
Enhanced electrochemical sensing of polyphenols by an oxygen-mediated surface†
Rui Gusmãoab,
Vanesa López-Puentec,
Isabel Pastoriza-Santosc,
Jorge Pérez-Justec,
Maria Fernanda Proençaa,
Fátima Bento*a,
Dulce Geraldoa,
Maria Conceição Paivab and
Elisa González-Romero*d
aCentro de Química, Universidade do Minho, Braga, Portugal. E-mail: fbento@quimica.uminho.pt; Tel: +351 253604399
bInstituto de Polímeros e Compósitos/I3N, Universidade do Minho, Guimarães, Portugal
cDepartamento de Química Física, CINBIO, Universidade de Vigo, Instituto de Investigación Biomédica de Vigo (IBIV), Vigo, Spain
dDepartamento de Química Analítica y Alimentaria, Universidad de Vigo, Vigo, Spain. E-mail: eromero@uvigo.es; Fax: +34 986 812 322; Tel: +34 986 812 240
First published on 11th December 2014
Abstract
We report a straightforward heat treatment in air of commercial screen-printed carbon electrodes (SPCE) at different temperatures and times (ht-SPCE) that produces considerable electrocatalytic effects. The active area and the presence of oxygen groups on the ht-SPCE surface increased upon thermal treatment, more than doubling and by 20%, respectively. The increase of oxygen-containing carbon surface groups results in strong interactions and substantial improvement in Faradaic currents, up to a 10 fold increase in the voltammetric response of relevant polyphenols (hydroquinone, catechol, pyrogallol and dopamine). Moreover, ht-SPCE displayed higher selectivity towards the oxidation of polyphenol mixtures than the untreated SPCE. Finally, good linear ranges were obtained by voltammetric determination of dopamine with detection and quantification limits 5 times lower than the values obtained for the untreated SPCE. The versatility of oxidation by air makes this activation procedure very attractive and easy to implement, which can be further used for numerous applications in the (bio)sensor field.
Introduction
The increasing demand for on-spot monitoring for clinical, environmental and industrial applications has driven the development of various kinds of disposable electrochemical sensors, such as those made using screen and inkjet printing.1 Carbon is one of the most used electrode materials for this kind of sensor2 mainly because of its well-known features: it is chemically inert, has low background currents and a wide potential window.3Screen-printed carbon electrodes (SPCE)4 are attractive miniaturized low cost electrochemical sensors that allow the performance of measurements with small sample volumes (typically 50 μL). Besides, SPCE can be readily modified by synthetic and/or biological compounds for improving sensor selectivity.5
Commercial SPCE are fabricated with carbon inks composed by carbon black, mineral or polymer binders and other proprietary additives for the promotion of dispersion, printing and adhesion.6 The inks composition from distinct commercial suppliers can greatly vary from each other.7 The presence of non-electroactive components in the ink formulation and relatively low graphitic carbon content, imposed by the constraints of screen-printing6 and curing processes, can greatly affect the electrochemical properties of the sensor.4
The aforementioned issues can be minimized using proper surface pretreatment methods. Nevertheless in contrast with glassy carbon electrode (GCE), SPCE surface treatments are limited, as the physical abrasive polishing and sonication cannot be applied. Thus they currently include: electrochemical,8 chemical prior to electrochemical,9 UV light10 and plasma treatments.11 Similar treatments have been carried out for other carbon electrode materials,12–14 among them the thermal treatment is one of the commonly used. This method usually involves high temperatures, up to 1100 °C, in the presence of oxidizing/inert gases.15 Nevertheless the high temperatures used for carbon treatment are not compatible with SPCE and thus have not been explored up to date.16
Often the activation from the above mentioned treatments is explained in terms of increase of surface roughness and/or oxygenated functional groups on the electrode surface. Surface analysis showed that most of the oxygen functional groups created from the oxygen treatment were carbonyl or quinone type.17
In this work, we explore the activation of commercial SPCE through a mild heat treatment. Thus, we study the effect of temperature (from 100 to 400 °C) and heating time (from 30 minutes to 8 hours) under air or inert atmosphere in its electrochemical performances using the hydroquinone (HQ) redox probe by cyclic voltammetry (CV). Besides, we provide a detailed physicochemical characterization of the activated electrode surface via scanning electron microscopy (SEM) and Raman, and X-ray photoelectron spectroscopies (XPS). Heat treated SPCE double layer capacitance and electroactive area were also estimated. Finally, we also investigate the electrochemical behavior of the heat treated SPCE towards a set of biologically relevant analytes (catechol, pyrogallol and dopamine).
Experimental section
Chemicals
Sodium phosphate monobasic monohydrate, sodium phosphate dibasic heptahydrate, catechol, dopamine (DA), hydroquinone (HQ), potassium ferricyanide and pyrogallol were purchased from Sigma-Aldrich. All chemicals were used as received. Milli-Q water was used as solvent.Heat treatments
Commercial SPCE were subjected to different thermal treatments (see full details in Table S1, ESI†) comprising temperatures ranging from 100 to 400 °C and heating times from 30 minutes to 8 hours under air or Ar atmosphere in a muffle furnace (Heron 12-PR/200 BB series). SPCE were submitted to a control vacuum heat treatment using a Büchi glass oven B-580. An Ar atmosphere was created by three successive cycles of purge and pressurization.Thermal analysis
Electrode materials were separately scraped from as received SPCE and analyzed by thermogravimetric analysis (TGA). The TGA was performed on a modulated Q500 from TA Instruments at two operating modes, isothermal and gradient. In isothermal operating mode, the sample was heated from room temperature to 200 °C and then it was kept constant during 480 min under a constant air flow (60 mL min−1). In the gradient operating mode, the sample was heated from room temperature to 700 °C at 10 °C min−1 under a constant flow (air or N2 at 60 mL min−1). The weight loss was measured for each case.Electrochemical measurements
Commercially available SPCE from DropSens (reference DRP-110) were used. These include a traditional three electrode configuration printed on the same ceramic strip: a carbon disk working electrode with a radius of 2 mm, a counter electrode (same material as working electrode) and a silver pseudo-reference electrode. An insulating layer delimits the working area and electric contacts. The production characteristics of commercial SPCE are regarded by the manufacturers as proprietary information. Voltammetric measurements were performed with Autolab PGSTAT12 and PGSTAT30 potentiostats/galvanostats (Ecochemie), at room temperature, by placing a 50 μL drop to cover the three electrodes. All potentials were measured and reported versus Ag pseudo-reference electrode potential. Differential pulse voltammetry (DPV) experiments were performed at a voltage step of 5 mV, a pulse amplitude of 50 mV, a pulse duration of 0.05 s, a interval time of 1 s, and a sweep rate of 5 mV s−1.Physicochemical surface characterization: SEM, Raman and XPS
Scanning electron micrographs (SEM) of the bare SPCE were taken on a NanoSEM – FEI Nova 200 (FEG/SEM), equipped with Energy Dispersive X-ray (EDAX) analysis Pegasus X4M (EDS/EBSD) equipment using an accelerating voltage of 5 kV and a working distance of 5 mm, in secondary electron imaging (SEI) mode. SEM images of the heat treated SPCE were taken on a JEOL JSM-6700F microscope (Tokyo, Japan) using an accelerating voltage of 10 kV and a working distance between 3 mm and 8 mm, also in SEI mode.Raman experiments were conducted with a Renishaw InVia Reflex system. The spectrographs used high-resolution gratings (1200–1800 grooves per mm) with additional band-pass filter optics, a confocal microscope and a 2D-CCD camera. Raman spectra were recorded by exciting the sample with different laser lines: 532 nm, 633 nm and 785 nm. All measurements were collected using a 20× objective with accumulation times of 10 s. The power at the sample was 10% of the initial laser power.
