keto esters are key species in basic organic reactions and are extensively used as building blocks for the synthesis of natural products.4 In principle the methods used for the synthesis of aromatic β-keto esters via carbonylation are categorized as 3 types (Scheme 1): metal catalyzed esterification of α-halo ketones using CO and alcohols (type 1);5 carbonylation of aryl halides in the presence of ester enolates (type 2);6,7 and, carbonylation of α-halo esters in the presence of aryl anions (type 3). Among them, to the best of our knowledge, the type 3 strategy has not been investigated so far. We recently reported that carbamoyl acetates could be synthesized using α-iodo acetates, CO, and amines with a Pd/light-induced system.8 We believed that the use of arylboronic acids instead of amines would lead to the synthesis of aryl β-keto esters, representing the type 3 reaction. Herein, we report that the envisaged reaction did proceed smoothly.
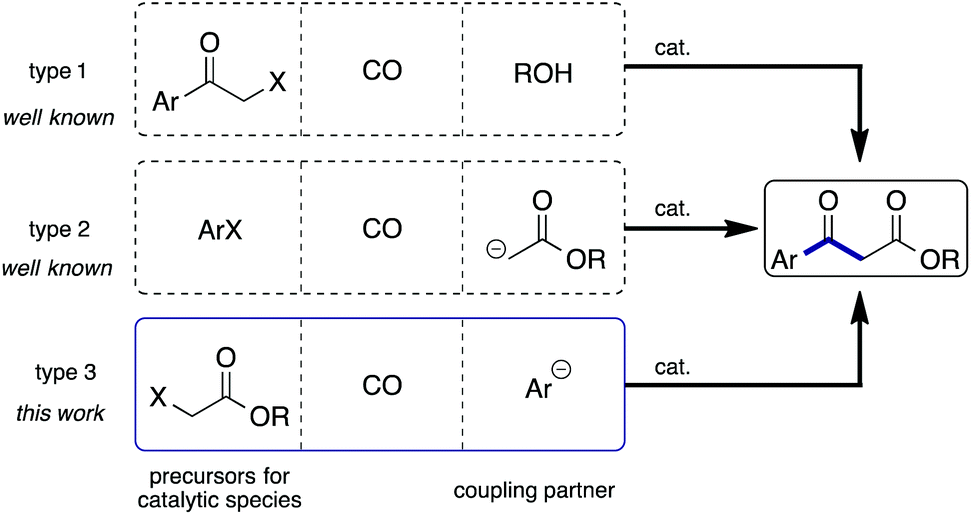 | ||
| Scheme 1 Possible strategies for aromatic β-keto esters via carbonylation reactions. | ||
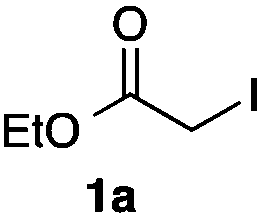
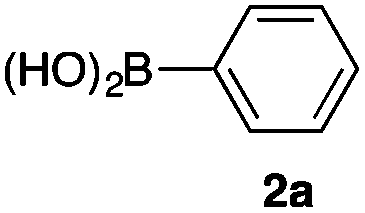
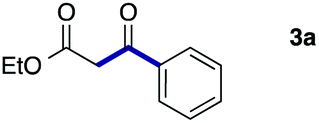
As a model, we chose the reaction of ethyl iodoacetate (1a) and phenylboronic acid (2a) (Table 1). When a mixture of 1a, 2a, K2CO3, and a catalytic amount of PdCl2(PPh3)2 (5 mol%) was irradiated under 10 atm of CO using a xenon lamp (500 W) through a Pyrex filter, ethyl benzoylacetate (3a) was obtained in 65% yield (entry 1). The reaction with atmospheric CO gave only 16% yield of 3a (entry 2). The use of a black light (15 W × 2) instead of a xenon lamp increased the yield to 76–78% (entries 3 and 4). We noticed that even under ambient temperature and dark conditions, 40% yield of 3a was obtained (entry 5). The reaction conducted at 40 °C gave 3a in 58% yield (entry 6). When we extended the reaction time to 8 h, 3a was obtained in 69% yield (entry 7).
