DOI:
10.1039/C4RA11374H
(Paper)
RSC Adv., 2015,
5, 7773-7780
Surface charge effect on the cellular interaction and cytotoxicity of NaYF4:Yb3+, Er3+@SiO2 nanoparticles†
Received
28th September 2014
, Accepted 22nd December 2014
First published on 23rd December 2014
Abstract
Taking advantage of the unique optical properties of upconversion nanoparticles (NP) and the feasibly modified nature of SiO2, NaYF4:Yb3+, Er3+@SiO2 core@shell (UCNP@SiO2) NPs with excellent morphology have been synthesized to investigate the surface charge effect on the interactions of NPs with cells. The cellular uptake behaviors, cytotoxicities and intracellular metal ion release of the as-prepared NPs have been investigated in two cell lines (HeLa and HepG-2) by fluorescence microscopy, MTT assay and inductively coupled plasma mass spectrometry (ICP-MS), respectively. It is found that the UCNP@SiO2 NPs with positive surface charge have much higher cell internalization ability and cytotoxicity than those with negative surface charge. The experimental results also demonstrate that the cytotoxicity of UCNP@SiO2 NP is closely related to the increase of intracellular reactive oxygen species (ROS) and metal ion release of internalized NPs.
Introduction
Functionalized and/or engineered nanoparticles (NPs) have been extensively employed as basic units to construct novel compounds/devices for many different fields ranging from clean energy to healthcare.1–10 However, there is a controversy surrounding the biological safety of nanomaterials which seriously hampers the translation of nanotechnological improvements into the field of biomedicine.11–27 Numerous factors, such as size, shape, surface charge and ligands have been associated with NP cytotoxicity and cellular responses.11–22 In addition, the elemental toxicity of NP composition can play a role in the case of heavy metal-containing nanoparticles because free metal ions could be released from NP surface in oxidative and/or acidic conditions.23 The amount of toxicological data produced is steadily increasing by studying the interactions of the wide variety of physicochemical properties of the NPs with the large range of different cell types.24,25 In general, cytotoxicity is dependent on the NP uptake rate and pathway of internalization since it takes place in the intracellular space. Many studies demonstrated that surface charge plays an important role in the interactions of NPs with cells.12,14,28 For example, Iyer et al. have demonstrated that quantum dots (QDs) elicited cytotoxicity is a function of a combination of factors where surface charge > surface functionalization (ligand) > NP size.28 In addition, NP's cytotoxicity is also closely related to the tested cell types because of different uptake behaviors of NPs for different cells.14,29,30 For instance, Ji et al. have demonstrated that the cytotoxicity of gold NPs is quite different between nonphagocytic cells (HepG-2) and phagocytic cells (RAW 264.7).29
Compared to downconversion materials, UCNPs have many advantages including minimum photodamage to living cells, low autofluorescence, high signal-to-noise ratio and detection sensitivity, and high penetration depth in biological or environmental samples, which make the UCNPs particularly useful for bioimaging.4,31 Recently, silica coated lanthanide-doped upconversion NPs (UCNP@SiO2) have been given a lot of attention in the development of new generation of imaging agents for bioimaging.32–37 The wide interest in UCNP@SiO2 stems from a broad variety of enticing properties that UCNPs (core) and SiO2 (shell) possess, including: (1) high efficiency and multicolor emissions, (2) monodispersed size and uniform shape, (3) nanochemically engineered surface for phase transfer and for coupling to targeting ligands, and (4) the high biocompatibility. The results of MTT (methyl thiazolyl tetrazolium), MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, sodium salts), and CCK-8 mitochondrial metabolic activity assays have suggested that UCNP@SiO2 are noncytotoxic (cellular viabilities > 90%) to a broad range of tested cell lines within a certain range of concentrations and a limited incubation period.35,38,39 However, the precise behaviours underlying cytotoxicity of UCNP@SiO2 are still not completely understood. In particular, the effect of surface charge on cytotoxicity of UCNP@SiO2 should be carefully studied since the cellular internalization of NPs is strongly dependent on the surface charge.15
Here, we systemically study the impact of surface charge of UCNP@SiO2 on the interactions of cells with UCNP@SiO2 using both HeLa and HepG-2 cells. The 37 nm carboxy-terminated UCNP@SiO2 with negative surface charge are conjugated with positively charged NH2-PEG (UCNP@SiO2-PEG) or NH2-PEG-NH2 (UCNP@SiO2-PEG-NH2) for generating passivate UCNP@SiO2 with different surface charges. The uptake of the all NPs into two cells lines was studied by fluorescence microscopy and ICP-MS. The results of MTT and ROS experiments indicate that the cytotoxicity of UCNP@SiO2 are affected by the NP's surface charge.
Experimental section
Materials
Yttrium oxide (Y2O3, 99.99%), ytterbium oxide (Yb2O3, 99.99%), and erbium oxide (Er2O3, 99.99%) were bought from Alfa Aesar (Ward Hill, USA). YCl3, YbCl3 and ErCl3 aqueous stock solutions were obtained by dissolving corresponding lanthanide oxides in hydrochloric acid, respectively. 1-Octadecene (ODE, 90%), oleic acid (OA, 90%), tetraethyl orthosilicate (TEOS, 99.999%), polyoxyethylene (5) nonylphenyl ether (Igepal CO-520, Mn = 441), ammonia solution (28%) and poly(ethylene glycol)bis(3-aminopropyl) terminated (NH2-PEG-NH2, Mn = 1500) were purchased from Sigma-Aldrich Co. (St Louis, USA). Carboxyethylsilanetriol (CTES, sodium salt, 25% in water) was obtained from Gelest Inc. (Morrisville, USA). N-Hydroxysulfosuccinimide sodium salt (sulfo-NHS) and methoxypolyethylene glycol amine (NH2-PEG, Mn = 2000) were purchased from Aladdin Reagent Co. (Shanghai, China). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Beijing Dingguo Biotechnology Ltd. (Beijing, China). Reactive oxygen species (ROS) detection kit was purchased from Beyotime Company (Jiangsu, China). Other reagents (analytical grade) were purchased from Beijing Chemical Reagents Company (Beijing, China). All these reagents were used as received without further purification. Milli-Q water (18.2 MΩ cm) was used for all experiments.
Characterization
Transmission electron microscope (TEM) micrographs were performed with a JEM 2000FX (Jeol Ltd., Japan). Powder X-ray diffraction (XRD) analysis was carried out on a D8 advance diffractometer (Bruker Co., Germany) with Cu Kα (0.15406 nm) radiation. Energy-dispersive X-ray spectra (EDS) were carried out on a field emission scanning electron microscope (FESEM, S4800, Hitachi Co., Japan) equipped with an EDS (Jeol JXA-840). Dynamic light scattering (DLS) and zeta potential measurements were carried out on Malvern zetasizer Nano ZS (Malvern Instruments Ltd., UK). Upconversion luminescence (UCL) spectra were recorded from an F-4500 spectrophotometer (Hitachi Co., Japan) using an external CW NIR laser as excitation light source.
Synthesis of NaYF4:Yb3+, Er3+ upconverting NPs
OA-capped NaYF4:Yb3+, Er3+ UCNPs were prepared by a previously reported method with slight modifications.34 Typically, 1 mL aqueous solution of LnCl3 (1 M, Ln = Y, Yb, Er) was added to a 100 mL flask, and dried by heating. OA (6 mL) and 1-octadecene (15 mL) were then added to the flask and the reaction mixture was incubated at 150 °C under Ar atmosphere for 5 min. Following cooled to room temperature, 10 mL methanol solution containing NaOH (2.5 mmol) and NH4F (4 mmol) was added and vigorously stirred for 60 min. For evaporating the methanol, the solution was heated to 70 °C and degassed at 110 °C for 10 min. Subsequently, the reaction mixture was incubated at 300 °C for 60 min and then cooled down to room temperature. The product was precipitated and washed by addition of 15 mL ethanol (3 times). Finally, as-prepared OA-capped NaYF4:Yb3+, Er3+ (UCNPs) were collected by centrifugation and redispersed in cyclohexane. In the following experiments, the concentration of NP was defined by the amount of yttrium (Y) element.
