
Chemoselective hydration of nitriles to amides using hydrated ionic liquid (IL) tetrabutylammonium hydroxide (TBAH) as a green catalyst†
Hojat Veisi*a,
Behrooz Maleki*b,
Mona Hameliana and
Samaneh Sedigh Ashrafib
aDepartment of Chemistry, Payame Noor University, Tehran 19395-4697, Iran. E-mail: hojatveisi@yahoo.com
bDepartment of Chemistry, Hakim Sabzevari University, Sabzevar 96179-76487, Iran. E-mail: b.maleki@hsu.ac.ir; Fax: +98-51-4401300; Tel: +98-51-44013324
First published on 17th December 2014
Abstract
A transition metal-free process, catalyzed by tetrabutylammonium hydroxide (TBAH), has been developed for the convenient and selective hydration of nitriles to the corresponding amides. The present process converts aromatic, aliphatic, and heteroaromatic nitriles with a wide variety of functional groups into amides. The regioselective hydration of one nitrile moiety in the presence of another nitrile group gives the present protocol high impact.
Introduction
Nitriles are one of the most important and versatile functional groups of organic compounds, and can easily be diversified into numerous important products, such as aldehydes, ketones, amines, amides, acids, and nitrogen-containing heterocycles, such as tetrazoles and oxazoles. Among these, the conversion of nitriles to amides by hydrolysis has great synthetic significance in the preparation of the corresponding amides, due to the isohypsic nature of the transformation which is an important factor in redox-economy in organic synthesis.1 This reaction has been investigated widely in organic synthesis, owing to its broad industrial and pharmacological applications.2 For example, hydration of acrylonitrile produces more than 2 × 105 tons of acrylamide annually, and is the most important technology for the production of this chemical.3 The reaction proceeds in a sequence of distinct steps upon treatment with strong acid or base. Due to these harsh conditions, carboxylic acids can form as the major byproduct, and more importantly, acid or base labile functional groups and protecting groups cannot be tolerated.4 Although metal-catalyzed and especially transition metal-catalyzed nitrile hydrolysis reactions have offered mild reaction conditions and selective formation of amides without the formation of carboxylic acids, functional group tolerance during nitrile hydrolysis has not yet been addressed.5 Several homogeneous and heterogeneous metal and metal oxide/salt catalysts, such as Ru,6 Rh,7 K,8 Na,9 Au,10 Os,11 Pt,12 Pd,13 Ir,14 Mo,15 Mn,16 Fe,17 Co,18 Cu,19 Ag,20 Ce,21 and Ni,22 have also been reported for the hydration of nitriles. Unfortunately, each of these protocols is associated with certain debilitating disadvantages, which include (i) the formation of unwanted acids as side products, (ii) high reaction temperatures, (iii) the presence of toxic transition metal cations within the molecular structures of reagents, and most importantly, (iv) the failure to achieve selectivity with substrates that contain two or more nitrile groups. Thus, the development of an efficient and mild process for the synthesis of amides from nitriles even in the presence of other labile functional groups would be a valuable tool in organic synthesis.Results and discussions
Herein, as part of our ongoing efforts to develop more efficient and environmentally benign methods for organic synthesis using economic and eco-friendly materials as catalysts and reagents,23 we report a selective hydrolysis reaction of nitriles to amides in essentially aqueous media using hydrated ionic liquid (IL) tetrabutylammonium hydroxide (50% in aqueous media), which allows other functional groups to remain intact (Scheme 1).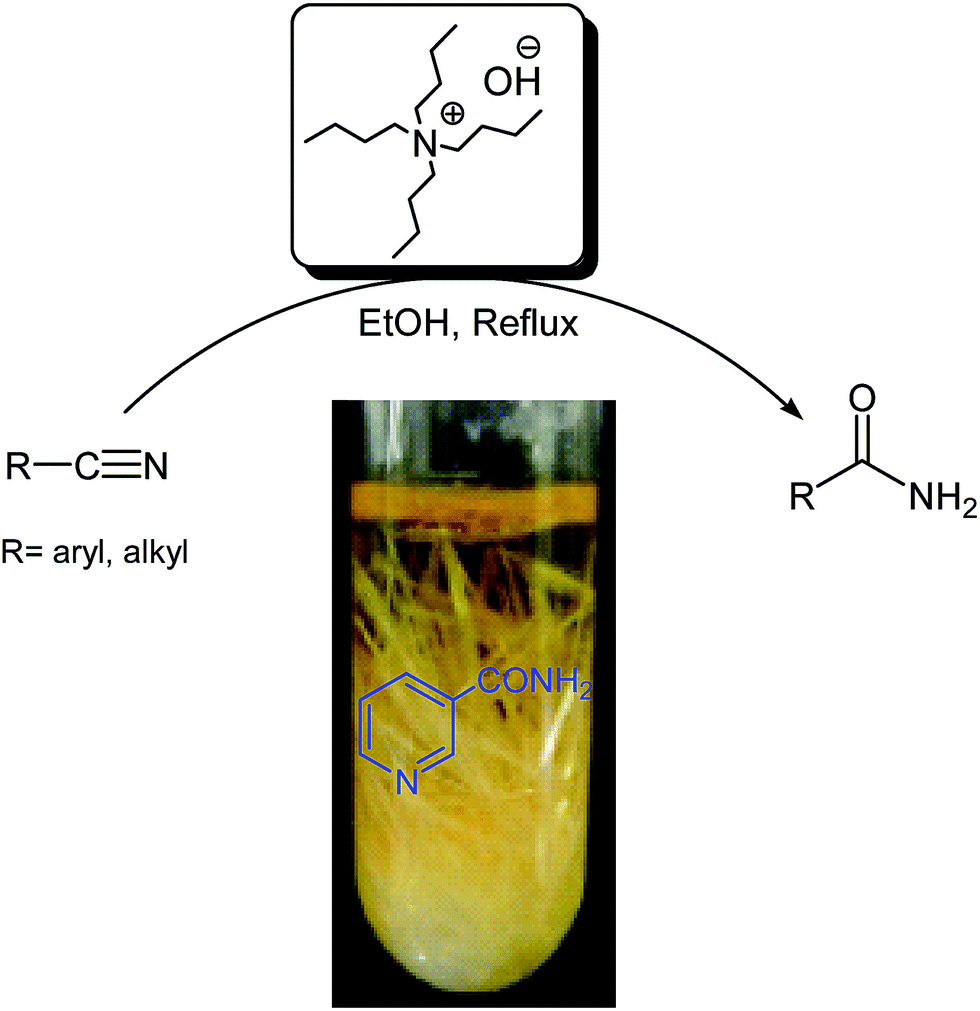 | ||
| Scheme 1 Selective hydrolysis of nitriles to amides. | ||
Ionic liquids (ILs) have received considerable interest as eco-friendly solvents, catalysts, and reagents in the context of green synthesis because of their solvating ability, low volatility, nonflammability, ability to dissolve a wide range of materials, easy recyclability and reusability.24
The reaction parameters were optimized using benzonitrile as a model substrate in the presence of hydrated ionic liquid (IL) tetrabutylammonium hydroxide (TBAH). Typical reaction parameters including various solvents, times and temperatures were screened, and the results are summarized in Table 1. The results show that EtOH is a better solvent (Table 1, entry 7, 96% yield). The hydrolysis of nitriles to amides in the presence of tetrabutylammonium hydroxide may be accomplished without the use of a diluent or solvent, provided that there is sufficient miscibility between the nitrile and water. In the event that there is not sufficient miscibility, a water-miscible solvent (for example, an alcohol) may be used so as to increase miscibility. In the reaction using H2O (entry 2) as solvent, the poor solubility of benzonitrile may have contributed to the lower yield (50%).
