DOI:
10.1039/C4RA12773K
(Paper)
RSC Adv., 2015,
5, 7766-7772
Theoretical prediction of detonation performance and stability for energetic polydinitroaminoprismanes
Received
20th October 2014
, Accepted 17th December 2014
First published on 17th December 2014
Abstract
Density functional theory simulations were performed to calculate the heats of formation in the gas state [HOF(g)] and in the solid state [HOF(s)], detonation performance and stability of twelve polydinitroaminoprismanes. Our results show that C2 has the best detonation properties of all the molecules, the detonation velocity is 9.56 km s−1 and the detonation pressure is 41.88 GPa, and detonation properties of C2, C3, D1, D2, and D3 are better than those of 1,3,5,7-tetranitro-1,3,5,7-tetraazacyclooctane (HMX). The stability of all the molecules was investigated by analyzing the energy gaps, bond dissociation energies, and characteristic heights of the molecules. The results show that the N–NO2 bonds of all the molecules are trigger bonds during the thermolysis initiation process, and seven molecules (A, B1–B3 and C1–C3) are less sensitive than 2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaazatetracyclo[5.5.0.0.0]dodecane (CL-20). The results of this study may provide basic information for the further study of this kind of compounds and molecular design of novel energetic materials.
1. Introduction
Energetic materials are more and more widely studied and applied due to their high energy density.1–6 In order to meet the requirements of future applications, continuous efforts have been made to develop new materials having good thermal stability, impact and shock insensitivity, better detonation performance, economic and environmentally friendly syntheses.7,8 Unfortunately, these characteristics are often contradictory to each other. For example, most explosives showing high performance were proved to be sensitive (like 2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaazatetracyclo[5.5.0.0.0]dodecane, also called CL-20),9,10 while explosives with good thermal stability and insensitivity usually exhibits poor explosive performance (as 2,4,6-trinitrotoluene). Recently, Maija M. Kuklja provided a brief overview of the current understanding of sensitivity of energetic materials to detonation. The result indicates that defects, deformations, and electronically excited or charged states play an imperative role in triggering chemical decomposition in energetics, pointed out that one can manipulate chemical composition of materials and their morphology to tune sensitivity to initiation in many different combinations.11 The explosives reported in the literature have been classified as follows in order to make the presentation more clear:12 thermally stable or heat-resistant explosives; high performance explosives, i.e. high molecular density, high detonation pressure and velocity of explosives; melt-castable explosives; energetic binder for explosive compositions; insensitive high explosives. So far, the way in improving detonation performance is introducing high-energy groups, such as –NO2, –N3, and –CN, and so on. Both formation of intramolecular or intermolecular hydrogen bonds and enhancement of conjugated effect are often employed to improve molecular stability. As has been reported that the introduction of N → O on nitrogen-heterocycles has been found to be useful for increasing the energetic performance of energetic material.13,14 For example, Jean' ne M. Shreeve15 reports the syntheses and full characterization of 3,3′-dinitroamino-4,4′-azoxyfurazan and its nitrogen-rich salts. Their results show that most of these compounds have high density and good performance properties. However, these new oxygen-rich groups have drawbacks, such as they may be difficult to synthesise or they may be sensitive to thermal conditions, impact and friction. Therefore, further research is required to develop new nitrogen-rich groups.
Cage molecules are also an important class of useful energetic materials due to their compact molecule skeleton and large strain energies. Typical substances are octanitrocubane (ONC), CL-20, tetranitrotetraazacubane,16 polynitratecubanes, and polydifluoroaminocubanes.17 In addition, two novel and super-high energy cage explosives: dodecanitrohexaprismane (DNH) and hexanitrohexaazaprismane (HNHAN) have also been studied by Zhu.18 The results indicate that DNH has much higher energetic properties than ONC, which may be the most powerful nonnuclear explosive known so far. Prismane derivatives have also been extensive attention recently. For example, relative bond strengths in prismane and some of its aza analogs have been studied by Politzer and Seminario in 1990.19 Their results showed that the strengths of C–C bonds decrease as three-sided faces of prismanes > bonds between three-sided faces of prismane, and the introducing of nitrogen has the effect of strengthening C–C and some of N–C bonds, while the presence of N–N bond introduces a decrease in relative thermodynamic in the aza prismane series. Furthermore, the thermodynamics properties of polyisocyanoprismanes were also studies by Xu.20 Their results showed that on average, the contribution of one isocyano group to the heat of formation is about 232.3 kJ mol−1 and 234.1 kJ mol−1 at the B3LYP/6-31G** and B3P86/6-31G** levels, respectively. And calculated bond dissociation energies values of C–NC bonds were not affected by the position and the number of isocyano group. However, they did not study the detonation performance of polyisocyanoprismanes. On the basis of previous research, we have also designed a series of polynitrosoprismanes and polynitramineprismanes, stability and detonation performance were studied in detail at B3LPY/6-311G** level.21,22 The results indicated that although they are excellent thermodynamic properties, their detonation performance does not meet requirement. To improve detonation property of prismane derivatives, dinitroamino group [–N(NO2)2] is introduced on prismane skeleton. It has higher nitrogen content than nitro, nitroso, and nitramine group. The high nitrogen content can lead to high crystal density, which is associated with increased denotation performance.
