
Templated synthesis of nitrogen-doped graphene-like carbon materials using spent montmorillonite†
Runliang Zhu*a,
Qingze Chenab,
Xin Wangc,
Shuangyin Wangc,
Jianxi Zhua and
Hongping Hea
aCAS Key Laboratory of Mineralogy and Metallogeny, Guangdong Provincial Key Laboratory of Mineral Physics and Material Research & Development, Guangzhou Institute of Geochemistry, Chinese Academy of Sciences, Guangzhou 510640, China. E-mail: zhurl@gig.ac.cn; Fax: +86-020-85297603; Tel: +86-020-85297603
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cState Key Laboratory of Chem/Bio-Sensing and Chemometrics, College of Chemistry & Chemical Engineering, Hunan University, Changsha 410082, China
First published on 19th December 2014
Abstract
Montmorillonite (Mt) as an environmentally-friendly, low-cost, and highly-efficient adsorbent for cationic dyes has a promising application in dye wastewater treatment. However, proper disposal of the spent Mt is still a challenge holding back the wide application of Mt. This article reports a simple method which can synthesize N-doped graphene-like carbon materials using the spent Mt after the adsorption of crystal violet (CV). The spent Mt was pyrolyzed under the protection of N2 to carbonize the adsorbed CV within the interlayer space of Mt, and the interlayer spacing of Mt decreased from 11.0 Å to approximately 3.6 Å, close to the thickness of a single graphene layer (3.4 Å). After demineralization (i.e., washing with a mixture of HF and HCl), the carbon material was released from the interlayer space of Mt. Raman spectra showed the presence of both the D-band and G-band on the obtained carbon materials, and transmission electron microscopy observed the thin layers of carbon material. X-ray photoelectron spectroscopy results indicated the simultaneous presence of pyridinic, pyrrolic, and quaternary N on the carbon materials. In addition, the percentage of pyridinic N increases with increasing pyrolysis temperature; whereas that of quaternary N decreases and of pyrrolic N remains relatively constant. The above results suggested the successful synthesis of N-doped graphene-like carbon material. Finally, the obtained materials show interesting electrocatalytic activity for the oxygen reduction reaction and show potential to be used as efficient metal-free electrocatalysts in fuel cells.
1. Introduction
Montmorillonite (Mt) is a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 type clay mineral comprised of one octahedral sheet sandwiched by two tetrahedral sheets. Due to isomorphous substitution, Mt layers contain negative charge, which needs to be balanced by inorganic cations (e.g., Na+, Ca2+). These inorganic cations are exchangeable, making Mt a potential adsorbent for various cationic contaminants such as heavy metal cations1,2 and cationic dyes.3 In addition, Mt as an adsorbent for cationic contaminants is low-cost, readily-available, and environmentally-friendly. Therefore, Mt has been considered as a promising adsorbent for wastewater treatment, notably dye wastewater.3–5 Numerous studies have shown that Mt can effectively adsorb various cationic dyes, e.g., crystal violet (CV) and methylene blue (MB), and the maximum adsorption amount generally can surpass the cation exchange capacity (CEC) of Mt.5 In addition, due to the high interaction affinity, the removal rate of cationic dyes by Mt can be very high when the adsorbed amount was below the CEC of Mt.4,5 However, proper disposal of the spent Mt is still a big challenge holding back the wide application of Mt in dye wastewater treatment. Because of the high interaction affinity between Mt and organic dyes, desorption of organic cations using inorganic cations can be really difficult. Other methods such as direct landfill disposal and incineration not only may cause secondary environmental problems (e.g., production of secondary contaminants, massive emission of carbon dioxide), but also is a huge waste of Mt resource.
:1 type clay mineral comprised of one octahedral sheet sandwiched by two tetrahedral sheets. Due to isomorphous substitution, Mt layers contain negative charge, which needs to be balanced by inorganic cations (e.g., Na+, Ca2+). These inorganic cations are exchangeable, making Mt a potential adsorbent for various cationic contaminants such as heavy metal cations1,2 and cationic dyes.3 In addition, Mt as an adsorbent for cationic contaminants is low-cost, readily-available, and environmentally-friendly. Therefore, Mt has been considered as a promising adsorbent for wastewater treatment, notably dye wastewater.3–5 Numerous studies have shown that Mt can effectively adsorb various cationic dyes, e.g., crystal violet (CV) and methylene blue (MB), and the maximum adsorption amount generally can surpass the cation exchange capacity (CEC) of Mt.5 In addition, due to the high interaction affinity, the removal rate of cationic dyes by Mt can be very high when the adsorbed amount was below the CEC of Mt.4,5 However, proper disposal of the spent Mt is still a big challenge holding back the wide application of Mt in dye wastewater treatment. Because of the high interaction affinity between Mt and organic dyes, desorption of organic cations using inorganic cations can be really difficult. Other methods such as direct landfill disposal and incineration not only may cause secondary environmental problems (e.g., production of secondary contaminants, massive emission of carbon dioxide), but also is a huge waste of Mt resource.
