
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
UV-A and UV-B excitation region broadened novel green color-emitting CaGd2ZnO5:Tb3+ nanophosphors†
G. Seeta
Rama Raju
,
E.
Pavitra
,
Goli
Nagaraju
,
Xiang-Yu
Guan
and
Jae Su
Yu
*
Department of Electronics and Radio Engineering, Institute for Laser Engineering, Kyung Hee University, 1 Seocheon-dong, Giheung-gu, Yongin-si, Gyeonggi-do 446-701, Republic of Korea. E-mail: jsyu@khu.ac.kr; Fax: +82-31-206-2820; Tel: +82-31-201-3820
First published on 19th February 2015
Abstract
Green color-emitting novel CaGd2ZnO5:Tb3+ (CGZO:Tb3+) nanophosphors were synthesized by a citrate sol–gel method. The structural and morphological properties were elucidated by X-ray diffraction and transmission electron microscope measurements. The photoluminescence properties of orthorhombic-phased CGZO:Tb3+ nanophosphors were studied as a function of Tb3+ ion concentration. The CGZO:Tb3+ nanophosphors revealed the enhanced broadband excitation between ultraviolet (UV)-B and UV-A regions. Under 317 nm excitation, even at dilute Tb3+ ion concentrations, only the emission transitions from 5D4 energy level were exhibited. This unusual behavior is due to the occurrence of nonradiative energy transfer via f–d transition rather than cross-relaxation process. The cathodoluminescence also showed similar behavior at low accelerating voltages. These luminescent powders are expected to find potential applications such as white light-emitting diodes and optical display systems.
Introduction
Rare-earth (RE) elements doped inorganic phosphors are unique components for solid-state lighting and biomedical applications because of their stable chemical and physical properties.1–6 Among the applications of solid-state lighting, phosphor-converted light-emitting diodes (pc-LEDs) have gained abundant interest and have been recognized as one of the most promising technologies. Also, pc-LEDs take advantage of direct conversion of electricity to light, resulting in a comparatively decreased electric power consumption. In general, white LEDs (WLEDs) are produced by combining a GaN-based blue LED chip with conventional yellow (YAG:Ce) phosphors. This type of WLED has a poor color rendering index (CRI) caused by the red color deficiency.4 However, a high CRI is one of the prerequisites for its application as a backlight for liquid crystal displays. Therefore, the alternative approach to achieving high CRI and high-power white light is an ultraviolet (UV) LED coated with red-green-blue (RGB) phosphors,7 or coupling a blue LED and RG phosphors.8 These pc-LEDs require efficient green phosphors that should have the excitation wavelength matching with the emission wavelength of the UV LEDs or the blue LEDs. Nowadays, UV LEDs with different emission wavelengths in the UV-A (315–400 nm), UV-B (280–315 nm) and UV-C (100–280 nm) parts of the electromagnetic spectrum have been developed.7,9–12 Hence, the availability of phosphors operating under UV excitation with better performance is of prime importance for such LEDs. In the case of the tricolor WLEDs, inorganic oxides would be the best candidate for RGB phosphors in terms of both chemical stability and luminescence efficiency. However, promising green oxide phosphors have not yet been found with the excitation wavelength between the UV-A and UV-B regions. Generally available green color-emitting phosphor, particularly Tb3+ ions doped phosphor, has the excitation wavelengths in the UV-C and UV-B regions or in the vacuum UV region.13–15 Thus, the development of a good green color-emitting phosphor having excitation wavelengths within the UV-A and UV-B regions is a key technology for achieving a tricolor WLED system. In this context, we developed a Tb3+ ions activated green color-emitting novel CaGd2ZnO5 (CGZO) nanophosphors with the enhanced broadened excitation region over the UV and near-UV (NUV) ranges. Xu et al. demonstrated this novel CGZO host lattice with GdO7, CaO11, and ZnO5 polyhedral units based on the ARE2MO5 structure,16 and our group have recently established the richness of this host lattice when doped with Eu3+ ions.17To the best of our knowledge, no reports have been found so far on the luminescent properties of Tb3+ ions activated CGZO host lattice. In this work, we reported the structural and detailed luminescent properties of CGZO:Tb3+ nanophosphors by a facile sol–gel synthesis. This phosphor showed the broadened excitation region and provided green emissions almost similar to the commercial green phosphor. In order to explore its suitability for field-emission displays (FEDs), cathodoluminescence (CL) properties have also been studied.
Experimental
CaGd2(1−x)ZnO5:xTb3+ (CGZO:Tb3+) nanophosphors were synthesized by a citrate based facile sol–gel method. Stoichiometric amounts of high-purity grade (purchased from Sigma-Aldrich) calcium nitrate tetrahydrate [Ca(NO3)2·4H2O], gadolinium nitrate hexahydrate [Gd(NO3)3·6H2O], terbium nitrate pentahydrate [Tb(NO3)3·5H2O], zinc nitrate hexahydrate [Zn(NO3)2·6H2O] and citric acid [HOC(COOH)(CH2COOH)2]. The solution was made by dissolving the 1 M of Ca(NO3)2·4H2O, 2(1 − x) M of Gd(NO3)3·6H2O, 2x M of Tb(NO3)3·5H2O, and 1 M Zn(NO3)2·6H2O in 200 ml de-ionized water. After the solution was stirred for few minutes, 8 M of citric acid (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio of metal ions to citric acid) was added to the metal ions solution. The stirring was continued for 2 h at room temperature to form a homogeneous solution. For homogeneous heating throughout the solution, the beaker was sealed with a cap and then it was heated on a hot plate at 80 °C of solution temperature under the continued magnetic stirring for an hour. After that, the cap was removed and the solution was evaporated gradually until the yellowish wet gel was formed. The wet gel was then dried at 120 °C in an oven for 12 h. As a result, a porous solid matrix called xerogel was formed due to the liquid expulsion from the pores (syneresis). This xerogel was decomposed, creating black-colored flakes with highly fine particles on further heating at 400 °C for 4 h. The resulting powder was further annealed at 1200 °C for 5 h. To convert Tb4+ to Tb3+ ion states, all the heat treated powders were then fired under a reducing atmosphere (95% N2/5% H2) at 1000 °C for 3 h, and the powder color changes from yellow to pure white, as shown in digital photographs in the inset of Fig. 1, which indicates that all terbium ions are in the trivalent state.