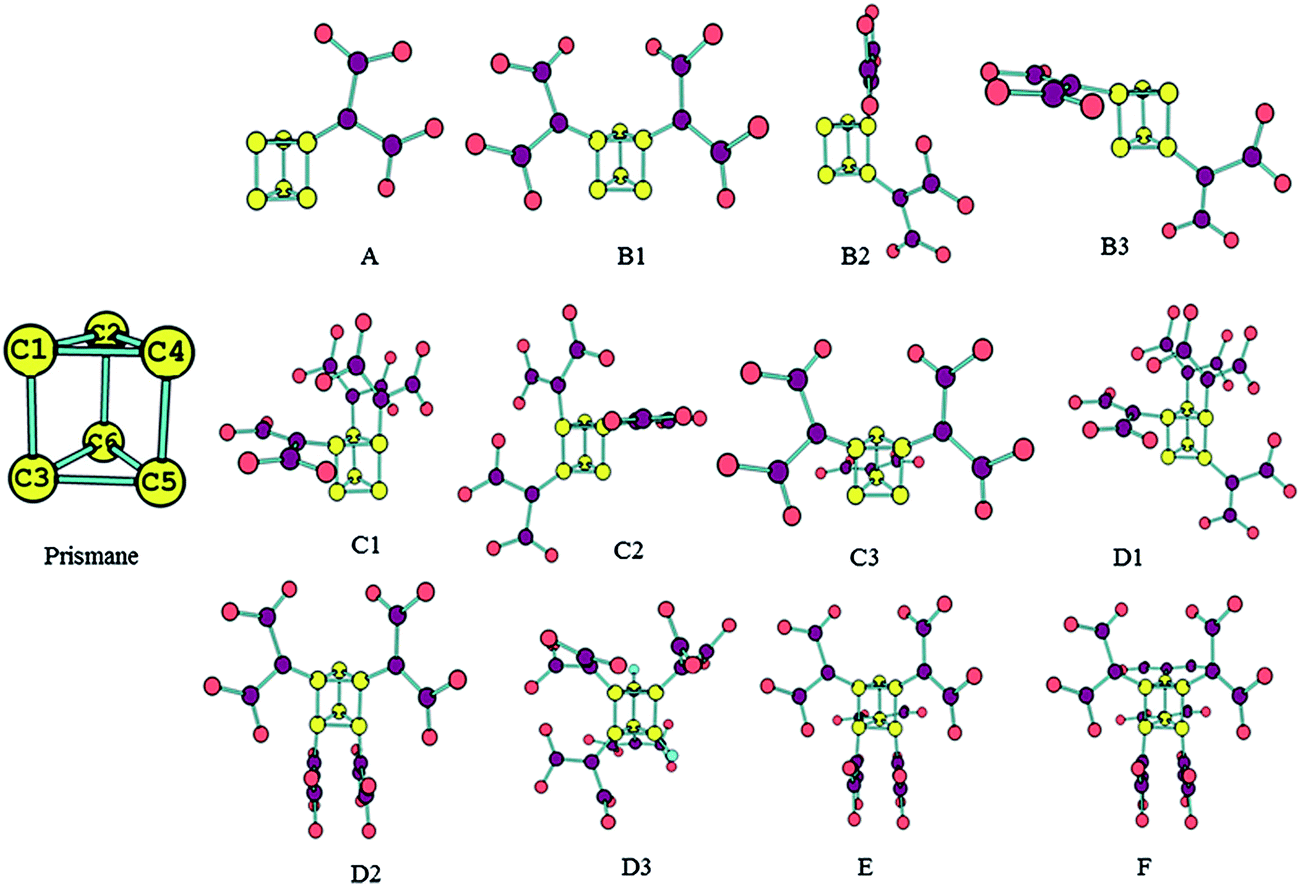
The main aim of present work is to design the molecules by replacement of the hydrogen atoms of prismane molecule with dinitroamino groups and make the new molecules (see Fig. 1) having well performance. We hope that our results can be helpful for further experiment to find novel high energy density materials (HEDMs).
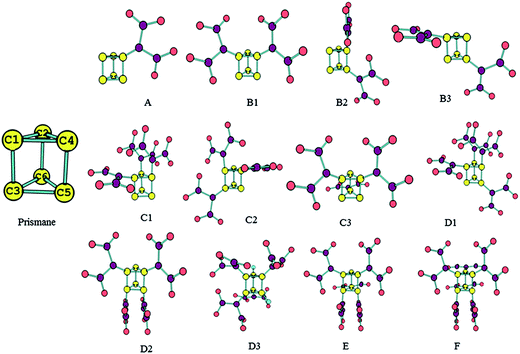 |
| | Fig. 1 Optimized the structures and names of prismane and polydinitroaminoprismanes. | |
2. Computational methods
Many studies have shown that the DFT-B3LYP method23 in combination with 6-311G** basis set can give accurate energies, molecular structures, and infrared vibrational frequencies. In this paper, the prismane derivatives are fully optimized at this level using the Gaussian 03 program package.24 In our work, we used the isodesmic reactions,25–27 in which the basic structural unit of prismane keeps invariable, and the big molecules are changed into small ones to obtain heat of formation (HOF). The isodesmic reactions for calculating the HOF of the title molecules at 298 K are the following:| | |
C6H6−n(N3O4)n + nCH4 = C6H6 + nCH3N3O4 (n = 1–6)
| (1) |
For the isodesmic reactions, heat of reactions ΔH298 at 298 K can be calculated form the following equation:
| | |
ΔH298 = ΔHf,P − ΔHf,R
| (2) |
where Δ
Hf,P and Δ
Hf,R are the HOF of the products and reactants at 298 K respectively. The experimental HOF of reference compound CH
4 and C
6H
6 are available, which are from NIST Chemistry Book Website.
28 Thus, the HOF of the prismane derivatives can be calculated when the heat of reaction Δ
H298 is known. The Δ
H298 is calculated using the following formula:
| | |
ΔH298 = ΔE + ΔZPE + ΔHT + nRT
| (3) |
Gas-phase HOF(g) usually misestimates the detonation properties of HEDMs. Therefore, solid-phase HOF(s) are calculated to determine the detonation properties of these molecules using the following equation:
| | |
ΔHOF(s) = ΔHOF(g) − ΔHsub
| (4) |
where Δ
Hsub is the heat of sublimation and can be evaluated by
eqn (5) suggested by Rice and Politzer
et al.29–31| | |
ΔHsub = α(SA)2 + β(γσtotal2)0.5 + λ
| (5) |
The values of α, β, and λ are from ref. 29.
Detonation velocity and detonation pressure are two important performance parameters for an energetic material, which can be calculated by Kamlet–Jacobs equations:15
| |
D = 1.01(N![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) 1/2Q1/2)1/2(1 + 1.30ρ0) 1/2Q1/2)1/2(1 + 1.30ρ0)
| (6) |
| | |
P = 1.558ρ02N1/2Q1/2
| (7) |
where
N is the moles of gaseous detonation products per gram of explosives,
is the average molecular weight of gaseous products,
Q is the chemical energy of detonation (cal g
−1) defined as the difference between the heats of formation of the products and that of reactants for the most exothermic reactants. Where
ρ0 is the density of explosives (g cm
−3).
As is known, accurate prediction of crystal density is of more difficulty for the unknown explosives. ‘‘Group or volume additivity” method,32 although simple and rapid, cannot give reliable results owing to its inherent drawbacks, while the ‘‘crystal packing” method,33,34 which is more reliable, has its limitation in routine calculation due to its extensive requirement in computational resources. However, the theoretical density was also obtained from the molecular weight divided by the average molecular volume. But the procedure used to estimate densities can lead to significant errors. Peter Politzer and coworker considered that the solid molecular density can be corrected by the electrostatic potential.35 The method is shown by eqn (8)–(12).
| | |
Crystal density (ρ0) = α2(M/Vm) + β2(νσtot2) + γ2
| (8) |
| |
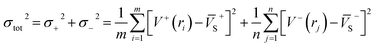 | (9) |
| |
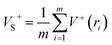 | (11) |
| |
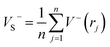 | (12) |
where
ν is balance parameter,
V(
r) is electrostatic potential,
V(
ri) is the value of
V(
r) at any point
ri on the surface,
VS+(
ri) and
VS−(
rj) represent the positive and negative value of
V(
r) on the surface.
![[V with combining macron]](https://www.rsc.org/images/entities/i_char_0056_0304.gif) S+
S+ and
S− are their averages,
σtot2 is the total variance. These parameters are obtained using Surface Analysis Suite compiled by Bulat.
