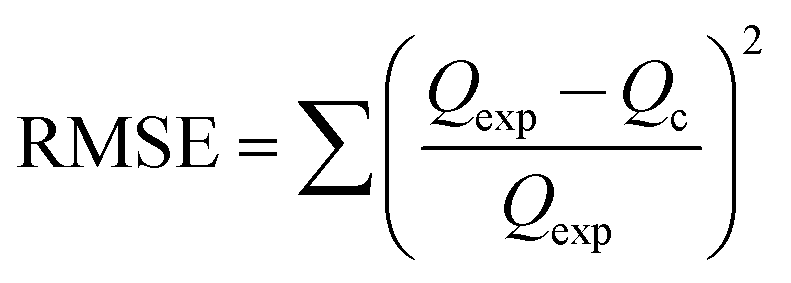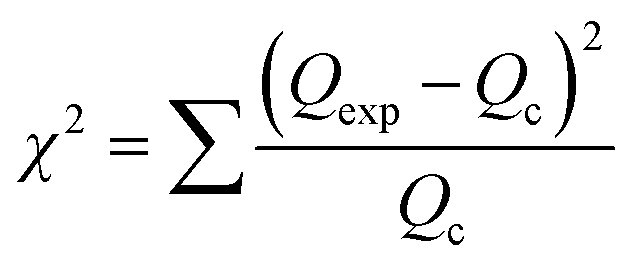DOI:
10.1039/C4RA12549E
(Paper)
RSC Adv., 2015,
5, 22224-22233
Dual application of facilely synthesized Fe3O4 nanoparticles: fast reduction of nitro compound and preparation of magnetic polyphenylthiourea nanocomposite for efficient adsorption of lead ions†
Received
16th October 2014
, Accepted 28th January 2015
First published on 29th January 2015
Abstract
Herein, magnetic Fe3O4 nanoparticles were synthesized by a relatively simple and fast sono-synthetic route within 2.0 min. The prepared nanoparticles and synergic effect of NaBH4 and Na2S2O4 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.3 mole ratio) were employed as a new magnetic recyclable catalyst system for fast (2–5 s) and effective reduction of nitroaniline to phenylenediamine. The progress of the catalytic reaction was monitored with UV-vis spectroscopy and the products were extracted with CHCl3 from an ethanol–water solution. Then, silica-coated magnetic nanoparticles were reacted with carbon disulfide and 1,4-phenylenediamine, which transformed from a monomeric form to a magnetic polyphenylthiourea nanocomposite. The prepared material was characterized with FT-IR, TGA, XRD, VSM, SEM, BET, TEM and EDS techniques. According to the characterization results, an average size of 50 nm was obtained for Fe3O4 nanoparticles. The specific surface area for nanocomposite and the saturated magnetization of the naked Fe3O4 and prepared composite were 47.9 m2 g−1, 37.5 emu g−1 and 21.2 emu g−1, respectively. The polymer nanocomposite shows relatively fast adsorption kinetics towards Pb2+ ions within 3 min and good adsorption capacity equal to 250 mg g−1. The results of an isotherm study and kinetic modelling showed that the Freundlich isotherm model and a pseudo-second order model could better describe the lead adsorption property of the composite. The prepared catalyst and composite had good stability as the reduction efficiency and recovery for trace lead adsorption remained constant after 10 and 5 cycles, respectively.
:0.3 mole ratio) were employed as a new magnetic recyclable catalyst system for fast (2–5 s) and effective reduction of nitroaniline to phenylenediamine. The progress of the catalytic reaction was monitored with UV-vis spectroscopy and the products were extracted with CHCl3 from an ethanol–water solution. Then, silica-coated magnetic nanoparticles were reacted with carbon disulfide and 1,4-phenylenediamine, which transformed from a monomeric form to a magnetic polyphenylthiourea nanocomposite. The prepared material was characterized with FT-IR, TGA, XRD, VSM, SEM, BET, TEM and EDS techniques. According to the characterization results, an average size of 50 nm was obtained for Fe3O4 nanoparticles. The specific surface area for nanocomposite and the saturated magnetization of the naked Fe3O4 and prepared composite were 47.9 m2 g−1, 37.5 emu g−1 and 21.2 emu g−1, respectively. The polymer nanocomposite shows relatively fast adsorption kinetics towards Pb2+ ions within 3 min and good adsorption capacity equal to 250 mg g−1. The results of an isotherm study and kinetic modelling showed that the Freundlich isotherm model and a pseudo-second order model could better describe the lead adsorption property of the composite. The prepared catalyst and composite had good stability as the reduction efficiency and recovery for trace lead adsorption remained constant after 10 and 5 cycles, respectively.
Introduction
Generally, magnetic nanoparticles (NPs) are accessible in a wide range of sizes, which consist of magnetic elements or their oxides and alloys with ferromagnetic or superparamagnetic properties.1 Among the magnetic NPs, the nanometer-sized iron oxides, especially Fe3O4, have attracted particular interest in a broad range of disciplines due to their electric and paramagnetic nature as well as their unique properties, such as chemical stability, biocompatibility, low toxicity, small size and high surface area, providing better physical and chemical characteristics. Moreover, their superparamagnetic property and easy manipulation under the influence of an external magnetic field allows the NPs to be reused or recycled. The simplicity of producing iron oxide nanoparticles in a large quantity as well as the ease of surface modification enables them to have a wide range of applications in pigments, recording materials, ferrofluids, immunosensors, drug-delivery carriers for antitumor therapy and magnetic resonance imaging.2–5 Furthermore, iron based materials have been widely employed as heterogeneous catalysts for the reduction of nitro functional groups to amines. The main drawbacks of the classical methods for nitro reduction are the formation of harmful azo and azoxy-derivatives, difficulty in separating and recovering of catalyst from the reaction systems, employment of more than a stoichiometric amount of iron compound and the requirement of a strong acidic medium that is not compatible with many organic functionalities. To overcome these limitations, magnetic iron materials, especially Fe3O4 was coated with metal nanoparticles, such as palladium and silver, and used as magnetically recyclable catalysts.6–11 Although these immobilization techniques are accessible for the fabrication of magnetic catalysts for organic reactions, the preparation of coated Fe3O4 with nanoparticles is time-consuming and is not economical, and thus exploring new catalytic systems for fast and efficient synthesis of aromatic amino-compounds, as a versatile chemical intermediates in the preparation of dyes, pigments, pharmaceuticals, agrochemicals and polymers, is an ongoing research topic.12–14
Another well-known area in the application of Fe3O4 is magnetic solid phase extraction that uses MNPs as an adsorbent to isolate cells and enzymes, and separate heavy metals, dyes and other environmental pollutants. The naked magnetic NPs are apt for the formation of large aggregates or oxidize/dissolve in acidic solutions, resulting in the changes of their surface properties; thus, the stability, selectivity and adsorption capacity of Fe3O4 NPs for samples with complex matrices can be increased by coating its surface with a layer of silica, alumina or titania, followed by functionalizing with specific active groups.15–19 Among the different types of coated magnetic nanoparticles, polymeric nanocomposites reinforced with Fe3O4 have attracted particular attention because of their tunable magnetic, mechanical, optical and electrical properties. Moreover, the shell of the polymer will increase repulsive forces to balance the magnetic and the attractive forces causing good dispersion, providing available surface functional groups.20–25
Based on these viewpoints, a fast and efficient method for the synthesis of Fe3O4 NPs and their employment for the reduction of nitroaniline and the fabrication of magnetic polymer nanocomposites have been developed. Magnetic Fe3O4 NPs were synthesized by a fast sono-synthetic route using FeSO4, NH3 and KBrO3 as the iron source, precipitant and oxidizing agent, respectively. Then, NPs were employed for very fast reduction of nitroaniline to 1,4-phenylenediamine using NaBH4–Na2S2O4 in a water–ethanol solution. The prepared diamine and carbon disulfide were used as the monomers for the preparation of a magnetic polyphenylthiourea nanocomposite (MPN). Finally, the prepared composite was utilized for the efficient and fast adsorption and preconcentration of Pb2+ ions from water samples within 3 min with a good adsorption capacity.