:2 ratio of metal ions to citric acid) was added to the metal ions solution. The stirring was continued for 2 h at room temperature to form a homogeneous solution. For homogeneous heating throughout the solution, the beaker was sealed with a cap and then it was heated on a hot plate at 80 °C of solution temperature under the continued magnetic stirring for an hour. After that, the cap was removed and the solution was evaporated gradually until the yellowish wet gel was formed. The wet gel was then dried at 120 °C in an oven for 12 h. As a result, a porous solid matrix called xerogel was formed due to the liquid expulsion from the pores (syneresis). This xerogel was decomposed, creating black-colored flakes with highly fine particles on further heating at 400 °C for 4 h. The resulting powder was further annealed at 1200 °C for 5 h. To convert Tb4+ to Tb3+ ion states, all the heat treated powders were then fired under a reducing atmosphere (95% N2/5% H2) at 1000 °C for 3 h, and the powder color changes from yellow to pure white, as shown in digital photographs in the inset of Fig. 1, which indicates that all terbium ions are in the trivalent state.
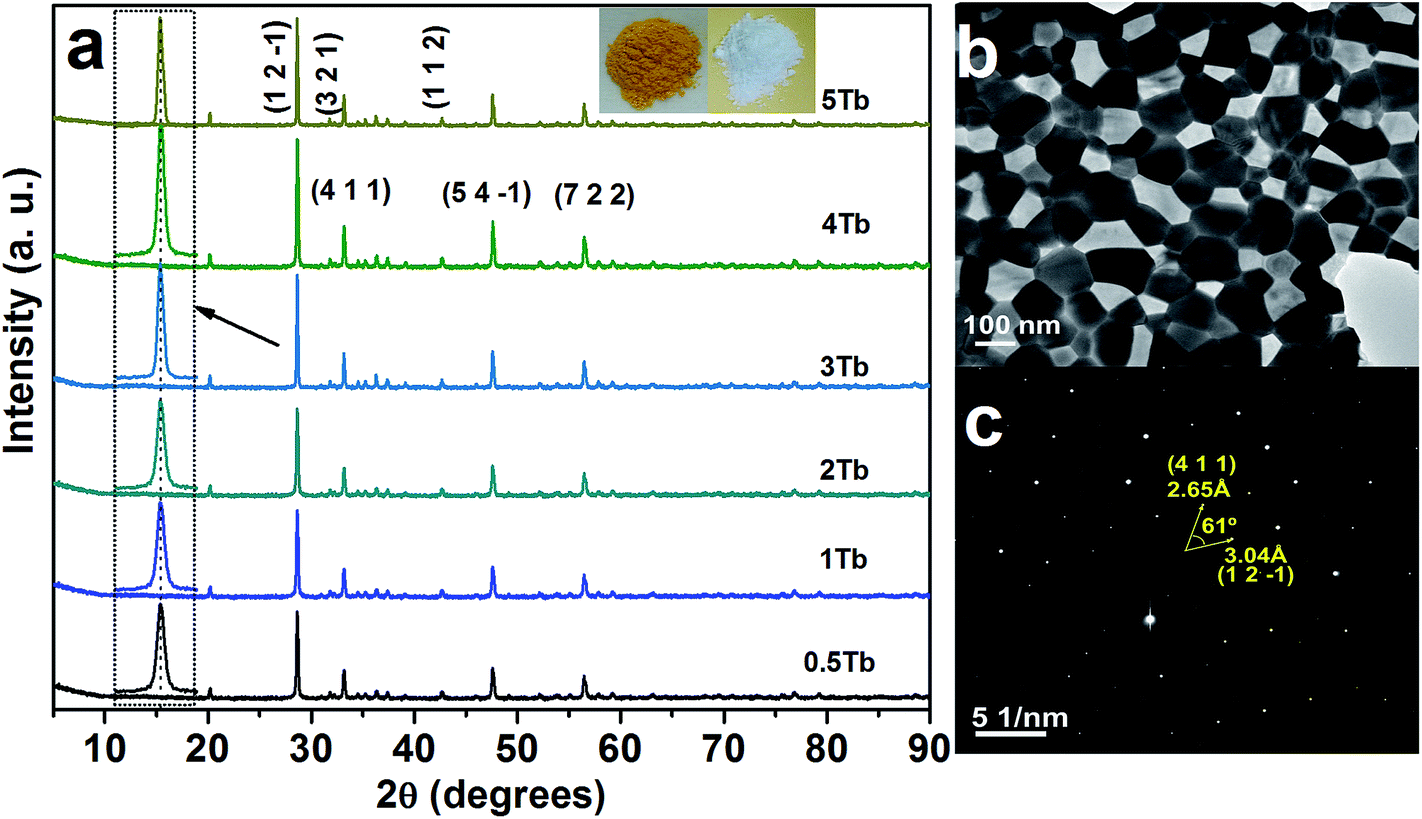 | ||
| Fig. 1 (a) XRD patterns of the CGZO nanophosphors doped with different concentrations of Tb3+ ions, (b) TEM image, and (c) SAED pattern of the CGZO:4Tb3+ nanophosphor. (In (a), the (h k l) planes are presented based on the ref. 16 and the inset shows the photographs of Tb3+ ions activated CGZO host lattices before and after annealing in reduced atmosphere). | ||
The XRD patterns of the CGZO:Tb3+ nanophosphors were recorded on Mac Science (M18XHF-SRA) X-ray powder diffractometer with CuKα = 1.5406 Å, and their morphological feature was observed by the field-emission transmission electron microscope (FE-TEM: JEOL JEM-2100F) images. Fourier transform infrared (FTIR) spectrum of the CGZO:Tb3+ nanophosphors was recorded on a thermo Nicolet-5700 spectrophotometer. The room-temperature photoluminescence (PL) spectra were measured by using a Photon Technology International (PTI, USA) fluorimeter with a Xe-arc lamp of 60 W power and the lifetime was measured with a phosphorimeter attachment to the main system with a Xe-flash lamp (25 watt power). For lifetime measurement, the start delay (0 μs) and end delay (4000 μs) with a count of 400 channels and 5 shots for each channel (average 3 times), an integration time of 50 μs, and a frequency of 10 Hz were used as parameters in the ph decay mode of PTI fluorimeter. The CL properties were measured by a Gatan (UK) MonoCL3 system attached with the scanning electron microscope (SEM: Hitachi S-4300 SE).