On the other side, templated synthesis of carbon materials using inorganic templates are drawing increasing interests recently.6–11 In this process, the adsorbed organic compounds serve as carbon sources and can be transformed into carbon materials within the pores of the inorganic template after carbonization (i.e., pyrolysis under the protection of inert gases such as N2/Ar).9 Then, pure carbon materials can be obtained after removing the inorganic templates by acid washing.12,13 Clearly, microstructure of the obtained carbon materials directly relates to the pore structure of the inorganic templates.12,13 For example, when clay minerals with layered structure are selected as templates, carbon materials with 2-dimensional structure (i.e., graphene-like materials) can be synthesized.14 These graphene-like carbon materials have found potential applications in various fields, notably as adsorbents10,11 and electrode materials.15,16 For example, Fernández-Saavedra et al.15 revealed that carbon materials derived from both Mt and sepiolite were applicable as electrode for rechargeable Li-batteries and supercapacitors. These studies enlightened us that the spent Mt may be used as precursor for synthesizing graphene-like carbon materials by simply carbonizing the adsorbed cationic dyes within the interlayer spaces of Mt.
However, one should notice that in most previous studies polymers were used as the carbon sources, which can be directly intercalated into clay minerals or obtained by in situ polymerizing the pre-intercalated monomers.9,14 Probably, small organic compounds are more readily to degrade into volatile organic compounds and escape from the interlayer space during the carbonization process. As such, the feasibility of using the adsorbed cationic dyes as the carbon source needs to be clarified. On the other side, cationic dyes always contain heteroatoms (e.g., N in CV, S in MB), which may be incorporated into the obtained carbon materials and then influence their microstructure and properties. As is well known, incorporating particular elements into carbon materials can lead to enhanced or even novel properties; and thus synthesizing and application of heteroatoms-doped carbon materials are drawing increasing interests nowadays, notably the heteroatoms-doped graphene.14,17–20 Wang et al.17 showed that in comparison with the pure graphene, the N-doped graphene nanosheets generally revealed better lithium storage properties. Yang et al.18 disclosed that the S-doped graphene exhibited better catalytic activity than the commercial Pt/C in alkaline media. As such, one may expect that using the spent Mt as precursor may be a facile method for synthesizing doped graphene-like carbon materials.
In this article, the spent Mt after the adsorption of CV was pyrolyzed at 600, 700, or 800 °C, respectively, under the protection of N2. The obtained carbon–Mt composites were demineralized by extensive washing with the mixture of HF and HCl to liberate the carbon materials from Mt. The structural characteristics of the obtained carbon materials were characterized using X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), Raman, X-ray photoelectron spectroscopy (XPS), and N2 adsorption–desorption. Finally, the oxygen reduction reaction (ORR) activity of the carbon materials was determined. Results of this study showed the successful synthesis of N-doped graphene-like carbon materials using spent Mt after the adsorption of CV, and the obtained materials showed impressive electrocatalytic activity for ORR. As such, this study provided a practical approach for the proper disposal of the spent Mt after the adsorption of cationic dyes and a facile method for synthesizing doped graphene-like carbon materials.
2. Materials and methods
2.1 Materials
Mt (purity > 95%) was from Inner-Mongolia, China. The chemical compositions (wt%) of Mt determined by X-ray fluorescence are SiO2 58.16%, Al2O3 16.95%, Fe2O3 5.26%, CaO 2.29%, MgO 3.57%, K2O 0.15%, Na2O 0.19%, MnO 0.027%, TiO2 0.2%, P2O5 0.08%, and the ignition loss is 13.23%. Its net charge is −0.82e per unit cell and CEC is 110.5 mmol 100 g−1. CV is of analytical grade and supplied by Shanghai Chemical Co. (China). HF (40 wt%) and HCl (37 wt%) are supplied by Guangzhou Chemical Reagent Factory (China). All the chemicals were used as received.2.2 Synthesis of carbon materials using the spent Mt
According to our previous study,21 the loading amount of CV on Mt was selected as 450 mg g−1. The collected Mt (CV–Mt) was air-dried and then pyrolyzed for 3 h at 600, 700, or 800 °C, respectively, under the protection of N2. This carbonization process was to carbonize the adsorbed CV within the interlayer space of Mt, and the resulting carbon–Mt composites were denoted as C600–Mt, C700–Mt and C800–Mt, according to the pyrolysis temperature.After that, the carbon–Mt composites were washed using the mixture of HF (20 wt%) and HCl (18 wt%) to liberate the carbon materials from Mt. In the washing process, the carbon–Mt composites were stirred in the acid solution for 3 h and repeated for 3 times. According to the pyrolysis temperature, the final products were denoted as C600, C700 and C800, respectively. XPS results showed that the obtained carbon materials mainly contain C, N, and O atoms, indicating a thorough removal of the Mt template from the carbon materials by acid washing.