XPS experiments were performed in a SPECS Sage HR 100 spectrometer with a non-monochromatic X-ray source Mg Kα line of 1253.6 eV energy and a power applied of 250 W and calibrated using the 3d5/2 line of Ag with a full width at half maximum (FWHM) of 1.1 eV. The selected resolution for the spectra was 30 eV of pass energy and 0.5 eV per step for the general survey spectra, 15 eV of pass energy and 0.15 eV per step for the detailed spectra of the different elements. All measurements were made in an ultra-high vacuum (UHV) chamber at a pressure around 5 × 10−8 mbar. In the fittings asymmetric and Gaussian–Lorentzian functions were used (after a Shirley background correction) where the FWHM of all the peaks were constrained while the peak positions and areas were set free.
Results and discussion
Hydroquinone voltammetry at heat-treated SPCE
Electrocatalytic properties of carbon electrodes from commercial SPCE submitted to different heat treatments were studied. Briefly, SPCE were subjected to different thermal treatments in aerobic or anaerobic conditions (see Table S1†). Thus, the temperatures and exposures times were varied from 100 to 400 °C and from 30 minutes to 8 hours, respectively (further details in Experimental section and Table S1†). After cooling down the electrocatalytic properties of SPCE were tested by hydroquinone (HQ) voltammetry. HQ is a well-known hydroxyl containing aromatic compound that oxidizes by an irreversible two electron redox mechanism on carbon electrodes.18 Fig. 1 shows CVs at 100 mV s−1 of 1.0 mM HQ in 0.1 M phosphate buffer solution (PBS) at pH 7.4 obtained for untreated and different heat-treated SPCE. The CV recorded using the untreated SPCE (dashed line) showed a characteristic irreversible redox process with a peak-to-peak separation (ΔEp) of 280 mV and with the anodic (Iap) and cathodic (Icp) peak currents of 36 μA and 40 μA, respectively. Up to 200 °C the voltammograms of the SPCE treated for 2 hours did not display any significant improvement, regarding either peaks current or potential (Fig. 1A).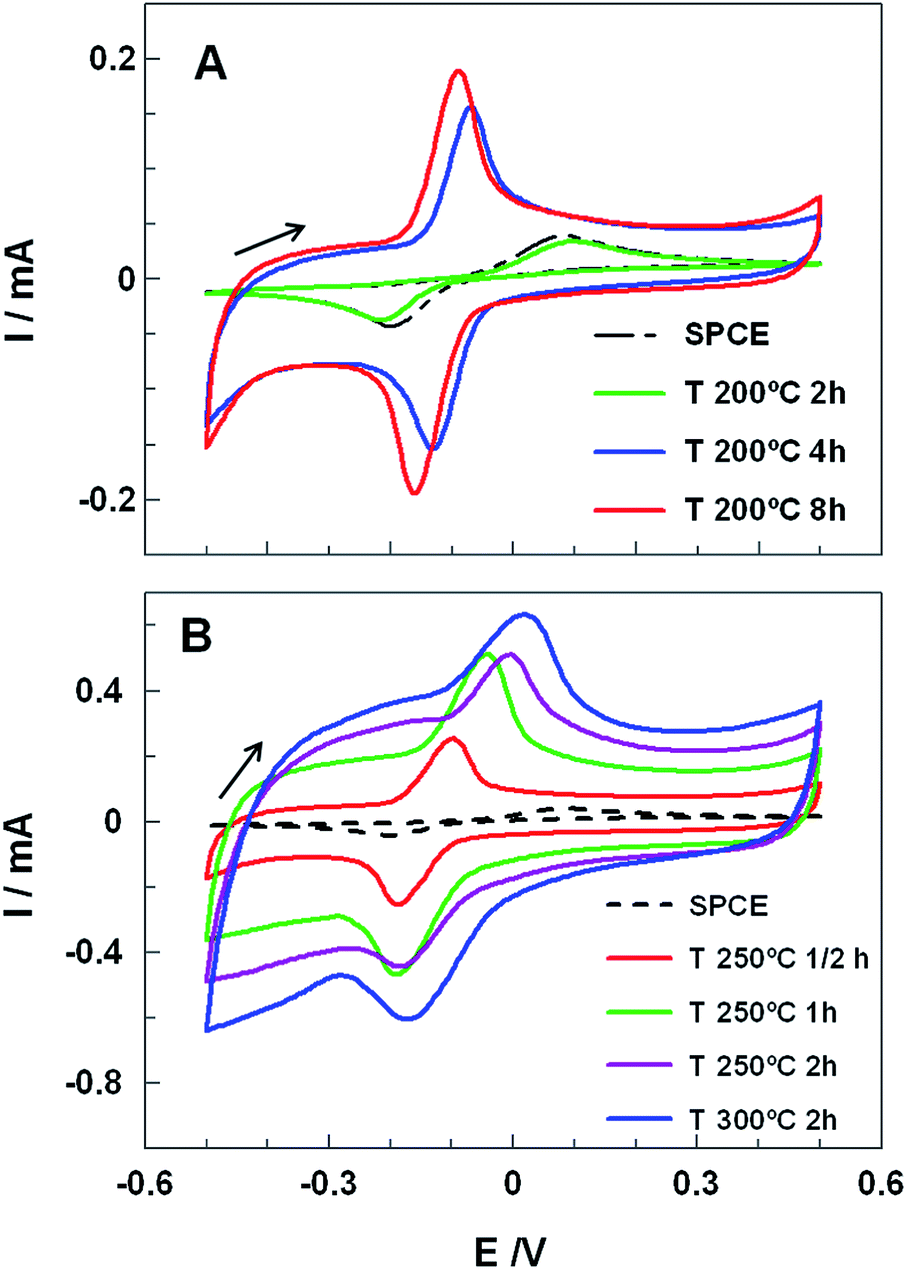 | ||
| Fig. 1 CVs obtained for 1.0 mM HQ in 0.1 M PBS pH 7.4, at an untreated (dashed black lines) and heat-treated SPCEs using different temperatures: (A) 200 °C and (B) 250 °C and 300 °C. The heating time at each temperature is indicated inset. All voltammograms are recorded at 100 mV s−1. | ||
Nevertheless, longer heat treatments at 200 °C (4 and 8 h) led to a very sharp increase of Iap and Icp (up to 4 times) and a remarkable decrease of ΔEp, from 280 mV to 70 mV or 59 mV for 4 or 8 h heat treatment, respectively. Moreover, SPCE treated for 8 h showed a formal potential, E0′, of (−56 ± 5) mV (calculated by averaging the anodic and cathodic peak potentials) and a ratio of anodic to cathodic current intensities of ca. 1.04, which indicated a quasireversible two-electron-transfer process. Nevertheless treatments at higher temperatures did not lead to good performances (Fig. 1B). While at 250 °C and 300 °C, higher capacitive currents, ΔEp, and great peak distortion were noticed, at 400 °C SPCE isolating material was damaged and the working electrode peeled off.