Experimental
Materials
All the reagents used in this study were of analytical grade and used as received without further purification. Aniline, acetic acid, ammonia, HNO3, H2SO4, FeSO4·7H2O, KBrO3, sodium hydroxide, 3-aminopropyltriethoxysilane (APTES), ethanol and carbondisulfide were obtained from Merck (Darmstadt, Germany). Standard solutions of Pb(II) ions (1000 mg L−1) were prepared by dissolving Pb(NO3)2 salt in a minimal amount of HNO3 and then diluting to the appropriate volume with distilled water. The pH adjustment was performed with 0.1 mol L−1 of HCl and 0.1 mol L−1 of NH3. All plastics and glassware were soaked in diluted HCl and rinsed with distilled water prior to use.
Instruments
The preparation of Fe3O4 NPs was assisted with an ultrasonic cleaning bath operating at 40 Hz with a power of 340 W (Elmasonic, D-78224 Singen/utw, type S, Germany). The prepared particles were characterized by powder X-ray diffraction analysis using a Phillips powder diffractometer, X′ Pert MPD, with Cu-Kα (λ = 1.540589 Å) radiation in a 2θ range of 2°–100°. TEM and SEM analysis were carried out using a Philips-EM 208 and HITACHI S 4160, respectively. Fourier transform infrared spectra (FT-IR) were measured with Equinox 55 Bruker with ATR method over the wavelength range of 400–4000 cm−1. TGA results and energy dispersive X-ray spectrometry (EDS) were recorded with a TA-Q-50 and Oxford ED-2000 (England), respectively. The N2 adsorption–desorption isotherms were measured on a Nova Station A system. A Lambda-25 UV-vis spectrophotometer was used for recording the progress of the catalytic reaction. A Varian model AA-400 flame atomic absorption (FAAS) spectrometer (Varian Australia Pty Ltd, Musgrave), equipped with a deuterium lamp background and hollow cathode lamp was used for the determination of lead ion concentration. A digital pH-meter (model 692, metrohm, Herisau, Switzerland) was used for pH adjustments. Separation was assisted using a neodymium–iron–boron (Nd2Fe12B) magnet.
Synthesize of Fe3O4 nanoparticles
For the synthesis of Fe3O4 NPs, 22 mmol of Fe2+ was dissolved in 50 mL of distilled water. To obtain a 1:2 mol ratio for Fe2+ and Fe3+, 3.0 mmol of BrO3− and 17 mmol of H+ was added to the iron solution (eqn (1) and (2)). The mixture was placed in an ultrasonic cleaning bath at room temperature, subjected to vigorous stirring and an ammonia solution was added. The black Fe3O4 NPs appeared immediately. After 2 min of reaction, the NPs were collected with a magnetic field, washed with distilled water, dried at 80 °C for 2 h, and then stored for subsequent application.| | |
BrO3− + 5Fe2+ + 6H+ → 5Fe3+ + Br− + 3H2O
| (1) |
| | |
2Fe3+ + Fe2+ + 8OH− → Fe3O4 + 4H2O
| (2) |
Catalytic reduction of nitroaniline
In a typical preparation route, 10 mg of 4-nitro aniline was dissolved in 5 mL of ethanol, which contained 16 mg of Fe3O4 nanoparticles. Then, 5 mL of an aqueous solution of NaBH4–NaS2O4 (1:0.3 with respect to nitroaniline) was quickly added to the nitro solution. The color of the solution immediately changed from yellow to colorless. The NPs were collected with an external magnetic field and the residual solution was decanted into a separating funnel. The nanocatalyst was washed with 5 mL of water and the effluents were added to the funnel. 1,4-Phenylenediamine was extracted with CHCl3 (two volume of 2.5 mL), the solvent was then removed with rotary evaporation and shiny white crystals (melting point 137–141 °C) were collected and stored for subsequent investigations.
Preparation of polymer nanocomposite
About 1.0 g of Fe3O4 NPs were ultrasonically dispersed in 50 mL of toluene for 10 min; then, 1.5 mL of APTES was added to the mixture and refluxed for 18 h with stirring. The APTES-coated Fe3O4 was collected with an external magnetic field, washed with ethanol and dried at 80 °C for 4 h.26 To prepare MPN, 0.5 g of silica coated NPs were dispersed in 50 mL of ethanol followed by the addition of 0.7 mL of carbondisulfide and 0.5 g of 1,4-phenylenediamine. The mixture was refluxed for 5 h and after cooling to room temperature, the solid mass was separated with a magnetic field, washed with ethanol, water and acetone and dried at 50 °C for 8 h. The same procedure was applied for polymer preparation in the absence of Fe3O4, and the obtained white powder (melting point: 271–280 °C) was collected with filter paper and dried in room temperature for 24 h. The process of magnetic nanocomposite preparation is schematically illustrated in Scheme 1.
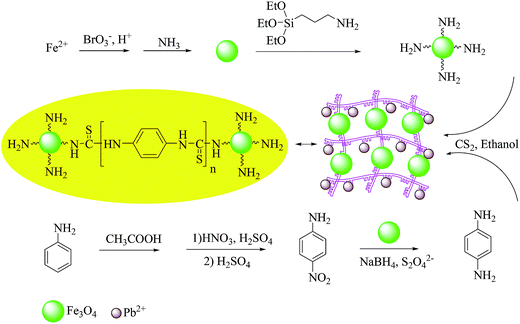 |
| | Scheme 1 The process of magnetic nanocomposite preparation. | |
Metal adsorption experiment
Metal adsorption experiments were performed for different concentrations of lead ions (0.05–50 mg L−1) in 50 mL volumetric flasks. The pH values of the solutions were adjusted to about 6.0, and then 20 mg of magnetic polymer was added to each flask. After shaking for 3 min, the solid mass was collected with an external magnetic field and the concentration of target ions in the liquid phase or in 5 mL of HNO3 (0.3 mol L−1) as eluent was determined by FAAS. The same procedure was performed for the blank solutions.
Results and discussion
Characterization of the magnetic nanocomposite
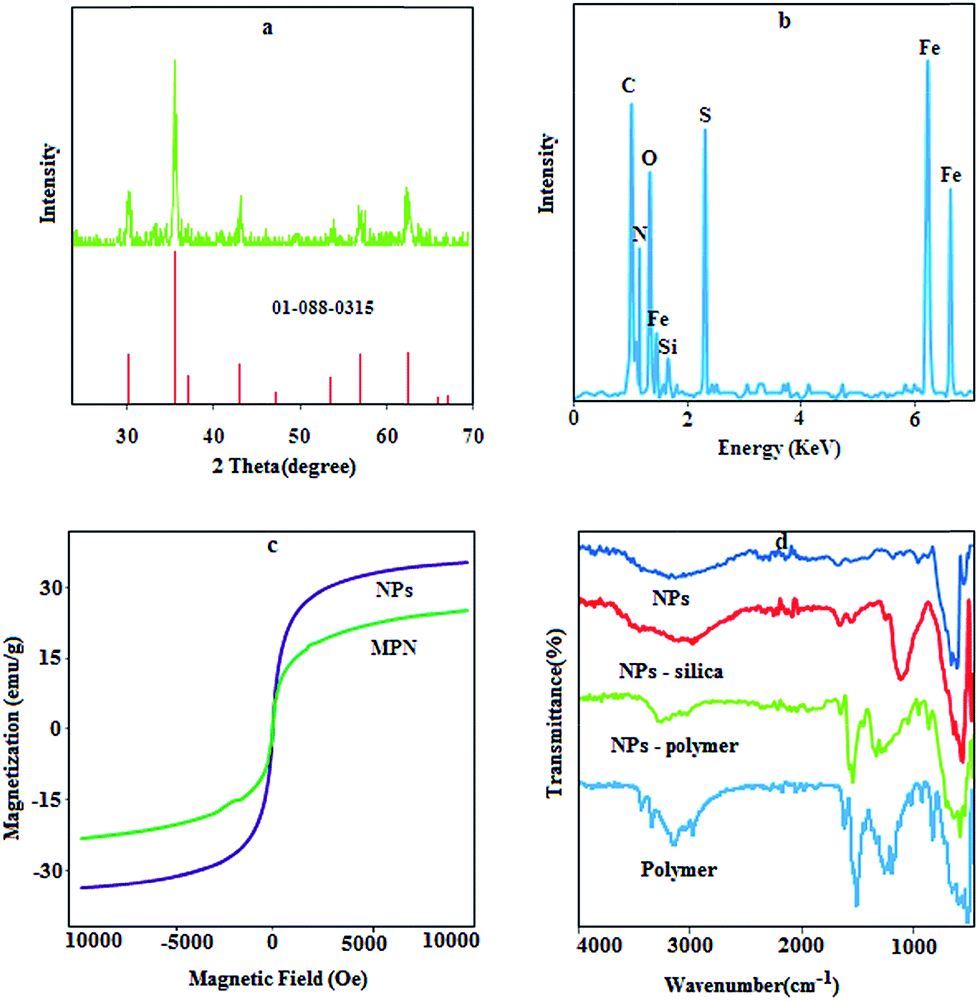
X-ray powder diffraction analysis was used to verify the composition of Fe3O4 NPs. Fe3O4 crystal structure, as shown in Fig. 1a, exhibits peaks at 2θ values of 30.36°, 35.85°, 43.63°, 53.52°, 57.48° and 63.2°, which can be indexed to the (220), (311), (400), (422), (511), and (440) planes of a cubic cell, respectively. This result efficiently matches with the standard spectra of Fe3O4 (01-0880315.CAF). The average crystallite size of NPs was determined using the Scherrer equation as follows:| | |
D = 0.9λ/βcosθ
| (3) |
where D is the average crystalline size, λ (1.540589 Å) is the X-ray wavelength used, β (0.3735° or 0.0065 r) is the angular line width of half maximum intensity in radians, and θ (17.9281) is Bragg's angle expressed in degree.27 According to this equation, an average size of 50 nm was obtained for the synthesized Fe3O4.