2.3 Structure characterization of the carbon materials
The XRD patterns of Mt, CV–Mt, C–Mt, and carbon materials were recorded on a Bruker D8 ADVANCE X-ray diffractometer. The measurements were operated at 40 kV and 40 mA with Cu Kα radiation, and the 2θ range between 1° and 25° was recorded with a scanning speed of 2° min−1.Raman spectra were acquired using a LabRAM Horiba JobinYvon spectrometer equipped with a CCD detector and a He–Ne laser (532 nm) at 15 mW in order to avoid any damaging of samples. All measurements were recorded in the wavelength range of 100–4000 cm−1 under the same conditions (24 s acquisition time) using a 50× magnification objective and a 300 μm pinhole.
SEM micrographs were observed using a field emission scanning electron microscopy (SUPRA 55/55VP, ZEISS Ltd. Germany), with accelerating voltage of 15 kV. Samples were anchored on the surface of the conducting resin, and then were sputter-coated with gold layer before examination.
TEM images were obtained on a FEI-Tecnai F20 transmission electron microscope operated at an acceleration voltage of 200 kV. The specimens for TEM observation were prepared by ultrasonicating in ethanol for 10 min, and then a drop of the sample suspension was dispersed onto a carbon-coated copper grid, which was allowed to stand for 10 min and transferred into the microscope.
XPS measurements were carried out using an X-ray photoelectron spectrometer (K-Alpha from Thermo Fisher Scientific, UK) with a monochromatic Al Kα X-ray source (excitation energy = 1468.6 eV). The XPS analysis chamber was evacuated to an ultra-high vacuum (a pressure of 5 × 10−8 mbar or lower) before analysis. Spectra were collected from 0 to 1350 eV using an X-ray spot size of 400 μm. The overall energy resolution was better than 0.5 eV.
N2 adsorption–desorption isotherms for the samples were determined on a Micromeritics ASAP 2020 analyzer (Micromeritics Co. Norcross, USA) at liquid nitrogen temperature (77 K). The samples were previously degassed under vacuum at 423 K for 12 h at the degas port and then transferred to the analysis port to degas further for 4 h below a relative pressures of 0.01 before measurement. The SBET value was calculated using the multiple-point Brunauer–Emmett–Teller (BET) method.
2.4 Electrochemical measurement
To prepare the working electrode, 4 mg sample was ultrasonically dispersed in 2 ml ethanol, followed by adding 100 μl Nafion solution (5 wt%) as a binder into the catalyst suspension. Then 5 μl of the resulting catalyst suspension was dropped onto the surface of a pre-polished glassy carbon electrode (GCE) and fully dried at room temperature. All the electrochemical measurements, including cyclic voltammograms and liner sweep voltammetry (LSV) were carried out using an electrochemical workstation (CHI 760E, CH Instrument, USA) with a typical three-electrode cell. A platinum mesh and a saturated calomel electrode (SCE) were used as counter electrode and reference electrode, respectively. The catalyst casted GCE was used as the working electrode in oxygen-saturated 0.1 M KOH solution to measure its ORR activity. Rotating ring-disk electrode (RRDE) was employed at a rotation rate of 1600 rpm for the measurement of LSV. All the measurements were performed at room temperature (25 ± 1 °C).3. Results and discussion
3.1 Structural characteristics of the carbon materials
XRD has quite often been used to track the structural evolution of intercalated Mt at different stages.22–24 The XRD patterns of Mt, CV–Mt, carbon–Mt composites and carbon materials were first compared in this work (Fig. 1). The XRD pattern of Mt indicates a typical Ca2+ form Mt containing two layers of water within the interlayer space.21 Adsorption of CV caused evident interlayer expansion and the basal spacing of CV–Mt reached 20.6 Å. As the thickness of one Mt layer is approximately 9.6 Å, the interlayer spacing of CV–Mt will be 11.0 Å, implying a tilt or multilayer arrangement of CV within the interlayer space of CV–Mt. Pyrolysis of CV–Mt led to significant decrease of basal spacing, and all the three carbon–Mt composites had a basal spacing of approximately 13.2 Å. As such, the interlayer spacing of the carbon–Mt composites will be 3.6 Å (13.2–9.6 Å), close to the thickness of a single graphene layer (3.4 Å).25 As such, one may expect that the pyrolyzed CV should have formed a carbon monolayer (i.e., graphene-like carbon material) within the interlayer space of the Mt template. After demineralization with acid washing, the liberated carbon materials showed a broad X-ray reflection at approximately 3.5 Å, which could be attributed to the 002 reflection of a disordered graphite.26,27 Similar results were obtained in previous studies using sugar as carbon sources and clay minerals as template.26 | ||
| Fig. 1 XRD patterns of the obtained materials. The XRD patterns of Mt, CV–Mt, C–Mt600 and C–Mt800 were adopted from ref. 18. | ||
SEM and TEM results can be used to show the morphology of the obtained carbon materials. The SEM micrograph of C800 showed a porous structure that was composed by the aggregation of thin carbon sheets (Fig. 2a). The TEM micrograph of C800 showed highly transparent texture and crumpled-sheet morphology (Fig. 2b), which further proved the presence of graphene-like sheets. Carbon materials with similar morphology were reported as well in previous studies using polymer as carbon sources.15,26 Not too much difference could be told from the SEM and TEM micrographs of C600 and C700 as compared with those of C800, and thus they were not further discussed.