36
The strength of bonding, which can be evaluated by bond dissociation energy (BDE), is fundamental to understanding chemical processes. The energy required for bond homolysis at 298 K and 1 atm corresponds to the enthalpy of reaction, A–B (g) → A˙ (g) + B˙ (g) which is the bond dissociation of enthalpy of the molecule A–B by definition. Therefore, at 0 K, the homolytic bond dissociation energy can be given in terms of eqn (13):
| | |
BDE(A–B) = E(A˙) + E(B˙) − E(A–B)
| (13) |
The bond dissociation energy with zero-point energy (ZPE) correction can be calculated by eqn (14).
| | |
BDE(A–B)ZPE = BDE(A–B) + ΔZPE
| (14) |
In addition, the characteristic height (h50) that can also reflect the impact sensitivity and stability of the molecules can be estimated using the eqn (15):37
| | |
h50 = α3σ+2 + β3γ + λ3
| (15) |
3. Results and discussion
3.1 Heats of formation (HOF)
In order to further investigate performance of polydinitroaminoprismanes, the structure parameters of the title compounds are given. Fig. 1 shows the structures of prismane and its derivatives. The types of C–C bonds for vicinal (e.g. C1–C4 bond) and distal bond (e.g. C1–C3 bond) can be distinguished in prismane. The vicinal bond with 1.522 Å is slight shorter than distal bond with 1.559 Å at B3LYP/6-311G** level. Our theoretical result is confirmed experimentally by X-ray diffraction.38 The polydinitroaminoprismanes are generated when the H atoms of prismane are replaced by dinitroamino group. HOF(s) reflects energy content of a compound. High positive HOF(s) is usually required for effective energetic materials. The method of isodesmic reactions is very successful to calculate HOF(s) from total energies obtained from ab initio calculations.39 We design isodesmic reactions in which the numbers of all kinds of bonds remain invariable to decrease the calculation error of HOF(s). Because the electronic circumstances of reactants and products are very similar in isodesmic reactions, the errors of electronic correction energies can be counteracted, and then the errors of the calculated HOF(s) can be greatly reduced. The related values of reference molecules are listed in Table 1. Table 2 lists the total energies, zero-point energies, thermal corrections, and calculated HOF(g) and HOF(s) of all molecules. The HOF(s) of all molecules are larger positive relative to prismane and increase with the increasing number of substituent group. In addition, there is a good linear relationship between HOF(s) and the number of substituent group (n): HOF(s) = 120.99n + 471.88 (n = 1–6, R2 = 0.989). Thus, the contributions of the dinitroamino groups to the HOF(s) of the polydinitroaminoprismanes clearly comply with the group additivity rule, and introducing an extra dinitroamino group increases the HOF(s) by 120.99 kJ mol−1. This means that the dinitroamino group plays an important role in increasing the HOF(s) for energetic materials. The reason is that dinitroamino group is an electron-withdrawing group, as the number of dinitroamino group on the prismane framework increases, two opposing factors are existed increasing the amount of electron withdrawal and the steric hindrance resulting from the additional functional group. In addition, abundant N–N bonds have also positive effect for increasing HOF(s) value. The relative positions of the dinitroamino groups have also some effect on the HOF(s). Such as B series, the two substituent groups are located on the same three-membered ring for B1, leading to a relatively high HOF(s). While the substituent group of B2 and B3 are bonded to the same four-membered ring, they are further apart, leading to a lower HOF(s) than that of B1. That is to say that the closer the dinitroamino groups are to one another, the higher the HOF(s), in other words, the higher energy density. Similar conclusion also can be found for other series.
Table 1 The related parameters of reference compounds
| Compounds |
E (a.u) |
ZPE (kJ mol−1) |
HT (kJ mol−1) |
HOF (kJ mol−1) |
| CH4 |
−40.53374 |
117.09 |
10.03 |
−74.60 |
| CH3N(NO2)2 |
−504.94238 |
179.03 |
22.55 |
53.50 |
| C6H6 |
−232.11118 |
253.89 |
13.23 |
567.71 |
Table 2 Calculated total energies (E0, a.u.), zero-point energies (ZPE, a.u), thermal corrections (HT, kJ mol−1), and heat of formation (HOF, kJ mol−1) of the prismane derivatives at the B3LYP/6-311G** levels
| Compounds |
E0 |
ZPE |
HT |
HOF(g) |
HOF(s) |
| A |
−696.531790 |
0.116868 |
30.17 |
649.42 |
620.43 |
| B1 |
−1160.943920 |
0.136820 |
47.41 |
753.13 |
714.06 |
| B2 |
−1160.946834 |
0.137021 |
47.05 |
745.65 |
706.07 |
| B3 |
−1160.946829 |
0.136665 |
47.79 |
745.47 |
704.66 |
| C1 |
−1625.346827 |
0.156703 |
64.40 |
880.63 |
832.79 |
| C2 |
−1625.355435 |
0.156798 |
64.64 |
858.52 |
808.43 |
| C3 |
−1625.353575 |
0.156275 |
65.53 |
862.92 |
810.10 |
| D1 |
−2089.753101 |
0.176516 |
81.58 |
999.30 |
938.18 |
| D2 |
−2089.759719 |
0.176752 |
81.70 |
982.66 |
921.55 |
| D3 |
−2089.754436 |
0.176258 |
82.61 |
1037.68 |
986.05 |
| E |
−2554.154513 |
0.196441 |
98.46 |
1130.72 |
1068.71 |
| F |
−3018.536232 |
0.216062 |
114.95 |
1312.65 |
1228.76 |
3.2 Detonation performance
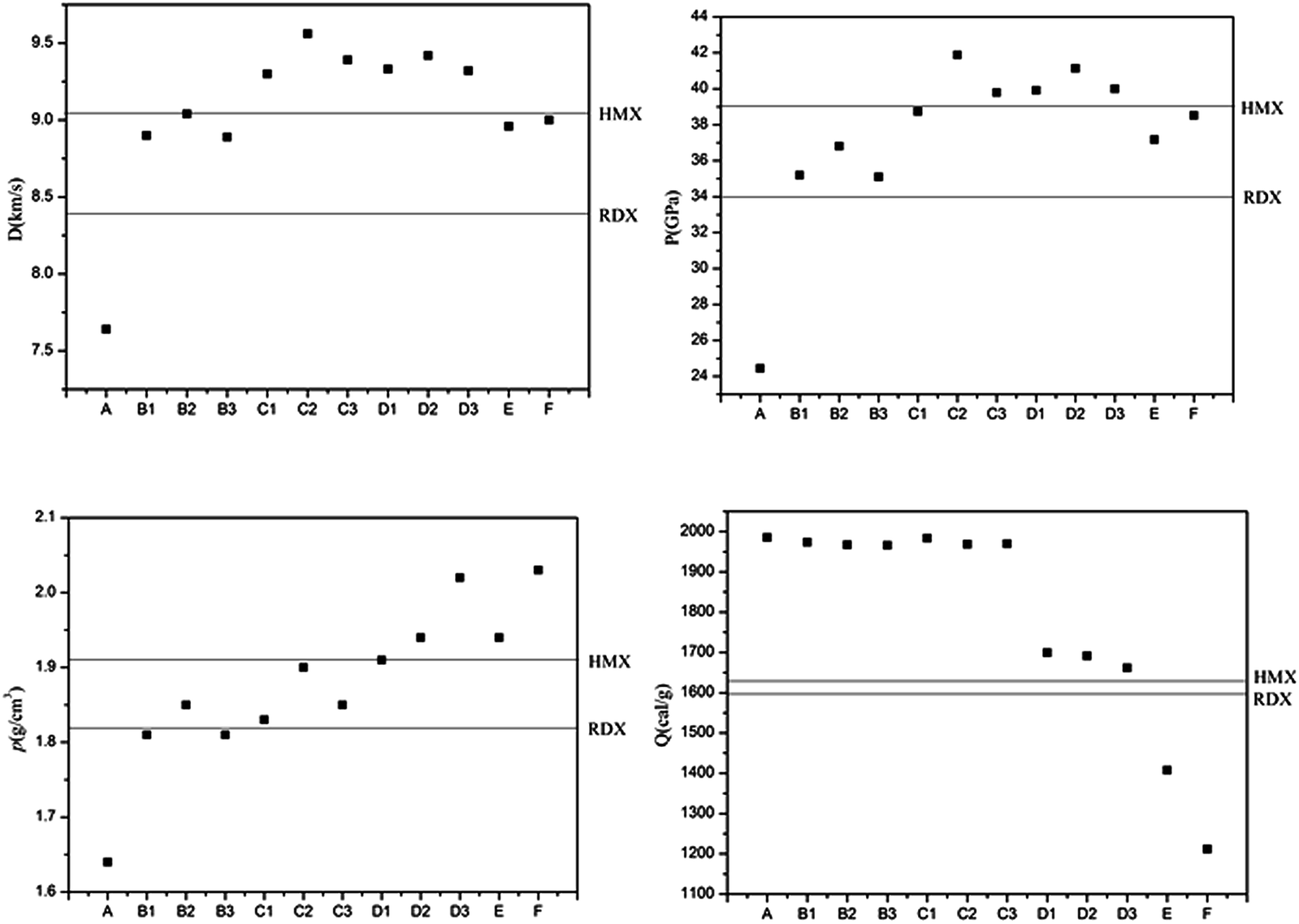