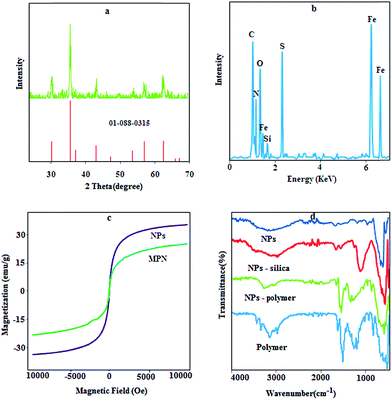 |
| | Fig. 1 XRD pattern (a), EDS spectra (b), VSM graph (c) and FT-IR spectra (d) for NPs and the prepared composite. | |
The results for the EDS analysis (Fig. 1b) show the weight ratios of Fe (49.33%), Si (0.85%), P (0.12%) and S (8.2%) with a LOI (loss on ignition) value of 39.1%, which proves that the polymer has been synthesized on the surface of the Fe3O4 NPs with a high density.
The magnetic hysteresis loops of the prepared NPs and MPN are shown in Fig. 1c. The saturated magnetizations of the naked Fe3O4 and prepared composite were 37.5 and 21.2 emu g−1, respectively. The quenching of the magnetic moment for MPN is due to the existence of organic layers on the surface of Fe3O4 nanoparticles and electron exchange between the functional group of polymer and the surface atoms of the Fe3O4 nanoparticles.28 The remanence value of the uncoated sample was 0.70 emu g−1, while for the magnetic polymer, it was 0.27 emu g−1, which indicates that the naked and modified NPs are superparamagnetic particles with a single magnetic domain.
In the FT-IR spectrum of NPs and silica coated NPs (Fig. 1d), the characteristic bands of Fe–O (569 cm−1), N–H bending mode for primary amine (1620 cm−1), stretching vibration of Si–O (1100 cm−1) and stretching vibration of –NH2 (3300–3500 cm−1) are observable. Certain absorption features of all the polymer components are visible in the spectra of MPN. The peaks around 2929 cm−1, 1570 cm−1, 1400 cm−1 and 1019 cm−1 can be assigned to C–H stretching, N–H bending units, aromatic backbone vibration and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S stretching, respectively.
S stretching, respectively.
The SEM images of Fe3O4 NPs are shown in Fig. 2a. The synthesized NPs have diameter in the range of 40–60 nm. After coating process, some agglomeration and changes in the particle size was observed (Fig. 2b). The TEM image (Fig. 2c) exhibits a relatively homogenous distribution of magnetic nanoparticles in polymer matrices because the diameters of NPs are about 50 nm and reveal that the coating process does not significantly change the size of the particles.
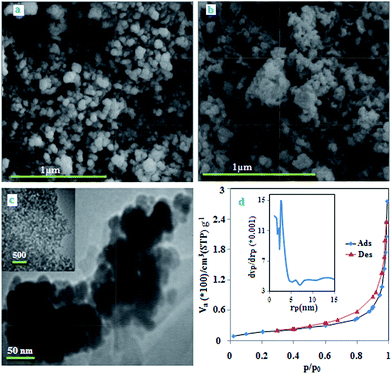 |
| | Fig. 2 SEM image of NPs and MPN (a and b), TEM image of MPN (c), the N2 adsorption–desorption and pore-size distribution (inset) curves of MPN (d). | |
The N2 adsorption–desorption isotherm and pore-size distribution curve of nanocomposite are shown in Fig. 2d. The BET isotherm belongs to the type II with a minor hysteresis loop, which can be due to the filling and emptying of the mesopores by capillary condensation.29 The specific surface area, average pore size and pore volume for the nanocomposite were 67.02 m2 g−1, 2.71 nm and 0.42 cm3 g−1, respectively. This result indicates that the magnetic polymer is mainly macroporous with a small amount of mesopores as a result of the spaces between the magnetic nanoparticles.
Thermal gravimetric analysis for NPs and MPN are shown in Fig. 3. The weight loss for the samples in the range of 50–200 °C is considerably small (5%) because of the removal of water and other solvents. The NPs do not show more weight loss after 200 °C, and the total decrease in weight is 7.75%. Significant weight loss for APTES-Fe3O4 occurred by increasing the temperature to 600 °C to initiate silica compound degradation. The results show that the total weight loses over the temperature range was about 13.74% for silica-coated NPs and reveals that about 8.74% of APTES are present on the surface of the NPs. Moreover, the curve of MPN shows total weight loss of 50.67%, and from the result for NPs and silica compound, it can be concluded that the polymer composed about 37% of the MPN structure. The DTA curve for NPs shows one peak around 200 °C due to the elimination of solvent and some impurities.30,31 The curve for APTES-NPs shows three peaks around 80, 220 and 450 °C as a result of solvent elimination and APTES degradation. The DTA curve of MPN shows one peak around 270 °C due to the polymer degradation, which is in good agreement with melting point result (271–280 °C). These results reveal that the polymer had been assembled on the surface of Fe3O4 with a high density.
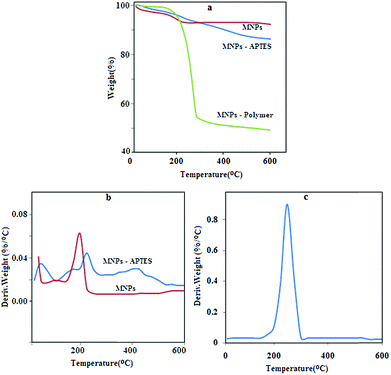 |
| | Fig. 3 TGA and DTA graph for NPs and MPN. | |
Evaluation of catalyst performance
The catalytic performance of the NPs was monitored by UV-vis spectroscopy. Fig. 4a displays the absorbance of nitroaniline, nitroaniline after catalytic reaction and phenylenediamine extracted with CHCl3. The result shows that after the addition of reducing agents, the peak for nitroaniline at 380 nm disappeared immediately and a new peak appeared at 303 nm, which can be assigned to phenylenediamine. Moreover, to obtain high efficiency, the amount of reducing agent was optimized. For this purpose different amount of NPs, NaBH4 and S2O42− (10, 5, 2 and 1 equivalent with respect to nitroaniline) were added to the reaction vessel and the same result was observed again. It was found that with 1.0 equivalent of Fe3O4 and NaBH4 and 0.3 equivalent of S2O42−, complete conversion can be achieved. Moreover, the catalytic activity of Fe3O4, NaBH4, S2O42− and Fe3O4 in the presence of one reducing agent (10 equivalents respect to the nitro compound) was investigated; the results exhibit that after 20 min, no conversion was observed and the reaction mixture shows the same UV spectrum of nitroaniline (Fig. 4b). The catalytic activity of the NaBH4–Na2S2O4 solution in the absence of Fe3O4 nanoparticles was also investigated. It was found that after 30 min, no reduction occurred with 1:1 mol ratio of nitroaniline and reducing agents. Reduction was observed after 10 min with 10:1 mol ratio of each reducing agent with respect to the nitro compound. It is known that with a molar ratio of 50 and more, reduction can be completed after one min. These results prove that the Fe3O4–NaBH4–Na2S2O4 system is considerably more efficient for the reduction of nitro compound because it needs a smaller amount of reducing agent and the reaction progresses very quickly.