Synthesis of carboxy-functionalized UCNP@SiO2 core@shell NPs
The carboxy-functionalized SiO2, UCNP@SiO2 core@shell NPs (UCNP@SiO2-COOH) were synthesized by our previously reported strategy.34 Generally, 0.1 mL Igepal CO-520, 8.6 mg OA-capped UCNPs and 10 mL cyclohexane were mixed and stirred for 10 min. 0.4 mL Igepal CO-520 and 0.08 mL ammonium hydroxide (28 wt% in water) were then added into the mixture and sonicated for 20 min. After sonicating, 0.05 mL TEOS was added dropwise and stirred for 5.75 h. Subsequently, 0.02 mL CTES was added into the solution and continually stirred for 2 days. The UCNP@SiO2-COOH were precipitated, washed and collected as previously described. The as-prepared UCNP@SiO2-COOH were redispersed in water and stored at room temperature.
Synthesis of amino-PEG-functionalized UCNP@SiO2 core@shell NPs
0.5 mL EDC (2.5 mg mL−1) and 0.5 mL sulfo-NHS (10 mg mL−1) were mixed with 1 mL UCNP@SiO2-COOH (1 mg mL−1) in MES buffer (0.1 M, pH 6.0). After gently stirred for 1 h at room temperature, NPs were centrifuged and washed with 2 mL MES buffer (0.1 M, pH 6.0). Subsequently, the NPs were redispersed in 5 mL HEPES buffer solution (0.01 M, pH 7.2) containing 7 mg NH2-PEG-NH2. The reaction mixture was incubated for 3 h at 37 °C with reciprocating oscillation (150 rpm). The amino-PEG-functionalized UCNP@SiO2 core@shell NPs (UCNP@SiO2-PEG-NH2) were purified by centrifugation (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 10 min, 3 times). The as-prepared UCNP@SiO2-PEG-NH2 were redispersed in water and stored at room temperature.
000 rpm, 10 min, 3 times). The as-prepared UCNP@SiO2-PEG-NH2 were redispersed in water and stored at room temperature.
Synthesis of PEG-functionalized UCNP@SiO2 core@shell NPs
The PEG-functionalized UCNP@SiO2 core@shell NPs (UCNP@SiO2-PEG) were synthesized by the same synthesis procedure of UCNP@SiO2-PEG-NH2, except for the use of NH2-PEG.
Measurements of cellular internalization of NPs
UCL imaging. HeLa and HepG-2 cells (1 × 105 cell per mL in 1 mL fresh culture medium) were seeded in 12 well cell-culture plate at 37 °C in a humidified atmosphere with 5% CO2 for 24 h, respectively. Then, the culture medium was discharged and the cells were washed with 1 mL PBS and fresh culture medium, respectively. Subsequently, cells were incubated in 1 mL fresh culture medium containing 25 μg mL−1 for another 24 h at 37 °C under 5% CO2. After incubating with NPs, the cells were washed with 1 mL fresh culture medium (3 times) and PBS (3 times) to remove any excess NPs. Finally, the UCL images of HeLa and HepG-2 cells were recorded on a reconstructive Nikon Ti–S fluorescent microscope under the excitation of CW 980 nm laser (BWT Beijing Ltd., China).
Measurements of the intracellular amounts of NPs
After treated with 25 μg mL−1 NPs as described above, the NPs stained cells were washed with 1 mL fresh culture medium (3 times) and PBS (3 times), detached from the culture plates by 0.2 mL trypsin, concentrated by centrifugation (500 g), freeze-dried, redispersed with concentrated nitric acid and analyzed the intracellular yttrium content by ICP-MS.
Measurements of intracellular metal ions release of internalized NPs
The HeLa and HepG-2 cells were incubated with 25 μg mL−1 NPs, washed, detached and concentrated as previously described. Then, the NPs stained cells were resuspended in 1 mL lysate buffer (Beijing Dingguo Biotechnology Co., Ltd., China) and incubated at 0 °C for 15 min. The homogenates were centrifuged at 19000 rpm for 20 min using an H-2050R centrifuge (Xiang Yi centrifuge instrument Co., Ltd., China) at 4 °C. Finally, the supernatants (cell lysates) were collected and analyzed by ICP-MS.
Cytotoxicity measurement
The in vitro cytotoxicities of NPs were studied by traditional MTT assay.40 Briefly, HeLa and HepG-2 cells (1 × 104 cells per well) were seeded in 96 well cell-culture plate and cultured at 37 °C in a humidified atmosphere with 5% CO2 for 24 h. Then, the culture medium was discharged and the cells were washed with 100 μL fresh culture medium and PBS, respectively. Subsequently, the NPs with desired concentrations (0, 0.78, 1.56, 3.125, 6.25, 12.5 and 25 μg mL−1) in 100 μL fresh culture medium were added into the wells and incubated at 37 °C in a humidified atmosphere with 5% CO2 for 24 h, respectively. After washed as previously described, the cells were treated by MTT (5 mg mL−1 in 100 μL culture medium) and cultured at 37 °C in a humidified atmosphere with 5% CO2 for another 4 h. After discharged the supernatant, dimethyl sulfoxide (DMSO) were added into the wells (100 μL per well) and shaken for 10 min. Finally, the absorbance (λ = 510 nm) of each well was recorded on a microplate reader (BioTek Instrument Co., Ltd., USA). The relative cell viabilities (%) were calculated by using the optical densities contrast to the control value. In this case, HeLa and HepG-2 cells cultured without NPs were used as control samples.
Measurements of intracellular reactive oxygen species (ROS)
The intracellular level of ROS was monitored by oxidation-sensitive fluorescent probe, 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA)-based assay kit with manufacturer's instructions.41 Briefly, HeLa and HepG-2 cells were treated with 25 μg mL−1 NPs for 24 h as previously described. The NPs stained cells were washed as previously described, incubated with 10 μmol mL−1 DCFH-DA in 1 mL serum-free medium (DMEM) at 37 °C for 20 min, and washed with 1 mL PBS (pH 7.4, sterilized, 3 times). Then, the cells were detached by trypsin, collected and analyzed by Fluoromax-4 spectrofluorometer (Horiba Jobin Yvon Inc., USA) under an excitation wavelength of 488 nm and emission wavelength of 525 nm.