| Entry | Solvent | Temp. (°C) | Time (h) | Yieldb (%) |
|---|---|---|---|---|
| a Reaction conditions: benzonitrile (1 mmol), TBAH (0.5 ml, 50% in aqueous media), solvent (5 ml).b Yields are those of the pure isolated products.c No reaction. | ||||
| 1 | None | 80 | 24 | —c |
| 2 | H2O | Reflux | 24 | 50 |
| 3 | Toluene | Reflux | 12 | 30 |
| 4 | THF | Reflux | 12 | 35 |
| 5 | CH3CN | Reflux | 10 | 30 |
| 6 | CH2Cl2 | Reflux | 24 | —c |
| 7 | EtOH | Reflux | 3 | 96 |
| 8 | CH3OH | Reflux | 5 | 80 |
| 9 | n-Hexane | Reflux | 12 | —c |
| 10 | EtOH | 25 | 10 | 35 |
| 11 | EtOH | 50 | 10 | 60 |
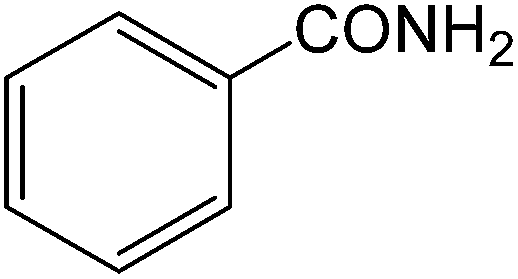
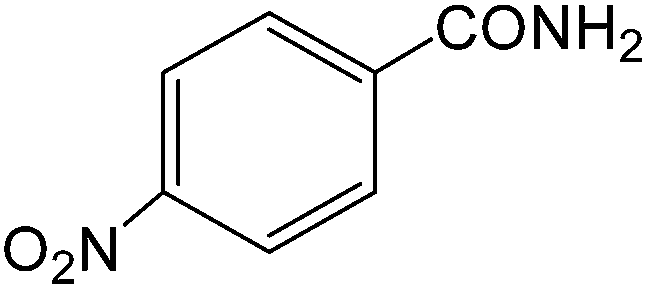
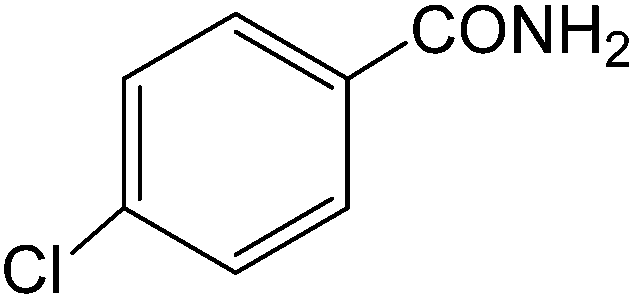
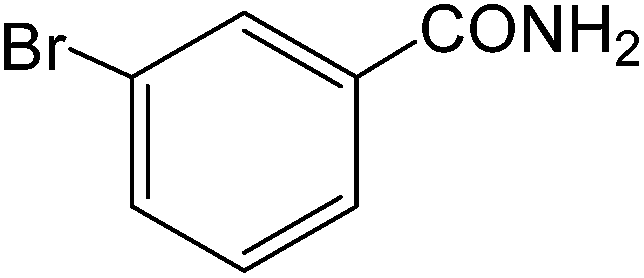
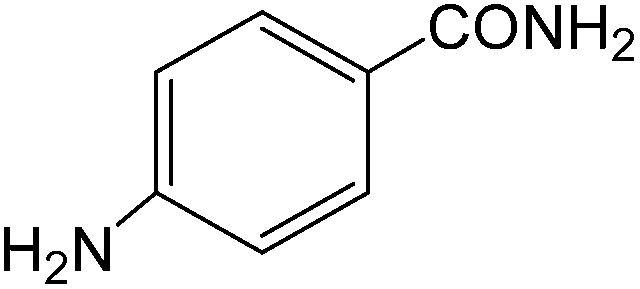
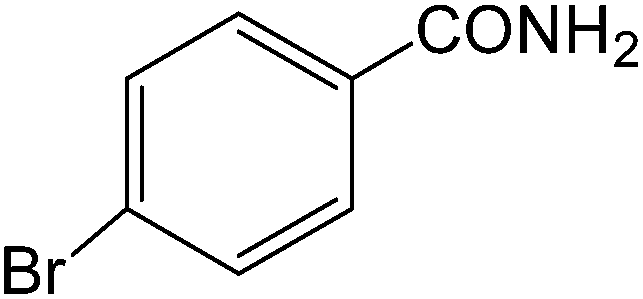
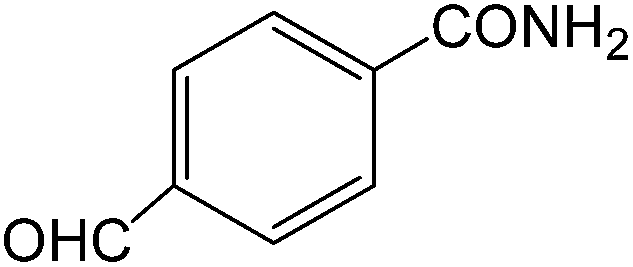
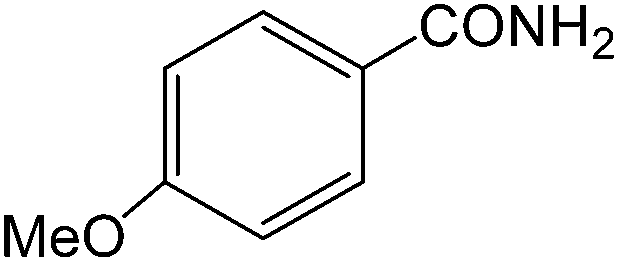
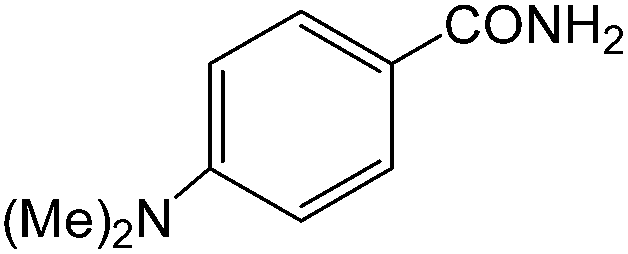
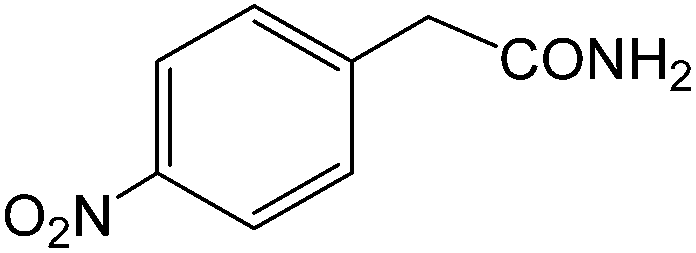
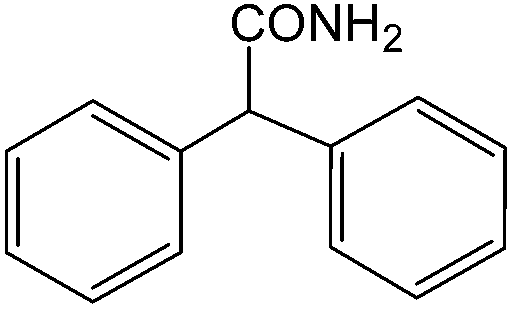
The present innovative procedure is applicable to the hydrolysis of a wide variety of nitriles to amides, including the hydrolysis of aliphatic nitriles, aromatic nitriles, and heterocyclic nitriles. As can be seen from Table 2, aromatic nitriles carrying either electron-donating groups such as p-methyl or p-methoxy, or electron-withdrawing groups such as p-bromo, p-chloro or p-nitro, exhibit comparable reactivity and react efficiently to yield the final product. In the case of 2-methyl- and 4-methyl-benzonitrile (entries 6 and 7), the hydration takes place within 5–6 hours and the yield is 90–92%; in the same way, 4-nitrobenzonitrile (entry 2) yields the corresponding 4-nitrobenzamide in 92% yield within 6 h. From this observation, it appears that electron-donating or electron-withdrawing groups do not significantly affect the rate of reaction. It was interesting to