Results and discussion
Fig. 1(a) shows the XRD patterns of the CGZO nanophosphors doped with different concentrations of Tb3+ ions, prepared by the sol–gel process and annealed at 1200 °C in ambient atmosphere and 1000 °C in reduced atmosphere. Recently, Xu et al.16 reported the novel CGZO host lattice, and so its JCPDS card is not available. The XRD pattern was in good agreement with the data of Xu et al., which confirms the orthorhombic phase of typical CGZO diffraction peaks with the space group of Pnma and the lattice constants are a = 12.325 Å, b = 5.545 Å, and c = 7.3225 Å. Within the given concentrations, the crystallite size was estimated by the well-known Scherrer equation, Dhkl = kλ/βcosθ, where D is the average grain size, k(0.9) is a shape factor, λ is X-ray wavelength (1.5406 Å), β is the full width at half maximum (FWHM) and θ is the diffraction angle of an observed peak, respectively. The strongest diffraction peaks were used to calculate the crystallite size of CGZO:Tb3+ nanophosphors, which yield average values of 89.5, 89.5, 89, 88.5, 88, and 88.5 nm for CGZO: 0.5 mol% Tb3+ (CGZO:0.5Tb3+), CGZO:1Tb3+, CGZO:2Tb3+, CGZO:3Tb3+, CGZO:4Tb3+, and CGZO:5Tb3+. By close observations, the differences in the crystallite sizes as a function of Tb3+ ion concentration are not considerable because very dilute concentrations of dopant (Tb3+) ions were used in the present work and the ionic radius of the Tb3+ (0.98 Å) ion is almost equal to the Gd3+ (1 Å) ion and also the energy supply was constant. Fig. 1(b) and (c) show the TEM image and selected area electron diffraction (SAED) pattern of the CGZO:4Tb3+ nanophosphor after annealing at 1000 °C in reduced atmosphere. The resulting particles acquired irregular shapes with nanometer size. The corresponding SAED pattern displays the regularly arranged diffraction spots, indicating their single crystalline nature with the twist angle (ϕ) of 61°. The pattern can be indexed to the reflection of an orthorhombic CGZO nanophosphor, which is consistent with the XRD results presented above.
The presence of metal ions in the CGZO:4Tb3+ nanophosphor was also been confirmed by the FTIR spectroscopy. Fig. 2 shows the FTIR spectrum of the CGZO:4Tb3+ nanophosphor, which consists of the vibrational bands at 3641, 1415, 873, 712, 545, and 433 cm−1. The sharp band at 3641 cm−1 corresponds to the O–H stretching vibrational mode of the non-hydrogen bonded hydroxyl groups in the Ca(OH)2 and the moderate broadband at 1415 cm−1 is related to the asymmetric stretching vibration of O–C–O.18 The absorption peaks at 875 and 712 cm−1 belong to the symmetric and asymmetric bending vibrations of CO32− groups and the characteristic stretching vibration of Gd–O appeared at 545 cm−1.19,20 The band centered at 433 cm−1 is related to the Zn–O stretching vibration.21–23 The absorption spectrum of the CGZO:4Tb3+ nanophosphor is also shown in Fig. 2(b). The spectrum revealed absorption bands in the visible to UV-A, UV-B, and UV-C regions due to the absorption of CGZO host lattice also called the host absorption band (HAB) and the 4f7–5d1 transition of Tb3+ ions. These broad absorption bands offer a vast selection of possible excitation wavelengths between the UV and visible regions.24–26 The absorption behaviors between 200–250 and 360–550 nm are due to the host absorption and the broad band between 250 and 360 nm is due to the 4f7–5d1 transition of Tb3+ ions. The observed absorption bands are well in agreement with the PL excitation (PLE) spectra of CGZO host lattice (Fig. S1†) and CGZO:Tb3+ nanophosphors (Fig. 3(a)).
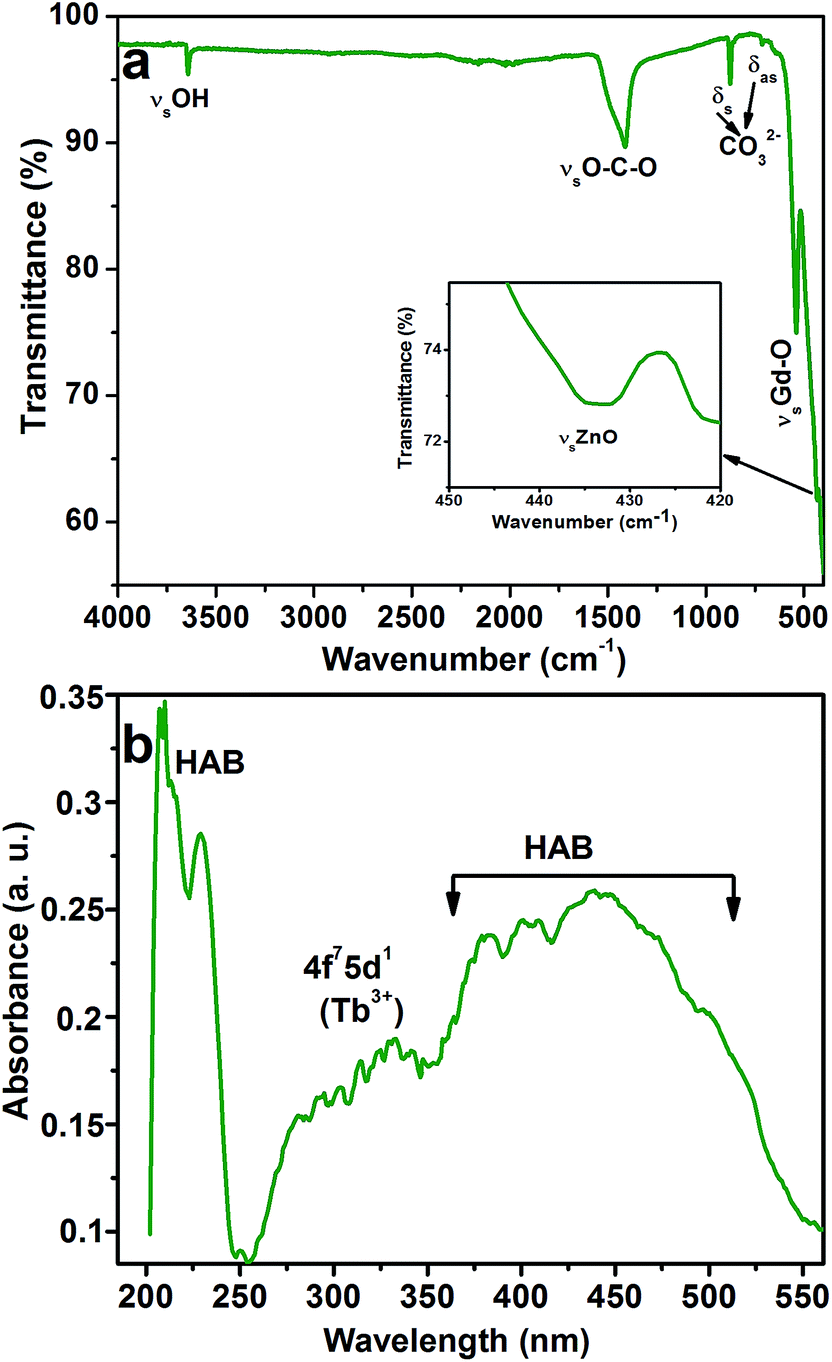 | ||
| Fig. 2 (a) FTIR and (b) absorption spectra of the CGZO:4Tb3+ nanophosphor after annealing at 1000 °C in reduced atmosphere. | ||