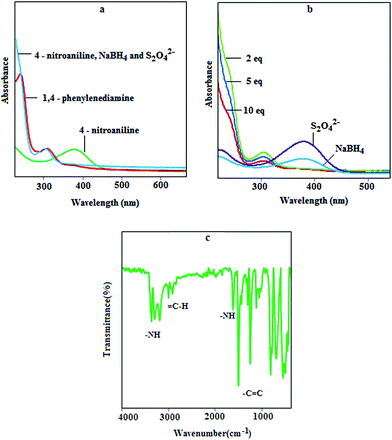 |
| | Fig. 4 UV-vis spectra for (a): nitroaniline and prepared diamine; (b): in the presence of one reductant and different equivalents of the reductant mixture in the presence of 1.0 equivalent of MNPs; (c): FT-IR spectra for synthesized phenylenediamine. | |
The reaction mechanism is not clear but the results suggest a hypothesis. The new catalyst system consists of three major components, including a metal fragment, NaBH4 and S2O42−, and the presence of each component is necessary for the reaction to proceed efficiently. In an aqueous solution, sodium dithionite decomposes to sodium bisulfate and sodium bisulfite, which can exhibit two different roles in nitro reduction. First, NaHSO4 acts as a proton source (H+) that can participate in the reduction process along with iron compounds. Second, NaHSO3 can directly act as a reducing agent in acidic solution. Another component is NaBH4, which releases hydrogen in aqueous solution or reacts with the nanoparticles to form the metal hydride followed by nitro adsorption on the metal surface.32,33 The rapid hydrolysis of borohydride occurs at higher temperatures or in the presence of acidic compounds and metal catalysts,34 and thus it can be concluded that dithionite and the NPs are an effective catalytic promoter, which can enhance the catalytic effect of the borohydride.
The FT-IR spectra for synthesized phenylenediamine are illustrated in Fig. 4c and the characteristic bands near 3200, 2900, 1600, and 1400 cm−1 can be assigned to the stretching of –NH and C–H, bending vibration of –NH and stretching of the CC groups, respectively.
The reusability of the catalyst for the reduction of nitro was also examined, and after 10 cycles, no decrease in the catalytic efficiency was observed. This situation can be observed due to the refreshing of the surface of the catalyst; in other words, the reduction of nitro compounds on the surface of the NPs can occur along with the oxidation of NPs; thus, the formation of oxide layer on the surface of NPs can be decreased by the reducing agents.
Effect of pH on lead adsorption
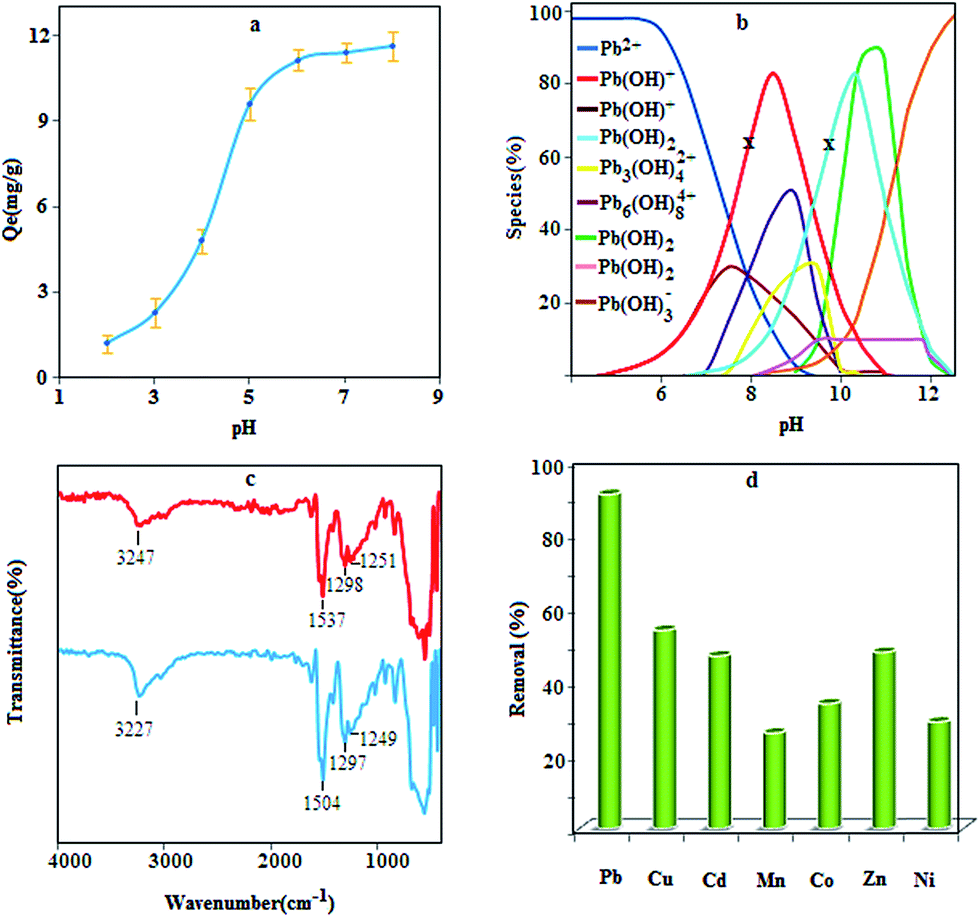
The pH of the solution is one of the most important parameters that affect metal adsorption efficiency. Therefore, the lead adsorption experiments were performed at various pH values by shaking 20 mg of the sorbent with 50 mL of lead ions solution (10 mg L−1) for 5.0 min, and then the adsorption capacity was calculated as follows:where Qe is adsorption capacity (mg g−1), C0 and Ce are the initial and equilibrium concentrations of the lead ions, respectively, V and m are the sample volume (in mL) and the amount of sorbent (mg), respectively. According to Fig. 5a, the adsorption of lead increases with increasing pH. In fact by decreasing the pH value, the functional groups of the sorbent are more protonated, and thus they are less available to retain the lead ions. Moreover, at pH values lower than 7, the removal of lead is mainly accomplished by the adsorption of Pb2+ ions,35,36 and at pH values greater than 7, the adsorption is due to the interaction of hydroxide species with the sorbent surface; therefore, to achieve high efficiency, a pH of 6.0 was selected for subsequent work.