observe that the selective oxidation of 4-formylbenzonitrile to the corresponding amide could be performed without oxidation of the formyl group (entry 9). Furthermore, benzyl nitriles bearing various functionalities are also tolerated, providing the corresponding amides in excellent yields irrespective of electronic effects under this newly developed protocol (entries 13–17). Similarly, aliphatic amides such as acrylonitrile and octanenitrile afforded the desired amides in excellent yields (entries 18 and 20). Most importantly, when 3-cyanopyridine (entry 12) was employed as a substrate, an excellent yield of the corresponding nicotinamide (95%) was obtained. A good yield was also obtained from the reaction of a sterically hindered nitrile (entry 17). The nature and position of substitution on the aryl ring does not make much difference to the reactivity. For example, 4-methylbenzamide (entry 7) is produced in 92% yield, and 2-methylbenzamide (entry 6) in 90% yield.
| Entry | Nitrile | Amide | Time (h) | Yielda (%) | Mp (°C) | |
|---|---|---|---|---|---|---|
| Found | Lit.b | |||||
| a All known products have been reported previously in the literature, and were characterized by comparison of IR and NMR spectra with authentic samples.b http://www.sigmaaldrich.com. | ||||||
| 1 | 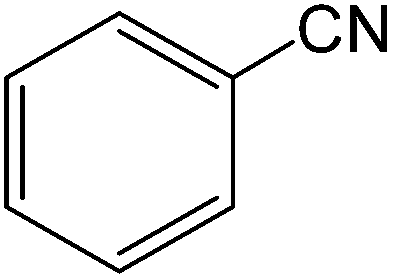 |
 |
3 | 96 | 124–126 | 123–125 |
| 2 | 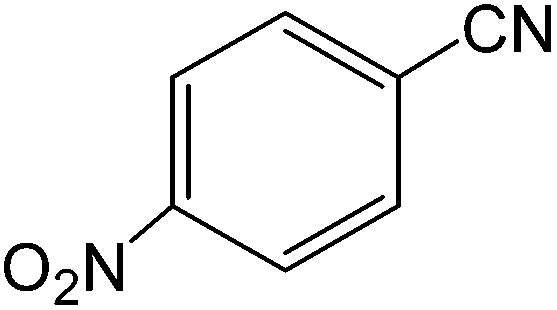 |
 |
6 | 92 | 197–198 | 198–200 |
| 3 | 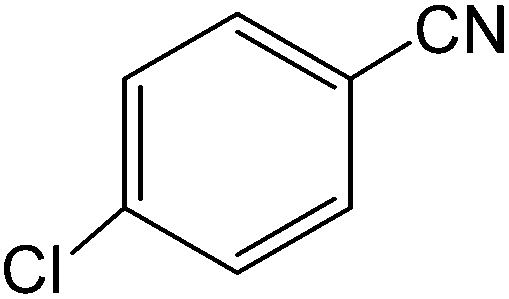 |
 |
5 | 95 | 178–179 | 177–179 |
| 4 |  |
 |
6 | 90 | 161–162 | 160–162 |
| 5 | 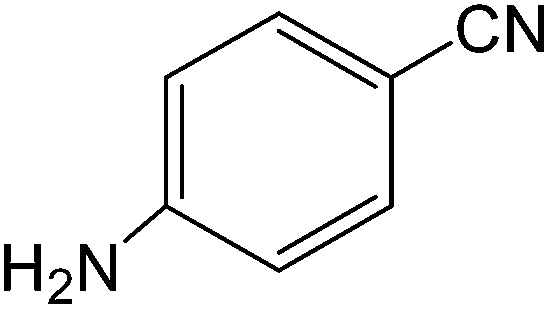 |
 |
7 | 85 | 183–184 | 182 |
| 6 | 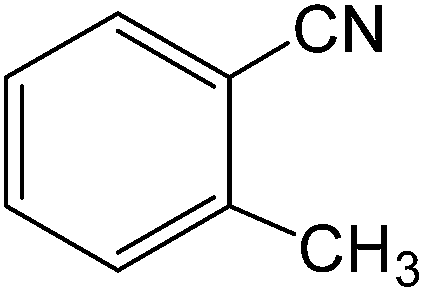 |
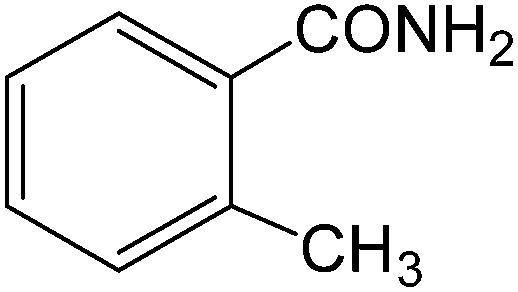 |
5 | 90 | 163–164 | 160–162 |
| 7 | 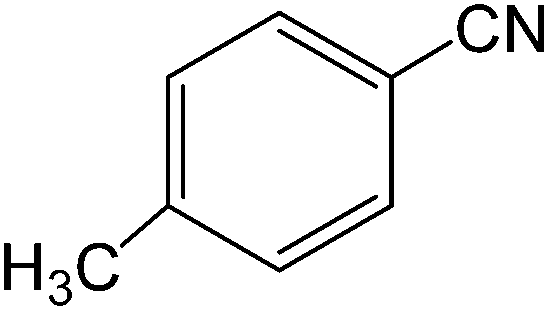 |
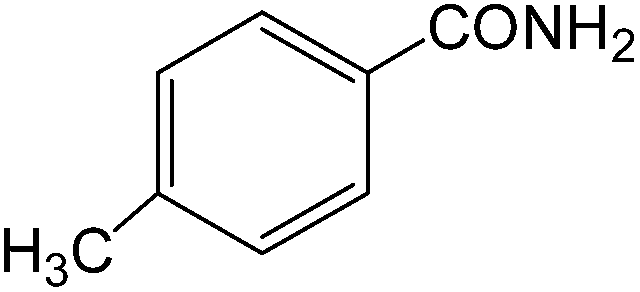 |
6 | 92 | 154–156 | 158–160 |
| 8 |  |
 |
7 | 95 | 191–193 | 190–193 |