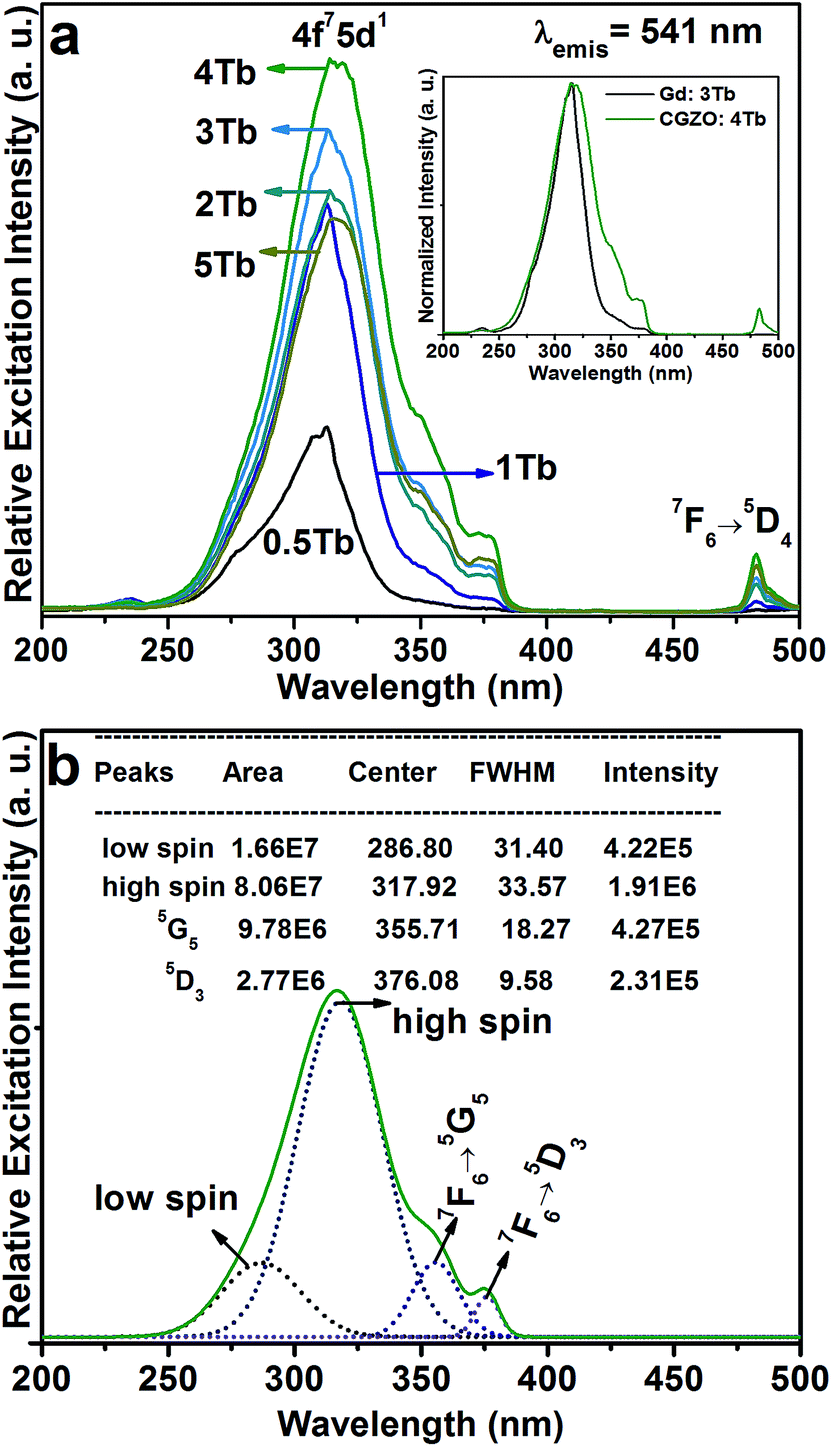 | ||
| Fig. 3 (a) PLE spectra of the CGZO:Tb3+ nanophosphors as a function of Tb3+ ion concentration (inset shows the comparison between Gd2O3:Tb3+and CGZO:Tb3+ nanophosphors), and (b) Gaussian fitting results for the CGZO:4Tb3+ nanophsophor. | ||
Fig. 3(a) shows the PLE spectra of the CGZO:Tb3+ nanophosphors as a function of Tb3+ ion concentration by monitoring the emission wavelength at 541 nm. The PLE spectra are made of two groups of excitation bands with inter-configurational 4fn → 4fn−15d1 (f–d transition) and 4f intra-configurational transitions of Tb3+ ions along with the weak HAB. The HAB appeared between 200 and 250 nm. At dilute concentration, the sharp edge of the f–d transition at 314 nm is related to the (8S7/2 → 6P7/2) Gd3+ transition. However, at 5 mol% Tb3+ ion concentration, the HAB disappeared totally and the f–d transition exhibited the smoother peak with a band maximum at 317 nm, indicating that the possibility of efficient energy transfer from Gd3+ to Tb3+ ions. Besides, the second group consists of 4f intra-configurational transitions of Tb3+ ions located in the higher wavelength region, which appeared more pronouncedly with increasing the Tb3+ ion concentration. It is noted that the f–d transition of Tb3+ ions seems unlikely as it is broadened upto 30 and 40 nm from the previously reported Gd2O3:3Tb3+ (inset of Fig. 3(a)) and Y2O3:Tb3+ phosphors, respectively.20,27 The reason is that the f–d transition is related to the Tb3+ local environment and the 5d orbital is strongly influenced by the strength and symmetry of crystal field experienced by the active ions.28,29 The enhanced broadness hints that the CGZO:Tb3+ nanophosphors are a promising material for UV and NUV based lighting or optical display applications. For better understanding, the f–d transition of the CGZO:4Tb3+ nanophosphor was deconvolved by the Gaussian fitting, as shown in Fig. 3(b). There are four bands between the UV and NUV regions. The bands centered at 287 and 317 nm are related to the spin allowed (low spin) and spin forbidden (high spin) f–d transitions of Tb3+ ions, respectively. The bands located at 356 nm (7F6 → 5G5) and 376 nm (7F6 → 5D3) are ascribed to the intra-configurational 4f transitions. The band centered at 483 nm is related to the (7F6 → 5D4) transition (Fig. 3(a) and (b)). It is known that the low spin transition is more intense than that of high spin transition, but the contrary result was observed in this work. The reason behind this change is that the f–f transitions of Gd3+ and Tb3+ ions at 309 and 314 nm due to the (8S7/2 → 6PJ=7/2,5/2) and (7F6 → 5HJ=6,7) transitions, respectively, overlaid with each other and the high spin f–d transition covered all these transitions including the (7F6 → 5D0) transition of Tb3+ ions. The f–f transitions in the higher energy side of the excitation spectrum is unusual, and the detailed explanation about this unusual appearance was done in the earlier report.30 Briefly, the intra 4f transitions of Tb3+ ions in the shorter wavelength region obtain intensity when uneven components are mixed with the opposite parity wave functions (such as 5d) into the 4f wave functions. Therefore, the intensity of Gd3+ ion transitions increased due to the increased intensity of these Tb3+ ion transitions. As a result, the high spin f–d transition gained higher intensity than that of the low spin f–d transition of Tb3+ ions. This kind of result based on the f–d transition of Tb3+ ions was not reported so far. In view of the above result, we selected the 317 nm as the excitation wavelength for the emission measurements.