|
|
||||
|---|---|---|---|---|
| Entry | Conditions | CO (atm) | C6H6 (mL) | Yieldb (%) |
| a Reaction conditions: 1a (0.5 mmol), 2a (1.5 equiv.), PdCl2(PPh3)2 (5 mol%), K2CO3 (2 equiv.), C6H6 (2 or 4 mL), H2O (2 mL), CO (1 or 10 atm), 4–8 h. b NMR (isolated) yield. c Black light (peak wavelength at 352 nm). | ||||
| 1 | hν (Xe, Pyrex), 4 h | 10 | 4 | 65 |
| 2 | hν (Xe, Pyrex), 4 h | 1 | 4 | 16 |
| 3 | hν (black light,c Pyrex), 5 h | 10 | 4 | 76 |
| 4 | hν (black light,c Pyrex), 5 h | 10 | 2 | 78 (72) |
| 5 | r.t., 5 h | 10 | 2 | 40 |
| 6 | 40 °C, 5 h | 10 | 2 | 58 |
| 7 | 40 °C, 8 h | 10 | 2 | 69 (65) |
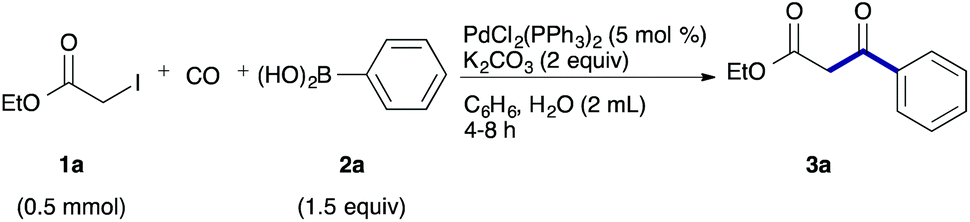
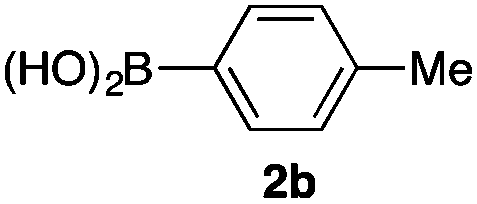
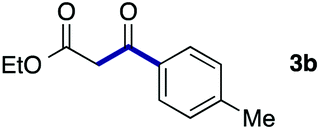
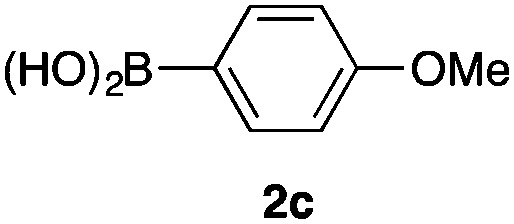
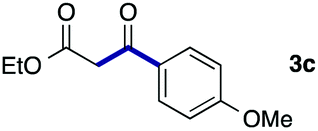
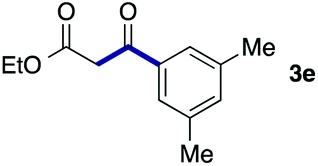
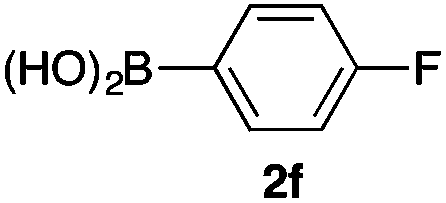
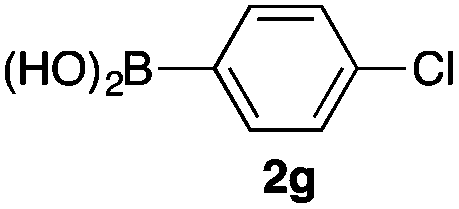
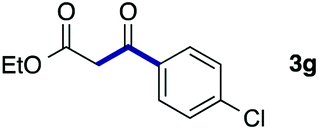
We then examined the generality of the carbonylative coupling reaction using a variety of α-iodo esters 1 and arylboronic acids 2. Both black light irradiated conditions (conditions A) and heated conditions (conditions B) were examined, and the results are summarized in Table 2. The reaction of 1a with 2- and 4-substituted arylboronic acids 2b–2d gave the corresponding β-keto esters 3a–d in good yields irrespective of the reaction conditions (entries 2–4). The reaction of 1a with (3,5-dimethylphenyl)boronic acid (2e) gave the corresponding product 3e in 61% yield (entry 5). When 1a was reacted with arylboronic acids 2f and 2g having a fluorine or chlorine atom, 3e and 3f were obtained in modest yields (entries 6 and 7). The reaction of bromo-substituted