In order to clarify the role of oxygen in the SPCE activation, the electrodes were treated under Ar atmosphere. The corresponding CVs obtained at 200 °C for 8 hours and 250 °C for 0.5 hours revealed a similar performance as the untreated SPCEs (Fig. S1, ESI†) indicating that the activation did not take place. The presence of oxygen might lead to its physisorption/chemisorption promoting a partial oxidation of the carbon surface, yielding oxygen-containing surface functional groups,19,20 simultaneously contributing to the degradation of the carbon ink binders. Taking into account the above results, subsequent studies were made using the SPCE treated at 200 °C for 8 hours (ht-SPCE).
CVs of HQ obtained at different scan rates, namely from 10 to 500 mV s−1, are displayed in Fig. 2A (similar study with untreated SPCE is shown in ESI, Fig. S2A†). The slight variations in the positions of peak currents with scan rates suggested a quasireversible electrode process. Furthermore, anodic and cathodic peak currents showed a good linear correlation with the scan rate (R2 = 0.998, Fig. 2B), indicating that the electrochemical redox processes is adsorption-controlled. It might be ascribed to the strong interaction between HQ and oxygen-containing surface functional groups at the ht-SPCE,20 strengthened by the fact that for the untreated SPCE the process was controlled by diffusion (ESI, Fig. S2B†).
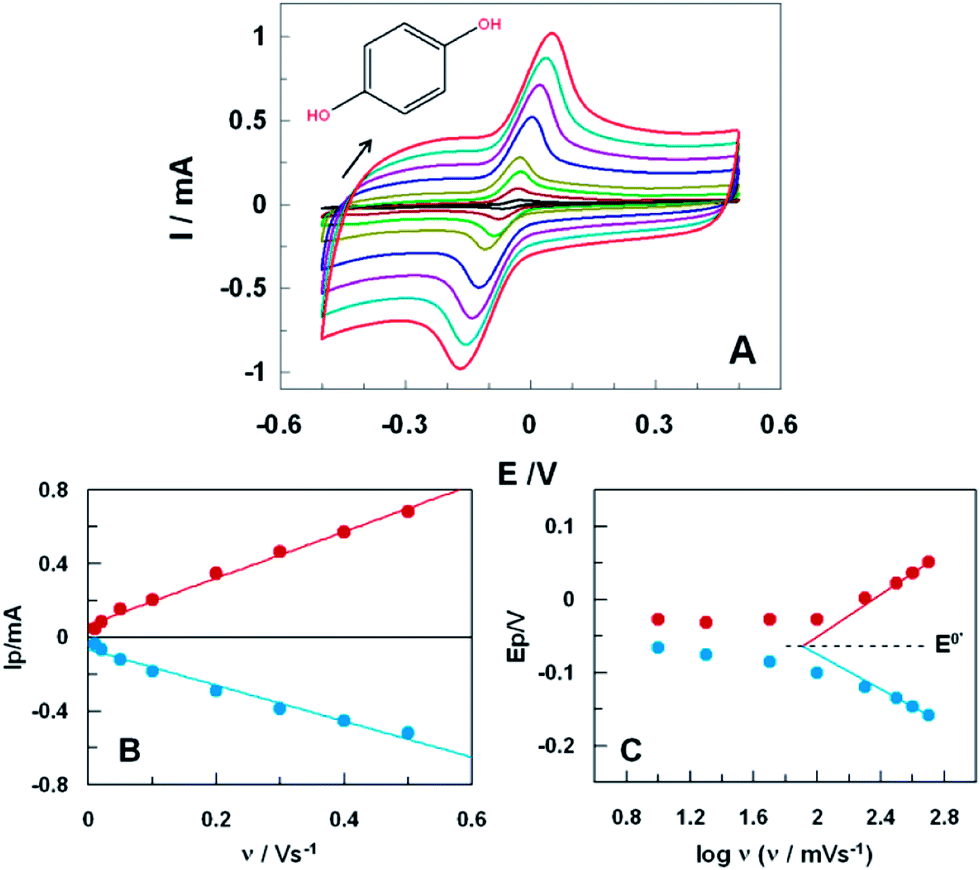 | ||
| Fig. 2 (A) CVs of 1.0 mM HQ in 0.1 M PB at pH 7.4, with a scan rates from 10 to 500 mV s−1 from inner to outer curves, at the ht-SPCE (200 °C for 8 h). (B) Linear relationship between the anodic and cathodic peak currents with the scan rate. (C) Semilogarithmic dependence of the peaks potential and the scan rate. | ||
The apparent rate constant of electron transfer (kET) and the transfer coefficient (αa) for HQ oxidation at ht-SPCE were estimated using Laviron analysis.21 By plotting anodic and cathodic peak potentials against the logarithmic of the scan rate, as shown in Fig. 2C, values of 0.20 and 1.24 s−1 were estimated for αa and for kET, respectively. This last value indicates a higher ability of the ht-SPCE for promoting electron transfer reactions with HQ, in contrast with untreated SPCE where a low value of kET (ca. 0.58 s−1) was obtained. Moreover, Laviron plotting for ht-SPCE showed a formal potential, E0′, of (−63 ± 2) mV. The electrochemical process of HQ on ht-SPCE is positively affected with promotion of electron transfers and 5 fold improvements of the peak currents by the surface catalytic activity. Considering the collected results, the background current increase might be correlated with increase of the oxygen functionalities on the surface after the treatment.