The PL spectra of the CGZO:Tb3+ nanophosphors as a function of Tb3+ ion concentration under 317 nm excitation are shown in Fig. 4(a). In the case of Tb3+ ions, usually, two groups of emission transitions appeared from the 5D3 and 5D4 energy levels, and the position of terbium ions is barely influenced due to the screen effect of 4f electrons, which are protected by its outer electron layer. However, in the present work, the PL spectra of the CGZO:Tb3+ nanophosphors exhibited the emission transitions at 482, 541, 580 and 620 nm corresponding to the 5D4→7FJ (J=6,5,4 and 3) transitions.31 No 5D3 emissions were observed even at dilute concentrations. This sort of result was also observed by Blasse et al.32 in the case of monoclinic Gd2O3:Tb3+ phosphors, indicating that the nonradiative transition occurs from 5D3 to 5D4 level via f–d transition instead of cross-relaxation process. This is possible only if the f–d transition appears in the low energy region (longer wavelength side). Due to this kind of nonradiative transition, the PL spectra exhibited mainly 5D4 emission bands even at dilute Tb3+ ion concentrations. Therefore, the CGZO:Tb3+ nanophosphors show the predominant green color from the magnetic dipole transition (5D4 → 7F5) at 541 nm, which can be explained by the large values of the reduced matrix element at J = 5 and the Judd–Ofelt theory.33,34 The Stark splitting in the emission peaks of Tb3+ ions is attributed to the effect of ligand field on terbium ions. It can also be found that the (5D4 → 7Fj) emission intensities of Tb3+ ions increased upto 4 mol% of Tb3+ ion concentration in CGZO host lattice due to the reduced loss of excitation energy. While the Tb3+ ion concentration increased over 4 mol%, the emission intensities decreases due to the concentration quenching, as shown in the inset of Fig. 4(a). The concentration quenching might be due to the excitation energy migration to the quenching centers. Furthermore, the decay profile for the 5D4 → 7F5 emission level of the CGZO:4Tb3+ nanophosphor under 317 nm excitation wavelength is shown in the Fig. 4(b). The results revealed that the decay curve was well fitted to a single exponential function of I = I0exp(−t/τ), where τ is the decay time. From the fitted curve, the calculated decay time of the CGZO:4Tb3+ was 1.37 ms.
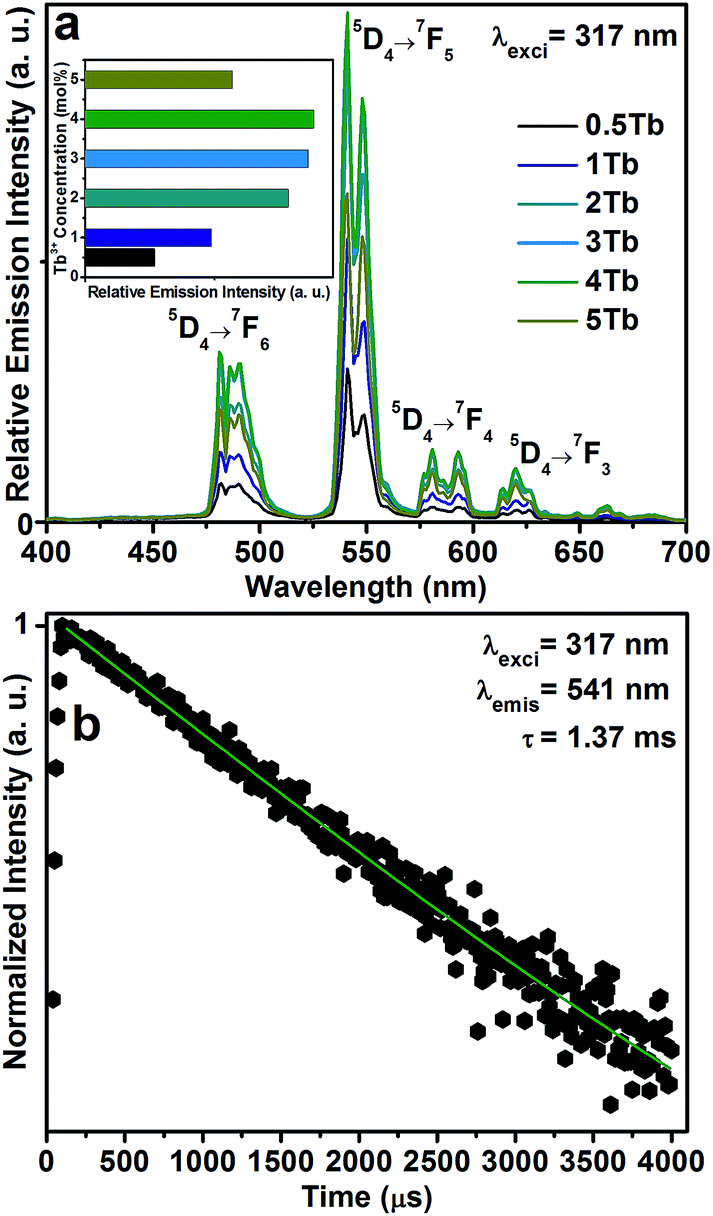 | ||
| Fig. 4 (a) PL spectra of the CGZO:Tb3+ nanophosphors as a function of Tb3+ ion concentration (inset shows the concentration quenching effect), and (b) decay curve of the CGZO:4Tb3+ nanophsophor. | ||
Fig. 5 shows the CL spectra of the CGZO:4Tb3+ nanophosphor as a function of (a) accelerating voltage and (b) filament current. The CL spectra revealed almost similar behavior except intensity. However, the CGZO:4Tb3+ nanophosphor showed weak emission bands in the blue wavelength region at the accelerating voltage of 5 kV (inset (i) of Fig. 5(a)). These bands were not observed in the PL spectra. This can be explained in view of the different mechanisms between CL and PL processes. In the PL process, UV or visible light based photons with the energy of only 4–6 eV are used. However, in the CL process, the fast energetic electrons are used from the anode voltage, which