 | ||
| Fig. 2 SEM micrograph (a) and TEM micrograph (b) of C800. | ||
The N2 adsorption–desorption isotherms of all the carbon materials have similar shape, except that C600 has slightly lower adsorption capacity of N2 (Fig. 3). Compared with Mt, carbon materials have much better N2 adsorption capacity, particularly in the relatively low pressure range, which indicated a much better developed pore system for the carbon materials. In addition, the carbon materials could evidently adsorb N2 at different relative pressure, which suggested that the obtained carbon material simultaneously contains micro-, meso-, and macropores. On the other side, the desorption isotherms of both Mt and the carbon materials showed H3-type hysteresis loop, indicative of the narrow slit-like pores created by the stacking of microparticles.28 As such, the carbon materials should be of layered structure, consistent with the Mt template. According to the obtained adsorption isotherms, the BET-N2 surface areas of the carbon materials were calculated (Table 1), which are much larger than the corresponding carbon–Mt composites (Fig. S1†). Elimination of Mt template from the carbon–Mt composites clearly could significantly enhance the specific surface areas of the resulting carbon materials. Increasing the pyrolysis temperature could lead to larger surface areas for the carbon materials. However, the difference between C700 and C800 was rather small. Similar BET-N2 surface area values were reported by Barata-Rodrigues et al.29 for the obtained carbon materials using furfuryl alcohol as carbon sources.
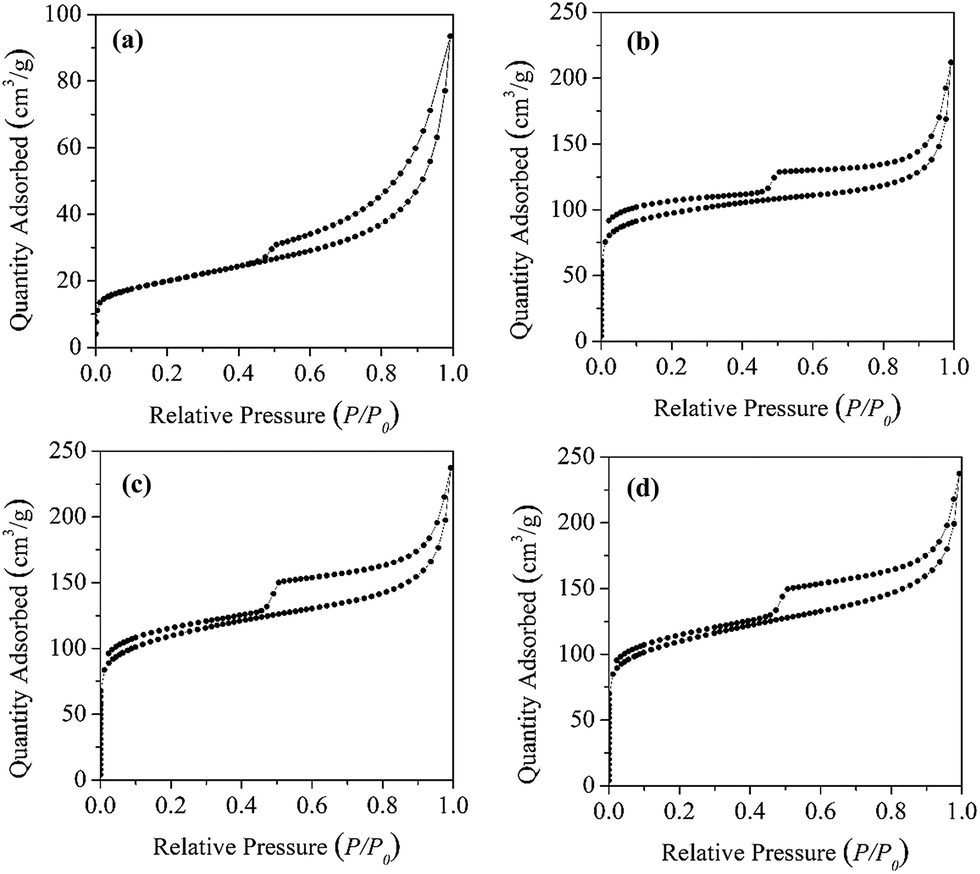 | ||
| Fig. 3 N2 adsorption–desorption isotherm of Mt (a), C600 (b), C700 (c) and C800 (d). | ||
| Sample | ID-band/IG-band | BET-N2 surface area (m2 g−1) |
|---|---|---|
| C600 | 0.88 | 366.97 |
| C700 | 0.92 | 403.72 |
| C800 | 0.94 | 405.36 |
The Raman spectra in the range of 800–2000 cm−1 for the carbon materials displayed two scattering bands at 1588 and 1350 cm−1 (Fig. 4a), which corresponds to the G-band and D-band, respectively. The former band is correlated with a graphitic structure (i.e., sp2 carbon); whereas the latter one is correlated to a sp3 carbon system.8,9 For carbon-based materials, the intensity ratio of the G-band and D-band can be used as an indicator of the relative contribution of sp2 and sp3 hybridized carbon atoms, or the structure ordering in a sp2-hybridized carbon system.8,9 The intensity ratio of D-band/G-band increased evidently with increasing pyrolysis temperature (Table 1), suggesting an increase of disordering in the graphitic structure. On the other side, in the absence of clay mineral templates, increasing pyrolysis temperature generally will lead to a more ordered graphitic structure for the resulting carbon materials.8 As such, the Mt template somehow showed a “hindering” effect for the graphitization of carbon sources (CV in this