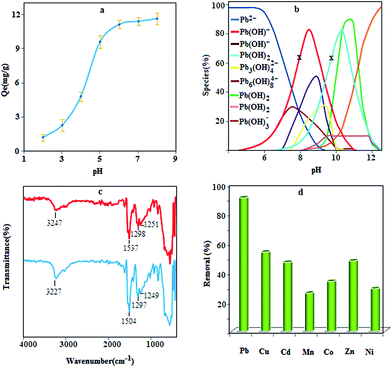 |
| | Fig. 5 Effect of pH on lead adsorption (a) conditions: sample volume 50 mL; contact time 5 min; Pb(II) concentration 10.0; sorbent 0.4 g L−1, species distribution of lead at different pH values and two concentration levels, 10 mg L−1 (for x) and 100 mg L−1, (otherwise), (b), FT – IR spectra of the prepared composite before and after adsorption of lead ions (c) and removal efficiency of some heavy metals using MPN (d). | |
Adsorption mechanism
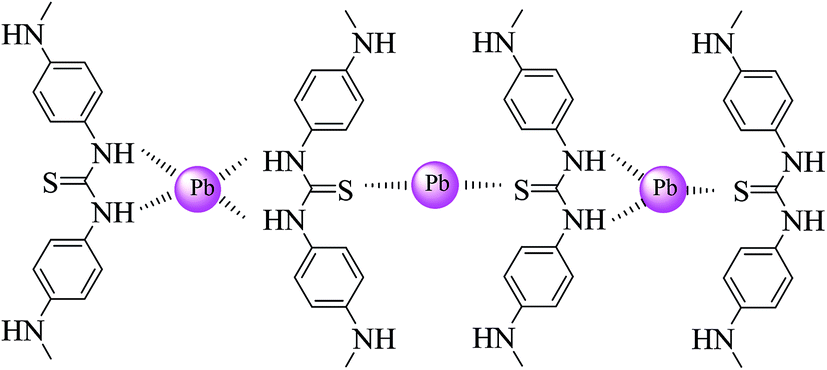
Lead ions are present as various species in the reaction media at different pH values. According to the species distribution diagram (Fig. 5b) for lead ions and based on the concentration of lead ions, in the pH range of 7–9, the main species are Pb(OH)+, Pb3(OH)42+, Pb6(OH)82+ and Pb(OH)2,37 which can probably be adsorbed by a complexation reaction or precipitation. Besides, at pH values lower than 7, the Pb2+ is dominant species in solution, which can interact with nanocomposites through three potential routes, i.e., complexation, ion exchange, and electrostatic interaction. With the precise inspection of nanocomposite structure, it can be seen that three main functional groups (N–H, –OH and CS) are present to capture lead ions. Among the mentioned functional groups, hydroxyl can interact with Pb2+ through ion exchange or electrostatic interaction. This concept can be concluded from the role of proton concentration on lead adsorption; as the concentration of H+ increases, the adsorption efficiency decreases, which confirms the competition between H+ and lead ions. Lead adsorption was further investigated by FT-IR technique (Fig. 5c). Closer inspection in the spectra shows a shift in the position of certain bands after being treated with lead due to the steric effect resulting from the interaction between lead and the sorbent fragments. According to the results, the band due to the stretching of amine and sulfur shifted to a lower wavenumber, which indicates that complexation may be the dominant adsorption mechanism (Scheme 2). Complex formation may occur between one Pb2+ ion and two sulfur atoms, sulfur–amine and four amine groups.
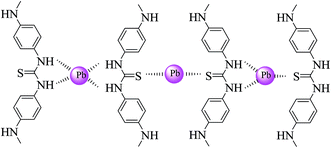 |
| | Scheme 2 Complex formation between lead ions and the functional groups of the polymer. | |
Effect of interfering ions
The effect of some ions, coexisting in natural samples, on the adsorption of lead ions was also investigated. For this purpose, 50 mL of sample solution containing 5 μg of the lead ions and a given amount of interfering ions were analyzed according to adsorption procedure. The results are exhibited in Table 1 and indicate that the ions normally present in water do not interfere under the employed experimental conditions. Moreover, to investigate the specificity of the MPN toward lead ions, the procedure was performed for different solutions with the concentration of 5 mg L−1 for nickel, cobalt, copper, cadmium, manganese and zinc ions. The result (Fig. 5d) indicates that the removal efficiency under optimal conditions is more than 90% for lead ions but for other ions it is less than 50%, demonstrating the specificity of this method for lead adsorption.
Table 1 Effect of interfering ions on lead adsorption
| Ions |
Interference/Pb ratio |
Recovery (%) |
| Cd2+ |
500 |
98 ± 1 |
| Mn2+ |
500 |
101 ± 1 |
| Co2+ |
500 |
99.1 ± 2 |
| Ni2+ |
500 |
97 ± 1 |
| Cr3+ |
1000 |
100 ± 2 |
| Zn2+ |
700 |
97 ± 2 |
| Cu2+ |
400 |
95 ± 1 |
| Ag+ |
200 |
95 ± 2 |
| Na+ |
5000 |
95 ± 1 |
| Mg2+ |
800 |
96 ± 2 |
| Ca2+ |
800 |
97 ± 1 |
| NO3− |
5000 |
98 ± 2 |
| PO43− |
5000 |
99 ± 1 |
| Cl− |
5000 |
96 ± 1 |
Kinetic models
By applying the appropriate kinetic model, quantifying the changes in adsorption with time can be investigated. The pseudo-first-order and pseudo-second-order models can be expressed by the following equations:| | |
ln(Qe − Qt) = lnQe − k1t
| (5) |
| | |
t/Qt = 1/(k2Qe2) + (1/Qe)t
| (6) |
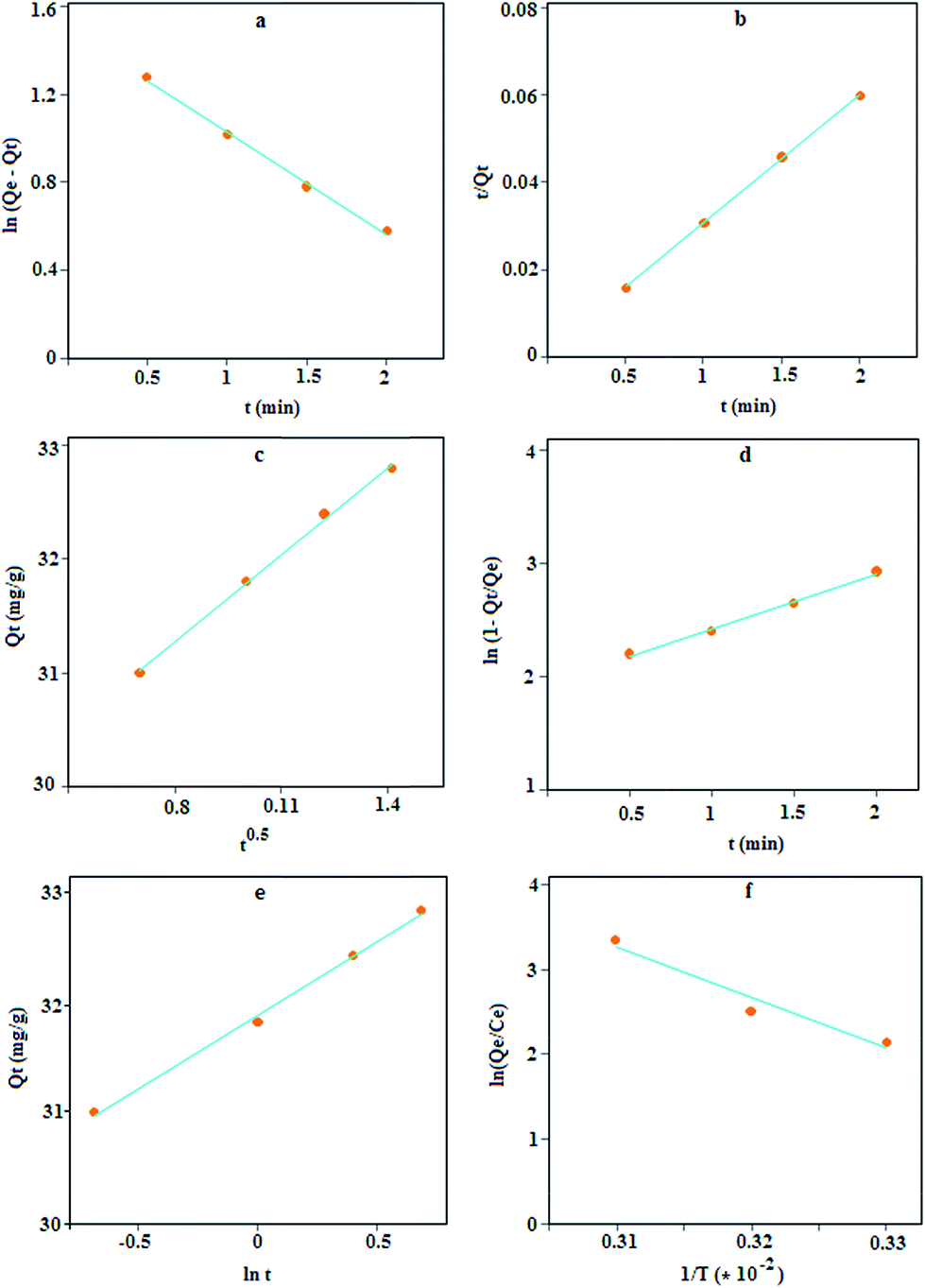
where k1, k2, Qe and Qt are the pseudo-first-order adsorption rate constant (min−1), the second-order rate constant (g mg−1 min−1), the values of the amount adsorbed per unit mass at equilibrium and at any time t, respectively.38 The plots of these kinetic models, Fig. 6a and b, indicate that the plots have good linearity for lead adsorption but according to the results in Table 2, the Qe obtained based on the first-order model deviates considerably from the experimental value but for the second-order model, the deviation is about 0.33%; thus, this model can be accepted as the kinetic mechanism for lead adsorption.