Two key factors of explosive performance are the detonation velocity (D) and the detonation pressure (P). They refer to the stable velocity of the shock front that characterizes detonation and the stable pressure that is developed behind the front, respectively. D and P can be evaluated by Kamlet–Jacobs empirical equations on the basis of their theoretical solid molecular density and calculated solid-phase heats of formation. D is proportional to the molecular density, while P is proportional to the square of the molecular density. The explosive reaction was identified by applying the “most exothermic” principle; that is, all of the N atoms turn into N2, while the O atoms initially react with H atoms to give H2O before forming CO2 with the C atom. The oxygen balance (OB100) influences detonation performance indirectly, so we also calculated OB100 following equation:40,41 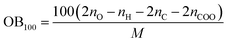 . where nO, nH, and nC represent the numbers of O, H, and C atoms in the molecule, nCOO is the number of carboxyl groups, and M is the molecular weight. Table 3 lists the OB100, heats of detonation, molecular volume, molecular solid density, detonation velocity, and detonation pressure of polydinitroaminoprismanes at B3LYP/6-311G** level. For comparative purposes, the experimental detonation performances of two known explosives (1,3,5-trinitroperhydro-1,3,5-triazine (RDX) and HMX) are also listed in this table, and more clear comparisons are showed in Fig. 2. Inspecting the Q values from Table 3, all molecules have larger Q, and the most of molecules possess higher Q than those of HMX and RDX except E and F. Furthermore, we find that Q decrease with the increasing of the number of substituent group. The change trend can be explained by oxygen balance. Positive high oxygen balance value shows that a large number of extra oxygen is produced in combustion reaction or explosive reaction, which is negative for the energy released. So, we should attach great importance to oxygen balance for designing novel energetic materials. For isomers, the relative positions of dinitroamino group also influence Q, the closer the dinitroamino groups are to one another, the higher the Q. For molecular volume and molecular density from Table 3, their change trend is the same that the values are larger with the increasing of the number of substituent group. A has the smallest molecular density (1.64 g cm−3) and F has the biggest molecular density (2.03 g cm−3) in all molecules. And the densities of the most of molecules are over 1.80 g cm−3. From Fig. 2, we conclude that C2, D1, D2, D3, E, and F can be used for future experiential tests as target molecules due to the fact that their densities are over 1.90 g cm−3. For D and P values given by Table 3, there is no linear relationship between D and P with the number of substituent group, the change trend does not comply with group additivity rule. The first increase and then decrease tendency of D and P is attributed to the gradually decreasing Q and increasing molecular density. C2 (D = 9.56 km s−1, P = 41.88 GPa), C3 (D = 9.39 km s−1, P = 39.79 GPa), D1 (D = 9.33 km s−1, P = 39.92 GPa), D2 (D = 9.42 km s−1, P = 41.14 GPa), and D3 (D = 9.32 km s−1, P = 40.00 GPa) have better detonation performance than HMX, and almost all the molecules have better performance than RDX except A. According to the usual energy criterion for HEDMs, i.e., D ≈ 9.0 km s−1, P ≈ 40.0 GPa,42 C2, C3, D1, D2, and D3 all satisfy the requirements. This indicates that they are all potential energetic molecules.
. where nO, nH, and nC represent the numbers of O, H, and C atoms in the molecule, nCOO is the number of carboxyl groups, and M is the molecular weight. Table 3 lists the OB100, heats of detonation, molecular volume, molecular solid density, detonation velocity, and detonation pressure of polydinitroaminoprismanes at B3LYP/6-311G** level. For comparative purposes, the experimental detonation performances of two known explosives (1,3,5-trinitroperhydro-1,3,5-triazine (RDX) and HMX) are also listed in this table, and more clear comparisons are showed in Fig. 2. Inspecting the Q values from Table 3, all molecules have larger Q, and the most of molecules possess higher Q than those of HMX and RDX except E and F. Furthermore, we find that Q decrease with the increasing of the number of substituent group. The change trend can be explained by oxygen balance. Positive high oxygen balance value shows that a large number of extra oxygen is produced in combustion reaction or explosive reaction, which is negative for the energy released. So, we should attach great importance to oxygen balance for designing novel energetic materials. For isomers, the relative positions of dinitroamino group also influence Q, the closer the dinitroamino groups are to one another, the higher the Q. For molecular volume and molecular density from Table 3, their change trend is the same that the values are larger with the increasing of the number of substituent group. A has the smallest molecular density (1.64 g cm−3) and F has the biggest molecular density (2.03 g cm−3) in all molecules. And the densities of the most of molecules are over 1.80 g cm−3. From Fig. 2, we conclude that C2, D1, D2, D3, E, and F can be used for future experiential tests as target molecules due to the fact that their densities are over 1.90 g cm−3. For D and P values given by Table 3, there is no linear relationship between D and P with the number of substituent group, the change trend does not comply with group additivity rule. The first increase and then decrease tendency of D and P is attributed to the gradually decreasing Q and increasing molecular density. C2 (D = 9.56 km s−1, P = 41.88 GPa), C3 (D = 9.39 km s−1, P = 39.79 GPa), D1 (D = 9.33 km s−1, P = 39.92 GPa), D2 (D = 9.42 km s−1, P = 41.14 GPa), and D3 (D = 9.32 km s−1, P = 40.00 GPa) have better detonation performance than HMX, and almost all the molecules have better performance than RDX except A. According to the usual energy criterion for HEDMs, i.e., D ≈ 9.0 km s−1, P ≈ 40.0 GPa,42 C2, C3, D1, D2, and D3 all satisfy the requirements. This indicates that they are all potential energetic molecules.