work). Within the nano-sized interspace of Mt, the mobility of carbon atoms will be restricted due to the confinement effect of Mt layers, which then may reduce the bonding chances among carbon atoms during the graphitization process, causing a “hindering” effect. As the lost carbon atoms cannot be readily supplemented by adjacent atoms due to the “hindering” effect, one can expect that the carbon materials derived from higher temperature may have a less graphitized structure. The higher-order peak appeared at 2700 cm−1 and a small broad peak at 2910 cm−1 were also observed, which can be assigned to a combination of D + D and D + G bands, respectively.30 Both the intensity and shape of the two peaks were quite similar for the obtained carbon composites.
 | ||
| Fig. 4 Raman spectra (a) and XPS survey scans (b) of the obtained carbon materials. | ||
XPS survey spectra showed that the carbon materials are mainly composed of C, N, and O atoms (Fig. 4b), and their contents decreased in the order C > O > N. Moreover, the N content of the carbon materials was in the range of 5.39–5.81%, and it gradually decreased as the pyrolysis temperature increased from 600 to 800 °C (Fig. 5b). Both C and N come from CV; whereas the source of O was complicated, which might originate from interlayer water molecules of CV–Mt, or directly from structural O of Mt. Additional work is necessary to clarify this issue.
 | ||
| Fig. 5 High resolution XPS patterns of N 1s spectrum of CV and the carbon materials (a); the content of three nitrogen species (pyridnic N, pyrrolic N and quaternary N) in the carbon materials (b); the composition of three different types of nitrogen on the carbon materials (c). | ||
Interestingly, the obtained carbon materials in this study contain N atoms in their structure. The XPS patterns in the range of 395–405 eV were recorded to evaluate the chemical environment of the N atoms (i.e., the doping atoms). The obtained N 1s peak for the carbon materials, which differed from the symmetric single peak of CV, was deconvoluted into three components (Fig. 5a), corresponding to pyridinic N, pyrrolic N, and quaternary N (Fig. 5c), respectively.19,31 The large contents of pyridinic N and pyrrolic N suggested a low crystallized graphitic structure for the carbon materials (Fig. 5b), consistent with above Raman characterization results. With increasing pyrolysis temperature, the content of pyridinic N decreased while that of quaternary N increased. As for pyrrolic N, its content remains relatively constant (∼1.1%). As such, pyridinic structure transformed into more stable quaternary nitrogen. Previous study by Stanczyk et al.32 showed that nitrogen structure (i.e., pyridinic N and pyrrolic N) of chars obtained from the nitrogen-containing compounds transformed to thermally more stable structures (i.e., quaternary N) with an increase of pyrolysis temperature, consistent with the result of this work.