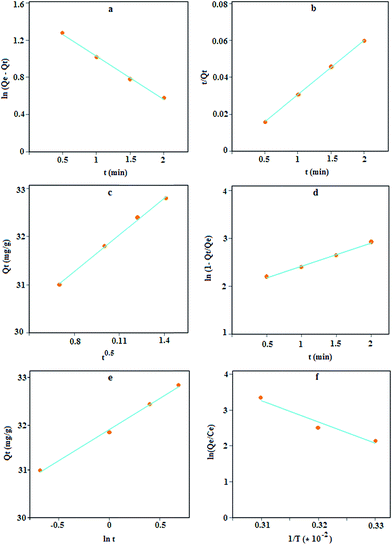 |
| | Fig. 6 Lagergren's pseudo-first order (a), second order (b), intra-particle diffusion (c), liquid film diffusion (d), Elovich model (e), and the effect of temperature on lead adsorption using MPN (f). | |
Table 2 The data of kinetic models for the adsorption of Pb2+ ions using MPN
| First order |
R2 |
0.996 |
Diffusion model |
R2 |
0.997 |
| k1 |
0.46 |
ki |
2.52 |
| Q |
4.48 |
C |
29.22 |
| Second order |
R2 |
0.999 |
Liquid film |
R2 |
0.994 |
| k2 |
0.48 |
kfd |
0.49 |
| Q |
34.48 |
C |
1.93 |
| |
|
|
Elovich |
R2 |
0.996 |
| |
|
α |
5.18 × 10−10 |
| Qexp |
34.6 |
β |
0.76 |
In the batch method, metal ions are transported from the aqueous phase to the surface of the adsorbent, and then subsequently diffused into the interior of the particles. The intra-particle diffusion is governed by the following equation:
The plot of Qt versus t1/2 (Fig. 6c), is approximately linear but with an intercept of 29.22 instead of zero; thus, this model is unlikely to be rate controlled.
The liquid film diffusion model (eqn (8)) explains the role of transport of the analyte from the liquid phase to the boundary of the solid phase.
| | |
ln(1 − Qt/Qe) = kfdt + C
| (8) |
where
kfd is the adsorption rate constant. A plot this model (
Fig. 6d) produces a linear curve, but with a small intercept (1.93) against the predictions of the model. This low value might point out the limited applicability of the model, thus indicating the role of liquid phase transport of target ions to the sorbent surface in controlling the kinetics.
The Elovich kinetic model39 can be expressed as follows:
| | |
Qt = 1/βln(αβ) + 1/βln(t)
| (9) |
where
α (mg g
−1 min
−1) and
β (g mg
−1) represent the initial adsorption rate and desorption constant, respectively. The application of this model resulted in a good linear fit (
Fig. 6e) with a high value of
α that shows a high adsorption rate. Thus, it can be concluded that the MPN has good affinity towards lead ions.
Thermodynamic study
The adsorption capacities of Pb(II) with an initial concentration of 10 mg L−1 and adsorbent dosage of 20 mg were 15.9, 17.87 and 21.27 mg g−1 at 298, 309 and 318 K, respectively. These results indicate that Pb(II) adsorption on MPN is endothermic. The thermodynamic parameters for the adsorption process, such as ΔH (kJ mol−1), ΔS (J K−1 mol−1) and ΔG (kJ mol−1), could be evaluated using the following equations:| | |
ΔG = −RTln(Qe/Ce)
| (10) |
| | |
ln(Qe/Ce) = ΔS/R − ΔH/RT
| (12) |
where R and T are gas constants (8.314 × 10−3 kJ K−1 mol−1) and absolute temperature (K), respectively. The plot of ln(Qe/Ce) versus 1/T is linear (Fig. 6f) with the slope and the intercept giving values of ΔH and ΔS.40 The results in Table 3 indicate that Pb(II)-MPN interactions were accompanied by a decrease in the Gibbs energy, which makes the interactions spontaneous and suggests that the process is feasible at higher temperatures. The endothermic enthalpy change, ΔH, for Pb(II) adsorption in 298 K, was calculated to be +49.46 kJ mol−1. Entropy increased for Pb(II) adsorption as the value of ΔS for this interaction was calculated to be +180.49 J K−1 mol−1, thus this adsorption process is entropy stabilized.
Table 3 The thermodynamic data for lead adsorption using MPN
| ΔG (kJ mol−1) |
ΔH (kJ mol−1) |
ΔS (J kmol−1) |
| 298 K |
309 K |
318 K |
|
|
| −4.18 |
−6.16 |
−7.78 |
+49.46 |
+180.49 |
Isotherm study and error analysis
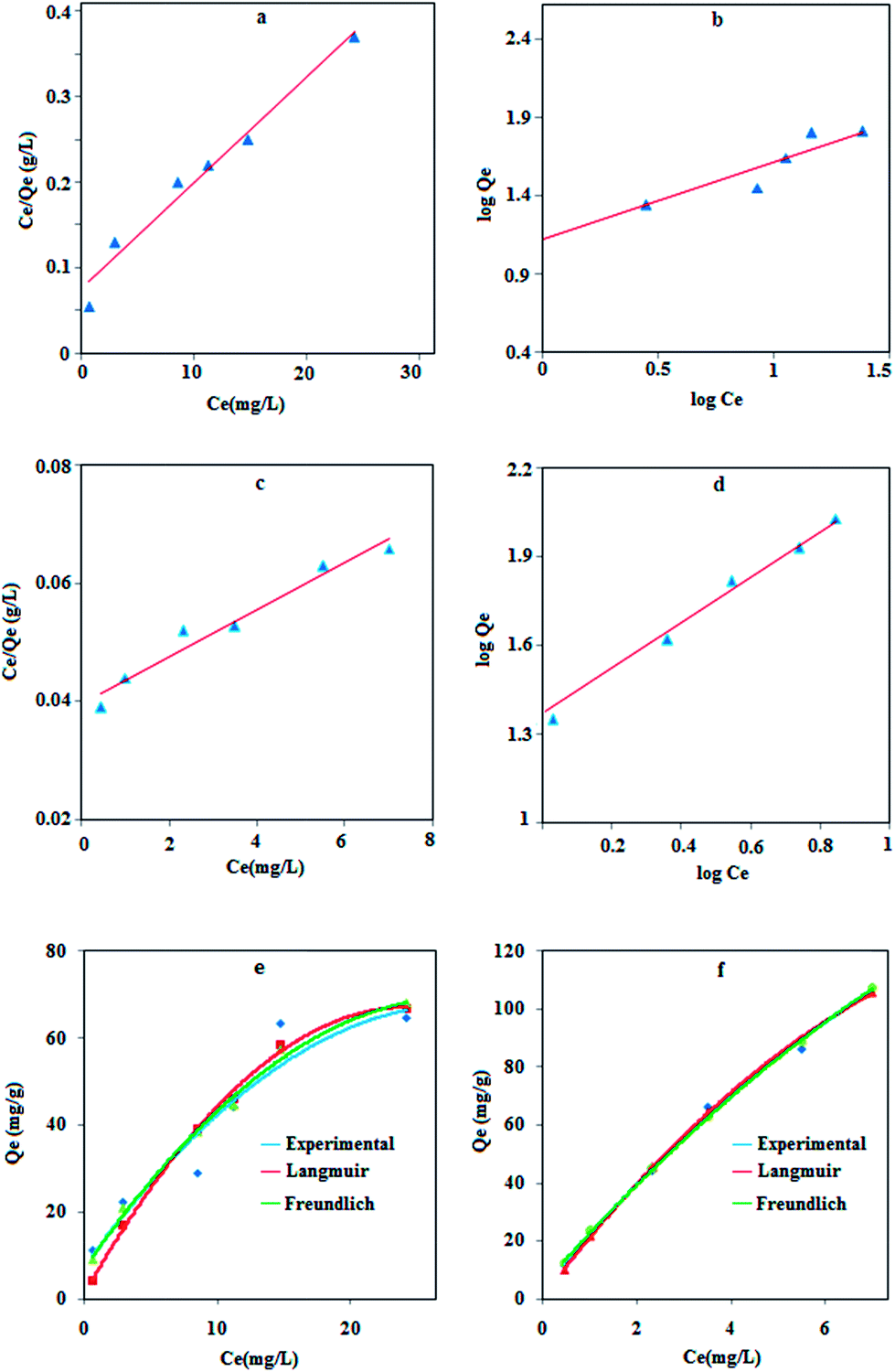
The equilibrium adsorption isotherm can describe the interactive behavior of the solution and the adsorbent. Therefore, the effect of Pb(II) concentrations on the adsorption process was analyzed in terms of Langmuir and Freundlich isotherms by Fe3O4 and MPN.