Table 3 Calculated the detonation performances of all molecules
| Compound |
OB100 |
Q (cal g−1) |
V (cm3) |
ρ (g cm−3) |
D (km s−1) |
P (GPa) |
| A |
−4.92 |
1985.35 |
114.67 |
1.64 |
7.64 |
24.45 |
| B1 |
0 |
1973.64 |
157.95 |
1.81 |
8.90 |
35.19 |
| B2 |
0 |
1967.01 |
157.23 |
1.85 |
9.04 |
36.81 |
| B3 |
0 |
1965.85 |
158.76 |
1.81 |
8.89 |
35.10 |
| C1 |
2.29 |
1983.46 |
206.33 |
1.83 |
9.30 |
38.74 |
| C2 |
2.29 |
1968.65 |
200.42 |
1.90 |
9.56 |
41.88 |
| C3 |
2.29 |
1969.66 |
204.45 |
1.85 |
9.39 |
39.79 |
| D1 |
3.61 |
1699.45 |
248.70 |
1.91 |
9.33 |
39.92 |
| D2 |
3.61 |
1691.47 |
245.60 |
1.94 |
9.42 |
41.14 |
| D3 |
3.61 |
1661.93 |
246.64 |
2.02 |
9.32 |
40.00 |
| E |
4.48 |
1407.33 |
294.28 |
1.94 |
8.96 |
37.18 |
| F |
5.08 |
1211.83 |
328.99 |
2.03 |
9.00 |
38.52 |
| RDX |
0 |
1591.03 |
— |
1.82 |
8.75 |
34.00 |
| HMX |
0 |
1633.90 |
— |
1.91 |
9.10 |
39.00 |
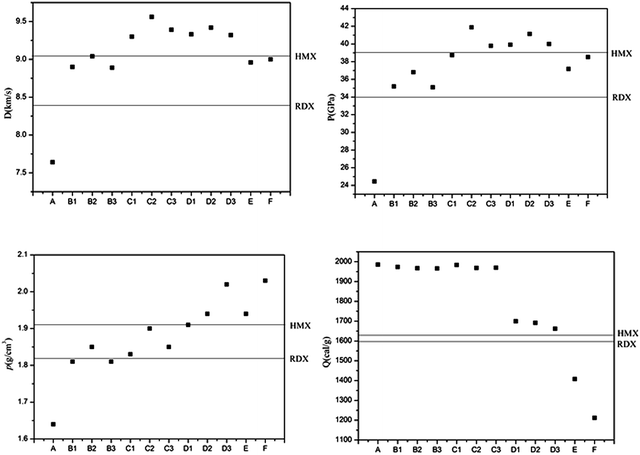 |
| | Fig. 2 Densities, heats of detonation, detonation velocities, and detonation pressures of polydinitroaminoprismanes. | |
3.3 Electronic structure and molecular stability
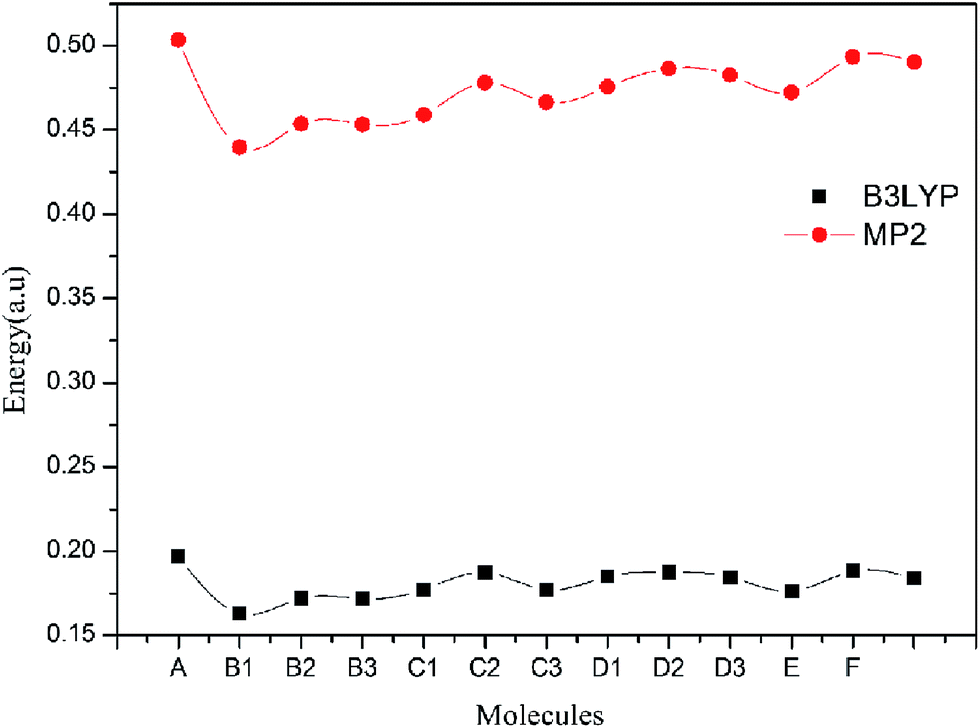
The energy gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) is important. Because energy gap closing may be the initiation step for the detonation of explosives.43 A larger energy gap implies high stability and small energy gap implies low stability, smaller the energy band between HOMO and LUMO, easier the electron transition and lesser the stability of explosive will be. Table 4 lists the energy gaps (ΔE) of all molecules at the B3LYP/6-311G** and MP2/6-311G** levels, respectively. ΔEB3LYP is obtained at B3LYP/6-311G** level, and ΔEMP2 is calculated at the MP2/6-311G** level. Fig. 3 shows the change trends of ΔE at two levels. From Table 4, it is found that all title molecules have lower energy gaps than that of unsubstituted prismane at the two levels. From Fig. 3, we know that although the ΔEB3LYP and ΔEMP2 have remarkable difference, the change trends of ΔEB3LYP and ΔEMP2 of all molecules are consistent. A has the smallest ΔE (0.1629 and 0.4398 a.u) and E has the biggest ΔE (0.1882 and 0.4933 a.u) in all molecules at B3LYP/6-311G** and MP2/6-311G** levels, respectively. The substituent group has little effect on the energy gaps for isomeride. It is noteworthy that ΔEB3LYP of all molecules are higher than 1,3,5-triamino-2,4,6-trinitrobenzene (TATB, 0.1621 a.u), which means that polydinitroaminoprismanes are less sensitive than TATB.