Above characterization results demonstrated that the adsorbed CV could be transformed into carbon monolayer within the interlayer space of Mt after pyrolysis treatment. Then, graphene-like carbon materials with doped N atoms could be obtained after removing the Mt template. In addition, the structure of the carbon materials may be significantly effected by the pyrolysis temperature.
3.2 Electrochemical properties analysis
One of the promising applications of the heteroatom-doped carbon materials (e.g. graphenes, carbon nanotubes) can be envisaged in fuel cells and air batteries as metal-free electrocatalyst for ORR.33–38 To evaluate the electrocatalytic activity of the resulting graphene-like materials, the ORR behavior on the electrode was investigated in 0.1 M KOH solution saturated with oxygen. As shown by the cyclic voltammogram curves (Fig. 6a), a quasi-rectangular voltammogram without any evident response was observed for C800 in the nitrogen-saturated solution. In contrast, when oxygen was introduced, a substantial reduction process occurred at about −0.27 V (the peak potential), which is more positive than that of N-doped graphene in Yang's report,31 indicating a more facile ORR process on this N-doped graphene-like materials.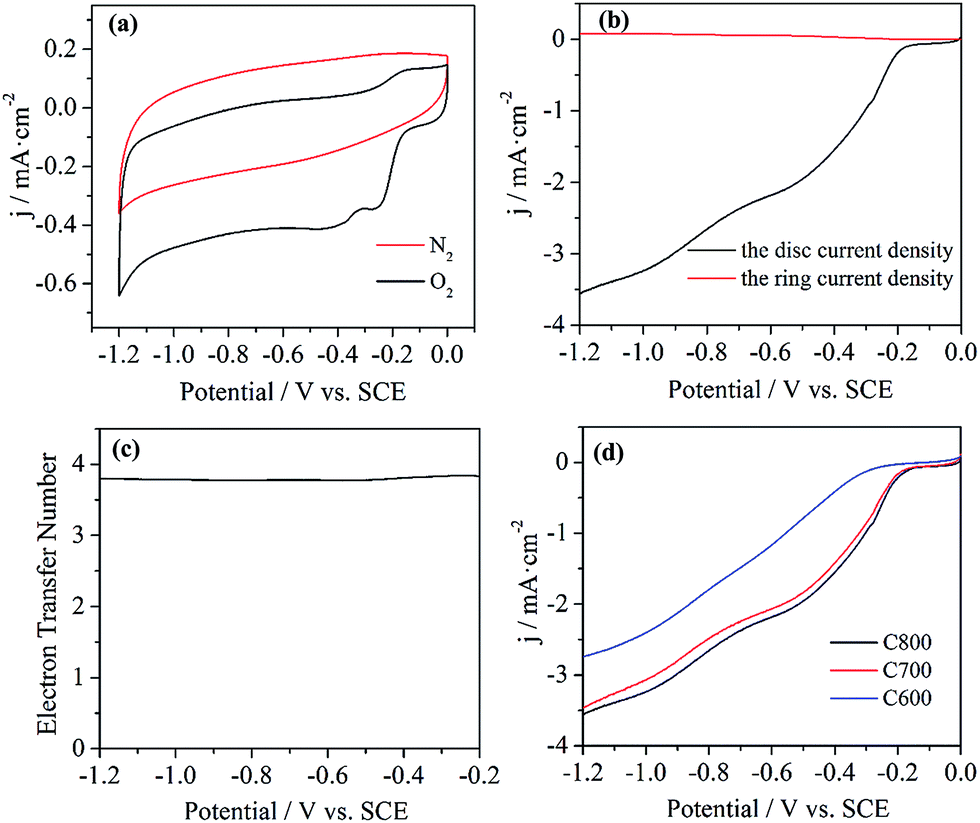 | ||
| Fig. 6 Cyclic voltammetry curves of ORR on C800 in nitrogen- and oxygen-saturated 0.1 M KOH solutions at a scan rate of 10 mV s−1 (a), RRDE testing on C800 in an oxygen-saturated 0.1 M KOH solution (b), and the corresponding electron transfer number of ORR on C800 (c), LSV curves of ORR on C600, C700 and C800 in an oxygen-saturated 0.1 M KOH solution (d). | ||
To further investigate the ORR electrochemical procedures on C800, LSV measurements was performed on the RRDE in oxygen-saturated 0.1 M KOH solution at the rotation rate of 1600 rpm and a scan rate of 10 mV s−1. The onset potential, the potential at which the ORR starts to occur, has been conveniently used to appraise the electrocatalytic activities of the catalysts toward ORR. The obtained onset potential for ORR on C800 is −0.17 V (Fig. 6b), close to that of the reported metal-free electrocatalysts such as NSG700.19 With the disc and ring currents in the LSV curve, the electron transfer number (n) per oxygen molecule involved in the ORR was calculated from the following equation:
| n = 4jD/(jD + jR/N) |
ORR activity of the obtained carbon materials on different pyrolysis temperatures was also investigated by the LSV curves (Fig. 6d). Obviously, C800 exhibited the best electrocatalytic activity among the three samples, as indicated by the most positive onset potential. Based on the XPS results (Fig. 5b), one might attribute the better ORR activity of C800 to its higher quaternary N content. Besides, the large surface area of C800 might be another important factor for the improved electrocatalytic performance by facilitating the electrolyte and reactant diffusion.33 According to the above results, one might expect that proper controlling the pyrolysis temperature could be a feasible approach for optimizing the electrocatalytic activity of the resulting carbon materials.