Results and discussion
Synthesis and characterization of UCNP@SiO2 core@shell NPs
The UCNP@SiO2-COOH were synthesized by previously reported water-in-oil microemulsion strategy.34 Generally, the hydrophobic OA-capped UCNP cores (Y:Yb:Er = 80%:18%:2%) were reacted with TEOS and CTES in the water-in-oil microemulsion. In particular, CTES was added to the reaction mixture following the addition of TEOS. The UCNP@SiO2-COOH were generated through the reactions of CTES with silanol groups on TEOS formed silica shell. We found that the morphologies of UCNP@SiO2-COOH were critically related to the reaction temperature and stirring rate (as shown in Fig. S1 in the ESI†). UCNP@SiO2-COOH with excellent morphology were obtained at 18 °C and 300 rpm. The TEM images show that the particles have uniform size and shape. In the following studies, the UCNP@SiO2-COOH were synthesized under this condition. The average size of UCNP@SiO2-COOH is about 37 nm in diameter, which is composed of a 18 nm UCNP core and a 9.5 nm silica shell (as shown in Fig. 1a and b). The zeta potential (−38.3 mV) of UCNP@SiO2-COOH suggests that the NPs have strongly negative surface charge (as shown in Table 1). In addition, the corresponding XRD pattern (as shown in Fig. S2 in the ESI†) of the sample shows that the UCNP cores are composed of pure hexagonal phase (Standard PDF card: 16-0334). The elemental composition of the UCNP@SiO2-COOH is further confirmed by XPS and EDS analysis (as shown in Fig. S3 and S4 in the ESI†).
 |
| | Fig. 1 TEM images of (a) OA-capped UCNPs, (b) UCNP@SiO2-COOH, (c) UCNP@SiO2-PEG and (d) UCNP@SiO2-PEG-NH2. | |
Table 1 UCNP@SiO2 core@shell NP physicochemical propertiesa
| |
Physical diameter [nm] |
Hydrodynamic diametera [nm] |
Hydrodynamic diameterb [nm] |
Zeta potentiala [mV] |
Zeta potentialb [mV] |
| The NPs were redispersed in watera and culture mediumb (DMEM + 10% FBS), respectively. The average physical diameters of NPs are obtained by TEM measurements and the hydrodynamic diameters of NPs are generated by DLS measurements. |
| UCNP@SiO2-COOH |
36.9 ± 1.6 |
104.7 ± 0.1 |
121.8 ± 1.1 |
−38.3 ± 0.5 |
−10.1 ± 1.3 |
| UCNP@SiO2-PEG |
37.8 ± 1.6 |
146.3 ± 0.6 |
238.5 ± 4.6 |
−13.7 ± 0.7 |
−10.2 ± 0.8 |
| UCNP@SiO2-PEG-NH2 |
37.7 ± 1.9 |
139.9 ± 0.6 |
149.7 ± 5.0 |
26.2 ± 0.5 |
−6.3 ± 0.2 |
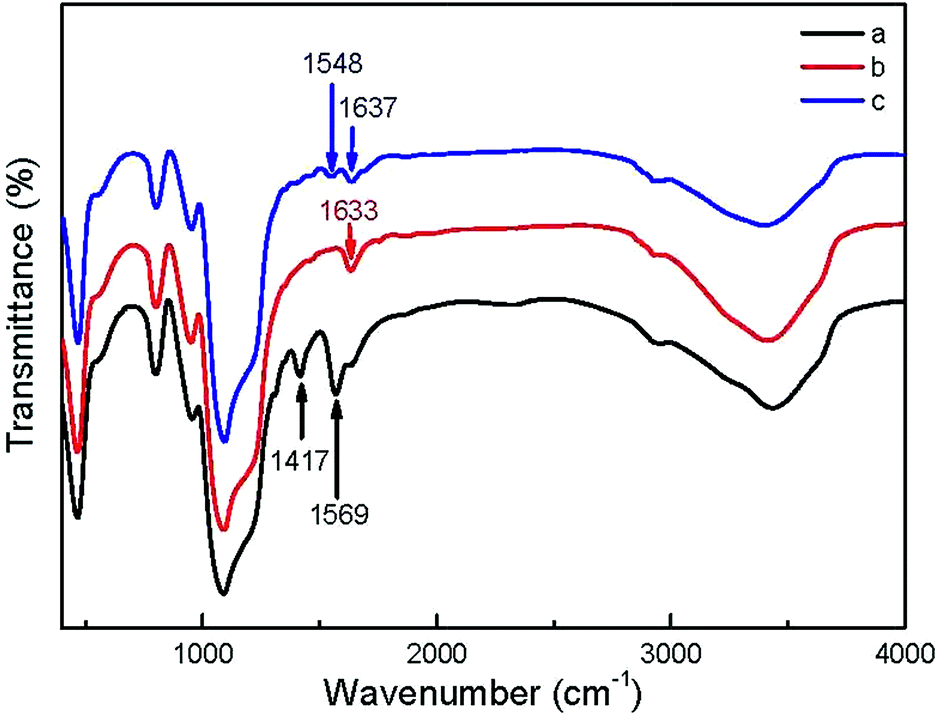
In order to adjust the surface charges of NPs, the as-prepared UCNP@SiO2-COOH were reacted with NH2-PEG and NH2-PEG-NH2, respectively. The NH2-PEG and NH2-PEG-NH2 were used in the study because the polyethylene glycol derivatives have good biocompativity.42,43 The FITR spectra of UCNP@SiO2-COOH, UCNP@SiO2-PEG and UCNP@SiO2-PEG-NH2 are shown in Fig. 2. The increasing of absorption at 1633 cm−1 and 1637 cm−1 (stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in amide I bands), 1548 cm−1 (stretching vibration of CO in amide II bands) and decreasing of absorption at 1417 cm−1 and 1569 cm−1 (asymmetric and symmetric stretching vibration bands of carboxy group) demonstrate that UCNP@SiO2-PEG and UCNP@SiO2-PEG-NH2 have been successfully synthesized. The TEM micrographs of the UCNP@SiO2-PEG and UCNP@SiO2-PEG-NH2 are shown in Fig. 1c and d. The TEM result indicates that the morphology and physical diameter of NPs are not affected by the polyethylene glycol derivative modification. However, the hydrodynamic diameters and zeta potentials of UCNP@SiO2-PEG and UCNP@SiO2-PEG-NH2 are changed significantly by the PEG derivative modification (as shown in Table 1). The hydrodynamic diameters of all NPs in culture medium are larger than their original sizes in water. The experimental result suggests that some serum proteins (e.g., serum albumin) adsorb on the NP surfaces in the culture medium. In the culture medium, the hydrodynamic diameter of UCNP@SiO2-PEG-NH2 is smaller than that of UCNP@SiO2-PEG. The phenomenon may due to that different PEG derivatives modified UCNP@SiO2 might adsorb different proteins in the cell culture media. Before and after incubation in culture medium, the zeta potential of UCNP@SiO2-PEG-NH2 change from positive (26.2 mV) to negative (−6.3 mV), while the zeta potential of UCNP@SiO2-COOH and UCNP@SiO2-PEG change to less negative. Similar phenomenon also has been observed of other NPs.17 The different zeta-potential changes of different NPs could be caused by different protein adsorbing behaviors.15,44,45 For instance, NPs with positive surface charge prefer to absorb negative proteins while NPs with negative surface charge like absorbing positive proteins. The differences in protein adsorption of NPs could result in the difference between endocytosis.15 In addition, the UCL spectra of NPs were not changed significantly after modification (as shown in Fig. S5 in the ESI†). In addition, the zeta potential and colloidal stability of UCNP@SiO2-PEG-NH2 is much higher than that of 3-aminopropyltrimethoxysilane (APS) modified UCNP@SiO2 (7.4 mV). And the UCNP@SiO2-PEG-NH2 and UCNP@SiO2-COOH have closed surface charge density (absolute value). Therefore, UCNP@SiO2-PEG-NH2 was used to study the effect of positive charge on the cytotoxicity of UCNP@SiO2.