Table 4 Calculated energy gaps ΔEB3LYP (a.u) and ΔEMP2 (a.u) of the title molecules at the B3LYP/6-311G** and MP2/6-311G** levels, respectively
| Compounds |
ΔEB3LYP |
ΔEMP2 |
| Prismane |
0.1968 |
0.5034 |
| A |
0.1629 |
0.4398 |
| B1 |
0.1722 |
0.4536 |
| B2 |
0.1718 |
0.4533 |
| B3 |
0.1773 |
0.4590 |
| C1 |
0.1874 |
0.4780 |
| C2 |
0.1772 |
0.4664 |
| C3 |
0.1849 |
0.4756 |
| D1 |
0.1876 |
0.4864 |
| D2 |
0.1846 |
0.4824 |
| D3 |
0.1763 |
0.4723 |
| E |
0.1882 |
0.4933 |
| F |
0.1838 |
0.4903 |
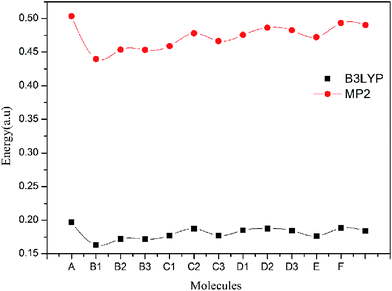 |
| | Fig. 3 The change trends of ΔEB3LYP and ΔEMP2 of the title molecules. | |
BDE provides useful information for understanding the stability of the title molecules. Generally speaking, the smaller the BDE, the more unstability the bond. Therefore, the BDE is directly related to the stability of energetic molecules. In our work, two possible bond dissociations are considered: (1) the C–N3O4 bond, (2) the N–NO2 bond. It should be pointed out that, among bonds of the same type, the weakest bond was selected as the breaking bond by the principle of the smallest bond order.44 A smaller bond order generally indicates a weaker bond. We obtained the bond orders of twelve polydinitroaminoprismanes by natural bond orbital (NBO) analyses. Table 5 lists the bond order, BDE, and characteristic height of the weakest bond of the title molecules at B3LYP/6-311G** level. We can find that all bond orders of C–N3O4 bonds are larger than that of N–NO2 bonds for same molecule and this indicates that the strength of C–N3O4 bonds is stronger than N–NO2 bonds. The same conclusion can be obtained by analyzing calculated BDE. For BDE0 and BDEZPE of C–N3O4 and N–NO2 bonds, the BDEZPE after zero point corrections is about 20 kJ mol−1 lower than that BDE0 without zero point energy corrections. However, the order of the BDEZPE and pyrolysis mechanism is not affected by the zero-point energies. The BDEZPE for the C–N3O4 bonds of all molecules are larger than 315 kJ mol−1, while those for N–NO2 bonds are smaller than 116 kJ mol−1. So, the trigger bond for polydinitroaminoprismanes is N–NO2 bond on the chain. However, the BDEZPE of all molecules are over 80 kJ mol−1, which meet the requirements as HEDMs. Furthermore, we should note that the B2 has relatively lower bond order but higher BDEZPE to B1. This result shows that to judge the thermal stability of prismane derivatives is not only by the bond order simply, but also depend on the BDEZPE.
Table 5 Calculated bond dissociation energies (BDE, kJ mol−1) and bond order (BO) of the weakest bonds, and characteristic height (h50, cm) for the prismane derivatives at the B3LYP/6-311G** level
| Compound |
BOC–N |
BDE0 |
BDEZPE |
BON–N |
BDE0 |
BDEZPE |
h50 |
| A |
1.0145 |
386.93 |
368.41 |
0.8149 |
116.93 |
98.33 |
55.54 |
| B1 |
1.0147 |
371.46 |
353.64 |
0.8120 |
121.63 |
102.74 |
30.83 |
| B2 |
1.0239 |
380.62 |
362.47 |
0.7922 |
125.16 |
104.76 |
39.91 |
| B3 |
1.0173 |
380.18 |
362.56 |
0.8305 |
136.26 |
115.95 |
31.79 |
| C1 |
1.0222 |
358.96 |
341.02 |
0.7323 |
113.81 |
94.64 |
14.33 |
| C2 |
1.0189 |
370.13 |
352.94 |
0.7960 |
129.07 |
109.03 |
16.92 |
| C3 |
1.0168 |
368.95 |
351.83 |
0.7677 |
125.98 |
107.49 |
13.67 |
| D1 |
1.0225 |
350.59 |
332.92 |
0.7344 |
118.70 |
99.48 |
8.75 |
| D2 |
1.0271 |
370.48 |
352.59 |
0.8043 |
134.15 |
113.83 |
8.89 |
| D3 |
1.0215 |
353.68 |
335.90 |
0.7963 |
125.84 |
108.91 |
7.96 |
| E |
1.0244 |
345.02 |
327.51 |
0.7425 |
124.59 |
105.36 |
7.51 |
| F |
1.0114 |
335.14 |
315.98 |
0.7511 |
107.98 |
94.15 |
6.01 |
The impact sensitivity also reflects the stability of a molecule, and it can be evaluated by the characteristic height (h50), which has been predicted in this work, the calculated results are listed in Table 5. It is found that the h50 decrease with increasing of the numbers of substituent group, which shows that the stabilities of molecules are more and more sensitivity. The conclusion from h50 is not very consistent with that from BDEZPE, such as the BDEZPE of N–NO2 bond of B3 is the biggest in all molecules, but it is not the case for h50, which supports the proposition of Politzer45 that the correlation between bond strength and impact sensitivity is not general but limited within certain classes of molecules. Compared with the h50 of famous explosive CL-20 (h50 = 12 cm), it is found that the most of derivatives are less sensitive than CL-20.
4. Conclusion
In conclusion, the molecular geometries, heat of formation, energy gap, bond dissociation energy, characteristic height, and detonation performance of polydinitroaminoprismanes have been explored at B3LYP/6-311G** level. From this theoretical study, the following conclusions can be drawn: the HOF(s) of polynitroaminoprismanes are larger and have a good linear relationship with increasing the numbers of dinitroamino group, introducing an extra dinitroamino group increases the HOF(s) by 120.99 kJ mol−1. The D and P have a binomial relationship with the number of substituent group. The calculated values of ρ, D and P for the isomers are close, implying that the position of dinitroamino groups has little influence on these parameters. According to the usual energy criterion for HEDMs, C2, C3, D1, D2, and D3 can become candidate of energetic materials. The energy gaps of all molecules have larger than that of TATB at B3LYP/6-311G** level. The trigger bond of all molecules is N–NO2 bond, and the BDEZPE of all molecules are over 80 kJ mol−1. The calculated impact sensitivity indicates that the most of molecules are more stability than CL-20. Overall consideration, C2 and C3 can become potential high energy with low sensitivity materials. We hope that these results are helpful for further developing now HEDMs.
Acknowledgements
This work is financially supported by the Major State Basic Research Development Programs of China (2011CBA00701), the National Natural Science Foundation of China (21473010, 20933001), This work is also supported by the opening project of State Key Laboratory of Explosion Science and Technology (Beijing Institute of Technology) (ZDKT12-03).
References
- O. S. Bushuyev, P. Brown, A. Maiti, R. H. Gee, G. R. Peterson, B. L. Weeks and L. J. Hope-Weeks, J. Am. Chem. Soc., 2012, 134, 1422–1425 CrossRef CAS PubMed.