4. Conclusions
N-doped graphene-like carbon materials were successfully synthesized through a simple, facile, and efficient pyrolysis method using the spent Mt after the adsorption of CV. Pyrolysis of the spent Mt under the protection of N2 led to the decrease of interlayer spacing of Mt from 11.0 Å to approximately 3.6 Å, close to the thickness of a single graphene layer (3.4 Å). Raman spectra showed the presence of both D-band and G-band on the as-prepared carbon materials released from the interlayer space of Mt, and TEM observed the thin layers of carbon material. XPS results indicated the simultaneous presence of pyridinic, pyrrolic, and quaternary N on the carbon materials. With increasing pyrolysis temperature, pyridinic N transformed into more stable quaternary N and the content of pyrrolic N remained relatively constant. Moreover, the resulting N-doped graphene-like carbon materials exhibited efficient electrocatalytic activity and showed the potential to be the metal-free ORR catalysts. Our work provided not only a feasible way for the disposal of the used Mt, but also an available and general approach to synthesize the heteroatom-doped graphene-like carbon materials as efficient metal-free electrocatalyst for ORR in fuel cells and metal–air batteries.Acknowledgements
This work was financially supported by the “One Hundred Talents program” of the Chinese Academy of Sciences (KZZD-EW-TZ-10), grants from the National Natural Science Foundation of China (41322014, 21177104), and Team Project of Natural Science Foundation of Guangdong Province (S2013030014241). This is contribution (no. IS-2002) from GIGCAS.Notes and references
- K. G. Bhattacharyya and S. S. Gupta, Adv. Colloid Interface Sci., 2008, 140, 114–131 CrossRef CAS PubMed
.
- H. P. He, J. G. Guo, X. D. Xie and J. L. Peng, Environ. Int., 2001, 26, 347–352 CrossRef CAS
- V. K. Gupta and Suhas, J. Environ. Manage., 2009, 90, 2313–2342 CrossRef CAS PubMed
- S. Nir, G. Rytwo, U. Yermiyahu and L. Margulies, Colloid Polym. Sci., 1994, 272, 619–632 CAS
- J. M. Wei, R. L. Zhu, J. X. Zhu, F. Ge, P. Yuan, H. P. He and C. Ming, J. Hazard. Mater., 2009, 166, 195–199 CrossRef CAS PubMed
- C. Ruiz-Garcia, M. Darder, P. Aranda and E. Ruiz-Hitzky, J. Mater. Chem. A, 2014, 2, 2009–2017 CAS
- M. Inagaki, H. Orikasa and T. Morishita, RSC Adv., 2011, 1, 1620–1640 RSC
- T. Kyotani, N. Sonobe and A. Tomita, Nature, 1988, 331, 331–333 CrossRef
- E. Ruiz-Hitzky, M. Darder, F. M. Fernandes, E. Zatile, F. J. Palomares and P. Aranda, Adv. Mater., 2011, 23, 5250–5255 CrossRef CAS
- A. Bakandritsos, E. Kouvelos, T. Steriotis and D. Petridis, Langmuir, 2005, 21, 2349–2355 CrossRef CAS PubMed
- J. Q. Nie, Q. Zhang, M. Q. Zhao, J. Q. Huang, Q. A. Wen, Y. Cui, W. Z. Qian and F. Wei, Carbon, 2011, 49, 1568–1580 CrossRef CAS PubMed
- T. J. Bandosz, J. Jagiello, K. Putyera and J. A. Schwarz, Chem. Mater., 1996, 8, 2023–2029 CrossRef CAS
- A. P. Wang, F. Y. Kang, Z. H. Huang and Z. C. Guo, Clays Clay Miner., 2006, 54, 485–490 CrossRef CAS
- C. G. Xu, G. Q. Ning, X. Zhu, G. Wang, X. F. Liu, J. S. Gao, Q. Zhang, W. Z. Qian and F. Wei, Carbon, 2013, 62, 213–221 CrossRef CAS PubMed