| 9 | 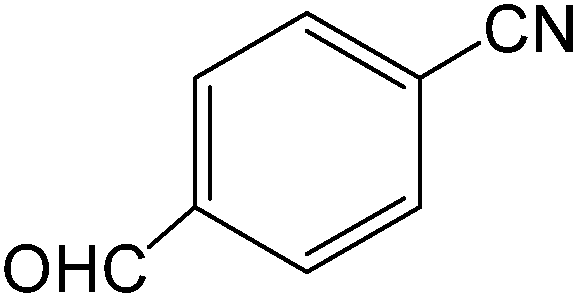 |
 |
6 | 90 | >290 dec | >290 dec |
| 10 | 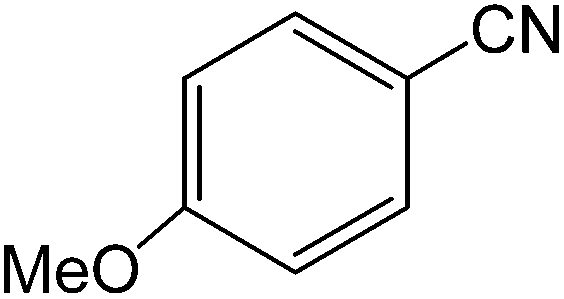 |
 |
7 | 95 | 166–167 | 163–165 |
| 11 | 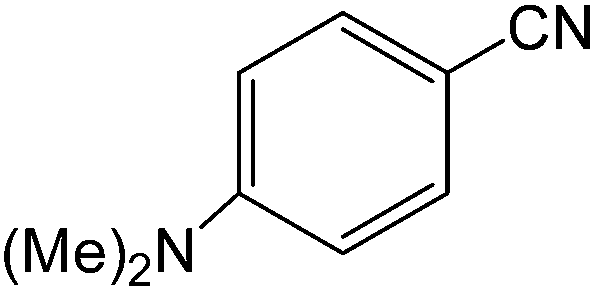 |
 |
7 | 88 | 206–208 | 207–209 |
| 12 | 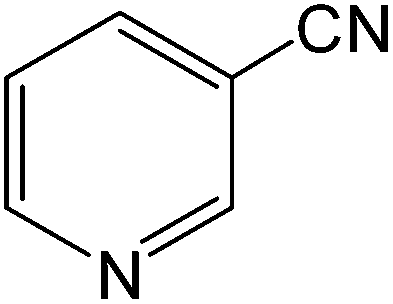 |
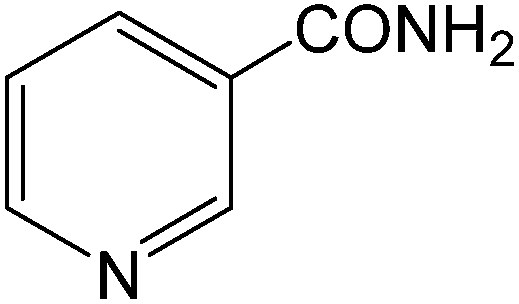 |
6 | 95 | 125–127 | 126–128 |
| 13 | 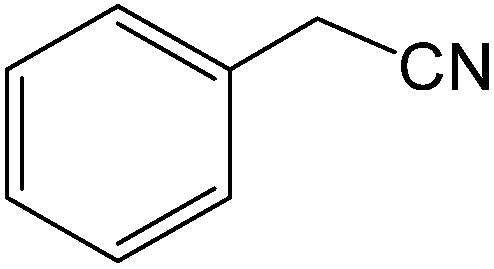 |
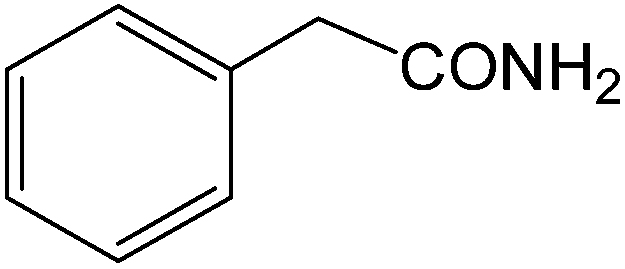 |
8 | 92 | 154–156 | 156–158 |
| 14 | 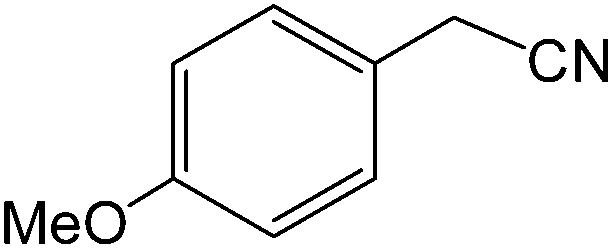 |
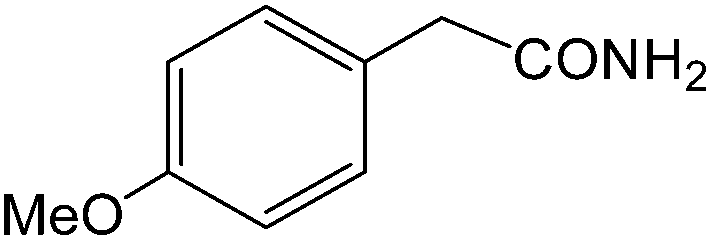 |
8 | 90 | 164–166 | 163–165 |
| 15 | 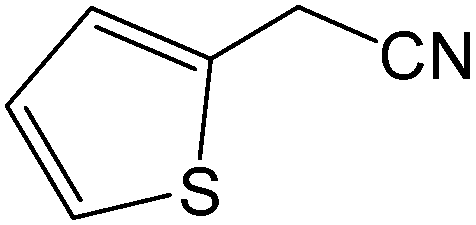 |
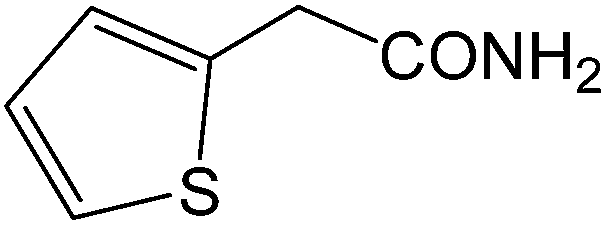 |
8 | 88 | 176–177 | 177–178 |
| 16 | 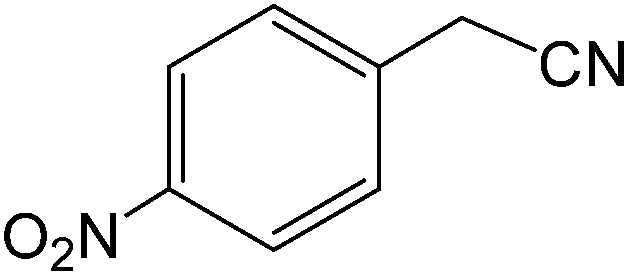 |
 |
10 | 90 | 214–216 | 213–215 |
| 17 | 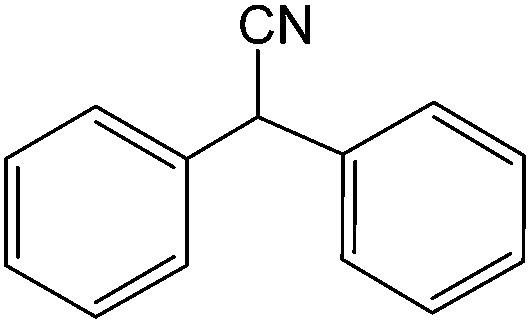 |
 |
12 | 92 | 165–167 | 164–167 |
| 18 | 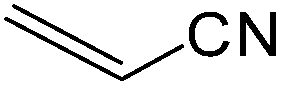 |
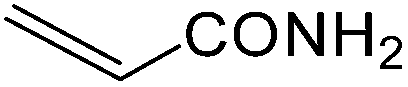 |
8 | 92 | 85–86 | 84–86 |
| 19 | 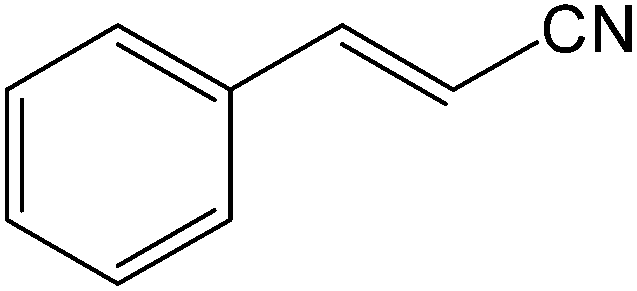 |
 |
8 | 92 | 148–150 | 148–150 |
| 20 | 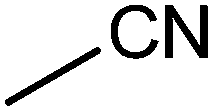 |
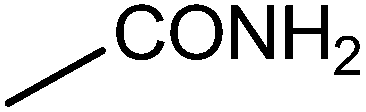 |
7 | 90 | 79–81 | 78–82 |
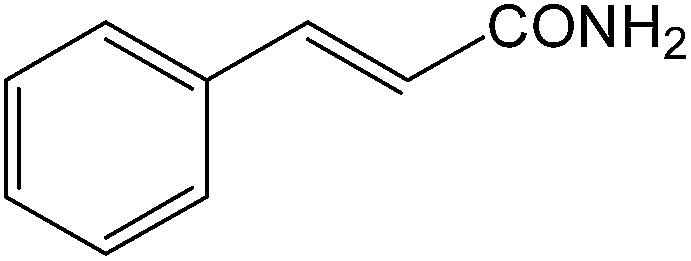
It is notable that nitriles were chemoselectively hydrated in the presence of other functional groups such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C. For example, in the reaction of acrylonitrile, hydration of the double bond was not observed and only the corresponding amide was obtained in 92–95% yields (entries 18 and 19).
C. For example, in the reaction of acrylonitrile, hydration of the double bond was not observed and only the corresponding amide was obtained in 92–95% yields (entries 18 and 19).