Thermal analysis of electrode materials
Thermogravimetric analysis (TGA) measures the changes in mass during oxidation processes and provides valuable information about materials degradation. The carbon part of the electrode was scraped from the ceramic template and analyzed by TGA under air and N2 atmosphere in temperature gradient mode (10 °C min−1, see Fig. 3A). The thermograms of the electrode materials displayed a similar behavior under nitrogen and air till 500 °C with a mass loss of 10% at 300 °C. Nevertheless, above 500 °C just the thermogram obtained in air shows a drastic mass loss, which is due to the carbon material combustion.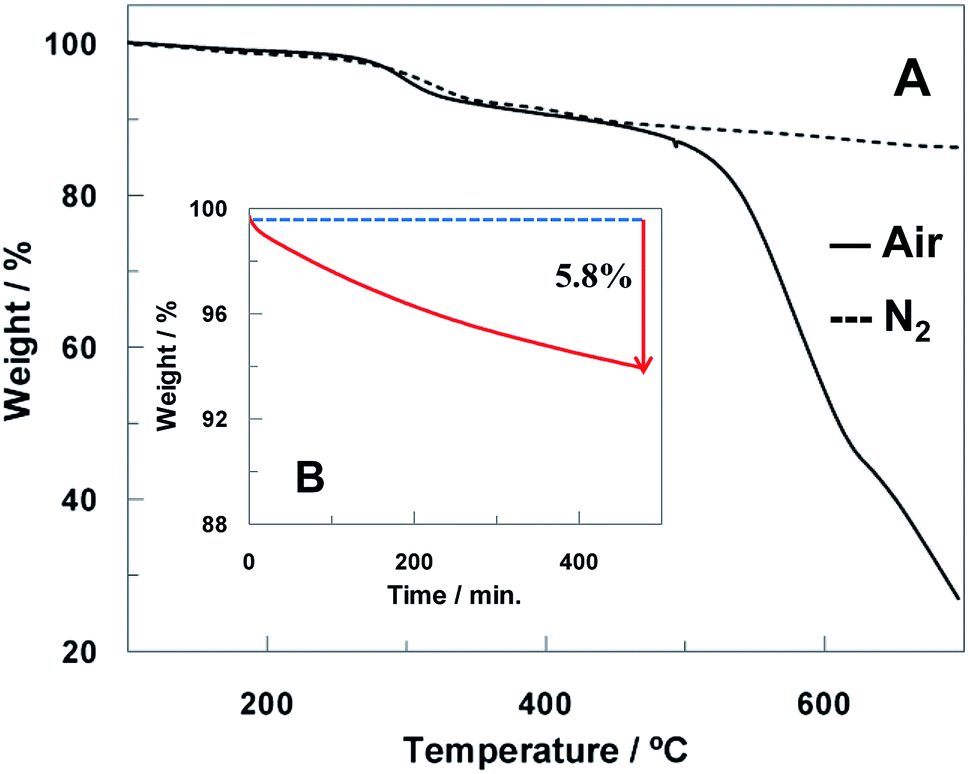 | ||
| Fig. 3 (A) TGA curves of scraped carbon electrode material in air (solid line) and N2 flow (dashed line) and (B) isothermal TGA curve at 200 °C of scraped carbon electrode material in constant air flow. | ||
SPCEs contain an insulating polymer layer which delimits the working area and the electrical contacts. In order to guarantee that the good electrical contact between electrodes was not compromised by temperature treatment, this polymer was also studied by TGA under N2 flow (shown in ESI, Fig. S3†) showing a major weight loss above 350 °C.
The scraped electrode carbon material was also heated isothermally in air at 200 °C for 8 h in order to simulate the heat treatment carried out on the ht-SPCE. Fig. 3B shows the isothermal TGA curve where a mass loss of 5.8% is observed after 8 h. Considering that no degradation process was observed in this temperature range when the temperature gradient mode is used, it can conclude that the heat-treatment performed in air induces primarily surface cleaning by desorption of molecules adsorbed at the carbon surface and thus it is not expected to compromise the electrode material integrity.
Characterization of heat treated SPCE
In order to clarify the improvement of the performance of the ht-SPCE, the treated and untreated SPCE were characterized using SEM, Raman and XPS spectroscopies. Besides the reversible redox couple Fe(CN)63−/Fe(CN)64− was used to electrochemically characterize the ht-SPCE surface by CV.The surfaces of the untreated and the treated SPCE were analysed by SEM (Fig. 4). Although both, the untreated and treated SPCE, presented a heterogeneous nanometric granular surface characteristic of carbon black with graphite flakes emerging sporadically, the ht-SPCE might display and apparent increase graphite flake exposure (Fig. 4B). Besides, the analysis of a wider surface area (ESI, Fig. S4A and B†) revealed an apparent increment of surface micro-cracks on the ht-SCPE micrographs. Overall, the untreated and ht-SPCE electrodes surface exhibit a reasonably similar morphology, thus the variations in electrochemical response may be mostly assigned to surface oxidation.