- R. Fernández-Saavedra, M. Darder, A. Gomez-Aviles, P. Aranda and E. Ruiz-Hitzky, J. Nanosci. Nanotechnol., 2008, 8, 1741–1750 CrossRef PubMed
- A. Gomez-Aviles, M. Darder, P. Aranda and E. Ruiz-Hitzky, Appl. Clay Sci., 2010, 47, 203–211 CrossRef CAS PubMed
- H. B. Wang, C. J. Zhang, Z. H. Liu, L. Wang, P. X. Han, H. X. Xu, K. J. Zhang, S. M. Dong, J. H. Yao and G. L. Cui, J. Mater. Chem., 2011, 21, 5430–5434 RSC
- Z. Yang, Z. Yao, G. F. Li, G. Y. Fang, H. G. Nie, Z. Liu, X. M. Zhou, X. Chen and S. M. Huang, ACS Nano, 2012, 6, 205–211 CrossRef CAS PubMed
- X. Wang, J. Wang, D. L. Wang, S. O. Dou, Z. L. Ma, J. H. Wu, L. Tao, A. L. Shen, C. B. Ouyang, Q. H. Liu and S. Y. Wang, Chem. Commun., 2014, 50, 4839–4842 RSC
- L. Sun, L. Wang, C. G. Tian, T. X. Tan, Y. Xie, K. Y. Shi, M. T. Li and H. G. Fu, RSC Adv., 2012, 2, 4498–4506 RSC
- Q. Z. Chen, R. L. Zhu, W. X. Deng, Y. Xu, J. X. Zhu, Q. Tao and H. P. He, Appl. Clay Sci., 2014, 100, 112–117 CrossRef CAS PubMed
- H. P. He, R. L. Frost and J. X. Zhu, Spectrochim. Acta, Part A, 2004, 60, 2853–2859 CrossRef
- Y. Xi, W. Martens, H. P. He and R. L. Frost, J. Therm. Anal. Calorim., 2005, 81, 91–97 CrossRef CAS
- W. P. Gates, Appl. Clay Sci., 2004, 27, 1–12 CrossRef CAS PubMed
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed
- A. Bakandritsos, T. Steriotis and D. Petridis, Chem. Mater., 2004, 16, 1551–1559 CrossRef CAS
- N. Yoshizawa, K. Maruyama, Y. Yamada and M. Zielinska-Blajet, Fuel, 2000, 79, 1461–1466 CrossRef CAS
- D. Liu, W. W. Yuan, L. L. Deng, W. B. Yu, H. J. Sun and P. Yuan, J. Colloid Interface Sci., 2014, 424, 22–26 CrossRef CAS PubMed
- P. M. Barata-Rodrigues, T. J. Mays and G. D. Moggridge, Carbon, 2003, 41, 2231–2246 CrossRef CAS
- S. Y. Wang, L. P. Zhang, Z. H. Xia, A. Roy, D. W. Chang, J. B. Baek and L. M. Dai, Angew. Chem., Int. Ed., 2012, 51, 4209–4212 CrossRef CAS PubMed
- S. B. Yang, L. J. Zhi, K. Tang, X. L. Feng, J. Maier and K. Mullen, Adv. Funct. Mater., 2012, 22, 3634–3640 CrossRef CAS
- K. Stanczyk, R. Dziembaj, Z. Piwowarska and S. Witkowski, Carbon, 1995, 33, 1383–1392 CrossRef CAS
- K. P. Gong, F. Du, Z. H. Xia, M. Durstock and L. M. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed
- Y. Zheng, Y. Jiao, L. Ge, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2013, 52, 3110–3116 CrossRef CAS PubMed
- Y. Zhao, C. G. Hu, L. Song, L. X. Wang, G. Q. Shi, L. M. Dai and L. T. Qu, Energy Environ. Sci., 2014, 7, 1913–1918 CAS
- H. B. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS
- H. B. Wang, M. S. Xie, L. Thia, A. Fisher and X. Wang, J. Phys. Chem. Lett., 2014, 5, 119–125 CrossRef CAS
- X. F. Fan, W. T. Zheng and J. L. Kuo, RSC Adv., 2013, 3, 5498–5505 RSC
- Y. W. Ma, L. Y. Sun, W. Huang, L. R. Zhang, J. Zhao, Q. L. Fan and W. Huang, J. Phys. Chem. C, 2011, 115, 24592–24597 CAS
- S. Y. Wang, E. Iyyamperumal, A. Roy, Y. H. Xue, D. S. Yu and L. M. Dai, Angew. Chem., Int. Ed., 2011, 50, 11756–11760 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra13732a |
| This journal is © The Royal Society of Chemistry 2015 |