To demonstrate the validity and regioselectivity of this reagent system, we studied the hydration of nitriles in the presence of oximes, acetals and sulfides. These studies would clearly show that this reagent system can be applied for the chemoselective hydration of nitriles in the presence of the above-mentioned functional groups in multifunctional compounds. Therefore, tetrabutylammonium hydroxide (0.5 ml, 50% in aqueous media) and ethanol (5 ml) were added to a mixture of 4-methylbenzonitrile (1 mmol) and 4-methylbenzaldehyde oxime (1 mmol) at room temperature. The reaction mixture was allowed to stir at 80 °C. The progress of the reaction was monitored by TLC analysis. After completion of the reaction (6 h), the crude product was purified by column chromatography (packed with silica gel, using n-hexane/ethyl acetate (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) as eluent) to obtain the desired 4-methylbenzamide in 92% yield, and 4-methylbenzaldehyde oxime was recovered (Scheme 2).
:2) as eluent) to obtain the desired 4-methylbenzamide in 92% yield, and 4-methylbenzaldehyde oxime was recovered (Scheme 2).
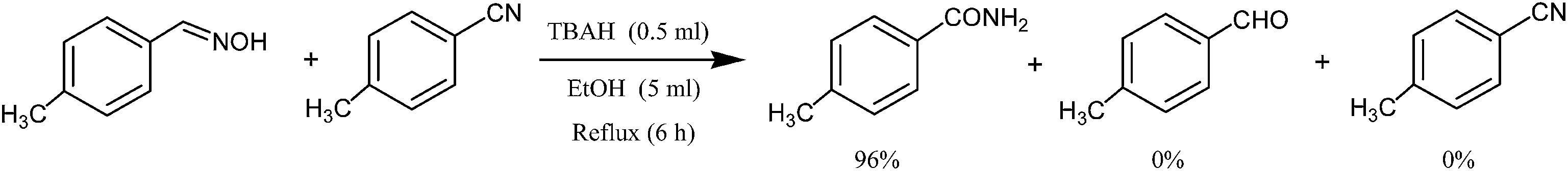 | ||
| Scheme 2 Selective synthesis of 4-methylbenzamide using tetrabutylammonium hydroxide in the presence of 4-methylbenzaldehyde oxime. | ||
In two separate reactions, 4-methylbenzaldehyde oxime was replaced with 2-(4-methylphenyl)-1,3-oxazoline and diphenyl sulfide. In all cases, 4-methylbenzamide was the only product of the reaction (Scheme 3).
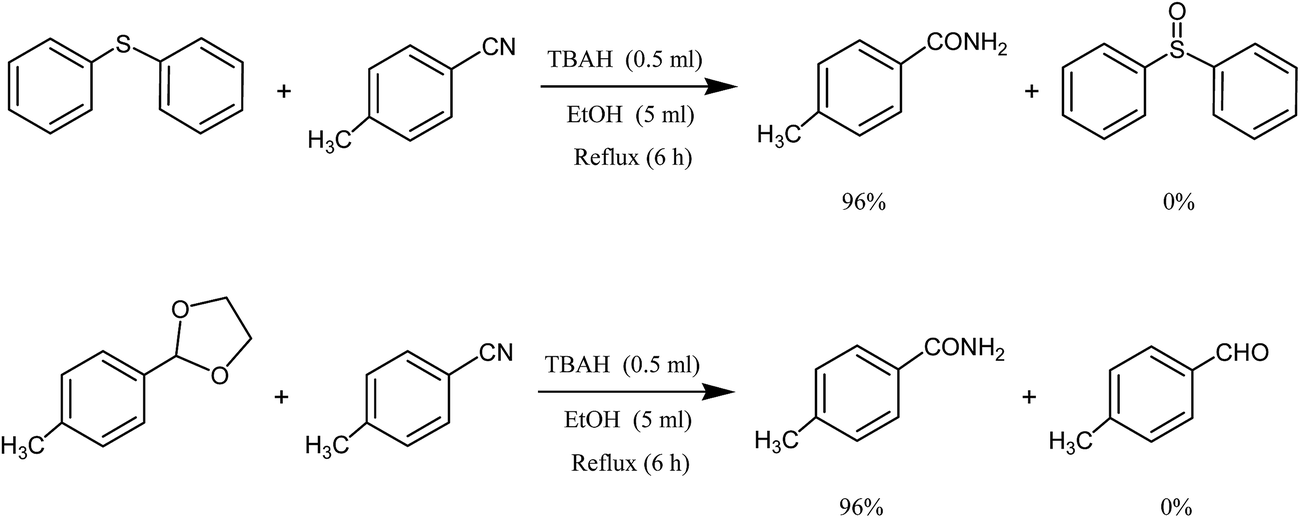 | ||
| Scheme 3 Selective synthesis of 4-methylbenzamide using tetrabutylammonium hydroxide in the presence of 2-(4-methylphenyl)-1,3-oxazoline or diphenyl sulfide. | ||
Interestingly, 1,4-dicyanobenzene could be hydrated over longer reaction times: a 6 h reaction time in refluxing EtOH led to the selective formation of the monohydrated derivative (93%), whereas a lengthened reaction time (9 h) resulted in the dihydration of 1,4-dicyanobenzene to give terephthalamide in 86% yield (Scheme 4).