 | ||
| Fig. 4 Representative SEM images of (A) untreated commercial SPCE surface and (B) after 8 hours at 200 °C treated ht-SPCE surface. | ||
In the case of a carbon based material, Raman spectroscopy is the commonly used tool to study structural properties, such as degree of bond disorder or sp2/sp3 ratio by monitoring the G band wavenumber or the intensity ratio for the bands D and G (ID/IG), respectively. Therefore Raman spectroscopy was used to monitor possible structural changes induced on the carbon layer of the working electrode by heat treatment. In order to neglect the effect of the laser line irradiation the 785 nm laser line was used. Fig. S5† in ESI shows clear changes in the Raman spectra of SPEC and ht-SPEC when 633 and 532 nm laser lines were used. Fig. 5 illustrates the Raman spectra obtained with 785 nm laser line for the working electrodes of a ht-SPCE and an untreated SPCE. Both Raman spectra show the similar features in terms of G band position and intensity ratio of band D to G (ID/IG) that indicate graphitic material presenting high structural disorder.22,23 Thus, it may be concluded that the heat treatment imposed to the SPCE did not affect the structure of the carbon deposited on the working electrode in terms of sp2 carbon content.24 Finally, an increase of the Raman intensity for D and G bands with the heat treatment (Fig. 5) was also noticed which might be related to charge transfer and conductivity σ (ṽ),25 but it is not conclusive.
 | ||
| Fig. 5 Raman spectra of untreated (blue line) and heat-treated (red line) SPCE recorded using a laser excitation line at 785 nm. The values of ID/IG ratio are showed inset. | ||
X-Ray photoelectron spectroscopy (XPS) was performed to obtain information about graphitic species and other functional groups at surface SPCE and ht-SPCE electrodes. As shown in Fig. S6,† the survey XPS spectra of SPCE (A) and ht-SPCE (B) present C1s and O1s as major components but also other minor peaks associated to chlorine (from the ink binder), silver (from the reference electrode and contacts) and aluminum (from the SPCE ceramic strip, 96% Al). The C1s and O1s peaks obtained in the high resolution spectra were deconvoluted to monitor possible chemical changes of the SPCE after heat treatment, resulting in several peaks. In the case of C1s (Fig. S6C and D†), the peaks with binding energies of 284.5 eV and 290.5 eV were assigned to C–C bonds and π–π* transition (shake-up peak) both characteristic of graphite-like materials, the peaks at 286.0 eV and 287.0 eV corresponded to C–O bonds, whereas the peak at 288.6 eV was assigned to carbonyl groups.26 In the case of O1s (Fig. S6E and F†), the peaks at around 534.2 eV and 531.8 eV were assigned to O–(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–O and O–CO bonds respectively.27 Comparing the intensity of the peaks in the deconvoluted spectra, atomic percentages for C and O as well as functional groups percentages were obtained, as listed in Table 1. From these values it is noteworthy that there is a higher O/C ratio for the ht-SPCE (0.30 versus 0.24 for SPCE) which might make the electrode more hydrophilic and thus more electrochemically active.22
O)–O and O–CO bonds respectively.27 Comparing the intensity of the peaks in the deconvoluted spectra, atomic percentages for C and O as well as functional groups percentages were obtained, as listed in Table 1. From these values it is noteworthy that there is a higher O/C ratio for the ht-SPCE (0.30 versus 0.24 for SPCE) which might make the electrode more hydrophilic and thus more electrochemically active.22
| Element | SPCE (at.%) | ht-SPCE (at.%) |
|---|---|---|
| Carbon | 67.3 | 66.2 |
| C–C | 32.6 | 36.7 |
| C–O | 29.9 | 24.2 |
| CO |
4.7 | 4.4 |
| Oxygen | 16.3 | 19.6 |
| O–(CO)–O |
15.3 | 17.7 |
| O–(CO) |
1.0 | 1.9 |
| Chlorine | 4.0 | 3.0 |
| Silver | 0.5 | 0.3 |
| Aluminium | 12.0 | 10.9 |
In order to fully characterize the effectiveness of the heat treatment, namely with respect to the increase of the current response, a careful electrochemical study was carried out using a potassium ferricyanide in 0.1 M KCl as redox probe (its diffusion coefficient, D, is 7.35 × 10−6 cm2 s−1).28 The active surface area of the ht-SPCE (200 °C for 8 hours) can be estimated from the Randle–Sevcik equation for a reversible electrochemical process under diffusive control. By performing cyclic voltammetric experiments at different scan rates (typically, from 10 to 500 mV s−1), linear relationships between the cathodic peak current and the square root of the scan rate were obtained (ESI, Fig. S7A†) and the surface area were evaluated.
Using this same methodology, the active area of the SPCE was calculated and a value of 0.06 cm2 with an equivalent radius of 1.40 mm was obtained. Taking into account that the geometric radius of the working electrode is 2 mm, this result shows that the rough carbon is partially passivated due to surface contaminants or due to the organic binder of the carbon ink. For the ht-SPCE the value obtained for the active surface area, 0.15 cm2, revealed a 2.5-fold increase after the heat treatment. The equivalent radius was 2.17 mm.