O in amide I bands), 1548 cm−1 (stretching vibration of CO in amide II bands) and decreasing of absorption at 1417 cm−1 and 1569 cm−1 (asymmetric and symmetric stretching vibration bands of carboxy group) demonstrate that UCNP@SiO2-PEG and UCNP@SiO2-PEG-NH2 have been successfully synthesized. The TEM micrographs of the UCNP@SiO2-PEG and UCNP@SiO2-PEG-NH2 are shown in Fig. 1c and d. The TEM result indicates that the morphology and physical diameter of NPs are not affected by the polyethylene glycol derivative modification. However, the hydrodynamic diameters and zeta potentials of UCNP@SiO2-PEG and UCNP@SiO2-PEG-NH2 are changed significantly by the PEG derivative modification (as shown in Table 1). The hydrodynamic diameters of all NPs in culture medium are larger than their original sizes in water. The experimental result suggests that some serum proteins (e.g., serum albumin) adsorb on the NP surfaces in the culture medium. In the culture medium, the hydrodynamic diameter of UCNP@SiO2-PEG-NH2 is smaller than that of UCNP@SiO2-PEG. The phenomenon may due to that different PEG derivatives modified UCNP@SiO2 might adsorb different proteins in the cell culture media. Before and after incubation in culture medium, the zeta potential of UCNP@SiO2-PEG-NH2 change from positive (26.2 mV) to negative (−6.3 mV), while the zeta potential of UCNP@SiO2-COOH and UCNP@SiO2-PEG change to less negative. Similar phenomenon also has been observed of other NPs.17 The different zeta-potential changes of different NPs could be caused by different protein adsorbing behaviors.15,44,45 For instance, NPs with positive surface charge prefer to absorb negative proteins while NPs with negative surface charge like absorbing positive proteins. The differences in protein adsorption of NPs could result in the difference between endocytosis.15 In addition, the UCL spectra of NPs were not changed significantly after modification (as shown in Fig. S5 in the ESI†). In addition, the zeta potential and colloidal stability of UCNP@SiO2-PEG-NH2 is much higher than that of 3-aminopropyltrimethoxysilane (APS) modified UCNP@SiO2 (7.4 mV). And the UCNP@SiO2-PEG-NH2 and UCNP@SiO2-COOH have closed surface charge density (absolute value). Therefore, UCNP@SiO2-PEG-NH2 was used to study the effect of positive charge on the cytotoxicity of UCNP@SiO2.
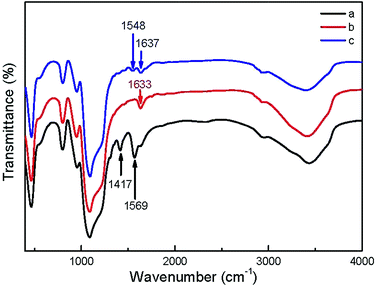 |
| | Fig. 2 FTIR spectra of (a) UCNP@SiO2-COOH, (b) UCNP@SiO2-PEG and (c) UCNP@SiO2-PEG-NH2, respectively. | |
Cellular internalization of NPs
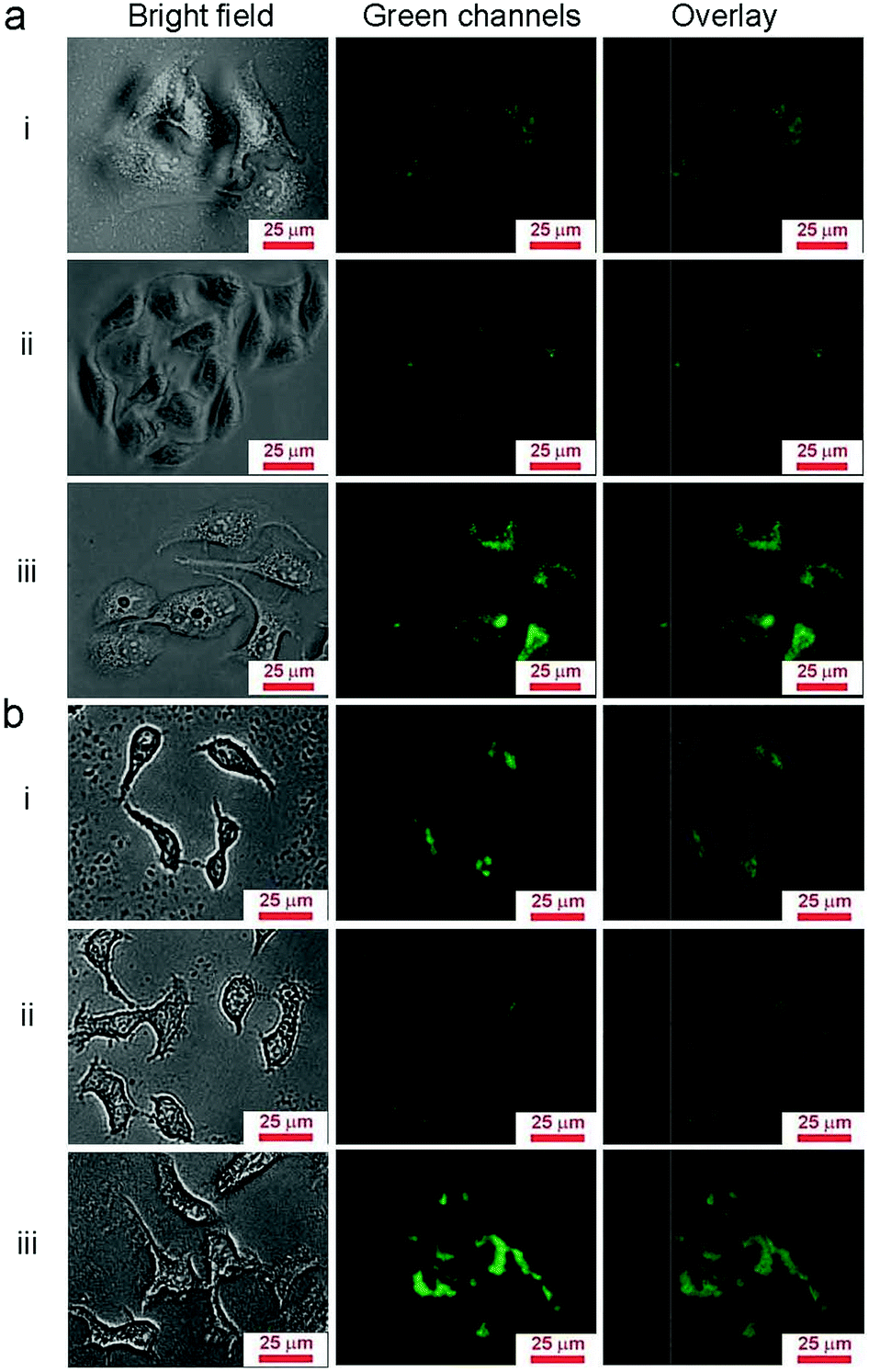
Due to the properties of upconversion luminescence, UCNP@SiO2 can be directly used as fluorescent probes for biological imaging. To investigate the cell uptake behavior of UCNP@SiO2 affected with different surface charges, the NPs were incubated in HepG-2 cells (human hepatoma cell line, as model cell of detoxification organ) and HeLa cells (cervix epithelioid carcinoma cell line, as model cell of non-detoxification organ), respectively. The different NPs were incubated with cells in the same experimental conditions. The NPs stained cells were observed by UCL imaging and cellular internalized NPs were quantified by ICP-MS with described as Y content in cells. It is clearly observed that the UCNP@SiO2-NH2 stained cells have the brightest UCL and UCL intensity of UCNP@SiO2-COOH stained cells is much stronger than that of UCNP@SiO2-PEG stained cells (as shown in Fig. 3). The UCL imaging of the cells provides direct evidence on that the NPs are uptaken by cells. To further quantitatively analyze the cellular uptaken amount of the NPs, the NPs stained cells are examined by ICP-MS measurements (as shown in Fig. 4). The result of ICP-MS measurement is consistent with that of UCL imaging experiment. For the same cell line, the uptake of NPs is following in the order: UCNP@SiO2-PEG-NH2 > UCNP@SiO2-COOH > UCNP@SiO2-PEG. The UCL imaging and ICP-MS experimental results demonstrate that the cellular internalization of NPs is not only strongly dependent on the surface charges of NPs but also affected by the cell types.