- G. K. Windler, M. X. Zhang, R. Zitterbart, P. F. Pagoria and K. P. C. Vollhardt, Chem.–Eur. J., 2012, 18, 6588–6603 CrossRef CAS PubMed.
- V. Thottempudi, H. Gao and J. N. M. Shreeve, J. Am. Chem. Soc., 2011, 133, 6464–6471 CrossRef CAS PubMed.
- T. Yan, G. Sun, W. Chi, L. Li, B. Li and H. Wu, C. R. Chim., 2013, 16, 765–772 CrossRef CAS PubMed.
- X. Jin, B. Hu, W. Lu, S. Gao, Z. Liu and C. Lv, RSC Adv., 2014, 4, 6471–6477 RSC.
- Y. Q. Wu, F. L. Huang and Z. Y. Zhang, RSC Adv., 2012, 2, 4152–4163 RSC.
- A. Elbeih, M. Jungová, S. Zeman, P. Vávra and Z. Akštein, Propellants, Explos., Pyrotech., 2012, 37, 329–334 CrossRef CAS.
- X. Jin, B. Hu, Z. Liu and C. Lv, RSC Adv., 2014, 4, 23898–23903 RSC.
- R. Simpson, P. Urtiew, D. Ornellas, G. Moody, K. Scribner and D. Hoffman, Propellants, Explos., Pyrotech., 1997, 22, 249–255 CrossRef CAS.
- P. Karakaya, M. Sidhoum, C. Christodoulatos, S. Nicolich and W. Balas, J. Hazard. Mater., 2005, 120, 183–191 CrossRef CAS PubMed.
- M. M. Kuklja, in Adv. Quantum Chem., ed. R. S. John, Academic Press, 2014, vol. 69, pp. 71–145 Search PubMed.
- J. P. Agrawal, Prog. Energy Combust. Sci., 1998, 24, 1–30 CrossRef CAS.
- M. Göbel, K. Karaghiosoff, T. M. Klapötke, D. G. Piercey and J. R. Stierstorfer, J. Am. Chem. Soc., 2010, 132, 17216–17226 CrossRef PubMed.
- P. Yin, D. A. Parrish and J. N. M. Shreeve, Angew. Chem., Int. Ed., 2014, 53, 12889–12892 CrossRef CAS PubMed.
- J. Zhang and J. N. M. Shreeve, J. Am. Chem. Soc., 2014, 136, 4437–4445 CrossRef CAS PubMed.
- P. Politzer, P. Lane and J. S. Murray, Cent. Eur. J. Energ. Mater., 2011, 8, 39–52 CAS.
- D. A. Hrovat, W. T. Borden, P. E. Eaton and B. Kahr, J. Am. Chem. Soc., 2001, 123, 1289–1293 CrossRef CAS.
- Q. Wu, W. Zhu and H. Xiao, RSC Adv., 2014, 4, 3789–3797 RSC.
- P. Politzer and J. M. Seminario, Struct. Chem., 1990, 1, 29–32 CrossRef CAS.
- W. G. Xu, X. F. Liu and S. X. Lu, J. Hazard. Mater., 2009, 162, 1317–1321 CrossRef CAS PubMed.
- W. Chi, G. Sun, T. Liu, B. Li and H. Wu, J. Mol. Model., 2012, 18, 4557–4563 CrossRef CAS PubMed.
- W. J. Chi, L. L. Li, B. T. Li and H. S. Wu, J. Mol. Model., 2013, 19, 1049–1057 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- M. J. Frisch, G. W. Trucks and H. B. Schlegel, Gaussian 03,Revision C.02., Gaussian Inc, Wallingford CT, 2004 Search PubMed.
- V. Ghule, P. Jadhav, R. Patil, S. Radhakrishnan and T. Soman, J. Phys. Chem. A, 2009, 114, 498–503 CrossRef PubMed.
- W. J. Hehre, R. Ditchfield, L. Radom and J. A. Pople, J. Am. Chem. Soc., 1970, 92, 4796–4801 CrossRef CAS.
- X. W. Fan and X. H. Ju, J. Comput. Chem., 2008, 29, 505–513 CrossRef CAS PubMed.
- http://webbook.nist.gov/chemistry/.
- P. Politzer, J. S. Murray, M. Edward Grice, M. Desalvo and E. Miller, Mol. Phys., 1997, 91, 923–928 CrossRef CAS.
- E. F. C. Byrd and B. M. Rice, J. Phys. Chem. A, 2005, 110, 1005–1013 CrossRef PubMed.
- B. M. Rice, S. V. Pai and J. Hare, Combust. Flame, 1999, 118, 445–458 CrossRef CAS.
- H. L. Ammon, Struct. Chem., 2001, 12, 205–212 CrossRef CAS.
- H. Karfunkel and R. Gdanitz, J. Comput. Chem., 1992, 13, 1171–1183 CrossRef CAS.
- D. C. Sorescu, B. M. Rice and D. L. Thompson, J. Phys. Chem. B, 1997, 101, 798–808 CrossRef CAS.
- P. Politzer, J. Martinez, J. S. Murray, M. C. Concha and A. Toro-Labbe, Mol. Phys., 2009, 107, 2095–2101 CrossRef CAS.
- F. A. Bulat, A. Toro-Labbé, T. Brinck, J. S. Murray and P. Politzer, J. Mol. Model., 2010, 16, 1679–1691 CrossRef CAS PubMed.
- M. Pospíšil, P. Vávra, M. C. Concha, J. S. Murray and P. Politzer, J. Mol. Model., 2010, 16, 895–901 CrossRef PubMed.
- H. Wingert, G. Maas and M. Regitz, Tetrahedron, 1986, 42, 5341–5353 CrossRef CAS.
- P. V. R. Schleyer, L. Radom, W. J. Hahre and J. A. Pople, Ab Initio Molecular Orbital Theory, New York, 1986 Search PubMed.
- M. Kamlet and H. Adolph, Propellants, Explos., Pyrotech., 1979, 4, 30–34 CrossRef CAS.
- C. Cao and S. Gao, J. Phys. Chem. B, 2007, 111, 12399–12402 CrossRef CAS PubMed.
- M. X. Xiao, X. J. Xu and L. Qiu, Theoretical Design of High Energy and Density Materials, Science Press, Beijin, 2008 Search PubMed.
- J. J. Gilman, Philos. Mag. B, 1993, 67, 207–214 CAS.
- F. Jianfen and X. Heming, J. Mol. Struct.: THEOCHEM, 1996, 365, 225–229 CrossRef.