As the capacitance of an electrode depends on the charge separation processes occurring at the interface, this parameter can be highly affected by electrode surface modifications. The double-layer capacitance (Cdl) of ht-SPCE was evaluated from the CVs of 0.1 M KCl solutions (ESI, Fig. S7B†). At a fixed potential, far from the switching potential (300 mV), the capacitive current |ic| is given by the equation:
| |ic| = ACdlv | (1) |
Application of ht-SPCE to other analytes of biological significance
The performance of the ht-SPCE was also demonstrated with other relevant hydroxyphenols of recognized biological importance. CVs of pyrogallol, catechol and dopamine (DA) obtained with SPEC and ht-SPCE, corrected by blank subtraction, are shown in Fig. 6. In the case of pyrogallol the one-electron irreversible oxidation peak at 212 mV with untreated SPCE (dashed black line), displayed a negative shift of 150 mV when the ht-SPCE (blue line) is used. Simultaneously, the values of Ip shows a 4 times increase (Fig. 6A).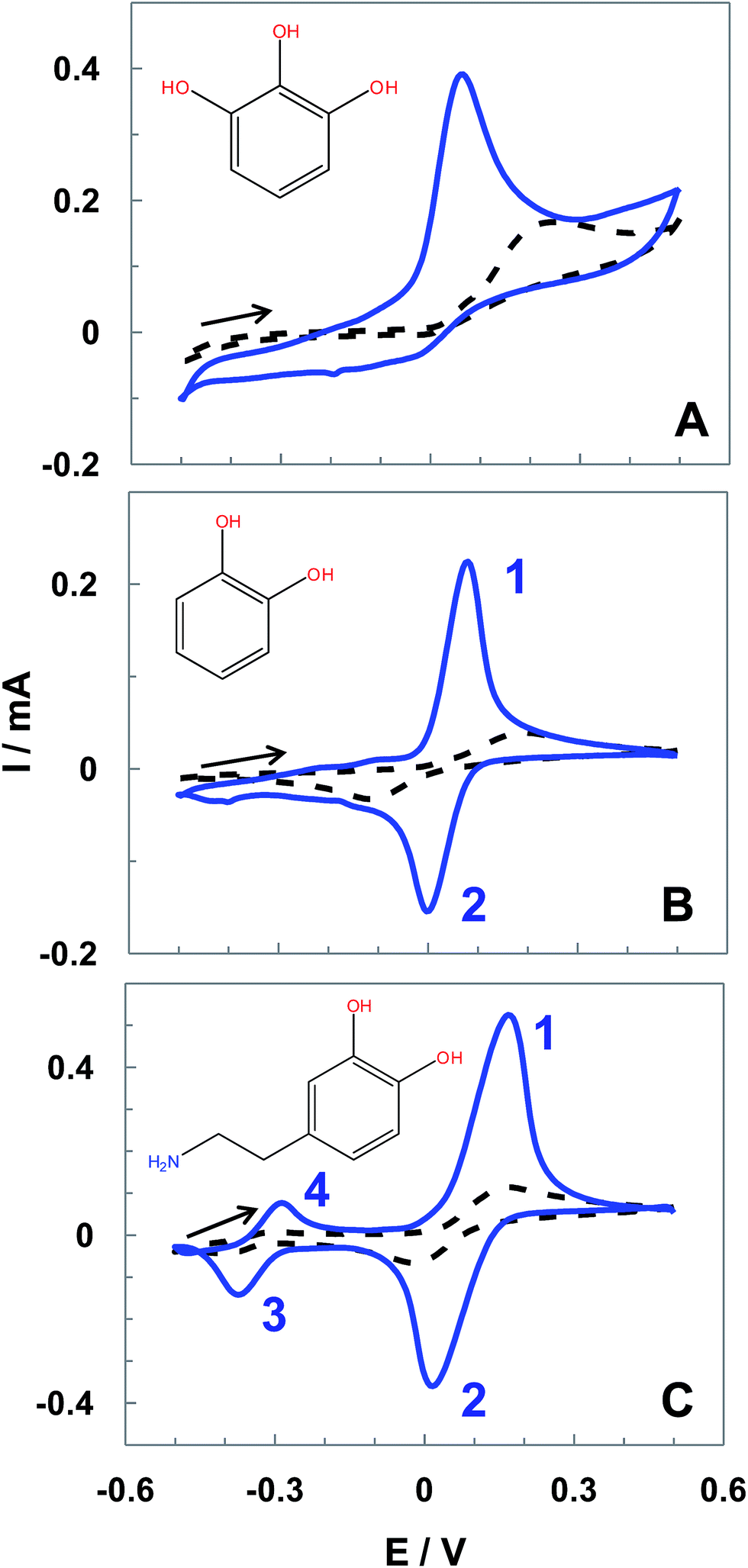 | ||
| Fig. 6 CVs with blank correction of 1.0 mM pyrogallol (A), catechol (B) and dopamine (C) in 0.1 M PBS pH 7.4, at untreated SPCE (dashed lines) and ht-SPCE (blue lines). All voltammograms were recorded at 100 mV s−1. | ||
Regarding the voltammetric response of catechol (Fig. 6B), besides the remarkable increase of the Ip (5 times increase), ΔE decreased dramatically from 290 mV for untreated SPCE (dashed black line) to 83 mV for ht-SPCE (blue line), being estimated an E0′ of 40 mV (vs. Ag pseudo-reference) with an Ipa/Ipc ratio of ca.1.01. The higher electron cloud density of catechol than HQ leads to its higher oxidized potential.21
In the same way, the voltammogram for DA displays a marked improvement (Fig. 6C). The two sets of peaks, corresponding to the redox couples dopamine/dopaminequinone (peaks 1 and 2 for the first process) and dopachrome/leucodopachrome (peaks 3 and 4 for the second process).31 For both, untreated SPCE and ht-SPCE, are shown the second scans of DA redox processes previously described. For the untreated SPCE (dashed black line) the E0′1 was ca. 85 mV and E0′2 of −315 mV, with an ΔE of 176 and 130 mV for the respective redox processes, whereas for ht-SPCE peak currents are 10 and 5-fold than that of untreated SPCE for the first and second process respectively. The E0′1 was ca. 93 mV and a E0′2 of −330 mV, with an ΔE of 131 mV for the first process and 88 mV for the second process.
It remains a challenge to electrochemically determinate DA by avoiding the interference of other hydroxyphenols. The addition of HQ does not affect the electrochemical behavior of DA at ht-SPCE (Fig. 7A), and well-defined, separated redox peaks of both DA and HQ can be observed, whereas for the untreated SPCE, the oxidation peaks of HQ and DA appear highly overlapped in a single process at ca. 0.2 V. The current intensities of DA and HQ at ht-SPCE are much higher than that at untreated SPCE (Fig. 7A), providing evidence that ht-SPCE has a greater gain in sensitivity and selectivity against the untreated SPCE.