phenylboronic acid 2e with 1a gave 3h retaining the carbon–bromine intact. The reaction was sluggish (31%, after 5 h), however the extended reaction time of 20 h gave 3h in 95% yield (entry 8). The reactions worked well for 2-naphthaleneboronic acid (2g) and 3-methoxyphenylboronic acid (2h) although the yields were moderate (entries 9 and 10). α-Iodo benzyl ester 1b reacted with 2a to give 3i in good yields under both conditions (entry 11). We then examined α-alkyl substituted iodo esters 1c and 1d, whose results were contrasting. The reaction of α-iodo-γ-butyrolactone (1c) with 2a was smooth under either conditions A or B to give the corresponding acylated product 3j in a good yield (entry 12). By contrast, only thermal conditions gave keto ester 3k in reactions with ethyl 2-iodooctanoate (1d) and 2a (entry 13). The failure of the reaction under photoirradiation conditions may be ascribed to the facile E2 elimination of linear iodide 1d. The carbonylative Suzuki–Miyaura coupling reaction can be successfully extended to iodomethyl phenyl sulfone (1e), which gave α-sulfonyl acetophenone 3k (entry 14).9 Whereas the reaction under photoirradiation conditions gave 3l in a moderate yield due to slow reaction, heated conditions (80 °C) gave 3l in 91% yield.10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
|
||||
|---|---|---|---|---|
| Entry | 1 | 2 | 3 | Yieldb (%) |
| a Reaction conditions: conditions A: 1 (0.5 mmol), 2 (0.75 mmol), PdCl2(PPh3)2 (5 mol%), K2CO3 (1.0 mmol), C6H6 (2 mL), H2O (2 mL), CO (10 atm), hν (black light, Pyrex), 5 h. conditions B: 1 (0.5 mmol), 2 (0.75 mmol), PdCl2(PPh3)2 (5 mol%), K2CO3 (1.0 mmol), C6H6 (2 mL), H2O (2 mL), CO (10 atm), 40 °C, 8 h. b Isolated yield. c 1 (1.0 mmol), 2 (1.5 mmol), PdCl2(PPh3)2 (5 mol%), K2CO3 (4.0 mmol), C6H6 (2 mL), H2O (2 mL), CO (10 atm), hν (black light, Pyrex), 8 h. d 20 h. e 80 °C. | ||||