- P. Politzer and J. S. Murray, J. Mol. Struct., 1996, 376, 419–424 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
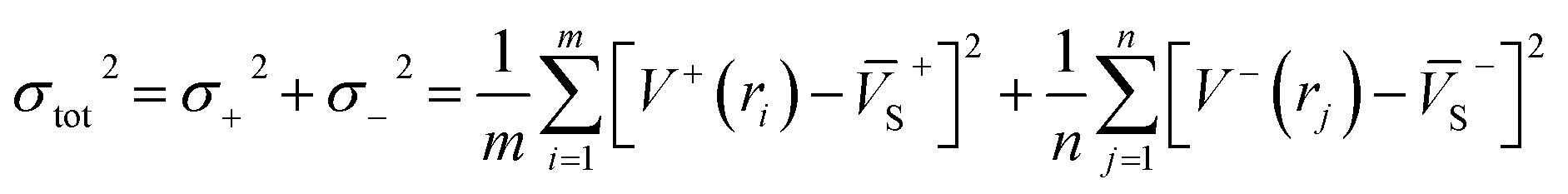
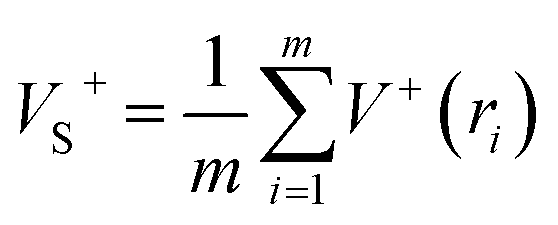
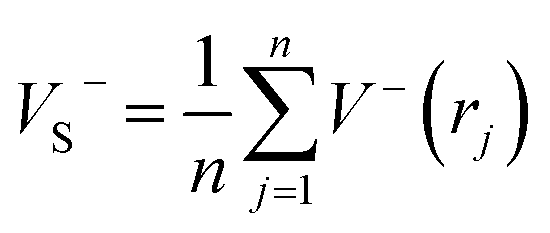
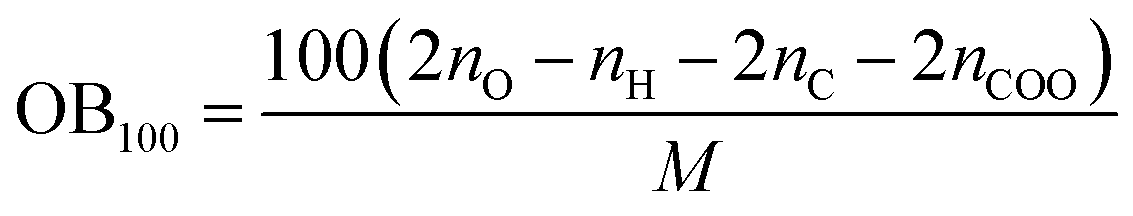 . where nO, nH, and nC represent the numbers of O, H, and C atoms in the molecule, nCOO is the number of carboxyl groups, and M is the molecular weight. Table 3 lists the OB100, heats of detonation, molecular volume, molecular solid density, detonation velocity, and detonation pressure of polydinitroaminoprismanes at B3LYP/6-311G** level. For comparative purposes, the experimental detonation performances of two known explosives (1,3,5-trinitroperhydro-1,3,5-triazine (RDX) and HMX) are also listed in this table, and more clear comparisons are showed in Fig. 2. Inspecting the Q values from Table 3, all molecules have larger Q, and the most of molecules possess higher Q than those of HMX and RDX except E and F. Furthermore, we find that Q decrease with the increasing of the number of substituent group. The change trend can be explained by oxygen balance. Positive high oxygen balance value shows that a large number of extra oxygen is produced in combustion reaction or explosive reaction, which is negative for the energy released. So, we should attach great importance to oxygen balance for designing novel energetic materials. For isomers, the relative positions of dinitroamino group also influence Q, the closer the dinitroamino groups are to one another, the higher the Q. For molecular volume and molecular density from Table 3, their change trend is the same that the values are larger with the increasing of the number of substituent group. A has the smallest molecular density (1.64 g cm−3) and F has the biggest molecular density (2.03 g cm−3) in all molecules. And the densities of the most of molecules are over 1.80 g cm−3. From Fig. 2, we conclude that C2, D1, D2, D3, E, and F can be used for future experiential tests as target molecules due to the fact that their densities are over 1.90 g cm−3. For D and P values given by Table 3, there is no linear relationship between D and P with the number of substituent group, the change trend does not comply with group additivity rule. The first increase and then decrease tendency of D and P is attributed to the gradually decreasing Q and increasing molecular density. C2 (D = 9.56 km s−1, P = 41.88 GPa), C3 (D = 9.39 km s−1, P = 39.79 GPa), D1 (D = 9.33 km s−1, P = 39.92 GPa), D2 (D = 9.42 km s−1, P = 41.14 GPa), and D3 (D = 9.32 km s−1, P = 40.00 GPa) have better detonation performance than HMX, and almost all the molecules have better performance than RDX except A. According to the usual energy criterion for HEDMs, i.e., D ≈ 9.0 km s−1, P ≈ 40.0 GPa,42 C2, C3, D1, D2, and D3 all satisfy the requirements. This indicates that they are all potential energetic molecules.
. where nO, nH, and nC represent the numbers of O, H, and C atoms in the molecule, nCOO is the number of carboxyl groups, and M is the molecular weight. Table 3 lists the OB100, heats of detonation, molecular volume, molecular solid density, detonation velocity, and detonation pressure of polydinitroaminoprismanes at B3LYP/6-311G** level. For comparative purposes, the experimental detonation performances of two known explosives (1,3,5-trinitroperhydro-1,3,5-triazine (RDX) and HMX) are also listed in this table, and more clear comparisons are showed in Fig. 2. Inspecting the Q values from Table 3, all molecules have larger Q, and the most of molecules possess higher Q than those of HMX and RDX except E and F. Furthermore, we find that Q decrease with the increasing of the number of substituent group. The change trend can be explained by oxygen balance. Positive high oxygen balance value shows that a large number of extra oxygen is produced in combustion reaction or explosive reaction, which is negative for the energy released. So, we should attach great importance to oxygen balance for designing novel energetic materials. For isomers, the relative positions of dinitroamino group also influence Q, the closer the dinitroamino groups are to one another, the higher the Q. For molecular volume and molecular density from Table 3, their change trend is the same that the values are larger with the increasing of the number of substituent group. A has the smallest molecular density (1.64 g cm−3) and F has the biggest molecular density (2.03 g cm−3) in all molecules. And the densities of the most of molecules are over 1.80 g cm−3. From Fig. 2, we conclude that C2, D1, D2, D3, E, and F can be used for future experiential tests as target molecules due to the fact that their densities are over 1.90 g cm−3. For D and P values given by Table 3, there is no linear relationship between D and P with the number of substituent group, the change trend does not comply with group additivity rule. The first increase and then decrease tendency of D and P is attributed to the gradually decreasing Q and increasing molecular density. C2 (D = 9.56 km s−1, P = 41.88 GPa), C3 (D = 9.39 km s−1, P = 39.79 GPa), D1 (D = 9.33 km s−1, P = 39.92 GPa), D2 (D = 9.42 km s−1, P = 41.14 GPa), and D3 (D = 9.32 km s−1, P = 40.00 GPa) have better detonation performance than HMX, and almost all the molecules have better performance than RDX except A. According to the usual energy criterion for HEDMs, i.e., D ≈ 9.0 km s−1, P ≈ 40.0 GPa,42 C2, C3, D1, D2, and D3 all satisfy the requirements. This indicates that they are all potential energetic molecules.