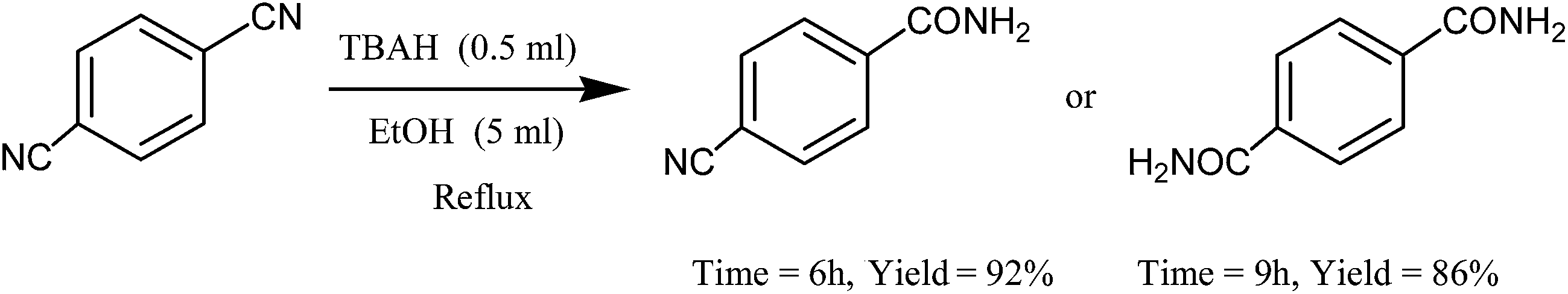 | ||
| Scheme 4 Selective synthesis of 4-cyanobenzamide and terephthalamide. | ||
From the mechanistic point of view, it is well reported that bases catalyze the hydration of nitriles.8,9,24j,k Therefore, we propose that the nitrile group is activated by coordination to quaternary ammonium hydroxide, and undergoes nucleophilic attack from an adjacent hydroxyl ion. It has been estimated that in this case the combination of the nucleophilic attack of the hydroxyl and the activation by coordination to the ammonium ion accelerates the rate of hydrolysis compared with the uncoordinated nitrile (Scheme 5).
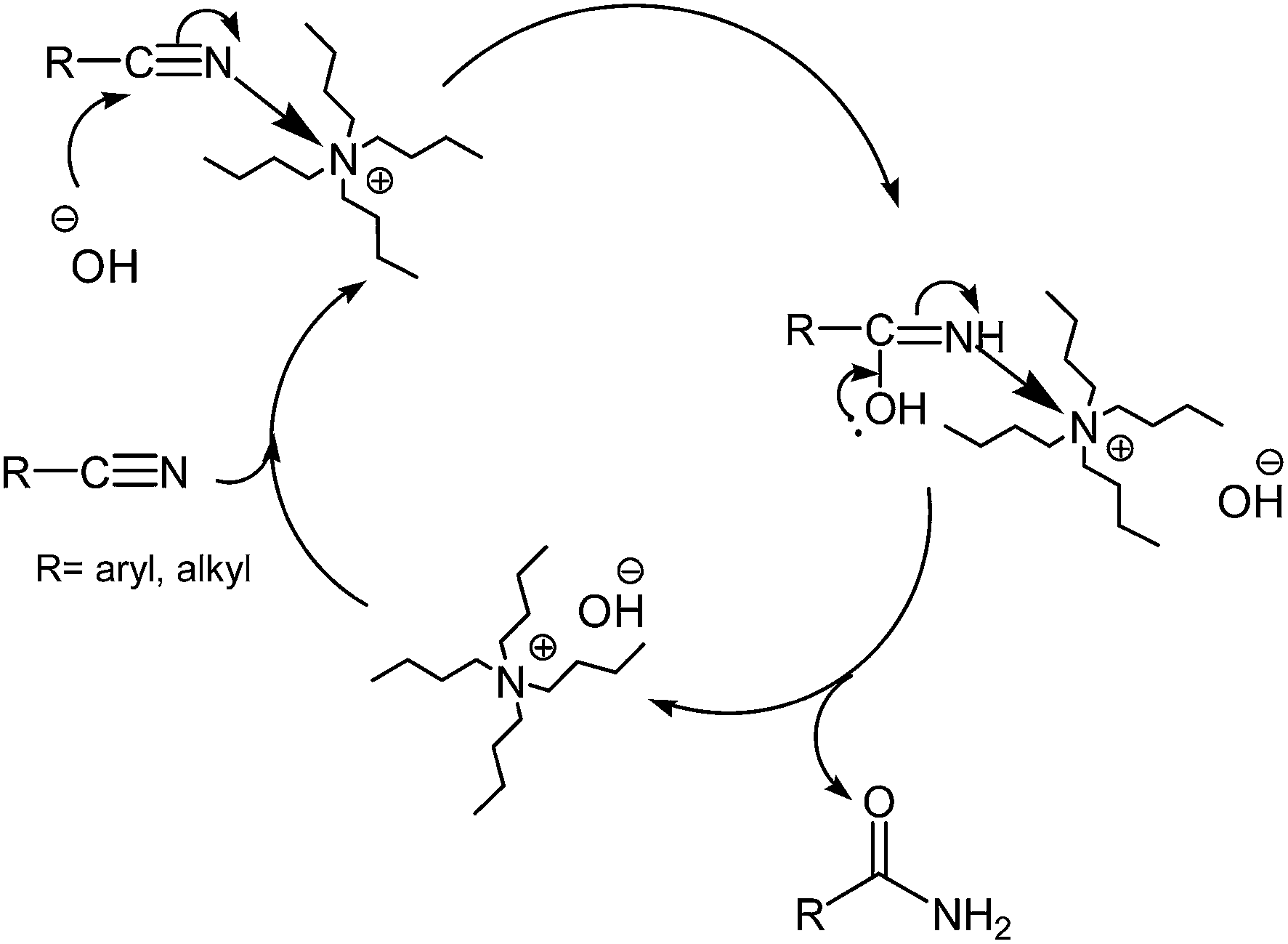 | ||
| Scheme 5 Suggested mechanism for the hydrolysis of nitriles using hydrated ionic liquid (IL) tetrabutylammonium hydroxide. | ||
After the completion of the reaction, the catalyst (TBAH) could be easily removed from the reaction mixture by adding Dowex 50WX8 hydrogen form, 50–100 mesh as an acidic ion exchange resin and stirring for 5 minutes at room temperature. The resin was separated by filtration and continuous removal of ammonium ion from the ion exchange resin, such that the catalyst could be recovered after another ion exchanging step for reuse in the hydrolysis reaction.