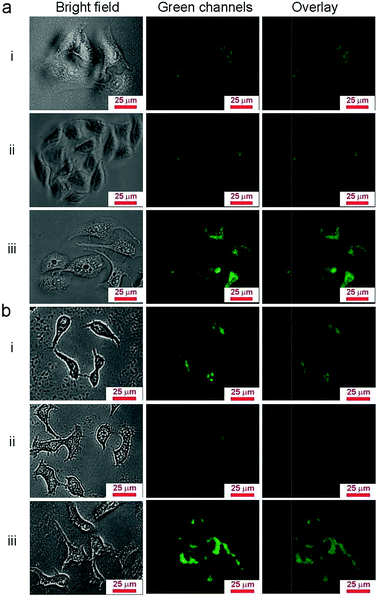 |
| | Fig. 3 UCL images of NPs stained HeLa cells (a) and HepG-2 cells (b). The cells are stained by UCNP@SiO2-COOH (i), UCNP@SiO2-PEG (ii) and UCNP@SiO2-PEG-NH2 (iii), respectively. The concentrations of NPs are 25 μg mL−1. | |
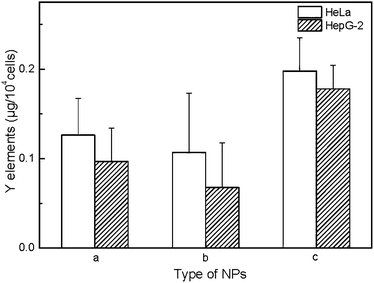 |
| | Fig. 4 ICP-MS measurements of cellular internalization of NPs. The cells are incubated with UCNP@SiO2-COOH (a), UCNP@SiO2-PEG (b) and UCNP@SiO2-PEG-NH2 (c), respectively. The concentrations of NPs are 25 μg mL−1. | |
Cytotoxicity of UCNP@SiO2 in HeLa and HepG-2 cells
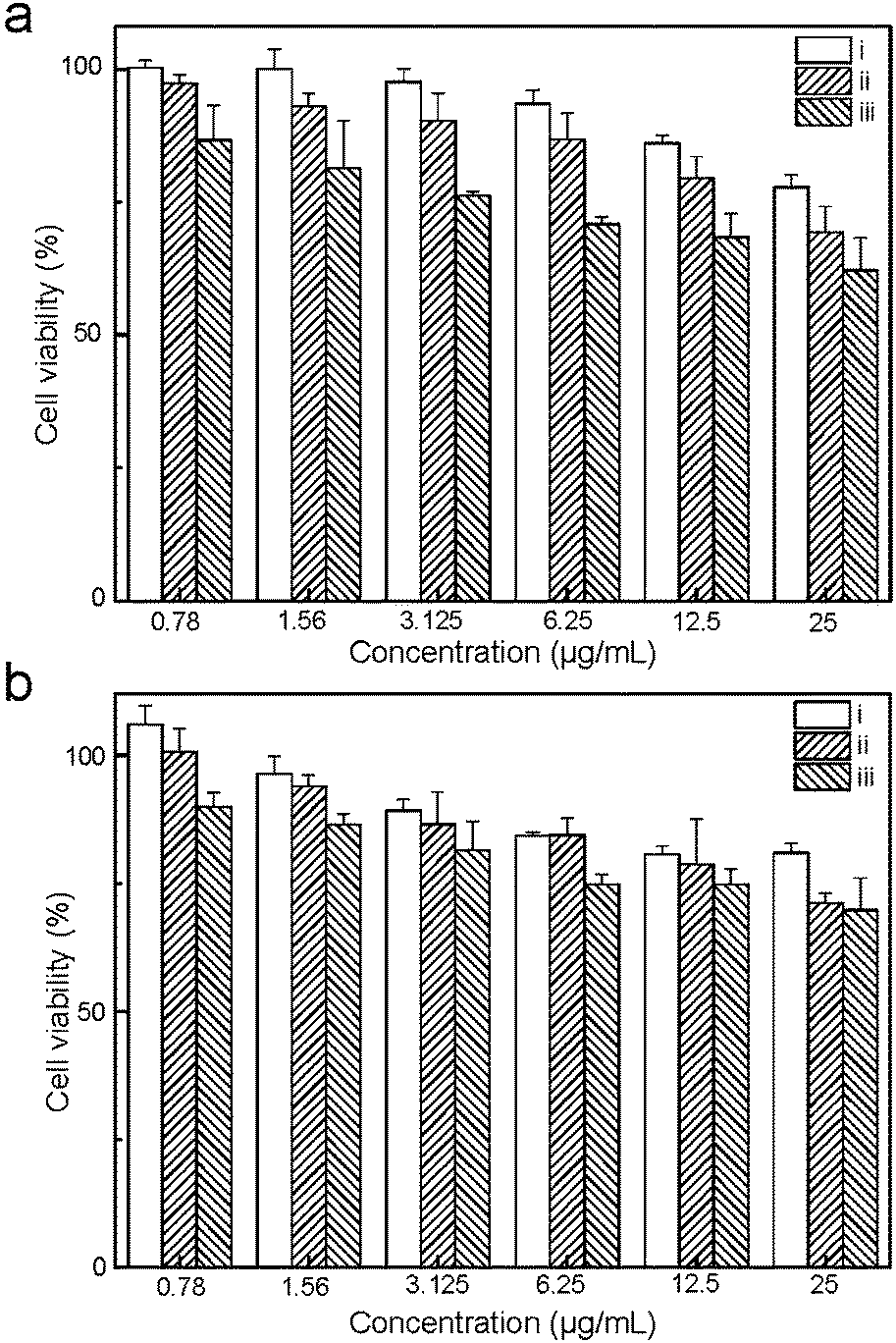
The cytotoxicity of the UCNP@SiO2 is examined by traditional MTT assay. Generally, all UCNP@SiO2 have moderate cytotoxicity against HeLa and HepG-2 cells when the concentrations of UCNP@SiO2 are more than 25 μg mL−1 (as shown in Fig. 5). For both HeLa cells and HepG-2 cells, UCNP@SiO2-PEG-NH2 exhibits the highest cytotoxicity and UCNP@SiO2-COOH has the lowest cytotoxicity. The MTT and ICP-MS experimental results indicate that the surface charge of UCNP@SiO2 plays a critical role in interactions of UCNP@SiO2 with their target cells. These interactions further determine intracellular uptake of UCNP@SiO2 and their effect on the cell functions. In principle the negative membrane potential of cells likely interacts more efficiently with cationic nanoparticles due to electrostatic interactions.17,46–48 This finding is consistent with the literature reports.15,44 In addition, the surface charge may also affect the protein corona (a multilayered cloud of proteins held together by weak noncovalent bonds) on UCNP@SiO2 surface, which ultimately impacts the cellular response.49 In culture medium, the weakly charged UCNP@SiO2-PEG may form larger and/or looser protein corona than those of highly charged UCNP@SiO2 because the hydrodynamic diameter of UCNP@SiO2-PEG is larger than those of UCNP@SiO2-PEG-NH2 and UCNP@SiO2-COOH. The larger hydrodynamic diameter increases the steric hindrance of the nanoparticle–cell interaction, resulting in low internalization efficiency of UCNP@SiO2-PEG and strong immune response of living cells. Therefore, the UCNP@SiO2-PEG shows lower cellular internalization efficiency and higher cytotoxicity than those of UCNP@SiO2-COOH.