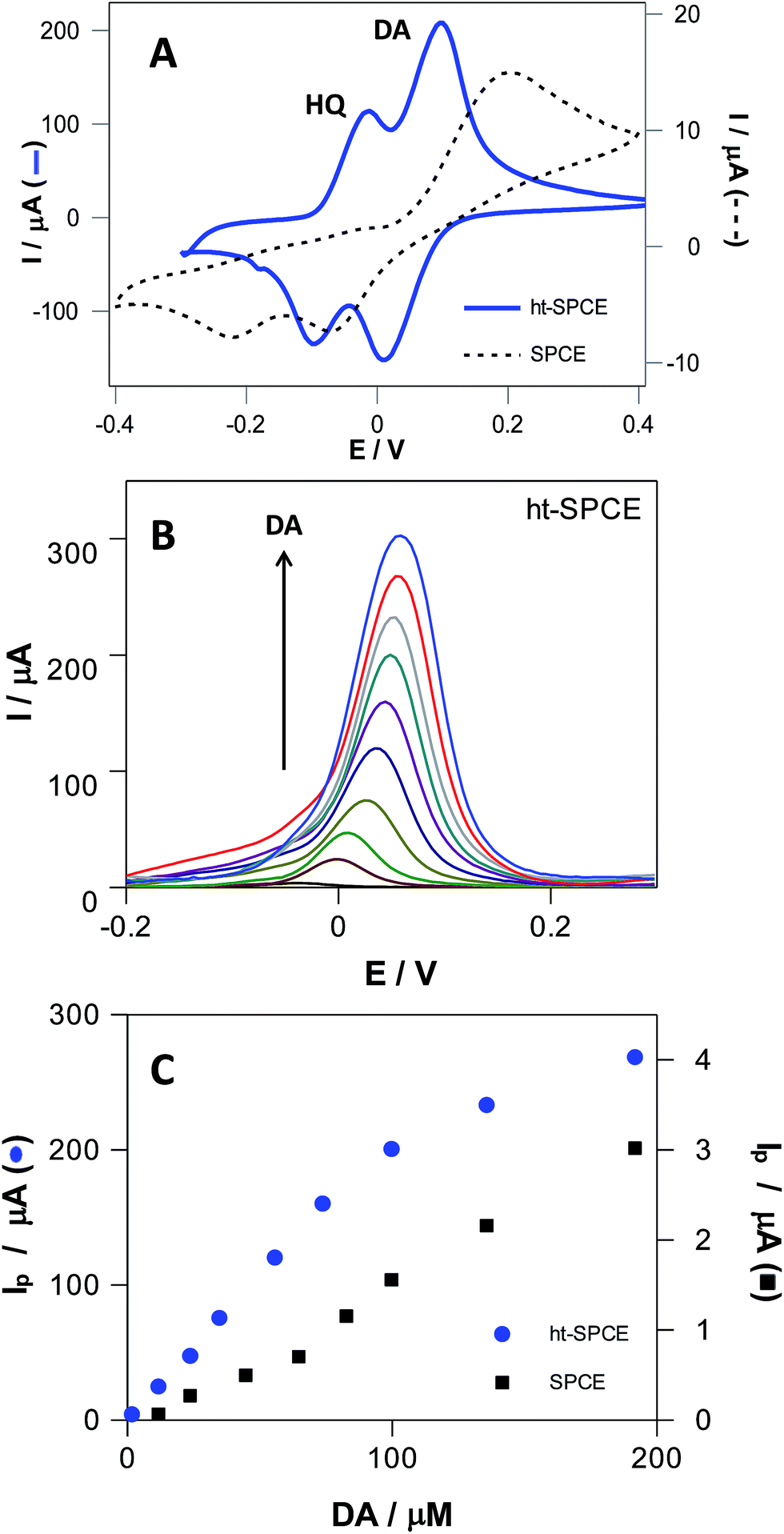 | ||
| Fig. 7 (A) CVs with blank correction of an equimolar 500 μM DA and HQ mixture at SPCE (dashed lines) and ht-SPCE (blue continuous line) in 0.1 M PBS pH 7.4, with a 100 mV s−1; (B) DPVs of DA in increasing concentrations at ht-SPCE, in 0.1 M PBS pH 7.4 with 5 mV s−1 scan rate. Linear relationship between the anodic peak currents with DA concentration (C) for SPCE (■, y = 0.02x − 0.18, R2 = 0.991) and ht-SPCE (●, y = 2.01x − 0.04, R2 = 0.9995). | ||
The increment in surface area and oxygen content on ht-SPCE can further enhance the electrochemical reaction of dihydroxyphenols, leading to the increased sensitivity. The different space resistances to these hydroxyphenols and the fast electrochemical reaction kinetics of ht-SPCE are beneficial to the separation of different hydroxyphenols voltammetric waves. Then, as a more sensitive technique than CV, differential pulse voltammetry (DPV) was employed for determination of linear range, detection (DL) and quantification limits (QL) of DA in both electrodes (Fig. 7B and C, Fig. S8†). The current intensities of DA at ht-SPCE increases linearly with concentration in the range of 1 to 100 μM with a DL and QL of 2.9 and 8.9 μM respectively, while the untreated SPCE has much higher values, DL of 16.2 and QL of 49.2 μM. For higher concentrations, there is a loss of linearity for both electrodes, although it is more accentuated for ht-SPCE. Nevertheless, SPCE has a one hundred times lower sensitivity than the ht-SPCE within the same concentration range.
In general, ht-SPCE has remarkable improvements in their electrochemical performance, with respect to untreated SPCE. The peak intensities, for a series of relevant hydroxyphenols increased up to 10 times, representing an increase of sensor sensibility and sensitivity. Relevant peak potential shifts occurred for the analyzed species. Furthermore, the change the peak shape demonstrated the modification in the nature of electrochemical process and more favored electron transfer kinetics, such as it was demonstrated for HQ. The improvements can be explained by the changes that occurred in the surface chemistry and by the increment in electrode effective electrochemical area.
Conclusions
Different activation procedures for SPCE may enhance the performance of electrodes, but may either require cost intensive instrumentation, which usually are unsuitable for mass production, or cause loss of stability in their performance. Therefore, a simple, low cost and yet effective strategy for the activation of SPCE without compromising the stability is continuously being sought. The current work has demonstrated an extremely simple and yet effective carbon electrode activation procedure of SPCE via oxygen adsorption and temperature activation.At optimized conditions, heat treatment shows an impressive improvement in the electrochemical performance of these SPCE, with electrochemical response for relevant analytes increasing up to 10 times, therefore increasing sensor sensibility. In addition, ht-SPCE displayed higher selectivity towards the oxidation of HQ and DA in a mixture than the untreated SPCE. The oxidation of these biomolecules showed well defined oxidation peaks with good peak separation, less positive oxidation potential and enhanced redox currents. Finally, good linear ranges were obtained by DPV determination of DA with detection and quantification limits 5 times lower than the values obtained for the untreated SPCE. A large temporal stability was also verified, heat treated SPCE hold their electrochemical performance after several months of usage, while untreated commercial SPCE usually have a short one week lifetime before pasivation.