Experimental
List of purchased materials and devices used
Nitriles: entries 1–10, 12–14 and 16–20 were purchased from Merck (Germany); entry 11 from Sigma Aldrich (United States); and entry 15 from Fluka (Switzerland). Dowex 50WX8 hydrogen form, 50–100 mesh was obtained from Sigma Aldrich (United States); EtOH from Merck (China); and tetrabutylammonium hydroxide (50% in aqueous media) from Sigma Aldrich (United States) in commercial grade. IR spectra were recorded on a Shimadzu 435-U-04 spectrophotometer (KBr pellets). 1H and 13C NMR spectra were obtained using a Bruker DRX-300 Avance spectrometer in DMSO-d6 or CDCl3, using TMS as an internal reference. Melting points were determined in open capillary tubes using a Stuart BI Branstead Electrothermal Cat no.: IA9200 apparatus, and are uncorrected.General procedure for the TBAH catalyzed hydration of nitriles to amides
5 ml ethanol was added to a stirred solution of nitrile (1 mmol) and tetrabutylammonium hydroxide (0.5 ml, 50% in aqueous media) at room temperature. The reaction mixture was stirred vigorously at 80 °C in an oil bath for the time specified in Table 2. The progress of the reaction in each case was monitored by TLC analysis. After completion of the reaction, the reaction mixture was extracted with ethyl acetate, and after the extraction, the catalyst was recovered by Dowex 50WX8 (hydrogen form, 50–100 mesh). The organic phase was cooled to 0 °C, and white crystals were precipitated from the filtrate. The crystalline product was obtained by simple filtration and dried under vacuum at room temperature to give the pure amide product. In cases where the product was not precipitated out, the reaction mixture was extracted with ethyl acetate, and subsequent purification by column chromatography on silica gel provided the amide product.All products are known and compounds were characterized by melting point (mp), 1H NMR, 13C NMR and FT-IR. Spectroscopic data of these compounds are presented below:
Conclusion
In conclusion, tetrabutylammonium hydroxide (TBAH) has high synthetic utility, because it is reusable and shows high activity for the selective hydration of aliphatic, aromatic, and heterocyclic nitriles to amides, without the formation of carboxylic acids. The simplicity and convenience of this oxidation procedure are attractive, and the fact that the reaction proceeds with excellent yields, the reusable catalyst, and its use in ethanol as a solvent hold promise as the basis for this process, which is environmentally safe and economical. The high reactivity combined with inexpensive, readily available reagents and the lack of side products in the reaction makes it a suitable practical alternative.Acknowledgements
We are thankful to Payame Noor University (PNU) and Hakim Sabzevari for partial support of this work.References
- N. Z. Burns, P. S. Baran and R. W. Hoffmann, Angew. Chem., Int. Ed., 2009, 48, 2854 CrossRef CAS PubMed.
- V. Y. Kukushkin and A. J. L. Pombeiro, Chem. Rev., 2002, 102, 1771 CrossRef CAS PubMed.
- R. Opsahl, Encyclopedia of Chemical Technology, ed. J. I. Kroschwitz, Wiley, New York, 1991, vol. 2, p. 346 Search PubMed.
- (a) V. Y. Kukushikin and A. J. L. Pombeiro, Inorg. Chim. Acta, 2005, 358, 1 CrossRef PubMed; (b) J. N. Moorthy and N. Singhal, J. Org. Chem., 2005, 70, 1926 CrossRef CAS PubMed; (c) H. R. Sydner and C. T. Elston, J. Am. Chem. Soc., 1954, 76, 3039 Search PubMed; (d) C. R. Hauser and C. J. Eby, J. Am. Chem. Soc., 1957, 79, 725 CrossRef CAS; (e) C. P. Wilgus, S. Downing, E. Molitor, S. Bains, R. M. Pagni and G. W. Kabalka, Tetrahedron Lett., 1995, 36, 3469 CrossRef CAS; (f) L. McMaster and F. B. Langreck, J. Am. Chem. Soc., 1917, 39, 103 CrossRef CAS; (g) N. Kornblum and S. Singaram, J. Org. Chem., 1979, 44, 4727 CrossRef CAS; (h) S. Caochi, D. Misiti and L. F. Torre, Synthesis, 1980, 243 CrossRef; (i) A. R. Katritzky, B. Pilarski and L. Urogdi, Synthesis, 1989, 949 CrossRef CAS.
- A. Goto, K. Endo and S. Saito, Angew. Chem., Int. Ed., 2008, 47, 3607 CrossRef CAS PubMed.
- (a) T. Oshiki, H. Yamashita, K. Sawada, M. Utsunomiya, K. Takahashi and K. Takai, Organometallics, 2005, 24, 6287 CrossRef CAS; (b) S. C. Ghosh, J. S. Y. Ngiam, A. M. Seayad, D. T. Tuan, C. L. L. Chai and A. Chen, J. Org. Chem., 2012, 77, 8007 CrossRef CAS PubMed; (c) E. Bolgy-Nagy, A. Udvardy, F. Joo and A. Katho, Tetrahedron Lett., 2014, 55, 3615 CrossRef PubMed; (d) V. Cadierno, J. Francos and J. Gimeno, Chem.–Eur. J., 2008, 14, 6601 CrossRef CAS PubMed; (e) R. B. Nasir Baig, M. N. Nadagouda and R. S. Varma, Green Chem., 2014, 16, 2122 RSC; (f) E. Tomas-Mendivil, F. J. Suarez, J. Diez and V. Cadierno, Chem. Commun., 2014, 50, 9661 RSC; (g) V. Polshettiwar and R. S. Varma, Chem.–Eur. J., 2009, 15, 1582 CrossRef CAS PubMed; (h) K. Yamaguchi, M. Matsushita and N. Mizuno, Angew. Chem., 2004, 116, 1602 CrossRef; (i) C. W. Leung, W. Zheng, Z. Zhou, Z. Lin and C. P. Lau, Organometallics, 2008, 27, 4957 CrossRef CAS; (j) C. S. Yi, T. N. Zeczycki and S. V. Lindeman, Organometallics, 2008, 27, 2030 CrossRef CAS.
- (a) A. Goto, K. Endo and S. Saito, Angew. Chem., 2008, 120, 3663 CrossRef; (b) A. Goto, H. Naka, R. Noyori and S. Saito, Chem.–Asian J., 2011, 6, 1740 CrossRef CAS PubMed.
- T. Tu, Z. Wang, Z. Liu and X. Feng, Green Chem., 2012, 14, 921 RSC.
- P. K. Verma, N. Kumar, U. Sharma, M. Bala, V. Kumar and B. Singh, Synth. Commun., 2013, 43, 2867 CrossRef CAS.
- R. S. Ramon, N. Marion and S. P. Nolan, Chem.–Eur. J., 2009, 15, 8695 CrossRef CAS PubMed.