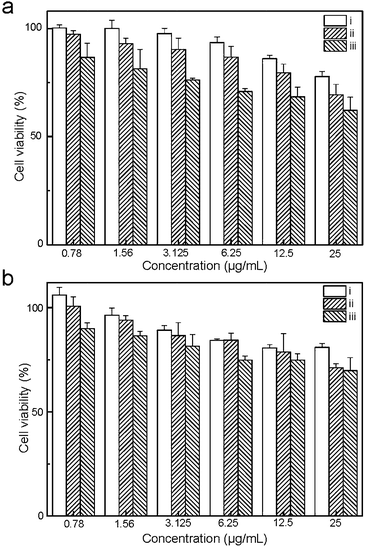 |
| | Fig. 5 MTT assay of HeLa (a) and HepG-2 (b) cells. The cells are treated with various concentrations of UCNP@SiO2-COOH (i), UCNP@SiO2-PEG (ii) and UCNP@SiO2-PEG-NH2 (iii), respectively. | |
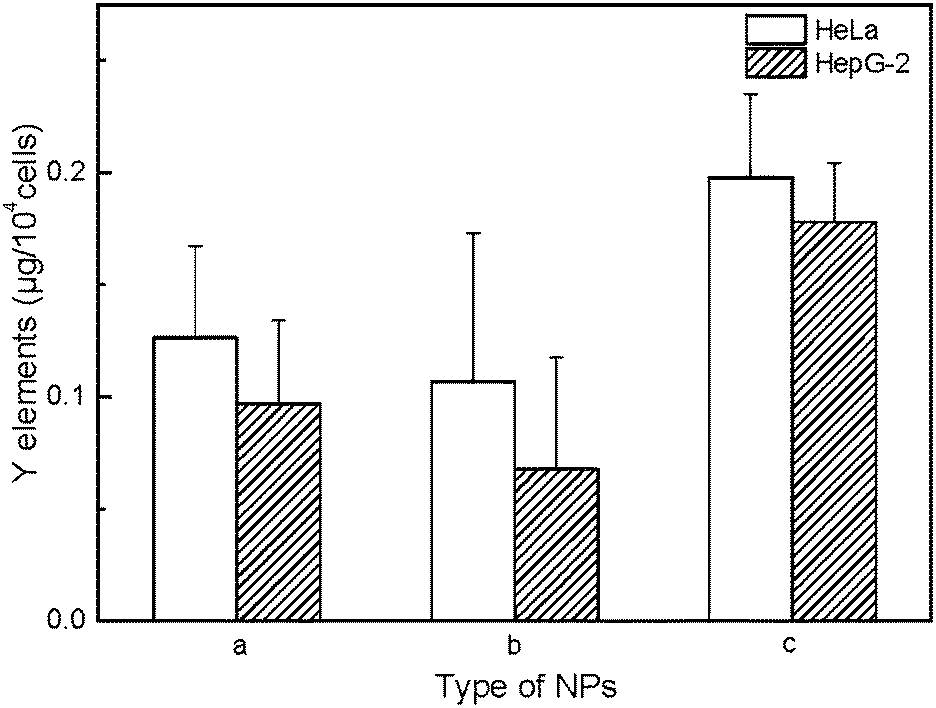
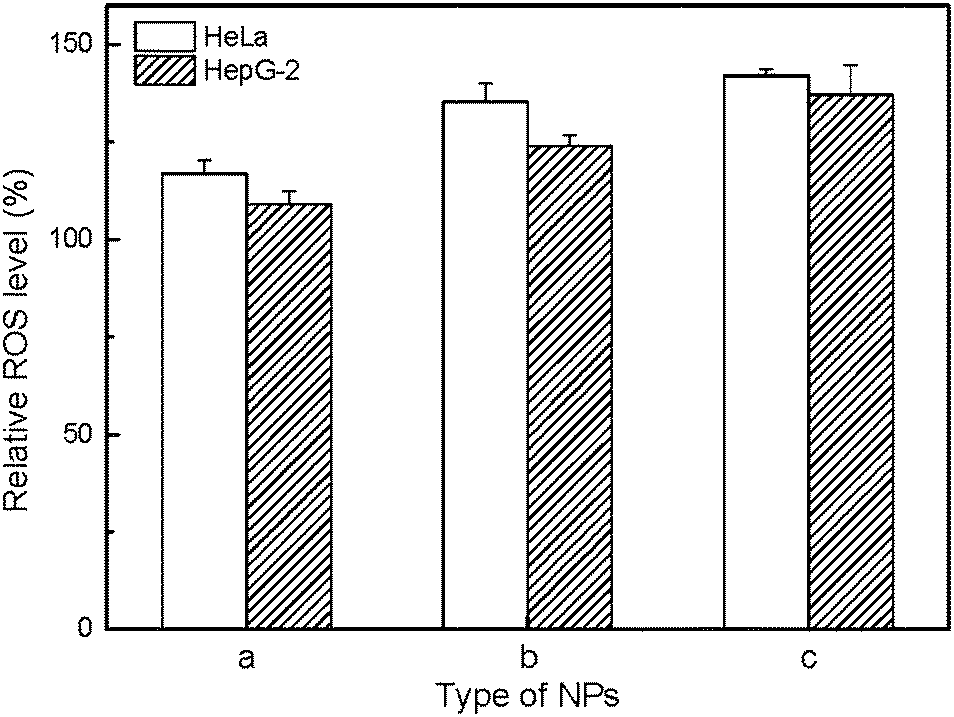
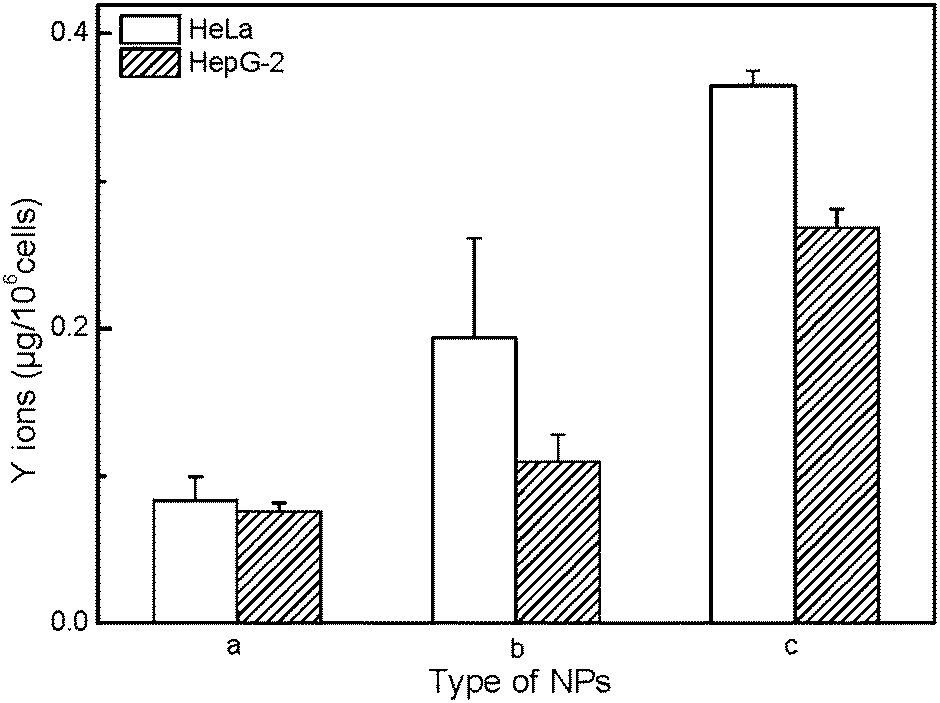
In order to evaluate the mechanism of cytotoxicity of UCNP@SiO2, we examined the redox status of cells on exposure to different UCNP@SiO2, intracellular releasing of metal ions and cytotoxicity of ligand. The UCNP@SiO2 induced redox stress is tested by DCFH-DA-based assay kit.41 DCFH-DA is redox-sensitive nonfluorescent dye, which fluoresces in response to intracellular ROS. The UCNP@SiO2-PEG-NH2 and UCNP@SiO2-PEG caused significant enhancement in intracellular ROS production compared to UCNP@SiO2-PEG-COOH treated cells (as shown in Fig. 6). The physiological function of cells could be disturbed by the accumulation of intracellular ROS.50,51 For instance, over redox stress can induce apoptosis of cells. Using Y as typical element, ICP-MS is employed to estimate the intracellular releasing of metal ions of UCNP@SiO2 stained cells. As shown in Fig. 7, the amounts of Y in cell lysates are following in the order: UCNP@SiO2-PEG-NH2 stained cells > UCNP@SiO2-PEG stained cells > UCNP@SiO2-COOH stained cells. The released metal ions could cause damage to cell function. Furthermore, the cytotoxicity of NH2-PEG-NH2 is higher than that of PEG-NH2 (as shown in Fig. S6 in the ESI†).