The Langmuir model can be expressed as follows:
| | |
Ce/Qe = 1/Qmb + Ce/Qm
| (13) |
where
Qe is the amount of metal ions sorbed per unit mass of the sorbent (mg g
−1) and
Ce is the amount of metal ions in the liquid phase at equilibrium (mg L
−1).
Qm is the maximum adsorption capacity and
b is the Langmuir coefficient.
41 This model is based on the assumption of a structurally homogeneous sorbent, where all the sorption sites are energetically the same and identical. The essential characteristics of the Langmuir isotherm can be explained in terms of a dimensionless constant separation factor (
RL), calculated using
eqn (14).
where
Ci is the initial concentration of metal ions and
RL describes the type of Langmuir isotherm, which can be irreversible (
RL = 0), favorable (0 <
RL < 1), linear (
RL = 1) or unfavorable (
RL > 1).
42
The Freundlich model is represented as follows:
where
n and
Kf are the Freundlich coefficients, which are evaluated from the slope and intercept of the linear plot.
43,44 The graphs for the linear and nonlinear isotherm model are depicted in
Fig. 7 and the results are listed in
Table 4, which show that the maximum adsorption capacity of MPN is 3.0 times greater than the naked NPs. This is due to the fact that lead ions have a soft Lewis acid character and its affinity to donor atoms should be in the order O < N < S based on the hard and soft acid–base (HSAB) theory. Therefore, the modification of MNPs with a sulfur functional group can increase the adsorption capacity.
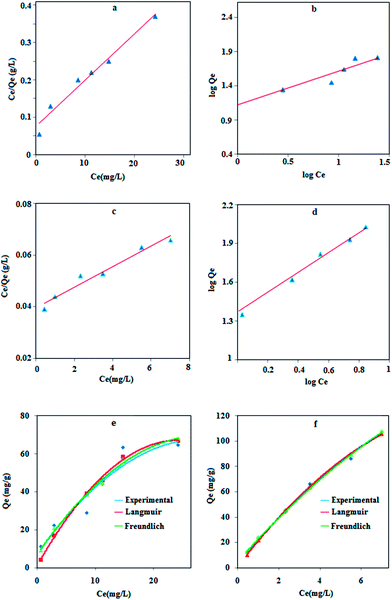 |
| | Fig. 7 Langmuir and Freundlich adsorption isotherm models for lead adsorption on Fe3O4 NPs (a and b) and MPN (c and d). The theoretical model fitted with experimental data using Fe3O4 (e) and MPN (f). | |
Table 4 The data of isotherm models for lead adsorption using NPs and MPN
| |
|
Fe3O4 NPs |
MPN |
| Langmuir |
Qm (mg g−1) |
83.3 |
250 |
| R2 |
0.97 |
0.96 |
| b |
0.16 |
0.1 |
| RL |
0.11–0.55 |
0.16–0.66 |
| χ2 |
15.84 |
0.45 |
| RSS |
208.89 |
21.94 |
| RMSE |
0.568 |
0.017 |
| Freundlich |
n |
2.01 |
1.37 |
| Kf |
13.15 |
23.44 |
| R2 |
0.94 |
0.99 |
| χ2 |
3.68 |
0.44 |
| RSS |
144.53 |
22.39 |
| RMSE |
0.157 |
0.016 |
The linear form of the isotherm equation does not represent the errors in the isotherm curves; therefore, it is necessary to fit the equilibrium adsorption data to analyze and design an adsorption process. In the present study, the sum of the squared residual (RSS), the root-mean-square error (RMSE) and the chi-square test (χ2) were used to test the accuracy of the isotherm models.45–47
These error functions are given as
| |
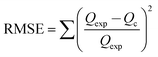 | (17) |
| |
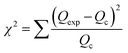 | (18) |
where
Qexp and
Qc are the experimental and calculated data from nonlinear models. According to the results (
Table 4), the
R2 value for both linear models is better than 0.90, indicating a low variance about the mean values, but the Langmuir and Freundlich models have better linearity for lead adsorption using NPs and MPN, respectively. An error analysis revealed that the Freundlich model could better describe the adsorption behavior of the lead ions on Fe
3O
4 and MPN.
Application for water sample
The developed procedure was applied for determining the concentration of lead ions in water samples. Moreover, the accuracy of the method was evaluated by real sample spiking. The results are depicted in Table 5 and show that the recovery of the spiked samples is in the range of 98–102%, which shows good efficiency of the method for the trace analysis of lead ions.
Table 5 The results for the analysis of lead ions from water samples
| Sample |
Added (μg L−1) |
Found (μg L−1) |
Recovery (%) |
| Not detected. Below the linear range. |
| Top water |
0 |
NDa |
— |
| 100 |
98 |
98 ± 2 |
| River water |
0 |
BLRb |
— |
| 100 |
102 |
102 ± 1 |
| Sea water |
0 |
10 |
— |
| 100 |
109 |
99 ± 2 |
Comparison with literature
In Table 6, the performance of the proposed method for the preparation of Fe3O4 nanocatalyst and the catalytic activity for nitro reduction are compared with the results from related reports. It is obvious that the catalyst preparation is fast, simple and cost-effective. The catalyst can reduce a nitro-compound in a few second; moreover, this catalytic system is more economic due to the presence of a less amount of reducing agents. A comparative study for Pb2+ adsorption time and adsorption capacity is presented in Table 7. Based on the results, the performance of this method was significantly better than the previous ones with respect to the adsorption time; furthermore, it shows a satisfactory adsorption capacity.
Table 6 Comparison of catalyst performance with other methods for nitro reduction
| Catalyst |
Preparation time (min) |
Reducing time |
Condition |
Ref. |
| Tetramethyldisiloxane Fe(acac)3 |
— |
24 min |
Fe(acac)3 (10 mol%) 60 °C |
7 |
| Fe3O4-supported Ag |
11 h |
125 min |
NaBH4 (300 equiv.), rt |
9 |
| γ-Fe2O3 |
60 |
5–7 min |
(CH3)2CHOH, KOH, 80 °C |
10 |
| Fe3O4 supported Pd |
240 |
1 min |
NaBH4 (75 equiv.), rt |
11 |
| Au–TiO2 |
1 h |
1 min |
NaBH4 (6 equiv.), rt |
12 |
| Iron nanoparticle |
5 |
2–3 min |
rt |
14 |
| Fe3O4 |
2 |
2–5 s |
NaBH4, K2S2O4(1, 0.3 equiv.), rt |
This work |
Table 7 Comparison of the lead adsorption property of the prepared composite with some other sorbents
| Adsorbent |
Maximum capacity (mg g−1) |
Equilibrium time (min) |
Ref. |
| Ethylene glycol-Fe3O4 |
35.1 |
10 |
21 |
| MWCNT-polyacrylamide |
37.44 |
4 h |
36 |
| Graphene oxide-nanosheets |
842 |
24 h |
37 |
| Basic oxygen furnace sludge |
92.5 |
60 |
39 |
| Soy protein hollow microspheres |
169.18 |
240 |
40 |
| Fe3O4 |
40.0 |
10 h |
41 |
| Fe3O4 |
83.3 |
10 |
This work |
| Polyphenylthiourea–Fe3O4 |
250 |
3 |
This work |
Conclusions
We have demonstrated a fast sono-chemistry based route for the preparation of magnetic Fe3O4 nanoparticles using one iron source. The NPs were applied as a magnetic separable catalyst for a rapid reduction of nitroaniline to phenylenediamine for the preparation of a magnetic polyphenylthiourea nanocomposite. The catalytic reaction was assisted by a synergic effect with NaBH4 and sodium dithionite as reducing agents. The magnetic nanocomposite shows relatively fast adsorption kinetics for Pb2+ ions with a high adsorption capacity and good reusability. The performance of this method was good with respect to efficiency, selectivity and adsorption capacity and showed its ability to be an efficient catalyst for nitro reduction and efficient adsorbent to solidify lead ions and reduce their hazardous impact.