| 1 |
|
|
|
72 (A) |
| 65 (B) | ||||
| 2 | 1a |
|
|
85 (A) |
| 60 (B) | ||||
| 3 | 1a |
|
|
78 (A) |
| 86 (B) | ||||
| 4 | 1a |
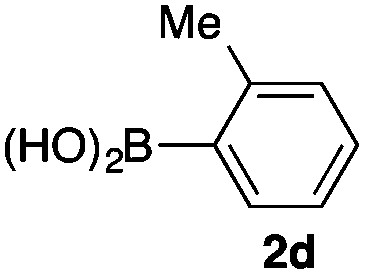
|
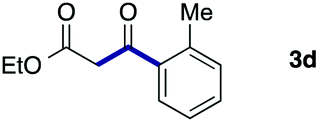
|
92 (A) |
| 89 (B) | ||||
| 5 | 1a |
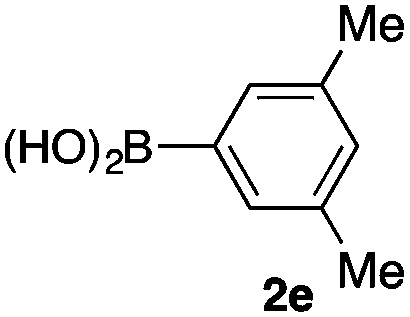
|
|
61 (B) |
| 6 | 1a |
|
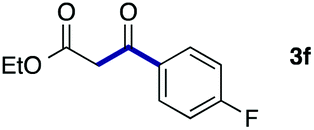
|
50 (A)c |
| 46 (B) | ||||
| 7 | 1a |
|
|
32 (A) |
| 37 (B) | ||||
| 8 | 1a |
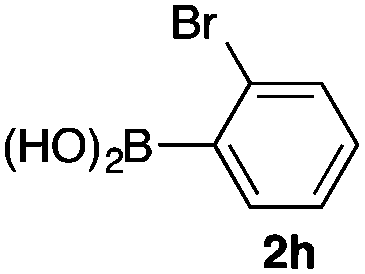
|
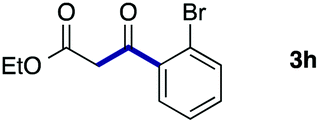
|
95 (A)d |
| 63 (B) | ||||
| 9 | 1a |
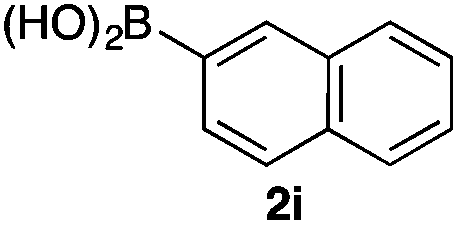
|
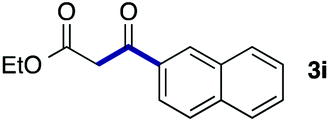
|
60 (A) |
| 47 (B) | ||||
| 10 | 1a |
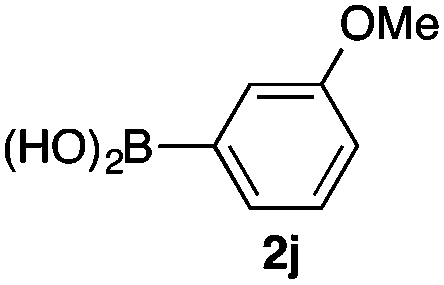
|
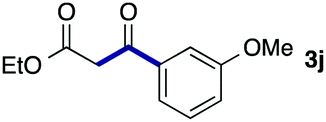
|
39 (A) |
| 34 (B) | ||||
| 11 |
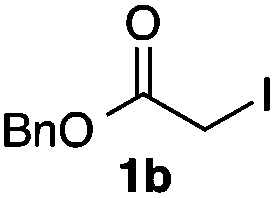
|
2a |
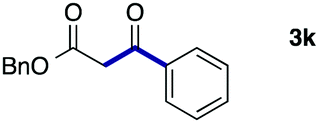
|
72 (A) |
| 82 (B) | ||||
| 12 |
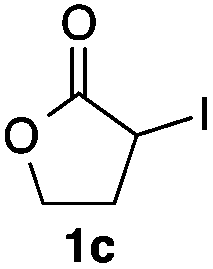
|
2a |
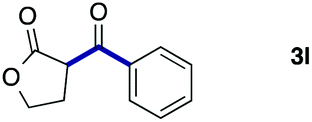
|
85 (A) |
| 70 (B) | ||||
| 13 |
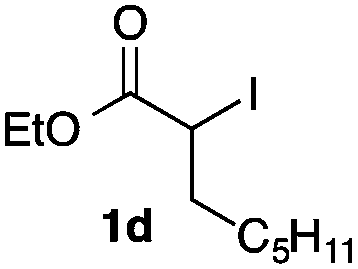
|
2a |
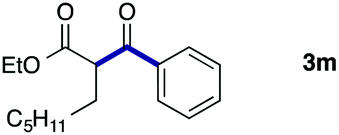
|
<1 (A) |
| 34 (B)e | ||||
| 14 |
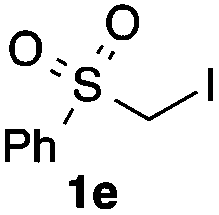
|
2a |
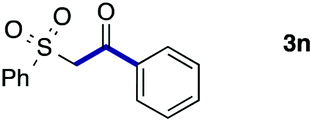
|
51 (A) |
| 91 (B)e | ||||
To examine the participation of radical species, we conducted the reaction in the presence of TEMPO (2,2,6,6-tetramethylpiperidine 1-oxy) as a radical scavenger (Scheme 2). Under both conditions, the desired reaction stopped and TEMPO adduct 4 was produced together with a largely recovered 1a.