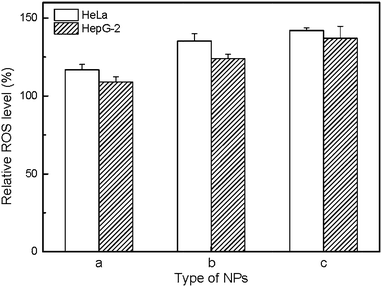 |
| | Fig. 6 ROS assay of HeLa and HepG-2 cells. The cells are treated with 25 μg mL−1 UCNP@SiO2-COOH (a), UCNP@SiO2-PEG (b) and UCNP@SiO2-PEG-NH2 (c), respectively. | |
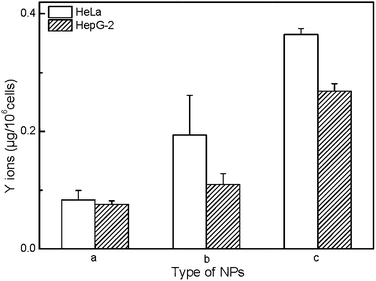 |
| | Fig. 7 ICP-MS measurements of intracellular releasing of Y ions. The cells are incubated with UCNP@SiO2-COOH (a), UCNP@SiO2-PEG (b) and UCNP@SiO2-PEG-NH2 (c), respectively. The concentrations of NPs are 25 μg mL−1. | |
In order to obtain NPs with higher colloidal stability, excess NH2-PEG-NH2 molecules are normally used in the process of PEG modification to prevent interparticle cross-linking.52,53 However, the excess NH2-PEG-NH2 molecules will be attracted to the surface of the NPs by electrostatic adsorption. Because of slightly acidic environments of some cell organelles (e.g., endosome, lysosome and mitochondria), the NH2-PEG-NH2 can be released from NP surface after UCNP@SiO2-PEG-NH2 uptaken by cells. Therefore, we surmise that cytotoxicity of UCNP@SiO2 is depends on the internalization amount, enhancement in intracellular ROS production, intracellular releasing metal ions and ligand of UCNP@SiO2.
Conclusions
In summary, UCNP@SiO2 core@shell NPs with different surface charges and PEG ligands have been synthesized for studying the interactions of NPs with cells. Through the systematic analysis of the interactions of UCNP@SiO2 NPs with two cell lines (HeLa and HepG-2), the experimental results demonstrate that positively charged UCNP@SiO2-PEG-NH2 exhibit higher cytotoxicity and cellular internalization efficiency than negatively charged UCNP@SiO2-COOH and UCNP@SiO2-PEG. We also posit that UCNP@SiO2-induced redox stress and releasing of metal ions and ligands might be the underlying mechanism of charge-based cytotoxicity. Therefore, we concluded that some factors including surface charge and physicochemical stability should be considered when engineering nanomaterials with excellent biocompatibility.
Acknowledgements
The authors thank National Basic Research Program of China (no. 2011CB935800) and National Natural Science Foundation of China (Grant no. 21205113) for financial support.
Notes and references
- L. Cheng, C. Wang, X. X. Ma, Q. L. Wang, Y. Cheng, H. Wang, Y. G. Li and Z. Liu, Adv. Funct. Mater., 2013, 23, 272–280 CrossRef CAS.
- T. W. Woolerton, S. Sheard, E. Reisner, E. Pierce, S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc., 2010, 132, 2132–2133 CrossRef CAS PubMed.
- L. Liao, Q. H. Zhang, Z. H. Su, Z. Z. Zhao, Y. N. Wang, Y. Li, X. X. Lu, D. G. Wei, G. Y. Feng, Q. K. Yu, X. J. Cai, J. M. Zhao, Z. F. Ren, H. Fang, F. R. Hernandez, S. Baldelli and J. M. Bao, Nat. Nanotechnol., 2014, 9, 69–73 CrossRef CAS PubMed.
- C. X. Li, J. L. Liu, S. Alonso, F. Y. Li and Y. Zhang, Nanoscale, 2012, 4, 6065–6071 RSC.
- Q. F. Xiao, X. P. Zheng, W. B. Bu, W. Q. Ge, S. J. Zhang, F. Chen, H. Y. Xing, Q. G. Ren, W. P. Fan, K. L. Zhao, Y. Q. Hua and J. L. Shi, J. Am. Chem. Soc., 2013, 135, 13041–13048 CrossRef CAS PubMed.
- H. S. Qian, H. C. Guo, P. C. L. Ho, R. Mahendran and Y. Zhang, Small, 2009, 5, 2285–2290 CrossRef CAS PubMed.
- N. M. Idris, M. K. Gnanasammandhan, J. Zhang, P. C. Ho, R. Mahendran and Y. Zhang, Nat. Med., 2012, 18, 1580–1585 CrossRef CAS PubMed.
- M. L. Yin, Z. H. Li, E. G. Ju, Z. Z. Wang, K. Dong, J. S. Ren and X. G. Qu, Chem. Commun., 2014, 50, 10488–10490 RSC.
- F. Wang, D. Banerjee, Y. S. Liu, X. Y. Chen and X. G. Liu, Analyst, 2010, 135, 1839–1854 RSC.
- C. Y. Liu, Z. Y. Gao, J. F. Zeng, Y. Hou, F. Fang, Y. L. Li, R. R. Qiao, H. Lei, W. S. Yang and M. Y. Gao, ACS Nano, 2013, 7, 7227–7240 CrossRef CAS PubMed.
- J. Zhu, L. Liao, L. N. Zhu, P. Zhang, K. Guo, J. L. Kong, C. Ji and B. H. Liu, Talanta, 2013, 107, 408–415 CrossRef CAS PubMed.
- J. L. Townson, Y. S. Lin, J. O. Agola, E. C. Carnes, H. S. Leong, J. D. Lewis, C. L. Haynes and C. J. Brinker, J. Am. Chem. Soc., 2013, 135, 16030–16033 CrossRef CAS PubMed.
- Y. J. Xiong, M. Brunson, J. Huh, A. Huang, A. Coster, K. Wendt, J. Fay and D. Qin, Small, 2013, 9, 2628–2638 CrossRef CAS PubMed.
- T. H. Chung, S. H. Wu, M. Yao, C. W. Lu, Y. S. Lin, Y. Hung, C. Y. Mou, Y. C. Chen and D. M. Huang, Biomaterials, 2007, 28, 2959–2966 CrossRef CAS PubMed.
- D. Hühn, X. K. Kantner, C. Geidel, S. Brandholt, I. D. Cock, S. J. H. Soenen, P. R. Gil, J. M. Montenegro, K. Braeckmans, K. Müllen, G. U. Nienhaus, M. Klapper and W. J. Parak, ACS Nano, 2013, 7, 3253–3263 CrossRef PubMed.