- I. N. Stepanenko, B. Cebrin-Losantos, V. B. Arion, A. A. Krokhin, A. A. Nazarov and B. K. Keppler, Eur. J. Inorg. Chem., 2007, 3, 400 CrossRef.
- X. B. Jiang, A. J. Minnaard, B. L. Feringa and J. G. DeVries, J. Org. Chem., 2004, 69, 2327 CrossRef CAS PubMed.
- (a) E. S. Kim, H. S. Kim and J. N. Kim, Tetrahedron Lett., 2009, 50, 2973 CrossRef CAS PubMed; (b) K. I. Shimizu, T. Kubo, A. Satsuma, T. Kamachi and K. Yoshizawa, ACS Catal., 2012, 2, 2467 CrossRef CAS.
- C. S. Chin, S. Y. Kim, K. S. Joo, G. Won and D. Chong, Bull. Korean Chem. Soc., 1999, 20, 535 CAS.
- (a) K. L. Breno, M. D. Pluth and D. R. Tyler, Organometallics, 2003, 22, 1203 CrossRef CAS; (b) K. L. Breno, M. D. Pluth, C. W. Landorf and D. R. Tyler, Organometallics, 2004, 23, 1738 CrossRef CAS.
- C. Battilocchio, J. M. Hawkins and S. V. Ley, Org. Lett., 2014, 16, 1060 CrossRef CAS PubMed.
- N. K. Thallaj, J. Przybilla, R. Welter and D. Mandon, J. Am. Chem. Soc., 2008, 130, 2414 CrossRef CAS PubMed.
- P. M. Angus and W. G. Jackson, Inorg. Chem., 1996, 35, 7196 CrossRef CAS PubMed.
- W. Schibler and T. A. Kaden, J. Chem. Soc., Chem. Commun., 1981, 603 RSC.
- (a) K. I. Shimizu, N. Imaiida, K. Sawabe and A. Satsuma, Appl. Catal., A, 2012, 421–422, 114 CrossRef CAS PubMed; (b) D. A. Buckingham, B. M. Foxman, A. M. Sargeson and A. Zanella, J. Am. Chem. Soc., 1972, 94, 8246 CrossRef CAS.
- M. Tamura, H. Wakasugi, K. Shimizu and A. Satsuma, Chem.–Eur. J., 2011, 17, 11428 CrossRef CAS PubMed.
- (a) T. Subramanian and K. Pitchumani, Catal. Commun., 2012, 29, 109 CrossRef CAS PubMed; (b) Y. Gangarajula and B. Gopal, Appl. Catal., A, 2014, 475, 211 CrossRef CAS PubMed.
- (a) B. Maleki, S. Hemmati, A. Sedrpoushan, S. Sedigh Ashrafi and H. Veisi, RSC Adv., 2014, 4, 40505 RSC; (b) B. Maleki and S. Sedigh Ashrafi, J. Mex. Chem. Soc., 2014, 58, 74 Search PubMed; (c) B. Maleki, S. Hemmati, R. Tayebee, S. Salemi, Y. Farokhzad, M. Baghayeri, F. Mohammadi Zonoz, E. Akbarzadeh, R. Moradi, A. Entezari, M. R. Abdi, S. Sedigh Ashrafi, F. Taimazi and M. Hashemi, Helv. Chim. Acta, 2013, 96, 2147 CrossRef CAS; (d) B. Maleki, S. Barzegar, Z. Sepehr, M. Kermanian and R. Tayebee, J. Iran. Chem. Soc., 2012, 9, 757 CrossRef CAS PubMed; (e) B. Maleki and F. Taimazi, Org. Prep. Proced. Int., 2014, 46, 252 CrossRef CAS; (f) H. Veisi, A. Khazaei, M. Safaei and D. Kordestani, J. Mol. Catal. A: Chem., 2014, 382, 106 CrossRef CAS PubMed; (g) H. Veisi, R. Masti, D. Kordestani, M. Safaei and O. Sahin, J. Mol. Catal. A: Chem., 2014, 385, 61 CrossRef CAS PubMed; (h) B. Maleki, S. Sedigh Ashrafi and R. Tayebee, RSC Adv., 2014, 4, 41521 RSC; (i) B. Maleki and S. Sedigh Ashrafi, J. Mex. Chem. Soc., 2014, 58, 159 Search PubMed; (j) B. Maleki and H. Veisi, Bull. Korean Chem. Soc., 2011, 32, 4366 CrossRef CAS; (k) H. Veisi, B. Maleki, F. Hosseini Eshbala, H. Veisi, R. Masti, S. Sedigh Ashrafi and M. Baghayeri, RSC Adv., 2014, 4, 30683 RSC; (l) B. Maleki and S. Sedigh Ashrafi, RSC Adv., 2014, 4, 42873 RSC; (m) H. Veisi, A. Maleki, Z. Omrani and S. Lotfi, RSC Adv., 2014, 4, 55313 RSC; (n) H. Veisi, M. Hamelian and S. Hemmati, J. Mol. Catal. A: Chem., 2014, 395, 25 CrossRef CAS PubMed; (o) M. Baghayeri, H. Veisi, H. Veisi, B. Maleki, H. Karimi-Maleh and H. Beitollahi, RSC Adv., 2014, 4, 49595 RSC; (p) M. Gholizadeh, M. Ebrahimpour, S. F. Hojati and B. Maleki, Arabian J. Chem., 2014, 7, 267 CrossRef CAS PubMed.
- (a) H. Veisi, A. Aminimanesh, N. Khankhani and R. Ghorbani-Vaghei, RSC Adv., 2014, 4, 25057 RSC; (b) M. A. Zolfigol, A. Khazaei, A. R. Moosavi-Zare, A. Zare, H. G. Kruger, V. Khakyzadeh and M. Kazem-Rostami, J. Org. Chem., 2012, 77, 3640 CrossRef CAS PubMed; (c) H. Veisi, A. Sedrpoushan, P. Mohammadi, A. R. Faraji and S. Sajjadifar, RSC Adv., 2014, 4, 25898 RSC; (d) R. Gusain, R. Singh, K. L. N. Sivakumar and O. P. Khatri, RSC Adv., 2014, 4, 1293 RSC; (e) Y. Zhang, A. Zhu, Q. Li, L. Li, Y. Zhao and J. Wang, RSC Adv., 2014, 4, 22946 RSC; (f) K. C. Lethesh, W. Dehaen and K. Binnemans, RSC Adv., 2014, 4, 4472 RSC; (g) A. Brandt, J. Gräsvik, J. P. Halletta and T. Welton, Green Chem., 2013, 15, 550 RSC; (h) L. L. Dong, L. He, G. H. Tao and C. w. Hu, RSC Adv., 2013, 3, 4806 RSC; (i) A. Zhu, S. Bai, W. Jin, R. Liu, L. Li, Y. Zhao and J. Wang, RSC Adv., 2014, 4, 36031 RSC; (j) H. Otaka, J. Ikeda, D. Tanaka and M. Tobe, Tetrahedron Lett., 2014, 55, 979 CrossRef CAS PubMed; (k) A. Khazaei, H. Veisi, M. Safaei and H. Ahmadian, J. Sulfur Chem., 2014, 35, 270 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra09864a |
| This journal is © The Royal Society of Chemistry 2015 |