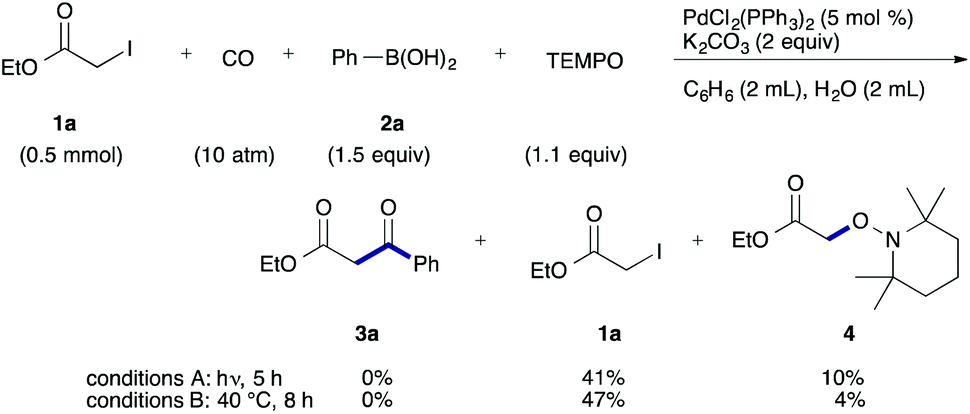 | ||
| Scheme 2 TEMPO-trapping experiment. | ||
Based on the above results, we can propose a reaction mechanism involving an α-ester radical (Scheme 3). In the first step, 1a reacts with Pd(0) to afford acetate radical A and Pd(I)I via a single-electron transfer.11,12 The resultant acetate radical A would couple with Pd(I)I to form α-pallado ester B.13 Then, insertion of CO into the C–Pd bond of B takes place to give acylpalladium species C. Transmetalation of C with phenylboronic acid (2a) followed by reductive elimination gives the product 3a with an accompanying liberation of Pd(0). An alterative path based on carbonylation of radical is highly unlikely because of ready backward decarbonylation.14
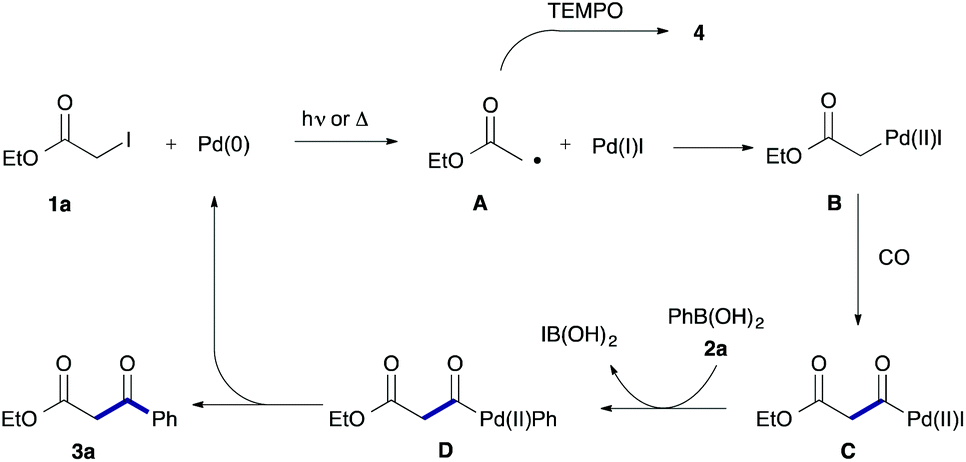 | ||
| Scheme 3 A cooperative radical and Pd-catalyzed mechanism. | ||
Conclusions
In summary, we have demonstrated that the carbonylative coupling reaction of readily available α-iodo esters, CO, and arylboronic acids proceeds effectively under either a combined Pd/light reaction system or Pd/thermal conditions. This method constitutes a valuable repertoire for the synthesis of aromatic β-keto esters. We proposed a reaction mechanism whereby radical reactions would lead to the effective formation of key organopalladium species for carbonylation.Acknowledgements
This work has been supported by a Grant-in-Aid for Scientific Research from the JSPS and MEXT. S. S. acknowledges a Research Fellowship of the JSPS for Young Scientists.Notes and references
- (a) M. Beller and X.-F. Wu, Transition Metal Catalyzed Carbonylation Reactions: Carbonylative Activation of C-X Bonds, Springer, Berlin, Heidelberg, 2013 Search PubMed; (b) L. Kollár, Modern Carbonylation Methods, Wiley-VCH, Weinheim, 2008 Search PubMed; (c) T. Fukuyama and I. Ryu, Carbon Monoxide, e-EROS Encycl. Reagents Org. Synth., 2006 DOI:10.1002/047084289X.rc013.pub2; (d) T. Fukuyama, S. Maetani and I. Ryu, Carbonylation and Decarbonylation Reactions, in Comprehensive Organic Synthesis, ed. G. A. Molander, and P. Knochel, Elsevier, Oxford, 2nd edn, 2014, vol. 3 Search PubMed.