- N. Oh and J. H. Park, ACS Nano, 2014, 8, 6232–6241 CrossRef CAS PubMed.
- C. Graf, Q. Gao, I. Schütz, C. N. Noufele, W. T. Ruan, U. Posselt, E. Korotianskiy, D. Nordmeyer, F. Rancan, S. Hadam, A. Vogt, J. Lademann, V. Haucke and E. Rühl, Langmuir, 2012, 28, 7598–7613 CrossRef CAS PubMed.
- H. Xu, F. Yan, E. E. Monson and R. Kopelman, J. Biomed. Mater. Res., Part A, 2003, 66, 870–879 CrossRef PubMed.
- W. Huang, W. Q. Liu, Z. D. She, H. K. Wu and X. T. Shi, Appl. Surf. Sci., 2011, 257, 9757–9761 CrossRef CAS PubMed.
- J. L. Shi, J. L. Choi, B. Chou, R. N. Johnson, J. G. Schellinger and S. H. Pun, ACS Nano, 2013, 7, 10612–10620 CrossRef CAS PubMed.
- J. N. Shan and Y. G. Ju, Appl. Phys. Lett., 2007, 91, 123103 CrossRef PubMed.
- N. J. J. Johnson, N. M. Sangeetha, J. C. Boyer and F. C. J. M. Veggel, Nanoscale, 2010, 2, 771–777 RSC.
- S. Sabella, R. P. Carney, V. Brunetti, M. A. Malvindi, N. A. Juffali, G. Vecchio, S. M. Janes, O. M. Bakr, R. Cingolani, F. Stellacci and P. P. Pompa, Nanoscale, 2014, 6, 7052–7061 RSC.
- J. S. Chang, K. L. Chang, D. F. Hwang and Z. L. Kong, Environ. Sci. Technol., 2007, 41, 2064–2068 CrossRef CAS.
- T. H. Tran, B. C. Bae, Y. K. Lee, K. Na and K. M. Huh, Carbohydr. Polym., 2013, 92, 1615–1624 CrossRef CAS PubMed.
- J. J. Peng, Y. Sun, Q. Liu, Y. Yang, J. Zhou, W. Feng, X. Z. Zhang and F. Y. Li, Nano Res., 2012, 5, 770–782 CrossRef CAS.
- Y. Hou, R. R. Qiao, F. Fang, X. X. Wang, C. Y. Dong, K. Liu, C. Y. Liu, Z. F. Liu, H. Lei, F. Wang and M. Y. Gao, ACS Nano, 2013, 7, 330–338 CrossRef CAS PubMed.
- A. Nagy, A. Steinbrück, J. Gao, N. Doggett, J. A. Hollingsworth and R. Iyer, ACS Nano, 2012, 6, 4748–4762 CrossRef CAS PubMed.
- X. S. Liu, N. Huang, H. Li, Q. Jin and J. Ji, Langmuir, 2013, 29, 9138–9148 CrossRef CAS PubMed.
- B. G. Trewyn, J. A. Nieweg, Y. N. Zhao and V. S. Y. Lin, Chem. Eng. J., 2008, 137, 23–29 CrossRef CAS PubMed.
- F. Wang, Y. Han, C. S. Lim, Y. H. Lu, J. Wang, J. Xu, H. Y. Chen, C. Zhang, M. H. Hong and X. G. Liu, Nature, 2010, 463, 1061–1065 CrossRef CAS PubMed.
- Z. Q. Li, Y. Zhang and S. Jiang, Adv. Mater., 2008, 20, 4765–4769 CrossRef CAS.
- H. Hu, L. Q. Xiong, J. Zhou, F. Y. Li, T. Y. Cao and C. H. Huang, Chem.–Eur. J., 2009, 15, 3577–3584 CrossRef CAS PubMed.
- F. Y. Liu, Q. Zhao, H. P. You and Z. X. Wang, Nanoscale, 2013, 5, 1047–1053 RSC.
- G. Y. Chen, H. L. Qiu, P. N. Prasad and X. Y. Chen, Chem. Rev., 2014, 114, 5161–5214 CrossRef CAS PubMed.
- H. S. Mader, P. Kele, S. M. Saleh and O. S. Wolfbeis, Curr. Opin. Chem. Biol., 2010, 14, 582–596 CrossRef CAS PubMed.
- C. Wang, L. Cheng and Z. Liu, Theranostics, 2013, 3, 317–330 CrossRef CAS PubMed.
- Q. B. Zhang, K. Song, J. W. Zhao, X. G. Kong, Y. J. Sun, X. M. Liu, Y. L. Zhang, Q. H. Zeng and H. Zhang, J. Colloid Interface Sci., 2009, 336, 171–175 CrossRef CAS PubMed.
- L. Q. Xiong, T. S. Yang, Y. Yang, C. J. Xu and F. Y. Li, Biomaterials, 2010, 31, 7078–7085 CrossRef CAS PubMed.
- L. N. Sun, Z. W. Wei, H. G. Chen, J. L. Liu, J. J. Guo, M. Cao, T. Q. Wen and L. Y. Shi, Nanoscale, 2014, 6, 8878–8883 RSC.
- L. Sun, Y. Li, X. M. Liu, M. H. Jin, L. Zhang, Z. J. Du, C. X. Guo, P. L. Huang and Z. W. Sun, Toxicol. in Vitro, 2011, 25, 1619–1629 CrossRef CAS PubMed.
- J. B. Liu, M. X. Yu, X. H. Ning, C. Zhou, S. Y. Yang and J. Zheng, Angew. Chem., Int. Ed., 2013, 52, 12572–12576 CrossRef CAS PubMed.
- J. V. Jokerst, T. Lobovkina, R. N. Zare and S. S. Gambhir, Nanomedicine, 2011, 6, 715–728 CrossRef CAS PubMed.
- N. P. Mortensen, G. B. Hurst, W. Wang, C. M. Foster, P. D. Nallathambye and S. T. Retterer, Nanoscale, 2013, 5, 6372–6380 RSC.
- A. Albanese, C. D. Walkey, J. B. Olsen, H. B. Guo, A. Emili and W. C. W. Chan, ACS Nano, 2014, 8, 5515–5526 CrossRef CAS PubMed.
- A. E. Nel, L. Mädler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- P. R. Leroueil, S. A. Berry, K. Duthie, G. Han, V. M. Rotello, D. Q. Mcnerny, J. R. Baker, B. G. Orr and M. M. B. Holl, Nano Lett., 2008, 8, 420–424 CrossRef CAS PubMed.
- T. Xia, M. Kovochich, M. Liong, J. I. Zink and A. E. Nel, ACS Nano, 2008, 2, 85–96 CrossRef CAS PubMed.
- M. A. Dobrovolskaia, A. K. Patri, J. Simak, J. B. Hall, J. Semberova, S. H. D. P. Lacerda and S. E. McNeil, Mol. Pharm., 2012, 9, 382–393 CrossRef CAS PubMed.
- R. M. Graff, R. Rumpker, M. Richter, T. V. Braga, F. Kjeldsen, J. Brewer, J. Hoyland, H. G. Rubahn and H. Erdmann, Toxicol. in Vitro, 2014, 28, 1280–1289 CrossRef PubMed.
- K. Apel and H. Hirt, Annu. Rev. Plant Biol., 2004, 55, 373–399 CrossRef CAS PubMed.
- A. Quarta, R. D. Corato, L. Manna, S. Argentiere, R. Cingolani, G. Barbarella and T. Pellegrino, J. Am. Chem. Soc., 2008, 130, 10545–10555 CrossRef CAS PubMed.
- R. A. Sperling, T. Pellegrino, J. K. Li, W. H. Chang and W. J. Parak, Adv. Funct. Mater., 2006, 16, 943–948 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra11374h |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.