can be tuned from few electron volts to thousands of electron volts. So, the excitation energy on the dopant ion is much larger in CL than that in PL. However, the fast electrons as a high-energy particle always excite the host lattice. After being penetrated into the host lattice of a luminescent material, the fast primary electron will cause ionization and create many secondary electrons. The secondary electrons can also give rise to ionization and create more secondary electrons. These secondary electrons excite the host lattice and create many electron–hole pairs. Therefore, the high energy electron beam easily excite the HAB or charge transfer bands (CTB) of Tb3+ ions (O2− → Tb3+ (≈152 nm)) and (O2− → Gd3+ (≈155 nm)), which may be overlapped with each other. Due to the strong interaction with the crystal lattice, the excited electrons tend to relax from CT states via a multiple-phonon emission process to the lowest levels of 4f–5d states. Hence, at higher accelerating voltage, the transition probability from 4f–5d configuration to 5D3, and 5D4 metastable states is expected to be large as compared to the PL process. Furthermore, the intensity was increased with increasing the accelerating voltage (Fig. 5(a)) or filament current (Fig. 5(b)) due to the improved penetration depth by the recombination of increased excitons.35 Because of the deeper penetration depth, more activator ions were excited at almost all parts of the particles including boundaries, surfaces and inside of particles. The electron penetration depth for the CGZO:Tb3+ nanophosphors was calculated by the following formula:20,36
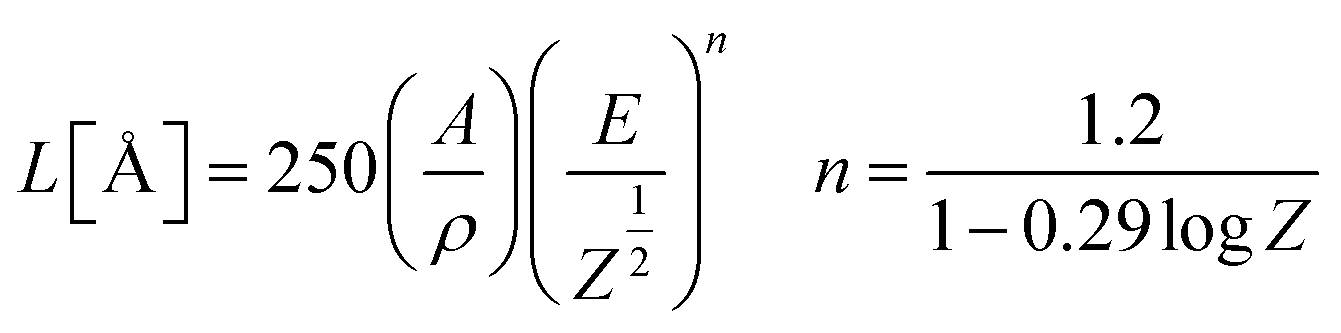
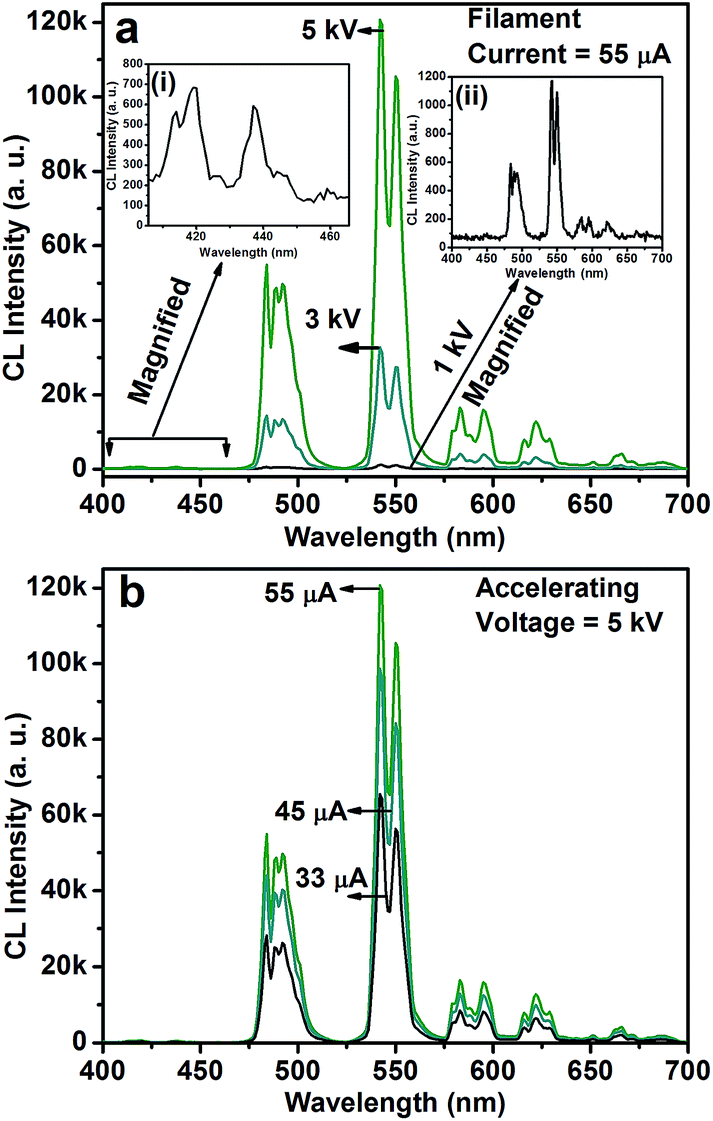 | ||
| Fig. 5 CL spectra of the CGZO:4Tb3+ nanophosphor as a function of (a) accelerating voltage at a fixed filament current of 55 μA and (b) filament current at a fixed accelerating voltage of 5 kV. The insets of (a) show (i) the magnified spectrum of the emissions from the 5D3 energy level and (ii) the magnified spectrum at 1 kV and 55 μA. | ||
The Commission International De l'Eclairage (CIE) chromaticity coordinates of the CGZO:Tb3+ nanophosphor at the optimized concentration at 4 mol% from PL the spectrum and from the CL spectra as a function of accelerating voltage and filament current were calculated (the calculations were presented in the ESI†) and the selected chromaticity coordinates were represented in Fig. 6. The PL spectrum exhibited the CIE chromaticity coordinate of (0.305, 0.556). From the CL spectra, the CIE chromaticity coordinates were (0.319, 0.469), (0.305, 0.567), and (0.309, 0.568) at 1, 3, and 5 kV of accelerating voltage, respectively, under a fixed filament current of 55 μA. Slight variations in the coordinates were observed as a function of filament current, indicating (0.308, 0.570), (0.306, 0.569), and (0.309, 0.568) for 33, 45, and 55 μA, respectively. The calculated CIE chromaticity coordinates of the CGZO:4Tb3+ nanophosphor indicate better green color purity than those of available green phosphors such as Y2O3:Tb3+ (0.319, 0.597)37 and SrGa2S:Eu2+ (0.32, 0.63)38 and are also in close proximity to the European broadcasting union (EBU) used green (0.29, 0.60) coordinate.