- (a) T. Fukuyama, S. Nishitani, T. Inouye, K. Morimoto and I. Ryu, Org. Lett., 2006, 8, 1383 CrossRef CAS PubMed; (b) A. Fusano, S. Sumino, T. Fukuyama and I. Ryu, Org. Lett., 2011, 13, 2114 CrossRef CAS PubMed; (c) A. Fusano, S. Sumino, S. Nishitani, T. Inouye, K. Morimoto, T. Fukuyama and I. Ryu, Chem. – Eur. J., 2012, 18, 9415 CrossRef CAS PubMed. Also see: (d) S. Sumino, A. Fusano, T. Fukuyama and I. Ryu, Acc. Chem. Res., 2014, 47, 1563 CrossRef CAS PubMed.
- S. Sumino, T. Ui and I. Ryu, Org. Lett., 2013, 15, 3142 CrossRef CAS PubMed.
- S. Benetti, R. Romagnoli, C. De Risi, G. Spalluto and Z. Vinicio, Chem. Rev., 1995, 95, 1065 CrossRef CAS.
- (a) B. Wahl, S. Giboulot, A. Mortreux, Y. Castanet, M. Sauthier, F. Liron and G. Poli, Adv. Synth. Catal., 2012, 354, 1077 CrossRef CAS PubMed; (b) B. Wahl, Y. Philipson, H. Bonin, A. Mortreux and M. Sauthier, J. Org. Chem., 2013, 78, 1547 CrossRef CAS PubMed.
- (a) T. Kobayashi and M. Tanaka, Tetrahedron Lett., 1986, 27, 4745 CrossRef CAS; (b) S. Korsager, D. U. Nielsen, R. H. Taaning and T. Skrydstrup, Angew. Chem., Int. Ed., 2013, 52, 9763 CrossRef CAS PubMed; (c) P. Baburajan and K. P. Elango, Tetrahedron Lett., 2014, 55, 3525 CrossRef CAS PubMed.
- For a variation of type 2 in which arylboronic acids act as an aryl source, see: W. Lu, Y. Li, C. Wang, D. Xue, J.-G. Chen and J. Xiao, Org. Biomol. Chem., 2014, 12, 5243 CAS.
- S. Sumino, A. Fusano, T. Fukuyama and I. Ryu, Synlett, 2012, 1331 CAS.
- For carbonylative Suzuki–Miyaura coupling of α-bromo sulfoxides, see: M. Medio-Simón, C. Mollar, N. Rodríguez and G. Asensio, Org. Lett., 2005, 7, 4669 CrossRef PubMed.
- Under both conditions A and B, bromoacetate did not work. On the other hand, the reaction of iodoacetophenone with 2a resulted in the major formation of acetophenone.
- (a) D. P. Curran and C.-T. Chang, Tetrahedron Lett., 1990, 31, 933 CrossRef CAS; (b) Z.-M. Qiu and D. J. Burton, J. Org. Chem., 1995, 60, 5570 CrossRef CAS.
- G. Manolikakes and P. Knochel, Angew. Chem., Int. Ed., 2009, 48, 205 CrossRef CAS PubMed.
- For isolable α-pallado esters, see: (a) E. R. Burkhardt, R. G. Bergman and C. H. Heathcock, Organometallics, 1990, 9, 30 CrossRef CAS; (b) D. A. Culkin and J. F. Hartwig, Organometallics, 2004, 23, 3398 CrossRef CAS.
- For reviews on radical carbonylation and acyl radical, see: (a) I. Ryu and N. Sonoda, Angew. Chem., Int. Ed. Engl., 1996, 35, 1050 CrossRef PubMed; (b) I. Ryu, N. Sonoda and D. P. Curran, Chem. Rev., 1996, 96, 177 CrossRef CAS PubMed; (c) C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1991 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00185d |
| This journal is © the Partner Organisations 2015 |