Acknowledgements
Support for this investigation by the Research Council of the University of Tehran through grants is gratefully acknowledged.
References
- S. Ni, X. Wang, G. Zhou, F. Yang, J. Wang, Q. Wang and D. He, Mater. Lett., 2009, 63, 2701 CrossRef CAS PubMed.
- G. Giakisikli and A. N. Anthemidis, Anal. Chim. Acta, 2013, 789, 1 CrossRef CAS PubMed.
- C. Huang, H. Zhang, Z. Sun, Y. Zhao, S. Chen, R. Tao and Z. Liu, J. Colloid Interface Sci., 2011, 364, 298 CrossRef CAS PubMed.
- P. I. Girginova, A. L. Daniel-da-Silva, C. P. Lopes, P. Figueira, M. Otero, V. S. Amaral, E. Pereira and T. Trindade, J. Colloid Interface Sci., 2010, 345, 234 CrossRef CAS PubMed.
- H. Yan, J. Zhang, C. You, Z. Song, B. Yu and Y. Shen, Mater. Chem. Phys., 2009, 113, 46 CrossRef CAS PubMed.
- R. Kaplánek and V. Krchnak, Tetrahedron Lett., 2013, 54, 2600 CrossRef PubMed.
- L. Pehlivan, E. Metay, S. Laval, W. Dayoub, P. Demonchaux, G. Mignani and M. Lemaire, Tetrahedron, 2011, 67, 1971 CrossRef CAS PubMed.
- L. Pehlivan, E. Métay, S. Laval, W. Dayoub, P. Demonchaux, G. Mignani and M. Lemaire, Tetrahedron Lett., 2010, 51, 1939 CrossRef CAS PubMed.
- H. Woo, K. Lee, S. Park and K. H. Park, Molecules, 2014, 19, 699 CrossRef PubMed.
- S. U. Sonavane, M. B. Gawande, S. S. Deshpande, A. Venkataraman and R. V. Jayaram, Catal. Commun., 2007, 8, 1803 CrossRef CAS PubMed.
- A. Baykal, E. Karaoglu, H. Sözeri, E. Uysal and M. S. Toprak, J. Supercond. Novel Magn., 2013, 26, 165 CrossRef CAS PubMed.
- I. Tamiolakis, S. Fountoulaki, N. Vordos, L. N. Lykakis and G. S. Armatas, J. Mater. Chem. A, 2013, 1, 14311 CAS.
- N. Pradhan, A. Pal and T. Pal, Langmuir, 2001, 17, 1800 CrossRef CAS.
- R. Dey, N. Mukherjee, S. Ahammed and B. C. Ranu, Chem. Commun., 2012, 48, 7982 RSC.
- J. Zhu, S. Wei, M. Chen, H. Gu, S. B. Rapole, S. Pallavkar, T. C. Ho, J. Hopper and Z. Guo, Adv. Powder Technol., 2013, 24, 459 CrossRef CAS PubMed.
- H. Bagheri, R. Daliri and A. A. Roostaie, Anal. Chim. Acta, 2013, 794, 38 CrossRef CAS PubMed.
- Q. Wang, L. Huang, P. Yu, J. Wang and S. Shen, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2013, 912, 33 CrossRef CAS PubMed.
- G. Bharath, D. Prabhu, D. Mangalaraj, C. Viswanathan and N. Ponpandian, RSC Adv., 2014, 4, 50510–50520 RSC.
- Z. Ma, D. Zhao, Y. Chang, S. Xing, Y. Wu and Y. Gao, Dalton Trans., 2013, 42, 14261 RSC.
- M. Zeng, Y. Huang, S. Zhang, S. Qin, J. Li and J. Xu, RSC Adv., 2014, 4, 5021 RSC.
- M. H. Mashhadizadeh, M. Amoli-Diva, M. R. Shapouri and H. Afruzi, Food Chem., 2014, 151, 300 CrossRef CAS PubMed.
- J. Zhu, S. Wei, J. Ryu, L. Sun, Z. Luo and Z. Guo, ACS Appl. Mater. Interfaces, 2010, 2, 2100 CAS.
- J. Li, Z. Shao, C. Chen and X. Wang, RSC Adv., 2014, 4, 38192 RSC.
- W. Wu, Q. He and C. Jiang, Nanoscale Res. Lett., 2008, 3, 397 CrossRef CAS PubMed.
- X. Liu, Q. Hu, Z. Fang, X. Zhang and B. Zhang, Langmuir, 2009, 25, 3 CrossRef CAS PubMed.
- M. Bayat, M. H. Beyki and F. Shemirani, J. Ind. Eng. Chem., 2015, 21, 912 CrossRef CAS PubMed.
- C. Li, Y. Wei, A. Liivat, Y. Zhu and J. Zhu, Mater. Lett., 2013, 107, 23 CrossRef CAS PubMed.
- G. Li, Y. Jiang, K. Huang, P. Ding and J. Chen, J. Alloys Compd., 2008, 466, 451 CrossRef CAS PubMed.
- P. Yuan, D. Liu, M. Fan, D. Yang, R. Zhu, F. Ge, J. X. Zhu and H. He, J. Hazard. Mater., 2010, 173, 614 CrossRef CAS PubMed.
- J. Chen, P. Xiao, J. Gu, D. Han, J. Zhang, A. Sun, W. Wang and T. Chen, Chem. Commun., 2014, 50, 1212 RSC.
- T. Poursaberi and M. Hassanisadi, Clean: Soil, Air, Water, 2013, 41, 1208 CrossRef CAS.
- S. Ozkar and M. Zahmakiran, J. Alloys Compd., 2005, 404–406, 728 CrossRef PubMed.
- S. Wunder, F. Polzer, Y. Lu, Y. Mei and M. Ballauff, J. Phys. Chem., 2010, 114, 8814 CAS.
- N. G. Gaylord, J. Chem. Educ., 1957, 34, 367 CrossRef CAS.
- S. Yang, J. Hu, C. Chen, D. Shao and X. Wang, Environ. Sci. Technol., 2011, 45, 3621 CrossRef CAS PubMed.
- G. Zhao, X. Ren, X. Gao, X. Tan, J. Li, C. Chen, Y. Huang and X. Wang, Dalton Trans., 2011, 40, 10945 RSC.
- V. M. Nurchi and I. Villaescusa, InTech, 2011, 409–424 Search PubMed.
- T. K. Naiya, A. K. Bhattacharya and S. K. Das, J. Hazard. Mater., 2009, 170, 252 CrossRef CAS PubMed.
- D. Liu, Z. Li, W. Li, Z. Zhong, J. Xu, J. Ren and Z. Ma, Ind. Eng. Chem. Res., 2013, 52, 11036 CrossRef CAS.
- S. H. Lin and R. S. Juang, J. Hazard. Mater., 2002, 92, 315 CrossRef CAS.
- L. Giraldo, A. Erto and J. C. Moreno-Pirajan, Adsorption, 2013, 19, 465 CrossRef CAS PubMed.
- L. Ai, Y. Zhou and J. Jiang, Desalination, 2011, 266, 72 CrossRef CAS PubMed.
- G. Z. Kyzas, P. I. Siafaka, D. Lambropoulou, N. K. Lazaridis and D. N. Bikiaris, Langmuir, 2014, 30, 120 CrossRef CAS PubMed.
- Y. Yang, Y. Xie, L. Pang, M. M. Li, X. Song, J. Wen and H. Zhao, Langmuir, 2013, 29, 10727 CrossRef CAS PubMed.
- S. Chowdhury and P. Saha, J. Environ. Eng., 2011, 137, 388 CrossRef CAS.
- B. Boulinguiez, P. L. Cloirec and D. Wolbert, Langmuir, 2008, 24, 6420 CrossRef CAS PubMed.
- P. Sampranpiboon, P. Charnkeitkong and X. Feng, WSEAS Trans. Environ. Dev., 2014, 10, 35 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra12549e |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.