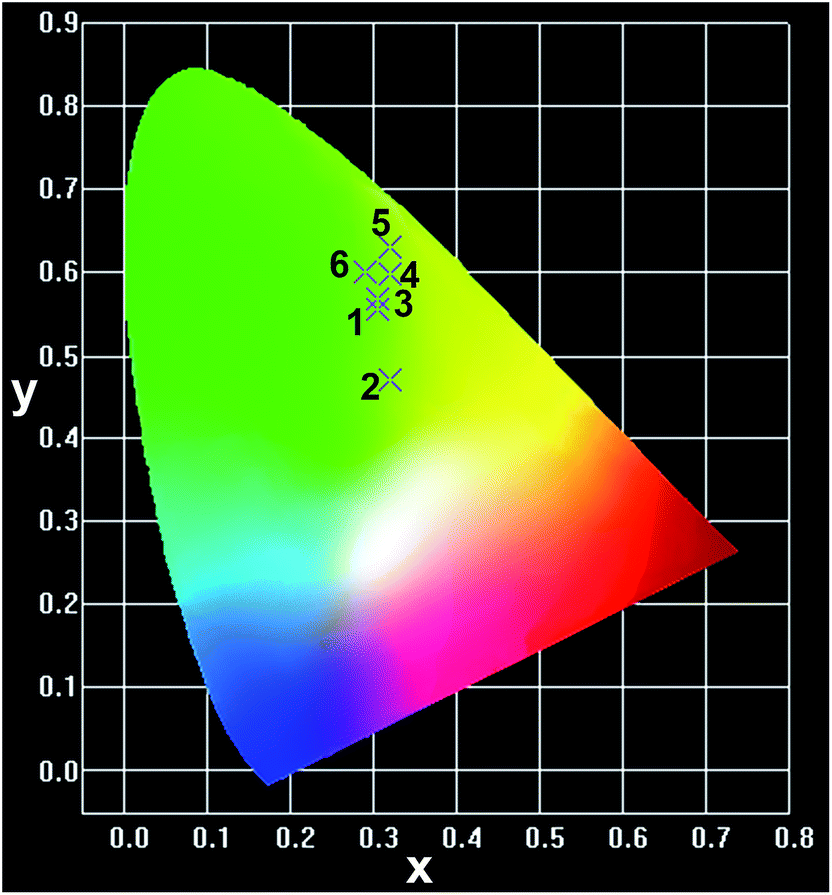 | ||
| Fig. 6 CIE chromaticity coordinates of the CGZO:4Tb3+ nanophosphor (1) from PL (0.305, 0.556), (2&3) from CL (0.319, 0.469), (0.305, 0.567) at the accelerating voltages of 1 and 5 kV, respectively, under a fixed filament current of 55 μA, and of (4) the Y2O3:Tb3+ (0.319, 0.597), (5) the SrGa2S:Eu2+ (0.32, 0.63), and (6) the EBU green (0.29, 0.60). | ||
Conclusions
CGZO:Tb3+ nanophosphors were successfully synthesized by a citrate based sol–gel method. The XRD patterns confirmed the orthorhombic phase of the CGZO:Tb3+ nanophosphors. The PLE spectra of the CGZO:Tb3+ nanophosphors exhibited the enhanced broadband excitation between 250–380 nm. Under 317 nm excitation, the emission transitions from only 5D4 energy levels were exhibited due to the nonradiative energy transfer between the 5D3 and 5D4 energy levels via f–d transition instead of cross-relaxation process. The CL spectra also showed this similar behavior at low accelerating voltages. From the obtained results, CGZO:Tb3+ nanophosphors are expected to have potential applications in the development of RGB phosphor based efficient WLEDs, optical display systems, and FEDs.Acknowledgements
This work was supported by a grant from the Kyung Hee University in 2013"(KHU-20131563)", and also supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (no. 2014-026864).References
- H. A. Höppe, Angew. Chem., Int. Ed., 2009, 48, 3572–3582 CrossRef PubMed.
- B. Moine and G. Bizarri, Opt. Mater., 2006, 28, 58–63 CrossRef CAS PubMed.
- C. Feldmann, T. Jüstel, C. R. Ronda and P. J. Schmidt, Adv. Funct. Mater., 2003, 13, 511–516 CrossRef CAS.
- H. Daicho, T. Iwasaki, K. Enomoto, Y. Sasaki, Y. Maeno, Y. Shinomiya, S. Aoyagi, E. Nishibori, M. Sakata, H. Sawa, S. Matsuishi and H. Hosono, Nat. Commun., 2012, 3, 1132 CrossRef PubMed.