Because oxidation in air is a controllable and environmentally friendly process, selective activation of carbon materials, such as graphite, graphene and CNT, in air is very attractive. In contrast to current chemical/electrochemical techniques for activation of electrode surfaces, air oxidation by heat-treatments does not require the use of toxic or aggressive chemicals, catalysts, or inhibitors and opens avenues for numerous new applications of carbon nanomaterials. The results reported here, make this one step and low cost effective approach very promising to facilitate the development of disposable and miniaturized electrochemical (bio)sensors.
Acknowledgements
The authors are highly indebted to Dr Luis Yate for XPS analysis and Dr Pablo Fanjul-Bolado for helpful comments on commercial SPCE. This work was supported by Xunta de Galicia/FEDER (GPC2013-006 and R2014/030), MINECO (CTQ 2010-16390, MAT2013-45168-R, CTQ2011-28157 and PRI-AIBPT-2011-1096) and by FCT/CRUP (Est-C/QUI/UI0686/2011; FCOMP-01-0124-FEDER-022716 and E-136/2012). VLP and RG acknowledge to PhD Grant (MAT2010-15374) and PostDoc Grant (SFRH/BPD/86690/2012), respectively.Notes and references
- M. Li, Y.-T. Li, D.-W. Li and Y.-T. Long, Anal. Chim. Acta, 2012, 734, 31 CrossRef CAS PubMed.
- Z. Taleat, A. Khoshroo and M. Mazloum-Ardakani, Microchim. Acta, 2014, 181, 865 CrossRef CAS.
- R. L. McCreery, Chem. Rev., 2008, 108, 2646 CrossRef CAS PubMed.
- N. Thiyagarajan, J.-L. Chang, K. Senthilkumar and J.-M. Zen, Electrochem. Commun., 2014, 38, 86 CrossRef CAS PubMed.
- J. P. Metters, R. O. Kadara and C. E. Banks, Analyst, 2011, 136, 1067 RSC.
- O. D. Renedo, M. A. Alonso-Lomillo and M. J. A. Martínez, Talanta, 2007, 73, 202 CrossRef CAS PubMed.
- R. O. Kadara, N. Jenkinson and C. E. Banks, Sens. Actuators, B, 2009, 138, 556–562 CrossRef CAS PubMed.
- J. Wang, M. Pedrero and H. Sakslund, Analyst, 1996, I, 345–350 RSC.
- G. Cui, J. H. J. Yoo, J. S. Lee, J. H. Uhm, G. S. Cha and H. Nam, Analyst, 2001, 126, 1399–1403 RSC.
- M. D. Osbome, B. J. Seddon, R. A. W. Dryfe, G. Lagger, U. Loyall, H. Schäfer and H. H. Girault, J. Electroanal. Chem., 1996, 417, 5–15 CrossRef.
- S. C. Wang, K. S. Chang and C. J. Yuan, Electrochim. Acta, 2009, 54, 4937–4943 CrossRef CAS PubMed.
- C.-M. Yoon, D. Long, S.-M. Jang, W. Qiao, L. Ling, J. Miyawaki, C.-K. Rhee, I. Mochida and S.-H. Yoon, Carbon, 2011, 49, 96–105 CrossRef CAS PubMed.
- J. Wang and E. Gonzalez-Romero, Electroanalysis, 1993, 5, 427 CrossRef CAS.
- X. He, Y. Geng, J. Qiu, M. Zheng, S. Long and X. Zhang, Carbon, 2010, 48, 1662 CrossRef CAS PubMed.
- A. G. Pandolfo and A. F. Hollenkamp, J. Power Sources, 2006, 157, 11 CrossRef CAS PubMed.
- C. W. Foster, J. P. Metters, D. K. Kampouris and C. E. Banks, Electroanalysis, 2014, 26, 262 CrossRef CAS.
- C.-T. Hsieh and H. Teng, Carbon, 2002, 40, 667 CrossRef CAS.
- I. Duo, C. Levy-Clement, A. Fujishima and C. Comninellis, J. Appl. Electrochem., 2004, 34, 935 CrossRef CAS.
- X. Ji, C. E. Banks, A. Crossley and R. G. Compton, ChemPhysChem, 2006, 7, 1337 CrossRef CAS PubMed.
- H. Boehm, Carbon, 2002, 40, 145 CrossRef CAS.
- E. Laviron, J. Electroanal. Chem. Interfacial Electrochem., 1979, 101, 19 CrossRef CAS.
- E. P. Randviir, D. A. C. Brownson, J. P. Metters, R. O. Kadara and C. E. Banks, Phys. Chem. Chem. Phys., 2014, 16, 4598 RSC.
- A. C. Ferrari, Solid State Commun., 2007, 143, 47 CrossRef CAS PubMed.
- P. Lespade, A. Marchand, M. Couzi and F. Cruege, Carbon, 1984, 22, 375 CrossRef CAS.
- G. Gouadec and P. Colomban, Prog. Cryst. Growth Charact. Mater., 2007, 53, 1 CrossRef CAS PubMed.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, 1979, vol. 3 Search PubMed.
- G. Beamson and D. Briggs, High Resolution XPS of organic polymers, The Scienta ESCA 300 database, John Wiley & Sons, 1992, vol. 15 Search PubMed.
- S. J. Konopka and B. McDuffie, Anal. Chem., 1970, 42, 1741 CrossRef CAS.
- P. Fanjul-Bolado, P. Queipo, P. J. Lamas-Ardisana and A. Costa-García, Talanta, 2007, 74, 427 CrossRef CAS PubMed.
- S. Yamazaki, Z. Siroma, T. Ioroi, K. Tanimoto and K. Yasuda, Carbon, 2007, 45, 256 CrossRef CAS PubMed.
- G.-Z. Hu, D.-P. Zhang, W.-L. Wu and Z.-S. Yang, Colloids Surf., B, 2008, 62, 199 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra12660b |
| This journal is © The Royal Society of Chemistry 2015 |