- G. S. R. Raju, E. Pavitra, G. P. Nagaraju, R. Kandimalla, B. F. El-Rayes and J. S. Yu, Cryst. Growth Des., 2013, 13, 4051–4058 CAS.
- G. S. R. Raju, E. Pavitra, G. Purnachandra Nagaraju, K. Ramesh, B. F. El-Rayes and J. S. Yu, Dalton Trans., 2014, 43, 3330–3338 RSC.
- A. J. Fernández-Carrión, M. Ocaña, J. García-Sevillano, E. Cantelar and A. I. Becerro, J. Phys. Chem. C, 2014, 118, 18035–18043 Search PubMed.
- N. Hirosaki, R.-J. Xie, K. Kimoto, T. Sekiguchi, Y. Yamamoto, T. Suehiro and M. Mitomo, Appl. Phys. Lett., 2005, 86, 211905 CrossRef PubMed.
- V. V. Tomaev, S. V. Egorov and T. V. Stoyanova, Glass Phys. Chem., 2014, 40, 208–214 CrossRef CAS.
- K. Mayes, A. Yasan, R. McClintock, D. Shiell, S. R. Darvish, P. Kung and M. Razeghi, Appl. Phys. Lett., 2004, 84, 1046–1048 CrossRef CAS PubMed.
- A. J. Fischer, A. A. Allerman, M. H. Crawford, K. H. A. Bogart, S. R. Lee, R. J. Kaplar, W. W. Chow, S. R. Kurtz, K. W. Fullmer and J. J. Figiel, Appl. Phys. Lett., 2004, 84, 3394–3396 CrossRef CAS PubMed.
- V. Adivarahan, S. Wu, J. P. Zhang, A. Chitnis, M. Shatalov, V. Mandavilli, R. Gaska and M. A. Khan, Appl. Phys. Lett., 2004, 84, 4762–4764 CrossRef CAS PubMed.
- M. Srinivas, B. A. Rao, M. Vithal and P. R. Rao, Luminescence, 2013, 28, 597–601 CrossRef CAS PubMed.
- H. Yu, W. Zi, S. Lan, S. Gan, H. Zou, X. Xu and G. Hong, Opt. Laser Technol., 2012, 44, 2306–2311 CrossRef CAS PubMed.
- D.-Y. Wang, Y.-C. Chen, C.-H. Huang, B.-M. Cheng and T.-M. Chen, J. Mater. Chem., 2012, 22, 9957–9962 RSC.
- D. Xu, D. Haranath, H. He, S. Mishra, I. Bharti, D. Yadav, B. Sivaiah, B. Gahtori, N. Vijayan, A. Dhar, J. Zhu, V. Shanker and R. Pandey, CrystEngComm, 2014, 16, 1652–1658 RSC.
- G. S. R. Raju, E. Pavitra, G. Nagaraju and J. S. Yu, Dalton Trans., 2015, 44, 1790–1799 RSC.
- M. Mizuguchi, M. Nara, K. Kawano and K. Nitta, FEBS Lett., 1997, 417, 153–156 CrossRef CAS.
- G. S. R. Raju, E. Pavitra and J. S. Yu, Phys. Chem. Chem. Phys., 2014, 16, 18124–18140 RSC.
- G. S. R. Raju, E. Pavitra and J. S. Yu, Dalton Trans., 2013, 42, 11400–11410 RSC.
- A. Al-Hajry, A. Umar, Y. B. Hahn and D. H. Kim, Superlattices Microstruct., 2009, 45, 529–534 CrossRef CAS PubMed.
- T. Prakash, R. Jayaprakash, G. Neri and S. Kumar, J. Nanopart., 2013, 2013, 8 Search PubMed.
- T. Gutul, E. Rusu, N. Condur, V. Ursaki, E. Goncearenco and P. Vlazan, Beilstein J. Nanotechnol., 2014, 5, 402–406 CrossRef CAS PubMed.
- M. Tomasulo, I. Yildiz and F. M. Raymo, J. Phys. Chem. B, 2006, 110, 3853–3855 CrossRef CAS PubMed.
- A. Wolcott, D. Gerion, M. Visconte, J. Sun, A. Schwartzberg, S. Chen and J. Z. Zhang, J. Phys. Chem. B, 2006, 110, 5779–5789 CrossRef CAS PubMed.
- A. Mandal, J. Nakayama, N. Tamai, V. Biju and M. Isikawa, J. Phys. Chem. B, 2007, 111, 12765–12771 CrossRef CAS PubMed.
- G. Alarcón-Flores, M. García-Hipólito, M. Aguilar-Frutis, S. Carmona-Téllez, R. Martinez-Martinez, M. P. Campos-Arias, M. Jiménez-Estrada and C. Falcony, ECS J. Solid State Sci. Technol., 2014, 3, R189–R194 CrossRef PubMed.
- P. C. Ricci, C. M. Carbonaro, R. Corpino, C. Cannas and M. Salis, J. Phys. Chem. C, 2011, 115, 16630–16636 CAS.
- P. Dorenbos, J. Lumin., 2000, 91, 91–106 CrossRef CAS.
- G. S. Raju, J. Y. Park, H. C. Jung, B. K. Moon, J. H. Jeong and J. H. Kim, J. Electrochem. Soc., 2011, 158, J20–J26 CrossRef CAS PubMed.
- W. T. Carnall, P. R. Fields and K. Rajnak, J. Chem. Phys., 1968, 49, 4447–4449 CrossRef CAS PubMed.
- G. Blasse, G. J. Dirksen, A. Meyerink, D. R. Terrell and L. Neyens, Mater. Chem. Phys., 1988, 19, 547–556 CrossRef CAS.
- G. S. Ofelt, J. Chem. Phys., 1962, 37, 511–520 CrossRef CAS PubMed.
- B. R. Judd, Phys. Rev., 1962, 127, 750–761 CrossRef CAS.
- X. G. Xu, J. Chen, S. Z. Deng, N. S. Xu and J. Lin, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.--Process., Meas., Phenom., 2010, 28, 490–494 CrossRef CAS.
- S. Shionoya, W. M. Yen and H. Yamamoto, Phosphor Handbook, Taylor & Francis, 2010 Search PubMed.
- J. Hao, S. A. Studenikin and M. Cocivera, J. Lumin., 2001, 93, 313–319 CrossRef CAS.
- S. Yang, C. Stoffers, F. Zhang, S. M. Jacobsen, B. K. Wagner, C. J. Summers and N. Yocom, Appl. Phys. Lett., 1998, 72, 158–160 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra15376f |
| This journal is © The Royal Society of Chemistry 2015 |